 Maohua Li
Maohua Li Jiandi Huang1†
Jiandi Huang1† Hong Guo
Hong Guo Xiaoyan Chen
Xiaoyan Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 27 January 2023
Sec. Neurogenetics
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1063090
This article is part of the Research Topic Neurogenetics – Case Report Collection 2022 View all 28 articles
Bethlem myopathy (BM) is a disease that is caused by mutations in the collagen VI genes. It is a mildly progressive disease characterized by proximal muscle weakness and contracture of the fingers, the wrist, the elbow, and the ankle. BM is an autosomal dominant inheritance that is mainly caused by dominant COL6A1, COL6A2, or COL6A3 mutations. However, a few cases of collagen VI mutations with bilateral facial weakness and Beevor's sign have also been reported. This study presents a 50-year-old female patient with symptoms of facial weakness beginning in childhood and with the slow progression of the disease with age. At the age of 30 years, the patient presented with asymmetrical proximal muscle weakness, and the neurological examination revealed bilateral facial weakness and a positive Beevor's sign. Phosphocreatine kinase was slightly elevated with electromyography showing myopathic changes and magnetic resonance imaging (MRI) of the lower limb muscles showing the muscle MRI associated with collagen VI (COL6)-related myopathy (COL6-RM). The whole-genome sequencing technology identified the heterozygous mutation c.6817-2(IVS27)A>G in the COL6A3 gene, which was in itself a novel mutation. The present study reports yet another case of BM, which is caused by the recessive COL6A3 intron variation, widening the clinical spectrum and genetic heterogeneity of BM.
Bethlem myopathy (BM) was reported first by Bethlem and Wijngaarden (1). The onset of symptoms of BM usually occurs in early childhood, but occasionally in adulthood. The main symptoms of a patient with BM are the weakness of the proximal muscles and contracture of the fingers, the wrist, the elbow, and the ankle. Nevertheless, the presentation of BM is quite variable, and either contractures or muscular weakness may be absent (2). Its benign and slowly progressive course may lead to misdiagnosis. Recent studies showed magnetic resonance imaging (MRI) to be a supportive tool to diagnose collagen VI-related myopathies. Fu found that the “sandwich” sign in the vastus lateralis (VL) and the “target” sign in the rectus femoris (RF) were common in patients with collagen VI-related myopathies and were specific to these conditions (3). These MRI changes showed a positive predictive value of 69% for BM (4).
Herein, this study describes a 50-year-old female patient presenting with a slow progression of muscle symptoms, including proximal limb and facial weakness, since childhood. Finally, the diagnoses of BM in the patient in this study were confirmed genetically.
The patient was a 50-year-old woman. She had no family history relevant to muscle weakness and was not from a consanguineous marriage. At the age of 5 years, she was unable to blow up balloons and gradually appeared to be unable to whistle or puff her cheeks. She had normal mental growth, and the progression of her disease became slower. At the age of 30 years, she presented with asymmetrical proximal muscle weakness in the lower limbs and milder asymmetrical proximal muscle weakness in the upper limbs, as well as mild weakness in the waist. The progression of muscle weakness was slow. She was unable to climb stairs, jump, run, and rise from the floor, but she had no difficulty in breathing. Her cardiovascular and respiratory examinations were normal. The bilateral eyelash sign was positive, and the right nasolabial fold was shallower than the left one. She was unable to grimace and puff her cheeks (Figures 1A, B). Mental function and other cranial nerve function tests showed normal values. Muscle weakness in the limbs was asymmetrical (Medical Research Council (MRC). Grades 4/5 proximally and 5/5 distally, and bilateral hip extensors 2/5) without limb muscle atrophy. Beevor's sign was positive. Her sensations were normal, and muscle stretch reflexes were not elicited.
