 Jianhui Deng1†
Jianhui Deng1† Xiaochun Zheng
Xiaochun Zheng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol., 08 September 2022
Sec. Dementia and Neurodegenerative Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.995747
This article is part of the Research TopicNeural Immuno-Inflammatory Response in Neurodegenerative DiseasesView all 6 articles
N6-methyladenosine (m6A), the most prevalent post-transcriptional RNA modification throughout the eukaryotic transcriptome, participates in diverse biophysiological processes including cell fates, embryonic development and stress responses. Accumulating evidence suggests that m6A modification in neural development and differentiation are highly regulated processes. As RNA m6A is crucial to protein translation and various bioprocesses, its modification dysregulation may also be associated with brain injury. This review highlights the biological significance of m6A modification in neurodegenerative disease and brain injury, including cerebrovascular disorders, is highlighted. Emphasis is placed on recent findings that elucidate the relevant molecular functional mechanism of m6A modification after brain injury and neurodegenerative disease. Finally, a neurobiological basis for further investigation of potential treatments is described.
N6-methyladenosine (m6A) modification, the most common RNA post-transcriptional modification, plays an important role in gene expression by regulating processes like RNA nuclear export, stability, degradation, splicing and translation (Figure 1). This dynamic nucleus-based RNA process occurs primarily on adenine in the RRACH sequence (R = G or A; H = A, C or U) (1, 2).
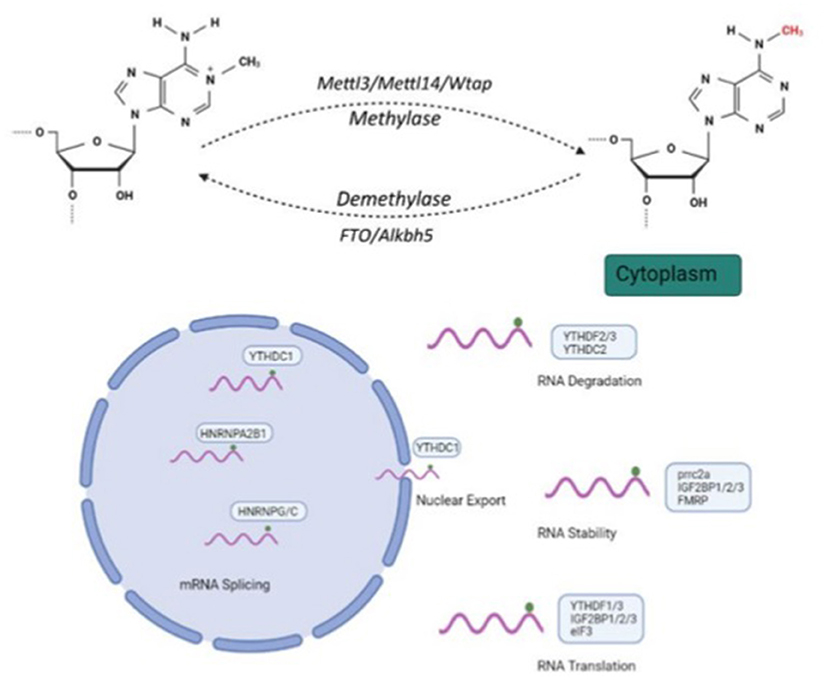
Figure 1. Molecular mechanism and three N6-methyladenosine(m6A) modification factors. m6A modification regulates RNA nuclear exports, stability, degradation, splicing and translation. Methyltransferase complexes, including METTL3, METTL14 and WTAP, as well as m6A RNA demethylases, FTO and ALKBH5, participate in m6A modification processes. Created with BioRender.com.
The distribution and motif of m6A modification vary across tissue type and are expressed in a spatiotemporal-specific pattern regulated by methyltransferase complex and demethylases in various neurodevelopment and pathological processes (3–5). m6A modification is widely enriched in specific tissues including the brain, liver and kidney; It is less abundant in the fetal brain but developmentally increases in brain tissue (6).
m6A modification plays a pivotal role in the diversification of embryonic and adult neurons, and in the regulation of neural progenitor capacity (5). m6A modification is essential for nervous system functioning and development (4, 7–9). Altered m6A modification levels in the brain are closely related to learning, long-term memory and brain-injuring diseases (10–12). This systematic review focuses on the research to date into the mechanism of epitranscriptional m6A modification in neurological diseases.
