 Pan Huang
Pan Huang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 08 February 2023
Sec. Neurological Biomarkers
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1083972
This article is part of the Research Topic Potential Biomarkers in Neurovascular Disorders, volume II View all 19 articles
Arachidonic Acid (AA) is the precursor of cerebrovascular active substances in the human body, and its metabolites are closely associated with the pathogenesis of cerebrovascular diseases. In recent years, the cytochrome P450 (CYP) metabolic pathway of AA has become a research hotspot. Furthermore, the CYP metabolic pathway of AA is regulated by soluble epoxide hydrolase (sEH). 1-trifluoromethoxyphenyl-3(1-propionylpiperidin-4-yl) urea (TPPU) is a novel sEH inhibitor that exerts cerebrovascular protective activity. This article reviews the mechanism of TPPU's protective effect on ischemic stroke disease.
Ischemic stroke is a serious condition that endangers human health. The prevalence and incidence of stroke in China is rising faster than in other countries due to an aging population, the continued high prevalence of risk factors such as hypertension and diabetes and irregular management. Studies have shown that the prevalence of stroke among Chinese residents aged ≥40 years is 2.58%, or ~17.5 million people. The prevalence of stroke in the adult population (≥18 years) is ~1.29%, with significantly higher increases in the male population and in urban areas (male vs. female: 18.1 vs. 7.3%; urban vs. rural: 18.6 vs. 9.9%), and overall China is facing the greatest stroke challenge in the world (1). Neuroprotective therapy has always been the primary choice in ischemic stroke treatment. In the past 30 years, various neuroprotective agents have been developed for the pathophysiology of cerebral ischemia, including antioxidants, calcium channel antagonists, excitatory amino acid receptor inhibitors, and neurotrophic factors. Experimental research and over 100 clinical studies, most of which are effective in animal experiments but ineffective in clinical trials, have resulted in clinical translation failure (2). Clinical guidelines have not yet suggested an effective neuroprotective agent; however, the exploration of an effective neuroprotective agent in humans continues (3).
Arachidonic acid (AA) is the precursor of cerebrovascular active substances in the human heart, and its metabolites are closely associated with the pathogenesis of cerebrovascular diseases (4). In recent years, the cytochrome P450 (CYP) metabolic pathway of AA has been a research hotspot (5). AA generates hydroxyeicosatetraenoic acid (HETEs) and epoxyeicosatrienoic acid (EETs) under the action of CYP hydroxylase and CYP epoxidase, respectively. Furthermore, EETs undergo the action of soluble epoxide hydrolase (sEH) to generate dihydroxyeicosatrienoic acids (DHETs) with weak biological activity (Figure 1). HETEs have potent cerebral vasoconstriction and pro-atherosclerotic effects. EETs possess various biological functions, including vasodilation, regulation of ion channels, and anti-atherosclerosis, as well as a protective effect on cardiovascular and cerebrovascular diseases. Previous studies have shown that the levels of CYP metabolic pathway metabolites (EETs and HETEs) of AA are closely associated with the deterioration of neurological function following acute ischemic stroke, implying that they may have a role in cerebral ischemia (6).
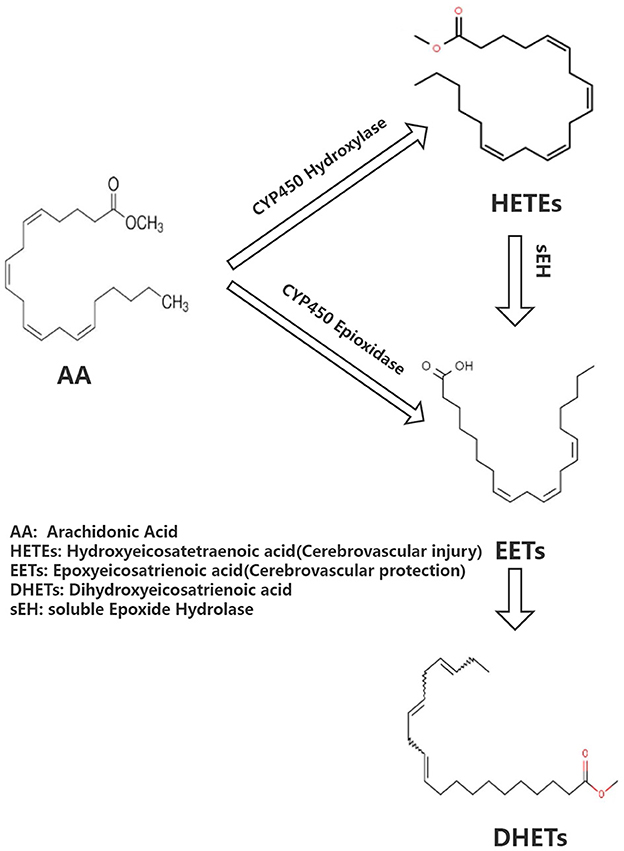
Figure 1. Brief overview of arachidonate acid cascade.