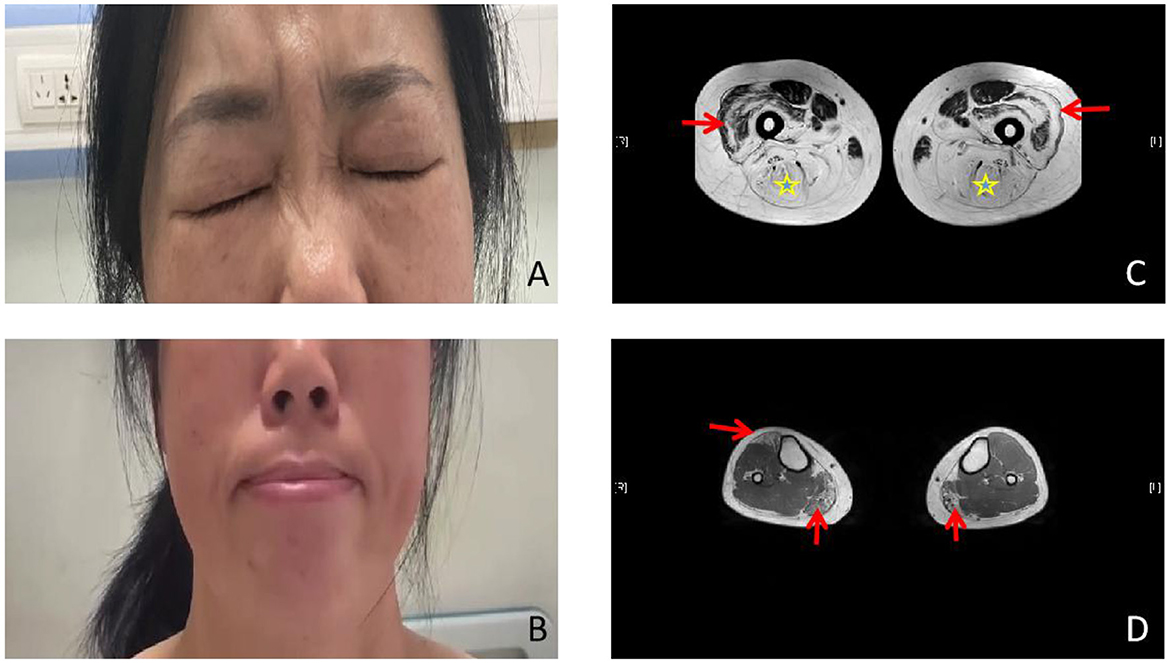
Figure 1. Photographs and lower limb muscle magnetic resonance imaging (MRI). (A, B) Photographs of the proband. Weakness of the eye and lip muscles. (C) The bilateral posterior thigh groups were markedly enlarged (star), especially of the lateral femoral muscle edges and central preservation (arrow). (D) Significant enlargement of the medial head of the gastrocnemius (arrow ↑) and the anterior tibial muscles in both lower legs.
Laboratory studies revealed a slightly elevated serum creatine kinase (CK) value of 291.5 international units per liter (IU/L; 26–174 IU/L). Respiratory parameters and cardiac evaluation [echocardiogram (echo) and electrocardiogram (ECG)] were unremarkable. Electromyography (EMG) showed myopathic changes, and nerve conduction was normal. The lower limb muscle MRI showed that the bilateral posterior thigh groups were markedly fattened, especially of the lateral femoral muscle edges and central preservation. A significant enlargement of the medial head of the gastrocnemius and anterior tibial muscles was observed in both lower legs (Figures 1C, D). Muscular pathological findings showed that the right tibial premuscle muscle fibers were clearly unequal in size, some of the muscle fibers were more clearly atrophied, some of the muscle fibers were mildly hypertrophied, a few muscle fiber nuclei were internally displaced, individual muscle clefts and nuclei aggregation were seen, muscle fiber degeneration and necrosis were not visible, and collagen VI immunostaining revealed a normal staining pattern (not shown; Figures 2A–C). Molecular studies, using the whole-genome sequencing technology, identified the heterozygous mutation c.6817-2(IVS27)A>G in the COL6A3 gene (Figures 2D–F). Sanger sequencing confirmed that the mutation was not detected in her mother or her son. Coenzyme Q10, inosine tablets, and vitamin E were administered to the patient. During hospitalization, rehabilitation doctors gave her rehabilitation training. After discharge, the patient was instructed to avoid strenuous exercise but to continue appropriate stretching, strength training, and muscle massage. Despite nerve nutrition and rehabilitation physiotherapy, there was no significant improvement in the patient's condition.