In 1974, m6A was detected in poly(A) RNA fractions; however, the lack of epitranscriptomic mapping methods for m6A reduced researcher interest. With the development of several methods incorporating next-generation sequencing techniques, regulation and functions of m6A modification have recently been revealed. The early mapping approach (termed “Me-RIP-seq”) provided the first comprehensive analysis of methylated mRNAs throughout the transcriptome (13), including application of methylated RNA immunoprecipitation and chromatin immunoprecipitation-sequencing, showing that m6A is abundant in coding sequences in long exons, particularly in the vicinity of the stop codon (14). Using mi-CLIP, individual m6A sites were detected in cellular mRNAs with high sensitivity and specificity (15). Similarly, mi-CLIP enabled differentiation between m6A and m6Am, which forms a 2-O-methylated residue adjacent to the 7-methylguanosine cap structure (16, 17). Yet there are constraints on existing methods, including high input RNA requirements and cross-reactivity to other RNA modifications. Thus, several advanced methods, including deamination adjacent to RNA modification targets (DART-seq), m6A-label-seq and m6A-SEAL, which detect mRNA m6A transcriptome-wide at base resolution through metabolic labeling, have been proposed (18–20). Development of these methods may allow a better understanding of the mechanism of m6A modification.
Compared with non-brain tissues, brain tissues show highly specific m6A methylome, suggesting its involvement in regulation of brain-specific functions. m6A modification shows ubiquitous presence in the cerebral cortex and remarkable increases during neural development. AlkB homolog 5 (ALKBH5), the main demethylase, exhibits tissue-specific regulation in the brain and is highly enrich in the cerebellum and olfactory bulb (7). Likewise, expression of fat mass and obesity associated protein (FTO), another demethylase of RNA, remains high in neurons and exerts its regulating effect on neurophysiological processes, including neuronal differentiation, learning and memory and neurogenesis (21, 22). m6A modification of brain tissue RNA stop codon in the forms of “writer,” “eraser,” and “reader” of are widely expressed in the cerebellum and cerebral cortex, where they are required for the control of delicate gene expressions and thus form tissue-specific regulators that play indispensable roles in nervous system activities (22).
m6A modification is dynamically regulated by a multicomponent methyltransferase complex, including methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14) and Wilms tumor 1-associated protein (WTAP), as well as the recently identified m6A RNA demethylases, FTO and ALKBH5 (1). METTL3 exhibits catalytic activity of the m6A methyltransferase, in which it transfers a methyl group from S-adenosylmethionine (SAM) to an adenosine in RNA (23). ALKBH5 is regarded as a major eraser of m6A modification, which has been found of m6A demethylase activity. The YTH family comprises numerous proteins containing the YTH domain, which selectively bind the m6A in RNA, and m6A exerts its effects by recruiting m6A-binding proteins (24). RNA methylation levels are finely balanced by an interplay between m6A methyltransferases (writers), demethylases (erasers) and binding proteins (readers).
It is reasonable to speculate that METTL3 is involved in mammalian brain development, based on immunostaining of various brain regions in which METTL3 is abundant. Conditional knockout of METTL3 results in smaller body size, ataxia-like movement disorders and early death in mice. In addition, abnormal upregulation of pivotal developmental genes, like Atoh1 and Cxcr4, and apoptosis-associated genes, like Dapk1 and FADD, account for regulation of mRNA stability induced by m6A depletion in CKO mice, which significant impacts survival and apoptosis of cerebellar cells. Further, Grin1, a synapse-related gene, is abnormally spliced in CKO mice, causing excessive calcium influx and ultimately apoptosis. This cumulative evidence confirms that METTL3 plays a critical role in cerebellar neuronal connection, survival and maturation (25). Furthermore, as it shown in the METTL3-knocked down cerebellum, apparent morphology changes are also observed in overexpression of METTL3 (5).
A heterodimeric enzyme complex composed of METTL3 and METTL14 lead to m6A modification of RNAs. In particular, the specific function of METTL14 is to support catalytic activity of methyltransferase of METTL3 in substrate recognition (23). Furthermore, the crucial role of METTL14 in the adult mammalian nervous system has been further confirmed by the significance of m6A in maintenance of normal nervous system functioning. As neural excitability is elevated by METTL14 deletion in the mouse striatum, striatal-mediated behaviors are significantly impaired, impacting learning and initial acquisition of response and sensorimotor learning (26). METTL14 is crucial for the regulation of gene expression during nervous system development; its deletion in neural progenitor cells and neural stem cells (NSC) can result in disruption of cortical development, in the forms of decreased proliferation and premature differentiation, respectively (27–29). Furthermore, embryonic mouse brains with knockdown METTL14 exhibit an extension of the cell cycle of radial glia cells and a period of cortical neurogenesis (27). Other studies have shown impaired injury-induced protein translation and functional axon regeneration in dorsal root ganglion (DRG), and CNS hypomyelination, from METTL14 knockdown (28–30). These studies suggest that METTL14 modulates diverse physiological and pathophysiological brain functions, including learning, cell proliferation and differentiation and CNS hypomyelination, providing new insights into the molecular mechanisms and treatments of brain diseases.
