 Himanshu Sharma1*†‡John P. McGinnis1†‡Katherine E. Kabotyanski2†Shankar P. Gopinath1§Jerry C. Goodman3§Claudia Robertson1§ǁJovany Cruz Navarro1,4§ǁ
Himanshu Sharma1*†‡John P. McGinnis1†‡Katherine E. Kabotyanski2†Shankar P. Gopinath1§Jerry C. Goodman3§Claudia Robertson1§ǁJovany Cruz Navarro1,4§ǁ- 1Department of Neurosurgery, Baylor College of Medicine, Houston, TX, United States
- 2Baylor College of Medicine, Houston, TX, United States
- 3Department of Pathology, Baylor College of Medicine, Houston, TX, United States
- 4Department of Anesthesiology, Baylor College of Medicine, Houston, TX, United States
Traditionally, intracranial pressure (ICP) and partial brain tissue oxygenation (PbtO2) have been the primary invasive intracranial measurements used to guide management in patients with severe traumatic brain injury (TBI). After injury however, the brain develops an increased metabolic demand which may require an increment in the oxidative metabolism of glucose. Simultaneously, metabolic, and electrical dysfunction can lead to an inability to meet these demands, even in the absence of ischemia or increased intracranial pressure. Cerebral microdialysis provides the ability to accurately measure local concentrations of various solutes including lactate, pyruvate, glycerol and glucose. Experimental and clinical data demonstrate that such measurements of cellular metabolism can yield critical missing information about a patient's physiologic state and help limit secondary damage. Glucose management in traumatic brain injury is still an unresolved question. As cerebral glucose metabolism may be uncoupled from systemic glucose levels due to the metabolic dysfunction, measurement of cerebral extracellular glucose concentrations could provide more predictive information and prove to be a better biomarker to avoid secondary injury of at-risk brain tissue. Based on data obtained from cerebral microdialysis, specific interventions such as ICP-directed therapy, blood glucose increment, seizure control, and/or brain oxygen optimization can be instituted to minimize or prevent secondary insults. Thus, microdialysis measurements of parenchymal metabolic function provides clinically valuable information that cannot be obtained by other monitoring adjuncts in the standard ICU setting.
Introduction
Cerebral microdialysis (CMD) is a popular tool for evaluating and managing traumatic brain injury (TBI) in the intensive care unit (ICU). Traditionally, the primary invasive measurements guiding TBI management in patients have been intracranial pressure (ICP) and brain tissue oxygenation. After injury, the brain's metabolic demand increases, which requires more oxidative metabolism of glucose. Simultaneously, however, metabolic and electrical dysfunction can lead to an inability to meet these demands, even in the absence of ischemia or increased ICP. CMD provides the ability to interrogate this derangement by measuring solutes including lactate, pyruvate, and glucose.
CMD involves placing an invasive intraparenchymal monitor into viable brain parenchyma to continuously sample the extracellular fluid milieu. It has been used in clinical settings to evaluate neurochemistry in TBI as well as subarachnoid hemorrhage, prognostication after spontaneous intracranial hemorrhage (1), hypoxic-ischemic encephalopathy (2), and to monitor delivery of chemotherapy (3).
These and other such studies demonstrate that cellular metabolism measurements yield important information about a patient's physiologic state and can help develop new treatment strategies. Targeting metabolic dysregulation could help significantly limit secondary damage because metabolic dysfunction in humans is associated with, and in some cases precedes seizures, ischemia, and increased ICP (4) with feedforward cycles leading to worsening excitotoxicity, mitochondrial dysfunction and impaired autoregulation.
Glucose management in traumatic brain injury is still an unresolved question. Indeed, as of this writing, the Brain Trauma Foundation does not make recommendations regarding serum glucose management in the setting of TBI due to insufficient and contradictory data (5). Metabolic dysfunction may result from derangements in cerebral extracellular glucose concentration, and this may be a biomarker of brain tissue at-risk for secondary injury and/or poor outcome post TBI. Here we review the role of cerebral microdialysis in traumatic brain injury, focusing on the role of cerebral glucose monitoring.
Cerebral microdialysis
Cerebral microdialysis is a technique that was initially developed as a basic science research tool in the 1960's (6) to more accurately assay alterations in neurochemistry in normal physiology and disease, and later to evaluate the ability of peripherally delivered medications to cross the blood-brain barrier [the early history and development are reviewed by Benveniste (7)].
In its modern clinical form, the technique involves the placement of a thin catheter with two concentric lumens into the brain parenchyma, with the inner cannula slightly shorter than the outer cannula (Figure 1). The outer cannula is capped by a semipermeable dialysis membrane which allows for certain metabolites (in clinical use, usually with an upper bound of 20 kDa molecular weight) in the extracellular fluid to enter the catheter. A perfusate fluid is then pumped through the outer cannula and taken up via the inner cannula, and metabolites thus sampled from the interstitial fluid are analyzed (8). A plethora of substrates, metabolites, and other chemicals may be measured via this method, though glucose, lactate, and pyruvate are currently considered the most clinically informative—“Tier 1”—measurements in the clinical neurocritical care setting (9). Particularly, as an adjunct to intracranial pressure and oxygen tension monitoring, the information from these measurements helps distinguish between ischemic and non-ischemic (e.g., mitochondrial dysfunction) pathophysiologies.