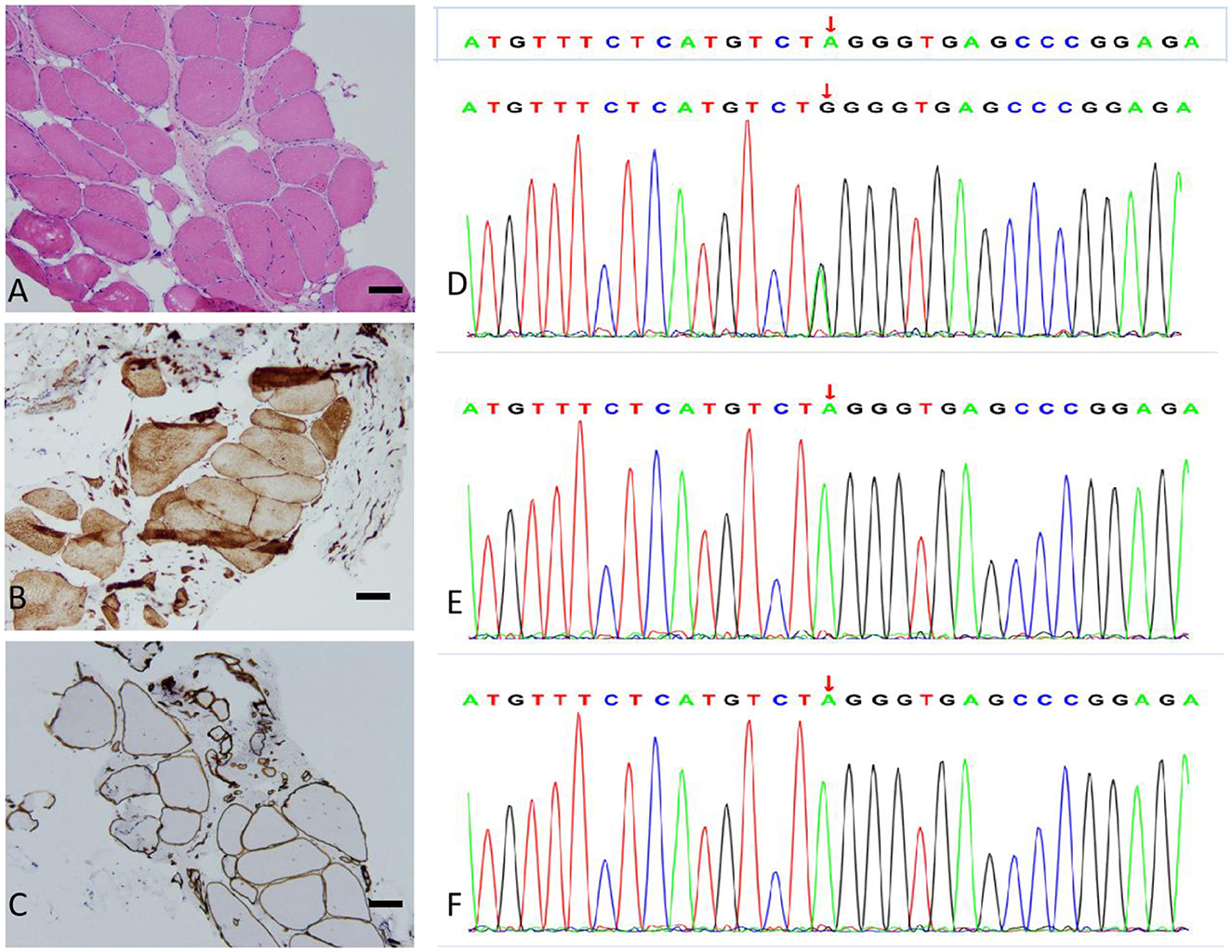
Figure 2. Muscle biopsy (bar = 100 μm) and genotype results. (A) Hematoxylin and eosin (H&E) staining: the skeletal muscle tissue of the right anterior tibial muscle exhibited a slight myogenic damage. (B) Dysferlin staining: the myofiber membrane and cytoplasm were positive. (C) α-Sarcoglycan staining: the muscle fiber membrane was positive, with a uniform and continuous expression. (D–F) Sequencing of the COL6A3 gene in the patient (D), patient's mother (E), and patient's son (F). The variation c.6817-2(IVS27)A>G is present only in the patient.