WTAP modulates gene expression and alternative splicing without catalytic activity through its interaction with the METTL3–METTL14 complex, which facilitates recruitment of the m6A methyltransferase to RNA targets, containing the consensus m6A motif, which affects cellular m6A deposition and localization into nuclear speckles (31). WTAP is required for normal differentiation processes, so that WTAP knockdown zebrafish embryos exhibit differentiation defects and increased apoptosis (32). Thus, there is strong evidence that WTAP may play a role as a regulatory subunit of the m6A methyltransferase complex, providing insight toward further investigating the fundamental mechanisms of the regulation functions of m6A.
Recent studies have advanced our understanding of the indispensable role of ALKBH5 in cerebellar development (5, 7). Brain and cerebellum sizes of ALKBH5-deficient mice are reduced, and in immunostaining analyses the numbers of Ki67+proliferating cells and phosphohistone3 (PH3+) mitotic cells in the EGL increased markedly compared with WT counterparts. Expanding cell numbers in the S phase of the cell cycle, reflected by immunostaining analysis, and mitotic cells have also been observed, denoting untimely and aberrant methylation induced by ALKBH5 deficiency, which hinders normal cerebellar cell proliferation and differentiation (5).
FTO is highly enriched in neurons and expressed in NSCs, which are also found most abundantly in the brain (33). Recently, FTO was revealed to be a regulator of proliferative and differentiated processes, but not apoptosis, in adult NSCs via modulating the Pdgfra/Socs5-Stat3 pathway, the products of which are involved in modulation of the stat3 pathway that participates in regulation of neurogenesis, regulation of which the BNDF pathway also participates (21, 34). Thus, FTO deficiency may lead to learning and memory impairments (21). A case report showed that homozygous mutations in FTO exhibits growth retardation and developmental delay, further confirming that FTO mutations may lead to early-onset neurodevelopmental disorders and suggesting that FTO regulates the activities of genes involved in key central nervous system development processes (35). After transient forebrain ischemia, FTO expression in the CA1 and CA2 is dramatically reduced, possibly contributing to hippocampal dysfunction (36).
Several recent studies have revealed that m6A RNA metabolism regulation effects, including mRNA splicing, RNA folding, translation, decay, stability and nuclear export, primarily depend on the function of YT521-B homology (YTH) domain-containing proteins which could be recruited to mRNA and directly recognize the m6A site through well-characterized YTH domains (37). Collectively, such YTH domain-containing proteins are: YTH domain-containing family protein (YTHDF), YTH domain-containing protein 1 (YTHDC1) and YTH domain-containing protein 2 (YTHDC2), which are evolutionarily conserved and involved in functional regulations such as translation efficiency and stability of m6A-containing RNAs (38–40). Moreover, YTH domain-containing proteins are required for pre-crossing axon guidance in spinal cord and neuronal survival improvement after ischemia brain injury and during neural development, raising the possibility that YTH proteins may play a critical role in brain structural integrity and physiological function (41–43).
Advanced research has confirmed Prrc2a as a novel m6A reader that controls OPC specification, differentiation and myelination by regulating expression of olig2, which has a oligodendroglial lineage determination function, in an m6A-dependent manner. GRE domains of Prrc2a are important for binding methylated RNA specifically, and for regulating olig2 mRNA stability, thus Prrc2a deficiency may interfere with the process of oligodendroglial specification and myelination, leading to hypomyelination. Furthermore, the interaction between Prrc2a and YTHDF2 competes for RNA bindings sites, implying that YTDHF2 may interact with Prrc2a (44). These discoveries open new avenues for investigating the fundamental mechanisms of myelination-related diseases and developing new therapeutics.
To gain insights into potential therapeutic strategies for traumatic brain injury (TBI), further investigations into its pathophysiological cascade and possible neuropathological mechanisms are needed. There is widespread public concern regarding TBI, particularly as it leads to increased mortality among young people and increases susceptibility to prolonged dysfunction, which also causes socioeconomic burdens (45, 46). TBI is grouped into primary and secondary injuries; the former manifests as cortical contusions and vascular injuries from direct hits, the latter as cerebral edema caused by a pathophysiological cascade, which may further exacerbate the primary injury and consequently aggravate neurological defects (47). m6A-modified transcripts have been implicated in the physiological and pathological consequences of TBI. Both expression of METTL3 and level of m6A in total RNA were significantly decreased in mouse hippocampus following traumatic brain injury (48). METTL3 deficiency in mouse hippocampus impairs spatial learning and memory (49). Additionally, an injury to the brain caused by TBI may result in disturbances in energy homeostasis and mitochondrial dysfunction, which may cause neurodegeneration and altered synaptic plasticity, resulting in cognitive dysfunction (50). Conjoint GO and pathway analyses of differentially methylated mRNA have shown that peaks in m6A-modified transcripts related to metabolic processes are significantly altered, implying that m6A modification is involved in altered metabolic hippocampal activity and participates in neuronal cell damage following TBI (50). Further, altered m6A methylation levels of some genes (e.g., cd14, Dll4, sox7) are associated with TBI pathophysiological processes (51). Lack of FTO also aggravates TBI-induced brain injuries, spatial memory problems, and neurological deficits (51).