sEH is a key rate-limiting enzyme regulating EETs. Previous research has shown that the reduction of peripheral blood EETs is not only closely associated with the deterioration of neurological function after ischemic stroke but also with the degree of carotid artery stenosis and plaque instability in patients with cerebral infarction and is regulated by the gene encoding sEH (epoxide hydrolase 2, EPHX2) (7–9). Another study has shown that EPHX2 gene knockout can increase cerebral blood flow, reduce infarct volume in rats with middle-arterial artery occlusion, and has a protective effect on cerebral ischemia. sEH is considered a novel target for ischemic stroke prevention and treatment (10). 1-trifluoromethoxyphenyl-3(1-propionylpiperidin-4-yl) urea (TPPU) is a novel sEH inhibitor that exerts a cerebrovascular protective effect. This article includes national and international basic original studies on TPPU intervention in ischaemic stroke, excluding: 1. studies that are more than 20 years old; 2. previous review studies on TPPU for ischaemic stroke. This paper provides a comprehensive understanding of the possible mechanisms of TPPU intervention in ischaemic stroke and hopefully contributes to the search for new targets for the treatment of ischaemic stroke.
Neuroprotection against sEH has been a hot topic in recent years. Although EPHX2 gene knockout has a protective effect on experimental cerebral ischemia, it is still distant from clinical prevention and treatment of ischemic stroke. Therefore, sEH inhibitors R&D has attracted much attention. In 2005, the she inhibitor 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA) was applied to the cerebral ischemic nerve. In protective experimental studies, it has been confirmed that AUDA has a protective effect on cerebral ischemia (11). Furthermore, in 2007, she inhibitor, trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB), improved the cerebral blood flow and had a protective effect on experimental cerebral ischemia. AUDA and t-AUCB have anti-apoptotic, anti-oxidative, and anti-inflammatory properties, inhibit Ca2+ influx, protect mitochondria, and antagonize N-Methyl-D-Aspartate Receptor (NMDAR)-mediated excitotoxicity, among other mechanisms in brain protection (12). However, these two traditional sEH inhibitors have shown poor absorption and bioavailability from the gastrointestinal tract. The drug dose needed to obtain the effective blood concentration must be substantial enough because the drugs are easily accumulated in the body, the side effects are severe, and animal tolerance is poor. These traditional sEH inhibitors were found to have short half-lives and unstable blood concentrations in vivo, resulting in the failure of clinical translation of traditional sEH inhibitors.
TPPU is a novel sEH inhibitor synthesized in 2012 by Professor Bruce from the Molecular Bioscience Center of the University of California Veterinary Medicine (13). Bruce Hamock is an entomologist whose research focuses on inhibitors of epoxides in rodents. In his basic research, Professor Bruce found that TPPu has therapeutic and protective effects against various diseases and may be a potential treatment for these diseases. TPPU has a molecular weight of 359.3, allowing it to easily cross the blood-brain barrier and bind to sEH in the central nervous system, inhibiting sEH activity (Figure 2). TPPU has also been proven in animal models of coronary atherosclerotic heart disease to prevent myocardial fibrosis following myocardial infarction and has various functions, including anti-apoptosis, anti-oxidation, and mitochondrial protection (14).
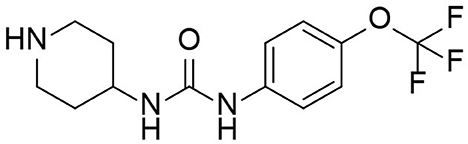
Figure 2. Molecular structure of TPPU.