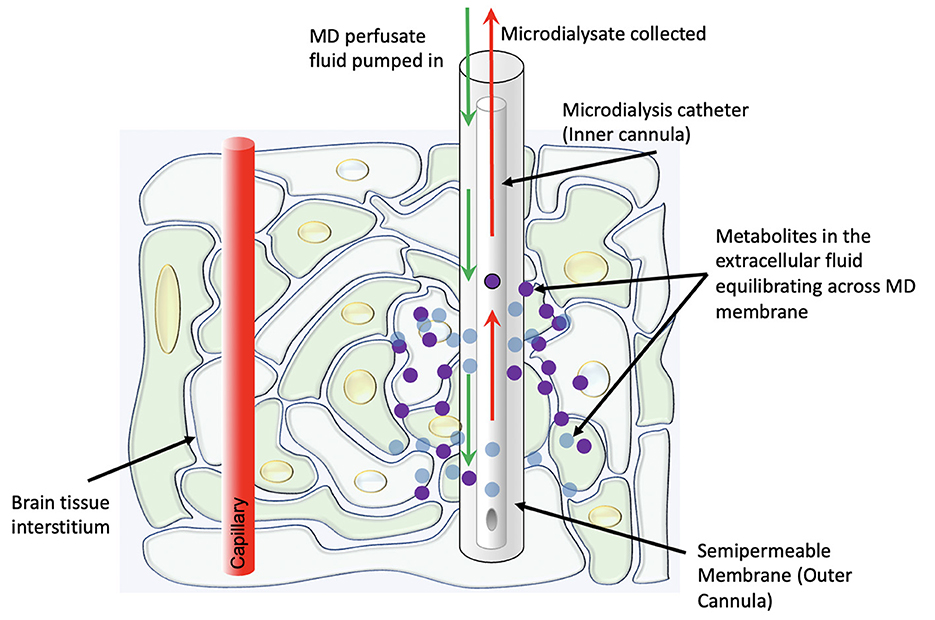
Figure 1. Schematic representation of the physiologic concept behind cerebral microdialysis.
The microdialysis catheter may be placed anywhere in the brain. Often, the catheter is placed in the contralateral hemisphere which can provide information on global metabolism adequacy, and in conjunction with the ICP monitor, can help guide decision making on medical management of ICP to improve oxygen and glucose delivery. However, in the setting of traumatic brain injury, studies have demonstrated that probes placement in “at risk” peri-lesional tissue (i.e., cortical tissue around a focal area of contusion or hematoma) is necessary to detect metabolic changes prior to the wave of global secondary injury (10). It is relevant that brain tissue which appears radiographically normal can nevertheless be significantly injured, remaining at risk, and that placement of a probe into brain tissue that is not salvageable is not useful. Thus, placement of a microdialysis catheter needs to be carefully considered, because while it can provide sustained readings over time, it does so only in a spatially confined region. Additional limitations of microdialysis include the sampling frequency (usually measured hourly) and the low sample volume—both in terms of dialysate (most commonly 0.3 uL/min) as well as spatial restriction.
Recent metabolic imaging studies evaluating blood flow, oxygen/glucose delivery, and oxygen and glucose utilization (11, 12) have demonstrated a great deal of spatial and temporal heterogeneity in metabolic derangements following traumatic brain injury; this work has shed light on ischemic changes that may be present even in the absence of intracranial hypertension and are only inconsistently detected by invasive brain oximetry. Thus, when access to advanced metabolic imaging [e.g., 15O and 18FDG PET, proton, and phosphorus spectroscopy (13)] is available, invasive and non-invasive monitoring can help reveal the extent of metabolic derangement seen after TBI, which can help guide clinical decision making as well as highlight potentially fruitful avenues for the development of therapeutics.
Relevance of invasive neurochemical monitoring
Cerebral microdialysis can provide several critical pieces of clinically relevant information that are not obtainable by other means in the standard ICU setting in TBI patients. These include a direct measurement of parenchymal metabolic state, under certain circumstances, global cerebral catastrophe (i.e., ICP elevation and metabolic failure) prior to onset, information about seizures and epileptiform discharges not detectable by surface electroencephalogram (EEG), and direct detection of metabolic dysfunction in the absence of ischemia. Combined with other forms of invasive monitors such as ICP and brain tissue oxygen monitoring, this information can help guide clinical decision-making when viable treatment options exist.
Glucose, lactate, and pyruvate as well as the secondarily calculated lactate:pyruvate ratio (LPR) convey information about the availability of substrate for adenosine triphosphate (ATP) generation as well as the redox state of the local tissue. Lactate conversion to pyruvate (an oxidative process) can be impaired by either mitochondrial injury or lack of oxygen availability and thus the LPR is a readout of metabolic stress.