A few studies have examined neuroinflammation and free radical production in brain ischemia, which lead to neurodegenerative disorders, destruction of blood-brain barrier integrity and even mortality (52). Neurons are more vulnerable than other cell types to hypoxic-ischemia damage, because of their lower energy compliments induced by glucose and oxygen brain reductions from relative or absolute hypoperfusion (53). Transient forebrain ischemia significantly reduces FTO expression 7–10 days after ischemia, which is associated with impaired hippocampal function (36). Further, FTO may play crucial roles in m6A methylation of mRNAs post-stroke, and differential m6A methylated RNA after focal ischemia were related with immune regulation, inflammatory signaling, and cell apoptosis (54). m6A methylation can be interfered with by SNPs altering the RNA sequences of target sites or key flanking nucleotides, it can affect the progression of disease consequently (55). The genome-wide association between m6A-SNPs and ischemic stroke by excavating data from a large-scale GWAS, rs2013162 and rs2273235 may exert its effects by altering the methylation level of m6A and regulating the expression of downstream gene IRF6 and NDST1 respectively, which were potential cause gene of ischemic stroke (56). Conjoint analysis of ischemic stroke genome-wide association study and mA-SNP list from the m6AVar database, dozens of m6A-SNPs were identified as functional polymorphisms and novel genetic biomarkers for ischemic stroke pathogenesis (57). YTHDC1 increases Akt phosphorylation by destabilizing PTEN mRNA and promoting degradation to assist neuronal survival, particularly after cerebral ischemia (43). These studies cumulatively show that m6A plays an important role in the maintenance of neurological function and that the regulation function of m6A may provide a potential therapeutic target for ischemic stroke.
Recent studies suggest that the mechanism of ischemia-reperfusion (I/R) damage involves intracellular calcium overload and reactive oxygen species (ROS) accumulation, which impair the integrity and function of mitochondria and, consequently, severely injured ischemic tissues (58). Hypothermia, the only treatment identified to ameliorate cerebral I/R injury, exerts its protective effect via activation of PTEN in a m6A-dependent manner. Additionally, through N6-methyladenosine modification of PTEN mRNA, hypothermia activates PI3K/Akt signaling, downregulates pyroptosis, and consequently plays a neuroprotective role (59). Another study showed a significant increase in m6A expression following OGD/R and MCAO (60). ALKBH5 and FTO selectively demethylate the anti-apoptotic protein Bcl2 transcripts, preventing degradation of Bcl2 transcripts and enhancing Bcl2 expression, and providing neuroprotective effects against I/R neuronal apoptosis (60). By targeting YTHDF1 with microRNA-421-3p, p65 mRNA is destabilized and its translation inhibited, thus preventing inflammatory responses in cerebral I/R (61).
Sepsis is caused by pathogenic infection that leads to a cascade of multi-organ inflammation and systemic immune dysfunction; progression can lead to development of severe sepsis and shock (62–64). Sepsis-related encephalopathy (SAE) can induce neuroinflammation and long-term cognitive dysfunction; its underlying mechanisms include blood-brain barrier damage and promotion of glial cell activation (65). The release of large numbers of inflammatory factors leads to inflammatory responses by the nervous system, oxidative stress and apoptosis (66). The SAE pathological process is primarily an early immune activation and late immune suppression, and is closely related to abnormal immune responses caused by dysfunction of neutrophils, macrophages/monocytes, dendritic cells and T lymphocytes (67). Mice with severe sepsis show a state of advanced immunosuppression, and immune cells (e.g., T cells, B cells) and inflammatory factors (e.g., IL-8, IL-6) are neurotoxic to the central nervous system (67), while T cell homeostasis is controlled by methylation of the m6A mRNA by targeting the pathways IL-7/STAT5/SOCS (68). JAK2/STAT3 expression is decreased in YTHDF1 knockout macrophages and increased IL-17 production, which is associated with the migration of inflammatory cells into the brain during sepsis. In addition, the methylation level of JAK2/STAT3 in cells reduces penetration of macrophages through the blood-brain barrier, improving neuroinflammation and reducing septic brain injury. Consequently, in YTHDF1 knockdown mouse brain tissue, the proportion of normal neurons (i.e., those that do not differ from cells exposed to severe sepsis) is significantly increase compared with mice with hyperemia (69). METTL3 inhibits the inflammation and pyroptosis through decreasing the mRNA stability of NLRP3 in LPS-treated 1321N1 cells, which exhibit a protective effect on SAE (70).