Inflammation can not only cause a cerebral infarction, but it can also further activate the inflammatory response, creating a vicious circle (15–18). Tu et al. developed a mouse model of cerebral infarction and found that TPPU could significantly promote the recovery of neurological function and reduce the infarct volume and the expression of inflammatory cytokines IL-1βmRNA and TNF-αmRNA, revealing that TPPU may reduce the inflammatory response after infarction and promote the recovery of neurological function (19). Yu et al. used endothelial human Nox4 dominant-negative (EDN) transgenic mice in an ApoE deficient background to mimic the dysfunction of endothelial Nox4 in atherosclerosis-prone conditions. sEH and the inflammatory marker vascular cell adhesion molecule 1 (VCAM1) were upregulated in EDN aortic endothelium. TPPU reduced atherosclerotic lesions in EDN mice. In EDN endothelial cells (ECs), the endoplasmic reticulum stress inhibitor, 4-phenyl butyric acid (4-PBA), downregulated the expression of sEH and VCAM1 and suppressed inflammation. Moreover, its application in vivo reduced atherosclerotic lesions of EDN mice (20). In a study by Schmelzer et al., lipopolysaccharide (LPS, 10 mg/kg) was injected into C57BL/6 mice to cause celiac inflammation 24 h before and after the subcutaneous TPPU injection (20 mg/kg) and at the same dose of processed palm in the control group. The findings revealed that all the mice in the experimental group survived, and their blood pressure was restored to pre-LPS levels 24 h later, while all the mice in the control group developed severe hypotension (systolic pressure <40 mmHg) and died within 4 days. Studies have shown that TPPU increases the EET indirect inhibition of nitric oxide synthase, reduces NO produce, hypotension due to reduce inflammation, and inhibits non-specific inflammation index COX-2 (Cyclooxygenase-2) expression. Simultaneously, suppressing TNF alpha, IL-6, and monocyte chemotactic factor may reduce inflammation, improve lipid oxygen element A4 production, and increase inflammation abreaction (21). TPPU can simulate the role of EET in hemodynamics and anti-inflammatory activities and regulate growth, utility, and the forecast for new atherosclerosis compounds (22) (Figure 3).
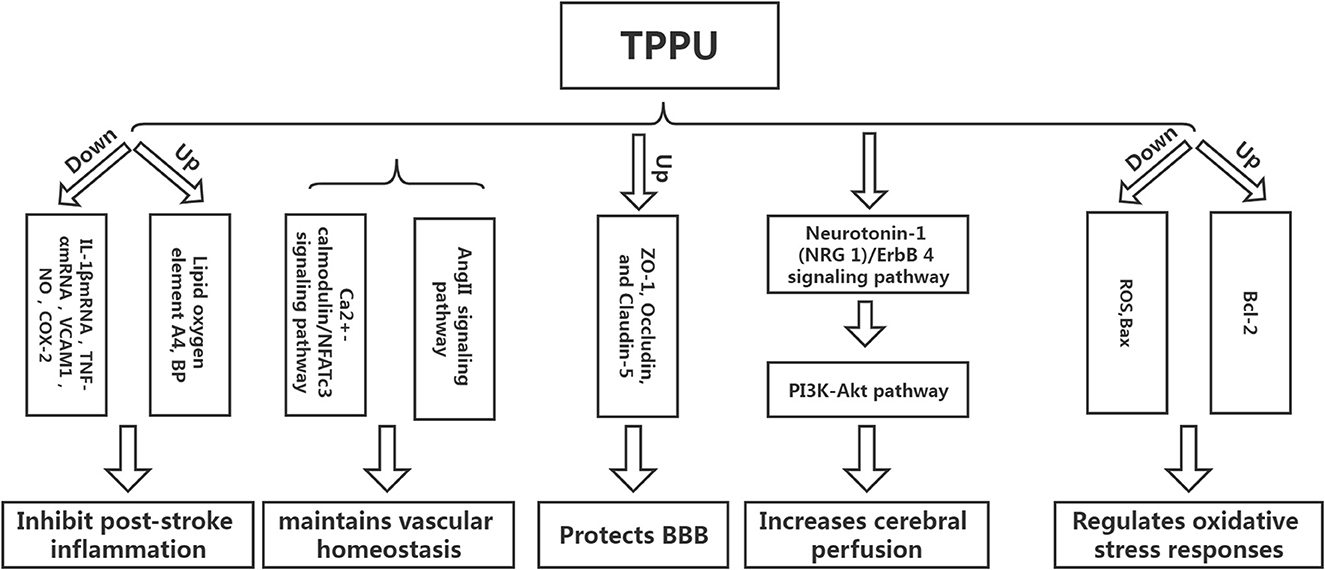
Figure 3. The protective mechanism of TPPU on stroke.