Interestingly, microdialysis of peri-contusional brain in patients with fatal traumatic brain injury (as opposed to “normal” brain distant from site of injury) demonstrated changes in cerebral energy metabolism prior to subsequent increase in intracranial pressure (10). Furthermore, in 43% of patients, epileptiform discharges were detectable only by depth electrode monitoring and not the clinically more frequently used surface EEG. Nonetheless, these episodes were associated with metabolic crisis [as defined by elevated LPR, and which have been associated with worsened outcomes (14–16)], detectable by microdialysis catheters, and patients who demonstrated seizures and epileptiform discharges could be distinguished from those who did not by their cerebral metabolic profiles (17). The relationship between epileptic activity and cerebral metabolic changes in the context of TBI is difficult to elucidate as metabolic changes can lead to epileptic activity, but the reverse is also certainly true. While the causal links between metabolic crises and epileptic activity remain an active area of investigation, it is possible that cerebral microdialysis may function as a less invasive measure than invasive EEG to identify patients who may benefit from additional antiepileptic treatment thereby potentially reducing secondary damage by improving ICP and the cerebral metabolic profile.
Additionally, experimental models in pigs have confirmed that various states of cerebral metabolic dysfunction—specifically with altered redox balance—can occur in the absence of cerebral ischemia. This dysfunction is detectable through elevations in lactate and the lactate/pyruvate ratio as measured by microdialysis (18). These studies have been validated in humans with TBI wherein the vast majority of these cerebral metabolic crises occur in the absence of a concomitant reduction in PbO2 readings (19). Additional clinical TBI studies using PET in conjunction with tissue PbO2 monitoring have noted evidence of tissue hypoxia in the absence of classical ischemia as defined by a reduction in cerebral blood flow and increase in oxygen extraction fraction (OEF). These findings are suggestive of an increased diffusion barrier preventing cellular oxygen delivery and are consistent with microvascular ischemia. Such findings are a further mechanism responsible for metabolic dysfunction consistent with metabolic crisis (20, 21).
Indeed, beyond its use in traumatic brain injury, cerebral microdialysis can give valuable information about neurologic compromise in other patients in the ICU setting. Elevated lactate levels and LPR can predict delayed cerebral ischemia in subarachnoid hemorrhage (22). Microdialysis has also been used to inspire and validate the use of new therapeutics and guide clinical treatment strategies in neurologic ICU patients (23–25).
Thus, focal microdialysis measurements of parenchymal metabolic function provide clinically valuable information continuously at the bedside that cannot be obtained by other monitoring adjuncts in the standard ICU setting.
Parenchymal glucose in traumatic brain injury
In the context of normal aerobic physiology, glucose is the predominant substrate (~95% of ATP generation) used to meet the metabolic requirements of the brain. An alternate energy source for neurons, particularly after injury, is lactate, which is produced by astrocytes and taken up by neurons as needed as per the astrocyte-neuron lactate shuttle hypothesis (26). Recent studies using CMD have also highlighted the use of ketone bodies as an energy source in the injured brain (27). Under anaerobic conditions on the other hand, glucose can no longer be oxidized to generate ATP, and instead glycolysis predominates with the generation of ATP, lactate, hydrogen ions, and pyruvate as glycolytic end products (Figure 2). Cerebral glucose metabolism in TBI is complex and, while beyond the scope of this review, have been reviewed elsewhere (13, 28). Certain key points bear discussion. Initially after TBI, cerebral glucose intake transiently increases without a rise in oxygen uptake—this has been suggested to be related to hyperglycolysis and/or the generation of reducing equivalents via the pentose phosphate pathway shunt which may provide alternate energy sources and reduce oxidative stress in the immediate post-injury period [reviewed by Zhou and Kalanuria (8)]. Subsequently, a global depression in glucose metabolism often predominates.