Alzheimer's disease (AD) is a chronic neurodegenerative disease characterized by the accumulation of insoluble neurotoxic aggregates, primarily abnormal extraneuronal β-amyloid deposition and excess intraneuronal tau protein (71). Phosphorylation forms neurofibrillary tangles, leading to synaptic dysfunction and cognitive impairment; patient quality of life is seriously impaired before these effects lead to death (72). Though beneficial results have been achieved in rat models, the role of m6A methylation cannot be ignored in the pathogenic factors of AD, as it is implicated in a variety of pathological processes (73). m6A modification exhibits temporal and spatial specificity during brain development and is closely related to aging and neurodegenerative diseases (74). METTL3 expression is increased and FTO expression decreased in the hippocampus of AD model mice, suggesting that RNA m6A methylation modification promotes AD progression (73). The methylases METTL3 and RBM15B are differentially expressed in the hippocampus of patients with AD, while in the insoluble fraction, the expression level of METTL3 is positively correlated with aberrantly phosphorylated tau (75). The increasing level of m6A in the hippocampus and cortex from AD patients, while the m6A-related regulator proteins, like METTL3, METTL14 and FTO, significantly decreased. Besides, neurodegeneration, spine loss, and gliosis are caused by METTL3 knockdown in the hippocampus, that shMettl3 induced cell cycle abnormalities and neurodegenerative changes, and overexpression of it may protect against the Aβ-induced synaptic toxicity (76). Familial AD (5XFAD) mice have elevated METTL3 expression levels, and GO analysis shows that differentially methylated genes, including synaptic transmission and ion transport regulation, are closely related to the physiological and pathological mechanisms of AD, while m6A controls the protein levels of key genes, its mRNA level shows no significant change (77).
Amyotrophic lateral sclerosis (ALS) is a mortal, progressive neurodegenerative disease involving upper and lower motor neurons, which causes muscle weakness, paralysis, and ultimately death from respiratory failure (78, 79). There is currently neither cure nor treatments to slow or reverse its progression. It is influenced by environmental and genetic factors, although the exact mechanisms of these remains unknown, epigenetic alteration may be crucially involved (79–81). RBP gene mutations have been associated with neurodegenerative and psychiatric diseases, suggesting that RBP may be a cause of ALS (82, 83). Mutations of translocated in liposarcoma/beta-catenin interacts with fusion (TLS/FUS), an RNA binding protein, located in the C-terminal region (84). This contains the nuclear localization signal (NLS), which alters localization and liquid-liquid phase separation (LLPS) of TLS/FUS in affected neurons, resulting in a further shift in the TLS/FUS into cellular aggregates involved in ALS pathogenesis (85). TLS/FUS binds intensely to m6A-modified RNA fragments, suggesting that TLS/FUS may serve as m6A readers (86). Additionally, m6A-modified RNA fragments inhibit aggregation of TLS/FUS in cytoplasmic and affect localization of TLS/FUS-interacting proteins, and thus enhancing cell viability (86). Better understanding the regulation of m6A-modified RNA fragments in TLS/FUS aggregation may contribute to development of novel ALS therapeutics.
A relatively common neurodegenerative disease with well-defined symptoms, Parkinson's disease (PD) is characterized by bradykinesia, muscular rigidity, resting tremor and postural instability (87). Patiens with PD have progressive loss of dopaminergic neurons in the substantia nigra pars compacta and accumulation of misfolded α-synuclein protein, which mediated neurotoxicity (88, 89). Behavioral response control and neuronal activity are also impaired by the inactivation of FTO gene (90). Arsenite exposure reduced dopamine content via impaired dopaminergic neurotransmission and learning and memory ability (91). Additionally, increasing m6A level induced by arsenite exposure in male adult mice, FTO may alleviate dopaminergic neurotransmission by regulating the expression of the key protein in dopaminergic neurotransmission like TH, DAT, COMT, and DRDs (91). Integrative analysis was used to screen for candidate m6A PD risk loci, and identified five m6A-SNPs (rs75072999 of GAK, rs1378602, rs4924839 and rs8071834 of ALKBH5, and rs1033500 of C6orf10) may be associated with PD risk (92), while GAK contributes to the deposition of neurotoxic a-synuclein aggregates in PD (93). Decreased global m6A modified levels were identified in 6-OHDA-induced PC12 cells and the striatum of a rat PD model. Furthermore, overexpression of m6A demethylase FTO significantly decreases the m6A methylation in striatum and induces the expression of NMDAR1, an ionotropic glutamate receptor; its overactivation can cause neuronal injury or apoptotic cell death, resulting in amplified Ca2+ influx and elevated oxidative stress. These in turn ultimately lead to increased apoptosis in dopaminergic neurons. These investigators speculated that FTO could reduce the m6A content and enhance the mRNA stability and expression of NDMAR1 (94). As a result of excessive manganese exposure, dopaminergic neurons are damaged and DA levels are reduced in the striatum, resulting in PD; it also causes decreased FTO mRNA and protein expressions. Axon guidance molecule ephrin-B2 expression can be regulated by FTO-mediated m6A modification via YTHDF2, causing motor dysfunction in a manganese-induced PD rat model (95).