Homeostasis of vascular structure and function is the basis of human physiological activities. If the vascular homeostasis is unbalanced, it may lead to the occurrence of various diseases (23). Vascular injury is one of the reasons for the imbalance of vascular homeostasis. After cerebral infarction, a vascular injury will inevitably occur, and the adventitia of blood vessels will undergo corresponding changes. Studies have shown that neutrophils can be detected in the adventitia 0.5 h following balloon stretch injury in the coronary arteries of the pig, while the intima-media can be detected later. The vascular endothelial growth factor can be detected in the adventitia at the earliest after balloon-stretch injury to the carotid artery of rats (24). The above findings suggest that the adventitia is the origin and active participant of vascular diseases and is one of the novel targets for treating abnormal vascular function. Protecting adventitia may reduce the imbalance of vascular homeostasis, thereby facilitating disease repair. Angiotensin II inhibitor competes with Ang II for AT1. Furthermore, it inhibits vasoconstriction, promotes aldosterone secretion, reverses hypertrophic cardiomyocytes, and lowers blood pressure. However, AngII-related preparations can result in vascular wall thickening and collagen deposition, affecting the remodeling of the vascular adventitia and causing an imbalance in vascular homeostasis. Researchers have found that TPPU intervention in AngII model mice could significantly prevent AngII-induced vascular adventitia damage. Furthermore, in vitro studies have also found that TPPU may affect collagen synthesis via the Ca2+-calmodulin/NFATc3 signaling pathway, suggesting that TPPU may be one of the novel ways to treat vascular adventitial injury (25).
Tight-junction proteins between endothelial cells, basement membrane, the foot process of astrocytes, and pericytes are the key structures that maintain the integrity of the blood-brain barrier (BBB). The main pathogenic changes in the early stages of cerebral ischemia are increased BBB permeability and damage of tight junction protein, which causes brain edema, directly or indirectly leading to neurological function damage. Hence, preventing ischemic stroke requires protecting the BBB, inhibiting increased permeability, and alleviating cerebral edema (26). Yi et al. found that TPPU can significantly reduce BBB damage in model rats following cerebral infarction by increasing the expression of ZO-1, Occludin, and Claudin-5, the subunits of tight junction proteins related to BBB (24). Claudin-5 regulates the normal and disturbed states of the BBB. The down-regulation of claudin-5 can directly cause an increase in BBB permeability. ZO-1, a regulator of tight junction proteins, plays an important role in maintaining cytoskeleton formation, cell polarity, and paracellular barrier (27). Multiple studies have reported that tight junction proteins are crucial in regulating BBB integrity and permeability, and the downregulation of tight junction protein expression is associated with increased BBB permeability (28, 29). Yi et al. found that a higher dose of TPPU (2 mg/kg) had a better protective effect on cerebral edema than a lower dose. However, the mechanism may be related to the pharmacokinetics of TPPU, as higher doses did not improve the protective effect (25). TPPU is readily absorbed and slowly eliminated, allowing it to remain in the bloodstream longer than other sEH inhibitors due to its metabolic stability. Previous studies have demonstrated that even at the lowest dose of 0.1 mg/kg, the concentration of TPPU in vitro is higher than the IC50 value. When the TPPU dose exceeds 1 mg/kg, the EET/DHET ratio is lower than the TPPU dose of 1 mg/kg, indicating that TPPU should not be overused (13). Furthermore, studies have shown that TPPU can also reduce the damage to the BBB by reducing the damage to vascular endothelial cells. However, there are few studies on this aspect, and the detailed mechanism is unclear (30).
The decrease in perfusion volume after cerebral infarction is the primary reason for the aggravation of neurological function. Low perfusion can not only stimulate the intensification of the local inflammatory response but also result in the activation of excessive oxygen free radicals, causing the aggravation of the disease (31). Hao et al. established a coil-type carotid artery stenosis model. They found that the neurological function of mice was significantly improved after TPPU intervention compared with the control group. Furthermore, basic research has shown that the neuroprotective effect of TPPU on cerebral hypoperfusion may be associated with the activation of the Neuregulin-1 (NRG 1)/ErbB 4 signaling pathway, which can further trigger the PI3K-Akt pathway, implying that TPPU can play a multi-targeted protective effect and reduce the degree of nerve damage in mice with chronic cerebral hypoperfusion (32). However, the increase in cerebral perfusion after cerebral infarction is a reason for the deterioration of neurological function; hence, it is particularly important to explore the appropriate dose of TPPU to achieve a balance between the two. Currently, there are limited reports on TPPU improving ischemic stroke perfusion, and more studies are required to demonstrate the specific mechanism in the future.