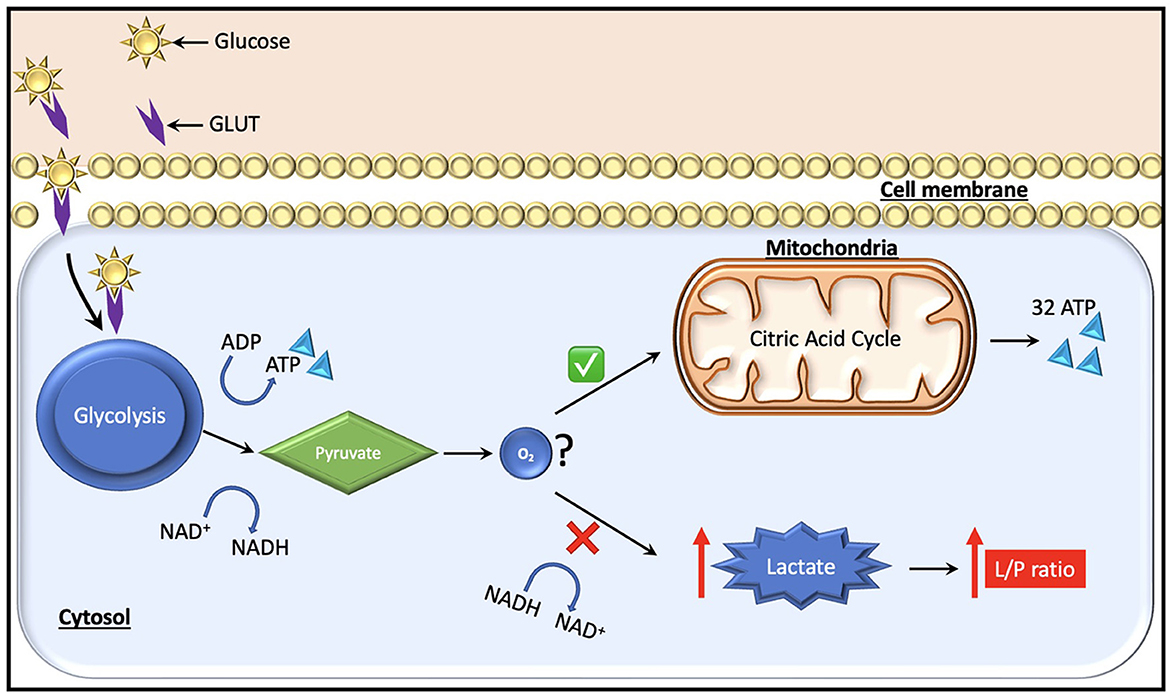
Figure 2. Glucose, which is the major fuel in the brain, is transported across the cell membranes by facilitated diffusion mediated by glucose transporter proteins (GLUT). Two types of glucose transporters (sodium dependent and facilitative transporters) are localized in the membranes of neurons, astrocytes and brain endothelial cells. Of these, GLUT-1 and GLUT-3 represent the two most important transporters in the brain. Under normal conditions, pyruvate is formed from glucose after entering the glycolytic process generating two molecules of ATP. Subsequently, it enters the citric acid cycle if oxygen is available, yielding 32 ATP molecules. During a state of cerebral hypoxia and low glucose (e.g., intracranial hypertension) the production of ATP from the citric acid cycle decreases. Then, cells attempt to compensate for the decrease in ATP production by increasing the turnover of glucose in the anaerobic part of the glycolysis. During this process it is necessary to regenerate NAD+ from NADH, which facilitates maintenance of glycolysis. The NAD+ regeneration causes an increase in lactate and the lactate/pyruvate ratio. The ratio is essentially the same in all tissues (i.e., about 20). A ratio above 25 is considered as an early warning of beginning metabolic crises.
The etiology of this global decrease in extracellular glucose availability remains a matter of debate, and may include a switch to lactate as a more preferred energy substrate in the injured brain [reviewed by Carpenter et al. (29)], but is regardless associated with poorer long-term outcomes (30). As mentioned previously, the role of clinical management of cerebral glucose levels remains unclear, since studies have demonstrated seemingly conflicting data. Table 1 highlights key papers studying the relationship between glucose control and clinical outcomes in TBI.
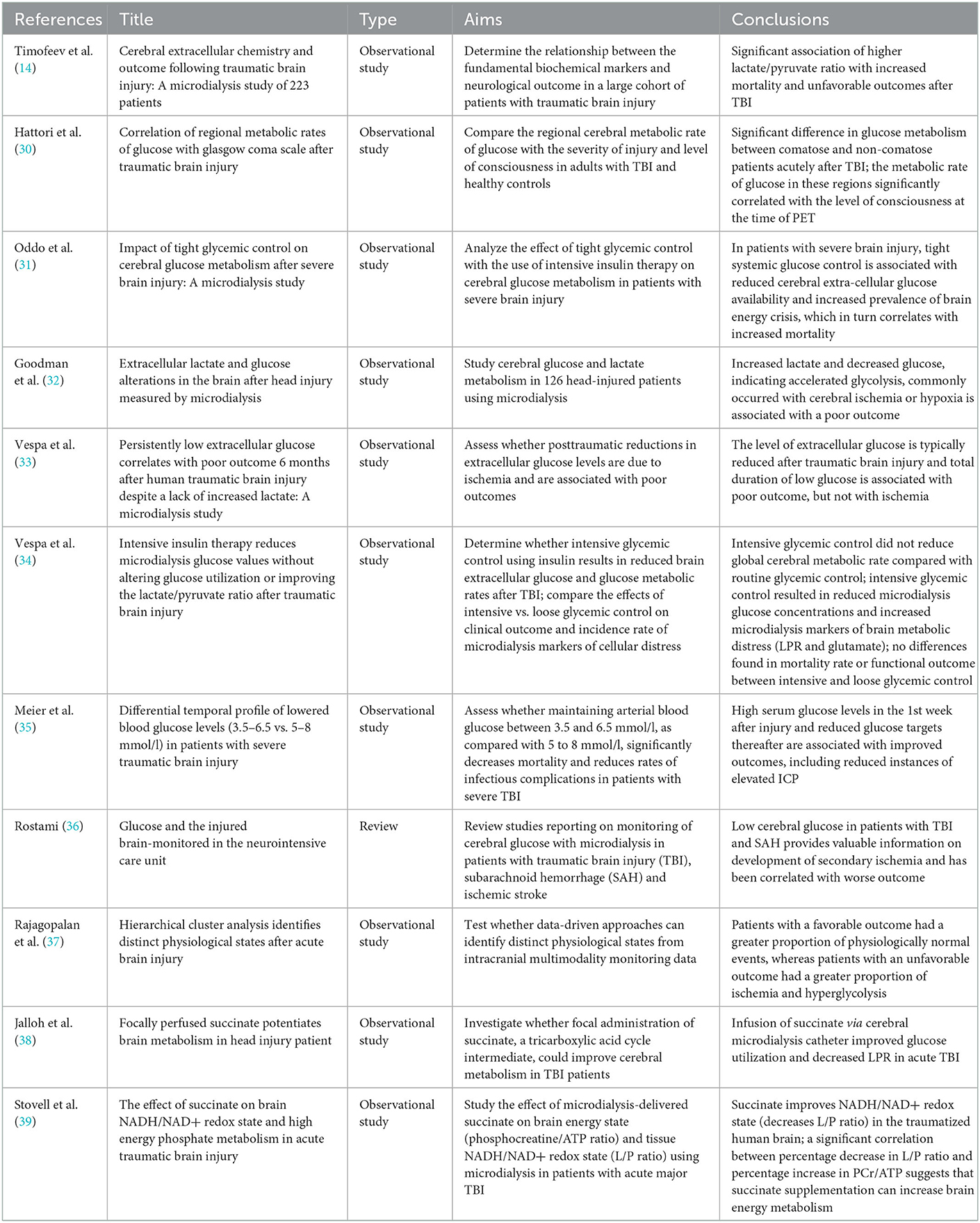
Table 1. Key papers studying the role of glucose levels and/or glycemic control parameters after traumatic brain injury and clinical and physiological outcome measures.
In traumatic brain injury, increased cerebral glucose is associated with increased serum glucose (40) and increased mortality (14) which is consistent with the currently accepted understanding that hyperglycemia in surgical patients is associated with poorer outcomes. However, a key caveat in neurologically injured patients is that when specifically evaluating injured and non-injured hemispheres in TBI patients, only the non-injured hemisphere demonstrated a correlation between serum and cerebral glucose levels, while the injured hemisphere had no correlation between the two (41). Furthermore, reduction of plasma glucose with insulin does, as expected, decrease cerebral glucose. However, an intensive approach to glucose control is associated with brain energy crises in TBI patients and has conflictingly been described to be (31–33), and not specifically to be (34), associated with worsened outcomes in TBI patients.