Collagen VI is a ubiquitous extracellular matrix protein that forms a microfibrous network. It is closely related to BM, Ullrich congenital muscular dystrophy (UCMD), and autosomal recessive myosclerosis myopathy. It is also closely associated with the basement membrane in the muscle, the cartilage, the periosteum, the ligaments, the cornea, the skin, the tendon, and the bone (5). Collagen VI consists of three different peptide chains: α1, α2, and α3. Each peptide chain has a triple helical (TH) region containing a single cysteine residue, with the defining Gly-X-Y amino acid sequence flanked by extensive N- and C-terminal globular domains that have subdomains with homology to the type A domains of the von Willebrand factor (6). The α3 (VI) chain has three additional C-terminal domains, a unique region (C3), a fibronectin type III repeat (C4), and a Kunitz-type protease inhibitor motif (C5), which can be cleaved extracellularly. The TH region folds in a zipper-like fashion from the C- to N-terminus to form a triple helix monomer and then assembles further into a tetramer. These tetramers are secreted into the extracellular matrix, where they assemble in an end-to-end fashion to form a microfiber network. The deficiency of collagen VI leads to an increase in apoptosis and oxidative stress, a decrease in autophagy, and an impairment of muscle regeneration (7). The genetics of collagen VI-related dystrophies (COL6-RD) is complex. COL6-RD is a disease entity that is caused by both dominant and recessive mutations in the three collagen VI-related genes, COL6A1, COL6A2, and COL6A3 (8). Patients with COL6-RD show a unique pattern of muscle involvement in MRI, which is an inward (outside-in) progression [starting from the fascia planes and progressing into the vastus lateralis (VL), the lateral gastrocnemius (LG), and central shadowing in the rectus femoris (RF)] of the fiber-fat material based on T1-weighted images. This inward progression of fiber-fat conversion causes the formation of a lipid-rich structural ring around the muscle whose thickness increases with an increasing severity of disease, and the muscle tissue is replaced by the fiber-fat material as the disease advances (9). The most common mutation in collagen VI myopathy affects the conserved Gly-X-Y motif in the TH domain (10).
The α3 chain is encoded by the COL6A3 gene and plays a determinant role in the monomer assembly. Mutations in the COL6A3 gene have been associated with UCMD and BM. Biallelic variants in COL6A3 have recently been suggested to be the cause of an early-onset isolated dystonia syndrome (DYT27) (11). Due to remarkable heterogeneity of the COL6 genes at the clinical and molecular levels, it is difficult to explain the clinical symptoms of the COL6A3 variants. The COL6A3 c.7447A>G variant in a homozygous state can lead to a mild Bethlem myopathy and/or the limb-girdle muscular dystrophy (LGMD) phenotype without respiratory involvement; however, a compound heterozygous mutation exhibits phenotypes ranging from a mild phenotype to an intermediate phenotype and a severe Ullrich-like phenotype (10). It has been reported that, in patients with other mutations in a compound heterozygous state, the phenotype severity depends entirely on the second mutation (10).