Apoptosis is one of the primary reasons for brain injury after ischemic stroke, and oxidative stress is the main pathway of apoptosis. Studies have shown that TPPU can reduce the production of reactive oxygen species (ROS) after ischemic stroke, increase the expression of Bcl-2 protein, and decrease the expression of Bax protein, implying that TPPU can inhibit cell apoptosis after ischemic stroke (33). ROS production plays a key role in the breakdown of the blood-brain barrier during cerebral ischemia/reperfusion (34). After cerebral ischemia, the expression of ROS and inflammatory cytokines increases, causing mitochondrial damage, activation of pro-apoptotic protein Bax, and stimulation of cytochrome c cascade reaction. Moreover, the Bcl-2 protein can inhibit the downstream apoptotic cascade and block the release of cytochrome C (35).
Due to the widespread existence of sEH in the human body, its metabolic pathway is involved in many diseases, including ischemic stroke, hypertension, heart disease, kidney disease, etc. In theory, the development of drugs that inhibit sEH would seem to reverse or treat the disease. Human drug exploration for sEH has never stopped. From the early discovery of epoxide sEH inhibitors to the design and synthesis of the third generation of urea human sEH inhibitors, the research on human sEH inhibitors has experienced more than 30 years of development. sEH inhibitors have progressed from the initial sEH inhibitors with only micromolar inhibitory activity in vitro and poor or no activity in vivo to the current sEH inhibitors with nanomolar activity in vitro and remarkable pharmacokinetic properties in vivo (36).
TPPU is a novel sEH with good activity in vivo. It has been used in basic research in various cardiovascular and cerebrovascular diseases. It can play a protective role through a variety of potential mechanisms. However, it has not yet entered the clinical research stage for cerebrovascular diseases. With the development of pharmacology, the effects and mechanisms of TPPU are constantly being discovered and clarified, further proving its rationality and effectiveness as a treatment for ischemic stroke. As the research progresses, TPPU and its derivatives could be novel drugs for treating ischemic stroke-related diseases.
In conclusion, TPPU can intervene in ischemic stroke in various ways and may be one of the new targets for treating acute ischemic stroke. However, the treatment of acute ischemic stroke with TPPU is still in the exploratory stage, no clinical research has been carried out yet, and more trials are needed to confirm its potential.
The author confirms being the sole contributor of this work and has approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tu WJ, Hua Y, Yan F, Bian H, Yang Y, Lou M, et al. Prevalence of stroke in China, 2013–2019: a population-based study. Lancet Reg Health West Pac. (2022) 28:100550. doi: 10.1016/j.lanwpc.2022.100550
2. Ward NC, Croft KD, Blacker D, Hankey GJ, Barden A, Mori TA, et al. Cytochrome P450 metabolites of arachidonic acid are elevated in stroke patients compared with healthy controls. Clin Sci. (2011) 121:501–7. doi: 10.1042/CS20110215
3. Dorrance AM, Rupp N, Pollock DM, Newman JW, Hammock BD, Imig JD. An epoxide hydrolase inhibitor, 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA), reduces ischemic cerebral infarct size in stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharmacol. (2005) 46:842–8. doi: 10.1097/01.fjc.0000189600.74157.6d
4. Lee JS, Yaffe K, Lui LY, Cauley J, Taylor B, Browner W, et al. Prospective study of endogenous circulating estradiol and risk of stroke in older women. Arch Neurol. (2010) 67:195–201. doi: 10.1001/archneurol.2009.322