Thus, it is likely that a more nuanced approach is necessary in managing cerebral glucose levels as the details of these studies indicate a heterogeneity in the causes and effects of cerebral glucopenia. Indeed, in one study glucopenia predicted poor outcomes even in the absence of increased lactate or lactate/pyruvate ratio, suggesting that the poorer outcomes predicted by cerebral glucopenia are not due to cerebral ischemia (33). Clinical TBI studies combining 15O and 18FDG PET in conjunction with microdialysis have noted that low extracellular glucose was related to dysfunctional glucose delivery, primarily due to reduced cerebral blood flow, leading to increases in the rate of glycolysis. This may act as a potent mechanism of secondary injury (12, 13). Additionally, while intensive insulin therapy reduced cerebral glucose in TBI patients, it worsened glutamate levels and the lactate/pyruvate ratio, and ultimately did not alter mortality or 6-month clinical outcome (34). Thus, cerebral glucose levels likely exert an impact on brain metabolism and function in both redox state dependent and independent manners. Additionally, a study evaluating various serum glucose targets (without cerebral microdialysis) suggested that high serum glucose levels in the 1st week after injury and reduced glucose targets thereafter are associated with improved outcomes including reduced instances of elevated ICP, suggesting that a temporal component may also be critical to understanding serum and cerebral glucose roles and optimal management strategies after traumatic brain injury (35).
Overall, as reviewed by Rostami (36), while hyperglycemia is associated with poorer outcomes in TBI patients, tight glycemic control does not consistently demonstrate a significant improvement in those outcomes and precipitates cerebral metabolic dysfunction, with some evidence that subsequent glucopenia and metabolic crisis may be associated with worse outcomes.
However, emerging data indicate that there are various causes of alterations in cerebral glucose metabolism after TBI, and a deeper understanding of these underlying factors needs to be sought. Variations in neuronal uptake of astrocyte-produced lactate as a preferred energy source as well as the presence of seizure activity need to be considered when managing TBI patients in the ICU. Clearly, however, without cerebral microdialysis information, the remaining balance of intra- and extracranial measurements of metabolism commonly used is insufficient to fully understand parenchymal tissue function and omits key predictive information from the physician's repertoire of data.