The present study reported a 50-year-old woman who had proximal limb weakness, bilateral facial weakness, and a positive Beevor's sign. Her CK was mildly elevated, and an electromyographic study revealed myopathic changes. The present study considered first facioscapulohumeral muscular dystrophy (FSHD), an inherited muscle disease that is characterized by progressive atrophy and weakness of the facial, shoulder limb-girdle, abdominal, and anterior leg muscles. FSHD has a tendency toward atypical findings, so it may be confused with other neuromuscular diseases. Muscle biopsy of FSHD most often shows non-specific chronic myopathic changes (12). Muscle MRI is a reliable tool to differentiate FSHD from other muscular dystrophies. Patients with FSHD exhibit a widespread involvement of the lower limb muscles, particularly the hamstring and the posterior calf muscles (13). However, in this patient, the bilateral posterior thigh groups were markedly enlarged, especially of the lateral femoral muscle edges and central preservation. Significant enlargement of the medial head of the gastrocnemius and anterior tibial muscles in both the lower legs was associated with COL6-RD-like muscles on MRI (4). Ultimately, the next-generation whole-genome sequencing revealed an intronic mutation in COL6A3, c.6817-2(IVS27)A>G. The heterozygous c.6817-2(IVS27)A>G mutation in the splice site of intron 27 led to exon skipping. The mutation was not present either in the unaffected mother or in the son, thereby indicating that it was a novel mutation that was present only in the patient. This mutation has also not been recorded in the Single Nucleotide Polymorphism Database (dsSNP) database, the Exome Aggregation Consortium (EXAC) database, or the 1,000 Genomes Project (1 KGP) database. According to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines, the variant was classified as a likely pathogenic variant [very strong evidence of the pathogenicity criterion (PVS1)+absent from controls (PM2)]. Because the patient's father has died, the patient refused to undergo Southern blotting or molecular combing and her sisters also refused to undergo the whole-genome sequencing; therefore, it was impossible to identify cosegregation with the phenotype within the family. Five patients mentioned in the report by Lee et al. (8) showed bilateral facial weakness, and all but one patient did show limb weakness. Therefore, the patient was diagnosed with probable BM (OMIM: 158810) based on her medical history, clinical examinations, muscle MRI, and genetic test results.
In conclusion, the present study describes a patient carrying a heterozygous COL6A3 c.6817-2(IVS27)A>G pathogenic splicing variant. The patient presented, since childhood, with facial weakness associated with proximal muscles, and the muscle MRI showed COL6-RM. Further analysis of molecular and genetic studies would help to elucidate whether this variant is of the disease-causing type. The muscle immunohistochemical marking for the COL6 protein is variable (14) and cannot be used as the basis for excluding the disease, while muscle MRI and genetic test are helpful in diagnosis. Currently, there are no pharmacological disease-modifying treatments for collagen VI-congenital muscular dystrophy (COL6-CMD). Genetic counseling and prenatal diagnosis are particularly important.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
MaL and JH wrote the manuscript draft. MiL, HG, and CD planned, designed, and analyzed the data. XC and YW organized and proofread the writing of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bethlem J, Wijngaarden GK. Benign myopathy, with autosomal dominant inheritance. A report on three pedigrees. Brain. (1976) 99:91–100. doi: 10.1093/brain/99.1.91
2. Panadés-de Oliveira L, Rodríguez-López C, Montenegro DC, Toledano MDMM, Fernández-Marmiesse A, Pérez JE, et al. Bethlem myopathy: A series of 16 patients and description of seven new associated mutations. J Neurol. (2019) 266:934–41. doi: 10.1007/s00415-019-09217-z
3. Fu J, Zheng YM, Jin SQ, Yi JF, Liu XJ, Lyn H, et al. “Target” and “sandwich” signs in thigh muscles have high diagnostic values for collagen VI-related myopathies. Chin Med J. (2016) 129:1811–6. doi: 10.4103/0366-6999.186638
4. ten Dam L, van der Kooi AJ, van Wattingen M, de Haan RJ, de Visser M. Reliability and accuracy of skeletal muscle imaging in limb-girdle muscular dystrophies. Neurology. (2012) 79:1716–23. doi: 10.1212/WNL.0b013e31826e9b73
5. Martins AI, Maarque C, Pinto-Basto J, Negrão L. Bethlem myopathy in a Portuguese patient - case report. Acta Myol. (2017) 36:178–81.