5. Huang H, Al-Shabrawey M, Wang MH. Cyclooxygenase-and cytochrome P450-derived eicosanoids in stroke. Prostaglandins Other Lipid Media. (2016) 122:45–53. doi: 10.1016/j.prostaglandins.2015.12.007
6. Yi X, Lin J, Li J, Zhou Q, Han Z. Epoxyeicosatrienoic acids are mediated by EPHX2 variants and may be a predictor of early neurological deterioration in acute minor ischemic stroke. J Atheroscler Thromb. (2017) 24:1258–66. doi: 10.5551/jat.41145
7. Yi X, Wu L, Liao D, Wang C, Zhang B. Interactions among CYP2C8, EPHX2, and CYP4A11 variants and CYP plasma metabolite levels in ischemic stroke. J Atheroscler Thromb. (2016) 23:1286–93. doi: 10.5551/jat.35279
8. Yi X, Liao D, Wang C, Cheng W, Fu X, Zhang B. Cytochrome P450 genetic variants and their metabolite levels associated with plaque stability in ischemic stroke patients. J Atheroscler Thromb. (2016) 23:330–8. doi: 10.5551/jat.31120
9. Yi X, Liao D, Wu L, Chen H, Li J, Wang C. Genetic variants, CYP metabolite levels, and symptomatic carotid stenosis in ischemic stroke patients. J Atheroscler Thromb. (2016) 23:621–31. doi: 10.5551/jat.32714
10. Zuloaga KL, Zhang W, Roese NE, Alkayed N. Soluble epoxide hydrolase gene deletion improves blood flow and reduces infarct size after cerebral ischemia in reproductively senescent female mice. Front Pharmacol. (2015) 15:290. doi: 10.3389/fphar.2014.00290
11. O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, et al. 1026 experimental treatments in acute stroke. Ann Neurol. (2006) 59:467–77. doi: 10.1002/ana.20741
12. Iliff JJ, Alkayed NJ. Soluble epoxide hydrolase inhibition: targeting multiple mechanisms of ischemic brain injury with a single agent. Future Neurol. (2009) 4:179–99. doi: 10.2217/14796708.4.2.179
13. Liu J, Lin YP, Qiu H, Morisseau C, Rose TE, Wang SH, et al. Substituted phenyl groups improve the pharmacokinetic profile and anti-inflammatory effect of urea-based soluble epoxide hydrolase inhibitors in murine models. Eur J Pharm Sci. (2013) 48:619–27. doi: 10.1016/j.ejps.2012.12.013
14. Sirish P, Li N, Liu JY, Lee Kin SS, Hwang SH, Qiu H, et al. Unique mechanistic insights into the beneficial effects of soluble epoxide hydrolase inhibitors in the prevention of cardiac fibrosis. Proc Natl Acad Sci U S A. (2013) 110:5618–23. doi: 10.1073/pnas.1221972110
15. Acar BA, Acar T, Vatan MB, Aras YG, Ulaş SB, Eryilmaz HA, et al. Predictive value of systemic immune-inflammation index for cerebral reperfusion and clinical outcomes in patients with acute ischemic stroke undergoing endovascular treatment. Eur Rev Med Pharmacol Sci. (2022) 26:5718–28. doi: 10.26355/eurrev_202208_29507
16. Mosconi MG, Paciaroni M. Treatments in ischemic stroke: current and future. Eur Neurol. (2022) 85:349–66. doi: 10.1159/000525822
17. Welsh P, Lowe GD, Chalmers J, Campbell DJ, Rumley A, Neal BC, et al. Associations of proinflammatory cytokines with the risk of recurrent stroke. Stroke. (2008) 39:2226–30. doi: 10.1161/STROKEAHA.107.504498
18. Tuttolomondo A, Pecoraro R, Pinto A. Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: a review of the evidence to date. Drug Des Devel Ther. (2014) 8:2221–38. doi: 10.2147/DDDT.S67655
19. Tu R, Armstrong J, Lee KSS, Hammock BD, Sapirstein A, Koehler RC. Soluble epoxide hydrolase inhibition decreases reperfusion injury after focal cerebral ischemia. Sci Rep. (2018) 8:5279–93. doi: 10.1038/s41598-018-23504-1
20. Yu W, Li S, Wu H, Hu P, Chen L, Zeng C, et al. Endothelial Nox4 dysfunction aggravates atherosclerosis by inducing endoplasmic reticulum stress and soluble epoxide hydrolase. Free Radic Biol Med. (2021) 164:44–57. doi: 10.1016/j.freeradbiomed.2020.12.450
21. Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci U S A. (2018) 102:9772–7. doi: 10.1073/pnas.0503279102
22. Imig J. Aspects of soluble epoxide hydrolase inhibitors. Cardiovas Ther. (2010) 24:169–88. doi: 10.1111/j.1527-3466.2006.00169.x