The degree of m6A methylation modification differs among brain injuries resulting from different diseases, but plays a crucial role in each of them. The regulatory role of m6A methylation modification has been confirmed in axon regeneration of the dorsal root ganglia and peripheral nerves; in addition multiple m6A modified genes that are differentially expressed after zebrafish spinal cord injury are all key nerve regeneration genes (28, 96).
m6A methylation modification is the most common epigenetic post-transcriptional modification in eukaryotes. It regulates gene expression via biological processes like RNA splicing, nuclear export, stability and degradation, and is involved in neurodevelopment and various brain functions. Further work is needed to fully elucidate the role of m6A in brain development and disease. The mechanism of action of the m6A RNA binding protein, and how the mRNA context affects neural development and neurological disease are remain unclear. Thus, much research remains to be accomplished.
Compared with other organs, m6A modification is very abundant in the brain and differentially expressed in multiple brain diseases, indicating that m6A methylation modification is closely related to its pathophysiological mechanism. The emergence of diverse, valid sequencing technologies has broadened our understanding of the specific impact of m6A on brain development and disease. In brain injury, m6A is involved in the regulation of multiple signaling pathways, in which it can intervene.
Therefore, studying the role of m6A methylation in regulating the pathogenesis of brain injury and regulating m6A-related genes may lead to strategies for treating various brain injuries. It has great potential for application in the clinical practice of brain injury prevention and treatment. Determining the impacts of m6A in the brain should be a strong priority in neurobiology research.
JD wrote and edited the article. AC searched for data. XC and XZ reviewed and edited the article. All authors contributed to the article and approved the submitted version.
This research was supported by National Natural Science Foundation of China (82001166 and 82171186) and the Joint Funds for the Innovation of Science and Technology (2019Y9028 and 2019Y9023).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.995747/full#supplementary-material
1. Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol. (2017) 33:319–42. doi: 10.1146/annurev-cellbio-100616-060758
2. Meyer KD, Jaffrey SR. The dynamic epitranscriptome: n6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. (2014) 15:313–26. doi: 10.1038/nrm3785
3. Zhang H, Shi X, Huang T, Zhao X, Chen W, Gu N, et al. Dynamic landscape and evolution of m6A methylation in human. Nucleic Acids Res. (2020) 48:6251–64. doi: 10.1093/nar/gkaa347
4. Xiao S, Cao S, Huang Q, Xia L, Deng M, Yang M, et al. The RNA N(6)-methyladenosine modification landscape of human fetal tissues. Nat Cell Biol. (2019) 21:651–61. doi: 10.1038/s41556-019-0315-4
5. Ma C, Chang M, Lv H, Zhang ZW, Zhang W, He X, et al. RNA m(6)A methylation participates in regulation of postnatal development of the mouse cerebellum. Genome Biol. (2018) 19:68. doi: 10.1186/s13059-018-1435-z
6. Xiong X, Hou L, Park YA-O, Molinie B, Gregory RI, Kellis MA-O. Genetic drivers of m(6)A methylation in human brain, lung, heart and muscle. Nat Genet. (2021) 53:1156–65. doi: 10.1038/s41588-021-00890-3
7. Du T, Li G, Yang J, Ma K. RNA demethylase Alkbh5 is widely expressed in neurons and decreased during brain development. Brain Res Bull. (2020) 163:150–9. doi: 10.1016/j.brainresbull.2020.07.018
8. Chang R, Huang Z, Zhao S, Zou JA-OX, Li YA-O, Tan SA-O. Emerging roles of FTO in neuropsychiatric disorders. BioMed Res Int. (2022) 2022:2677312. doi: 10.1155/2022/2677312