Current and ongoing directions
More recently, multimodal hierarchical cluster analyses utilizing cerebral microdialysis have begun to delineate neurologic “events” differentiating for example, between hyperglycolysis, ischemia, and pure ICP elevations each correlated with different outcome profiles (37).
Excitingly, the use of cerebral microdialysis has permitted direct interrogation and manipulation of parenchymal tissue metabolism. This opens the possibility of targeting, either by systemic or more focused interventions, certain levels of metabolism in a manner similar to our targeting of brain oxygenation. Further work will need to clarify whether such targeting can improve clinical outcomes. In one recent study, infusion of succinate (a TCA cycle intermediate) via the CMD catheter improved glucose utilization and decreased LPR as well as extracellular glutamate (38, 39).
There is burgeoning interest in the use of CMD to guide nutrition in TBI patients, though this field is still in its infancy (4, 42). Evidence of ketone body utilization and the possibility of ketogenic diet for TBI patients (27) and the development of hypertonic lactate as a therapeutic in TBI (23) are some ongoing avenues of research in this field, and likely with more data, more optimized nutritional strategies may be formulated for TBI patients.
Perhaps less intuitively, systemic complications may also be reflected by intracranial metabolism after traumatic brain injury. For example, early elevation of cerebral lactate levels correlated positively with the development of pneumonia in patients with aneurysmal subarachnoid hemorrhage, whereas delayed elevation in lactate correlated with delayed ischemic neurologic deficits, highlighting potential links with immune regulation (43).
Thus, while the field remains in its infancy at the moment, direct measurement of intraparenchymal metabolic function via cerebral microdialysis has shed light on a wide variety of hitherto unexplored mechanisms underlying dysfunction and recovery in traumatic brain injury. Glucose metabolism in particular is dysregulated in this state, and metabolic stress in patients with glucopenia is likely mediated through both ischemic and non-ischemic mechanisms. The relationship between cerebral glucose levels and recovery, systemic physiologic events, and long-term neurologic outcomes is complex, and merits further investigation.
Author contributions
All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nikaina I, Paterakis K, Paraforos G, Dardiotis E, Chovas A, Papadopoulos D, et al. Cerebral perfusion pressure, microdialysis biochemistry, and clinical outcome in patients with spontaneous intracerebral hematomas. J Crit Care. (2012) 27:83–8. doi: 10.1016/j.jcrc.2011.04.004
2. Rajagopalan S, Navarro JC, Baghshomali S, Kirschen M, Greer D, Kofke WA, et al. Physiological signatures of brain death uncovered by intracranial multimodal neuromonitoring. J Neurosurg Anesthesiol. (2021) 33:347–50. doi: 10.1097/ANA.0000000000000672
3. Pierce CF, Kwasnicki A, Lakka SS, Engelhard HH. Cerebral microdialysis as a tool for assessing the delivery of chemotherapy in brain tumor patients. World Neurosurg. (2021) 145:187–96. doi: 10.1016/j.wneu.2020.08.161
4. Kurtz P, Rocha EEM. Nutrition therapy, glucose control, and brain metabolism in traumatic brain injury: A multimodal monitoring approach. Front Neurosci. (2020) 14:190. doi: 10.3389/fnins.2020.00190
5. Hawryluk GWJ, Rubiano AM, Totten AM, O'Reilly C, Ullman JS, Bratton SL, et al. Guidelines for the management of severe traumatic brain injury. Neurosurgery. (2017) 80:6–15. doi: 10.1227/NEU.0000000000001432
6. Bito L, Davson H, Levin E, Murray M, Snider N. The concentrations of free amino acids and other electrolytes in cerebrospinal fluid, in vivo dialysate of brain, and blood plasma of the dog. J Neurochem. (1966) 13:1057–67. doi: 10.1111/j.1471-4159.1966.tb04265.x
7. Benveniste H. Brain microdialysis. J Neurochem. (1989) 52:1667–79. doi: 10.1111/j.1471-4159.1989.tb07243.x
8. Zhou T, Kalanuria A. Cerebral microdialysis in neurocritical care. Curr Neurol Neurosci Rep. (2018) 18:101. doi: 10.1007/s11910-018-0915-6
9. Hutchinson PJ, Jalloh I, Helmy A, Carpenter KLH, Rostami E, Bellander B-M, et al. Consensus statement from the 2014 International Microdialysis Forum. Intensive Care Med. (2015) 41:1517–28. doi: 10.1007/s00134-015-3930-y
10. Ståhl N, Mellergård P, Hallström A, Ungerstedt U, Nordström CH. Intracerebral microdialysis and bedside biochemical analysis in patients with fatal traumatic brain lesions. Acta Anaesthesiol Scand. (2001) 45:977–85. doi: 10.1034/j.1399-6576.2001.450810.x