6. Whittaker CA, Hynes RO. Distribution and evolution of von Willebrand/integrin A domains: Widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell. (2002) 13:3369–87. doi: 10.1091/mbc.e02-05-0259
7. Lamandé SR, Bateman JF. Collagen VI disorders: Insights on form and function in the extracellular matrix and beyond. Matrix Biol. (2018) 71–2:348–67. doi: 10.1016/j.matbio.2017.12.008
8. Lee JH, Shin HY, Park HJ, Kim SH, Kim SM, Choi YC. Clinical, pathologic, and genetic features of collagen VI-related myopathy in Korea. J Clin Neurol. (2017) 13:331–9. doi: 10.3988/jcn.2017.13.4.331
9. Batra A, Lott DJ, Willcocks R, Forbes SC, Triplett W, Dastgir J, et al. Lower extremity muscle involvement in the intermediate and bethlem myopathy forms of COL6-related dystrophy and duchenne muscular dystrophy: A cross-sectional study. J Neuromuscul Dis. (2020) 7:407–17. doi: 10.3233/JND-190457
10. Stavusis J, Micule I, Wright NT, Straub V, Topf A, Panadés-de Oliveira L, et al. Collagen VI-related limb-girdle syndrome caused by frequent mutation in COL6A3 gene with conflicting reports of pathogenicity. Neuromuscul Disord. (2020) 30:483–91. doi: 10.1016/j.nmd.2020.03.010
11. Panda PK, Sharawat IK. COL6A3 mutation associated early-onset isolated dystonia (DYT)-27: Report of a new case and review of published literature. Brain Dev. (2020) 42:329–35. doi: 10.1016/j.braindev.2020.01.004
12. Preston MK, Tawil R, Wang LH, . Facioscapulohumeral muscular dystrophy. In:MP Adam, DB Everman, GM Mirzaa, Pagon RA, Wallace SE, Bean LJH, et al., editors, GeneReviews®. Seattle, WA: University of Washington, Seattle (1999). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1443/
13. Gerevini S, Scarlato M, Maggi L, Cava M, Caliendo G, Pasanisi B, et al. Muscle MRI findings in facioscapulohumeral muscular dystrophy. Eur Radiol. (2016) 26:693–705. doi: 10.1007/s00330-015-3890-1
Keywords: Bethlem myopathy, COL6A3, muscular dystrophy, muscle MRI, COL6-RD
Citation: Li M, Huang J, Liu M, Duan C, Guo H, Chen X and Wang Y (2023) A novel variant of COL6A3 c.6817-2(IVS27)A>G causing Bethlem myopathy: A case report. Front. Neurol. 14:1063090. doi: 10.3389/fneur.2023.1063090
Received: 06 October 2022; Accepted: 05 January 2023;
Published: 27 January 2023.
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Jianhai Chen, The University of Chicago, United StatesCopyright © 2023 Li, Huang, Liu, Duan, Guo, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue Wang,  d3psMjM4QDE2My5jb20=; Xiaoyan Chen, Y3ExOTk3QDE2My5jb20=; Hong Guo, Z3VvaG9uZzAyQGdtYWlsLmNvbQ==
d3psMjM4QDE2My5jb20=; Xiaoyan Chen, Y3ExOTk3QDE2My5jb20=; Hong Guo, Z3VvaG9uZzAyQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.