9. Yang Y, Shuai P, Li X, Sun K, Jiang X, Liu W, et al. METTL14-mediated m6A modification is essential for visual function and retinal photoreceptor survival. BMC Biol. (2022) 20:140. doi: 10.1186/s12915-022-01335-x
10. Zhang Z, Wang M, Xie D, Huang Z, Zhang L, Yang Y, et al. METTL3-mediated N(6)-methyladenosine mRNA modification enhances long-term memory consolidation. Cell Res. (2018) 28:1050–61. doi: 10.1038/s41422-018-0092-9
11. Lv Z, Xu T, Li R, Zheng D, Li Y, Li W, et al. Downregulation of m6A methyltransferase in the hippocampus of tyrobp (-/-) mice and implications for learning and memory deficits. Front Neurosci. (2022) 16:739201. doi: 10.3389/fnins.2022.739201
12. Livneh IA-O, Moshitch-Moshkovitz S, Amariglio N, Rechavi G, Dominissini DA-O. The m(6)A epitranscriptome: transcriptome plasticity in brain development and function. Nat Rev Neurosci. (2020) 21:36-51. doi: 10.1038/s41583-019-0244-z
13. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse M6a RNA methylomes revealed by m6A-seq. Nature. (2012) 485:201–6. doi: 10.1038/nature11112
14. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRS and near stop codons. Cell. (2012) 149:1635–46. doi: 10.1016/j.cell.2012.05.003
15. Grozhik AV, Linder B, Olarerin-George AO, Jaffrey SR. Mapping M6A at individual-nucleotide resolution using crosslinking and immunoprecipitation (miCLIP). Methods Mol Biol (Clifton, NJ). (2017) 1562:55–78. doi: 10.1007/978-1-4939-6807-7_5
16. Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. (2015) 12:767–72. doi: 10.1038/nmeth.3453
17. Hawley BR, Jaffrey SR. Transcriptome-wide mapping of m6a and m6am at single-nucleotide resolution using miCLIP. Curr Protoc Mol Biol. (2019) 126:e88. doi: 10.1002/cpmb.88
18. Shu X, Cao J, Cheng M, Xiang S, Gao M, Li T, et al. A metabolic labeling method detects m6A transcriptome-wide at single base resolution. Nat Chem Biol. (2020) 16:887–95. doi: 10.1038/s41589-020-0526-9
19. Clyde D. New tools for transcriptome-wide mapping of m6A. Nat Rev Genet. (2020) 21:387. doi: 10.1038/s41576-020-0248-6
20. Liu Je, Li K, Cai J, Zhang M, Zhang X, Xiong X, et al. Landscape and regulation of m6A and m6Am methylome across human and mouse tissues. Mol Cell. (2020) 77:426–40. doi: 10.1016/j.molcel.2019.09.032
21. Li L, Zang L, Zhang F, Chen J, Shen H, Shu L, et al. Fat mass and obesity-associated (FTO) protein regulates adult neurogenesis. Hum Mol Genet. (2017) 26:2398–411. doi: 10.1093/hmg/ddx128
22. Gao X, Shin YH Li M, Wang F, Tong Q, Zhang P. The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS ONE. (2010) 5:e14005. doi: 10.1371/journal.pone.0014005
23. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A Mettl3-Mettl14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. (2014) 10:93–5. doi: 10.1038/nchembio.1432
24. Zhao Y, Shi Y, Shen H, Xie W. M(6)a-Binding proteins: the emerging crucial performers in epigenetics. J Hematol Oncol. (2020) 13:35. doi: 10.1186/s13045-020-00872-8
25. Wang CX, Cui GS, Liu X, Xu K, Wang M, Zhang XX, et al. METTL3-mediated m6A modification is required for cerebellar development. PLoS Biol. (2018) 16:e2004880. doi: 10.1371/journal.pbio.2004880
26. Koranda JL, Dore L, Shi H, Patel MJ, Vaasjo LO, Rao MN, et al. METTL14 is essential for epitranscriptomic regulation of striatal function and learning. Neuron. (2018) 99:283–92. doi: 10.1016/j.neuron.2018.06.007
27. Yoon K-J, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez-Cyrus D, et al. Temporal control of mammalian cortical neurogenesis by m6A methylation. Cell. (2017) 171:877–89. doi: 10.1016/j.cell.2017.09.003
28. Weng Y-L, Wang X, An R, Cassin J, Vissers C, Liu Y, et al. Epitranscriptomic m6A regulation of axon regeneration in the adult mammalian nervous system. Neuron. (2018) 97:313–25. doi: 10.1016/j.neuron.2017.12.036