11. Launey Y, Fryer TD, Hong YT, Steiner LA, Nortje J, Veenith TV, et al. Spatial and temporal pattern of ischemia and abnormal vascular function following traumatic brain injury. J Am Med Assoc Neurol. (2020) 77:339–49. doi: 10.1001/jamaneurol.2019.3854
12. Hermanides J, Hong YT, Trivedi M, Outtrim J, Aigbirhio F, Nestor PJ, et al. Metabolic derangements are associated with impaired glucose delivery following traumatic brain injury. Brain. (2021) 144:3492–504. doi: 10.1093/brain/awab255
13. Demers-Marcil S, Coles JP. Cerebral metabolic derangements following traumatic brain injury. Curr Opin Anaesthesiol. (2022) 35:562–9. doi: 10.1097/ACO.0000000000001183
14. Timofeev I, Carpenter KLH, Nortje J, Al-Rawi PG, O'Connell MT, Czosnyka M, et al. Cerebral extracellular chemistry and outcome following traumatic brain injury: A microdialysis study of 223 patients. Brain. (2011) 134:484–94. doi: 10.1093/brain/awq353
15. Xu Y, McArthur DL, Alger JR, Etchepare M, Hovda DA, Glenn TC, et al. Early nonischemic oxidative metabolic dysfunction leads to chronic brain atrophy in traumatic brain injury. J Cereb Blood Flow Metab. (2010) 30:883–94. doi: 10.1038/jcbfm.2009.263
16. Wright MJ, McArthur DL, Alger JR, Van Horn J, Irimia A, Filippou M, et al. Early metabolic crisis-related brain atrophy and cognition in traumatic brain injury. Brain Imaging Behav. (2013) 7:307–15. doi: 10.1007/s11682-013-9231-6
17. Vespa P, Tubi M, Claassen J, Buitrago-Blanco M, McArthur D, Velazquez AG, et al. Metabolic crisis occurs with seizures and periodic discharges after brain trauma. Ann Neurol. (2016) 79:579–90. doi: 10.1002/ana.24606
18. Nielsen TH, Bindslev TT, Pedersen SM, Toft P, Olsen NV, Nordström CH. Cerebral energy metabolism during induced mitochondrial dysfunction. Acta Anaesthesiol Scand. (2013) 57:229–35. doi: 10.1111/j.1399-6576.2012.02783.x
19. Merino MA, Sahuquillo J, Borrull A, Poca MA, Riveiro M, Expósito L. Is lactate a good indicator of brain tissue hypoxia in the acute phase of traumatic brain injury? Results of a pilot study in 21 patients. Neurocirugia. (2010) 21:289–301. doi: 10.4321/s1130-14732010000400001
20. Veenith TV, Carter EL, Geeraerts T, Grossac J, Newcombe VFJ, Outtrim J, et al. Pathophysiologic mechanisms of cerebral ischemia and diffusion hypoxia in traumatic brain injury. J Am Med Assoc Neurol. (2016) 73:542–50. doi: 10.1001/jamaneurol.2016.0091
21. Menon DK, Coles JP, Gupta AK, Fryer TD, Smielewski P, Chatfield DA, et al. Diffusion limited oxygen delivery following head injury. Crit Care Med. (2004) 32:1384–90. doi: 10.1097/01.CCM.0000127777.16609.08
22. Rostami E, Engquist H, Howells T, Johnson U, Ronne-Engström E, Nilsson P, et al. Early low cerebral blood flow and high cerebral lactate: Prediction of delayed cerebral ischemia in subarachnoid hemorrhage. J Neurosurg. (2018) 128:1762–70. doi: 10.3171/2016.11.JNS161140
23. Bernini A, Miroz J-P, Abed-Maillard S, Favre E, Iaquaniello C, Ben-Hamouda N, et al. Hypertonic lactate for the treatment of intracranial hypertension in patients with acute brain injury. Sci Rep. (2022) 12:3035. doi: 10.1038/s41598-022-07129-z
24. Brandi G, Stocchetti N, Pagnamenta A, Stretti F, Steiger P, Klinzing S. Cerebral metabolism is not affected by moderate hyperventilation in patients with traumatic brain injury. Crit Care. (2019) 23:45. doi: 10.1186/s13054-018-2304-6
25. Juhairiyah F, de Lange ECM. Understanding drug delivery to the brain using liposome-based strategies: Studies that provide mechanistic insights are essential. AAPS J. (2021) 23:114. doi: 10.1208/s12248-021-00648-z
26. Pellerin L. Brain energetics (thought needs food). Curr Opin Clin Nutr Metab Care. (2008) 11:701–5. doi: 10.1097/MCO.0b013e328312c368
27. Bernini A, Masoodi M, Solari D, Miroz J-P, Carteron L, Christinat N, et al. Modulation of cerebral ketone metabolism following traumatic brain injury in humans. J Cereb Blood Flow Metab. (2020) 40:177–86. doi: 10.1177/0271678X18808947
28. Magistretti PJ, Allaman I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat Rev Neurosci. (2018) 19:235–49. doi: 10.1038/nrn.2018.19
29. Carpenter KL, Jalloh I, Hutchinson PJ. Glycolysis and the significance of lactate in traumatic brain injury. Front Neurosci. (2015) 9:112. doi: 10.3389/fnins.2015.00112
30. Hattori N, Huang S-C, Wu H-M, Yeh E, Glenn TC, Vespa PM, et al. Correlation of regional metabolic rates of glucose with glasgow coma scale after traumatic brain injury. J Nucl Med. (2003) 44:1709–16.