23. Kim HS, Ullevig SL, Zamora D, Lee CF, Asmis R. Redox regulation of MAPK phosphatase 1 controls monocyte migration and macrophage recruitment. Proc Natl Acad Sci USA. (2012) 109:E2803–12. doi: 10.1073/pnas.1212596109
24. Yi XY, Xu CX, Huang P, Zhang LL, Qing T, Li J., et al. 1-Trifluoromethoxyphenyl-3-(1-Propionylpiperidin-4-yl) urea protects the blood-brain barrier against ischemic injury by upregulating tight junction protein expression, mitigating apoptosis and inflammation in vivo and in vitro model. Front Pharmacol. (2020) 11:1197. doi: 10.3389/fphar.2020.01197
25. Li XD, Chen J, Ruan CC, Zhu DL, Gao PJ. Vascular endothelial growth factor-induced osteopontin expression mediates vascular inflammation and neointima formation via Flt-1 in adventitial fibroblasts. Arterioscl Throm Vas. (2012) 32:2250–8. doi: 10.1161/ATVBAHA.112.255216
26. Engelhardt S, Patkar S, Ogunshola OO. Cell-specific blood–brain barrier regulation in health and disease: a focus on hypoxia. Br J Pharmacol. (2014) 171:1210–30. doi: 10.1111/bph.12489
27. Park SY. Expression of E-cadherin in epithelial cancer cells increases cell motility and directionality through the localization of ZO-1 during collective cell migration. Bioengineering. (2021) 8:65. doi: 10.3390/bioengineering8050065
28. Lochhead JJ, McCaffrey G, Quigley CE, Finch J, DeMarco KM, Nametz N, et al. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxiareoxygenation. J Cereb Blood Flow Metab. (2010) 30:1625–36. doi: 10.1038/jcbfm.2010.29
29. Ye ZY, Xing HY, Wang B, Liu M, Yuan LP. DL–n-butylphthalide protects the blood-brain barrier against ischemia/hypoxiainjury via upregulation of tight junction proteins. Chin Med J. (2019) 132:1344–53. doi: 10.1097/CM9.0000000000000232
30. Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. (2012) 92:101–30. doi: 10.1152/physrev.00021.2011
31. Ng CF, Churilov L, Yassi N, Kleinig JT, Thijs V, Wu YT, et al. Microvascular dysfunction in blood-brain barrier disruption and hypoperfusion within the infarct posttreatment are associated with cerebral edema. Stroke. (2022) 53:1597–605. doi: 10.1161/STROKEAHA.121.036104
32. Hao JH, Chen YX, Yao E, Liu XH. Soluble epoxide hydrolase inhibition alleviated cognitive impairments via NRG1/ErbB4 signaling after chronic cerebral hypoperfusion induced by bilateral carotid artery stenosis in mice. Brain Res. (2018) 1699:89–99. doi: 10.1016/j.brainres.2018.07.002
33. Chuang YC, Yang JL, Yang DI. Roles of sestrin 2 and ribosomal protein S6 in transient global ischemia-induced hippocampal neuronal injury. Int J Mol Sci. (2015) 16:26406–16. doi: 10.3390/ijms161125963
34. Jurcau A, Ardelean AI. Oxidative stress in ischemia/reperfusion injuries following acute ischemic stroke. Biomedicines. (2022) 10:574–574. doi: 10.3390/biomedicines10030574
35. Nhu NT Li Q, Liu Y, Xu J, Xiao SY, Lee SD. Effects of Mdivi-1 on neural mitochondrial dysfunction and mitochondria-mediated apoptosis in ischemia-reperfusion injury after stroke: a systematic review of preclinical studies. Front Mol Neurosci. (2021) 14:778569. doi: 10.3389/fnmol.2021.778569
Keywords: TPPU, ischemic stroke, blood-brain barrier, ischemia reperfusion, arachidonic acid
Citation: Huang P (2023) Research progress on the protective mechanism of a novel soluble epoxide hydrolase inhibitor TPPU on ischemic stroke. Front. Neurol. 14:1083972. doi: 10.3389/fneur.2023.1083972
Received: 29 October 2022; Accepted: 20 January 2023;
Published: 08 February 2023.
Edited by:
Wen-Jun Tu, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Raouf Hajji, University of Sousse, TunisiaCopyright © 2023 Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pan Huang,  MTAzMjg1Nzk3MEBxcS5jb20=
MTAzMjg1Nzk3MEBxcS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.