31. Oddo M, Schmidt JM, Carrera E, Badjatia N, Connolly ES, Presciutti M, et al. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: A microdialysis study. Crit Care Med. (2008) 36:3233–8. doi: 10.1097/CCM.0b013e31818f4026
32. Goodman JC, Valadka AB, Gopinath SP, Uzura M, Robertson CS. Extracellular lactate and glucose alterations in the brain after head injury measured by microdialysis. Crit Care Med. (1999) 27:1965–73. doi: 10.1097/00003246-199909000-00041
33. Vespa PM, McArthur D, O'Phelan K, Glenn T, Etchepare M, Kelly D, et al. Persistently low extracellular glucose correlates with poor outcome 6 months after human traumatic brain injury despite a lack of increased lactate: A microdialysis study. J Cereb Blood Flow Metab. (2003) 23:865–77. doi: 10.1097/01.WCB.0000076701.45782.EF
34. Vespa P, Boonyaputthikul R, McArthur DL, Miller C, Etchepare M, Bergsneider M, et al. Intensive insulin therapy reduces microdialysis glucose values without altering glucose utilization or improving the lactate/pyruvate ratio after traumatic brain injury. Crit Care Med. (2006) 34:850–6. doi: 10.1097/01.CCM.0000201875.12245.6F
35. Meier R, Béchir M, Ludwig S, Sommerfeld J, Keel M, Steiger P, et al. Differential temporal profile of lowered blood glucose levels (35 to 65 mmol/l versus 5 to 8 mmol/l) in patients with severe traumatic brain injury. Crit Care. (2008) 12:R98. doi: 10.1186/cc6974
36. Rostami E. Glucose and the injured brain-monitored in the neurointensive care unit. Front Neurol. (2014) 5:91. doi: 10.3389/fneur.2014.00091
37. Rajagopalan S, Baker W, Mahanna-Gabrielli E, Kofke AW, Balu R. Hierarchical cluster analysis identifies distinct physiological states after acute brain injury. Neurocrit Care. (2022) 36:630–9. doi: 10.1007/s12028-021-01362-6
38. Jalloh I, Helmy A, Howe DJ, Shannon RJ, Grice P, Mason A, et al. Focally perfused succinate potentiates brain metabolism in head injury patients. J Cereb Blood Flow Metab. (2017) 37:2626–38. doi: 10.1177/0271678X16672665
39. Stovell MG, Mada MO, Helmy A, Carpenter TA, Thelin EP, Yan J-L, et al. The effect of succinate on brain NADH/NAD(+) redox state and high energy phosphate metabolism in acute traumatic brain injury. Sci Rep. (2018) 8:11140. doi: 10.1038/s41598-018-29255-3
40. Diaz-Parejo P, Ståhl N, Xu W, Reinstrup P, Ungerstedt U, Nordström C-H. Cerebral energy metabolism during transient hyperglycemia in patients with severe brain trauma. Intensive Care Med. (2003) 29:544–50. doi: 10.1007/s00134-003-1669-3
41. Rostami E, Bellander BM. Monitoring of glucose in brain, adipose tissue, and peripheral blood in patients with traumatic brain injury: A microdialysis study. J Diabetes Sci Technol. (2011) 5:596–604. doi: 10.1177/193229681100500314
42. Tavarez T, Roehl K, Koffman L. Nutrition in the neurocritical care unit: A new frontier. Curr Treat Options Neurol. (2021) 23:16. doi: 10.1007/s11940-021-00670-8
Keywords: cerebral microdialysis, glucopenia, traumatic brain injury, lactate-to-pyruvate ratio, glucose
Citation: Sharma H, McGinnis JP, Kabotyanski KE, Gopinath SP, Goodman JC, Robertson C and Cruz Navarro J (2023) Cerebral microdialysis and glucopenia in traumatic brain injury: A review. Front. Neurol. 14:1017290. doi: 10.3389/fneur.2023.1017290
Received: 11 August 2022; Accepted: 09 January 2023;
Published: 27 January 2023.
Edited by:
Keri Linda Carpenter, University of Cambridge, United KingdomReviewed by:
Marek Czosnyka, University of Cambridge, United KingdomJefferson W. Chen, University of California, Irvine, United States
Patrick M. Chen, Massachusetts General Hospital and Harvard Medical School, United States
Copyright © 2023 Sharma, McGinnis, Kabotyanski, Gopinath, Goodman, Robertson and Cruz Navarro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Himanshu Sharma,  himanshu.sharma@bcm.edu
himanshu.sharma@bcm.edu
†These authors have contributed equally to this work
‡These authors have contributed equally to this work and share first authorship
§These authors have contributed equally to this work and share senior authorship
ǁThese authors have contributed equally to this work and share last authorship