29. Wang Y, Li Y, Yue M, Wang J, Kumar S, Wechsler-Reya RJ, et al. N(6)-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci. (2018) 21:195–206. doi: 10.1038/s41593-017-0057-1
30. Xu H, Dzhashiashvili Y, Shah A, Kunjamma RB, Weng Y-L, Elbaz B, et al. m6a mRNA methylation is essential for oligodendrocyte maturation and CNS myelination. Neuron. (2020) 105:293–309. doi: 10.1016/j.neuron.2019.12.013
31. Schöller E, Weichmann F, Treiber T, Ringle S, Treiber N, Flatley A, et al. Interactions, localization, and phosphorylation of the m(6)A generating METTL3-METTL14-WTAP Complex. RNA (New York, NY). (2018) 24:499–512. doi: 10.1261/rna.064063.117
32. Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. (2014) 24:177–89. doi: 10.1038/cr.2014.3
33. Du K, Zhang Z, Zeng Z, Tang J, Lee T, Sun T. Distinct Roles of FTO and METTL3 in controlling development of the cerebral cortex through transcriptional and translational regulations. Cell Death Dis. (2021) 12:700. doi: 10.1038/s41419-021-03992-2
34. Cao Y, Zhuang Y, Chen J, Xu W, Shou Y, Huang X, et al. Dynamic effects of FTO in regulating the proliferation and differentiation of adult neural stem cells of mice. Hum Mol Genet. (2020) 29:727–35. doi: 10.1093/hmg/ddz274
35. Daoud H, Zhang D, McMurray F, Yu A, Luco SM, Vanstone J, et al. Identification of a pathogenic FTO mutation by next-generation sequencing in a newborn with growth retardation and developmental delay. J Med Genet. (2016) 53:200–7. doi: 10.1136/jmedgenet-2015-103399
36. Kim W, Kang MS, Kim TH, Yoo DY, Park JH, Jung HY, et al. Ischemia-related changes of fat-mass and obesity-associated protein expression in the gerbil hippocampus. Metab Brain Dis. (2020) 35:335–42. doi: 10.1007/s11011-019-00513-1
37. Roundtree IA, Luo G-Z, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N-methyladenosine methylated mRNAs. eLife. (2017) 6:e31311. doi: 10.7554/eLife.31311.040
38. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. (2014) 505:117–20. doi: 10.1038/nature12730
39. Fu Y, Luo G-Z, Chen K, Deng X, Yu M, Han D, et al. N6-methyldeoxyadenosine marks active transcription start sites in chlamydomonas. Cell. (2015) 161:879–92. doi: 10.1016/j.cell.2015.04.010
40. Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 Destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. (2016) 7:12626. doi: 10.1038/ncomms12626
41. Li M, Zhao X, Wang W, Shi H, Pan Q, Lu Z, et al. YTHDF2-mediated m6A mRNA clearance modulates neural development in mice. Genome Biol. (2018) 19:69. doi: 10.1186/s13059-018-1436-y
42. Zhuang M, Li X, Zhu J, Zhang J, Niu F, Liang F, et al. The m6A reader YTHDF1 regulates axon guidance through translational control of Robo3.1 expression. Nucleic Acids Res. (2019) 47:4765–77. doi: 10.1093/nar/gkz157
43. Zhang Z, Wang Q, Zhao X, Shao L, Liu G, Zheng X, et al. YTHDC1 mitigates ischemic stroke by promoting AKT phosphorylation through destabilizing PTEN mRNA. Cell Death Dis. (2020) 11:977. doi: 10.1038/s41419-020-03186-2
44. Wu R, Li A, Sun B, Sun JG, Zhang J, Zhang T, et al. A novel m(6)A reader PRRC2A controls oligodendroglial specification and myelination. Cell Res. (2019) 29:23–41. doi: 10.1038/s41422-018-0113-8
45. Reis C, Wang Y, Akyol O, Ho WM Ii RA, Stier G, et al. What's new in traumatic brain injury: update on tracking, monitoring and treatment. Int J Mol Sci. (2015) 16:11903–65. doi: 10.3390/ijms160611903
46. Morganti-Kossmann MC, Semple BD, Hellewell SC, Bye N, Ziebell JM. The complexity of neuroinflammation consequent to traumatic brain injury: from research evidence to potential treatments. Acta Neuropathol. (2019) 137:731–55. doi: 10.1007/s00401-018-1944-6
47. Kubal WS. Updated imaging of traumatic brain injury. Radiol Clin North Am. (2012) 50:15–41. doi: 10.1016/j.rcl.2011.08.010
48. Wang Y, Mao J, Wang X, Lin Y, Hou G, Zhu J, et al. Genome-wide screening of altered m6A-tagged transcript profiles in the hippocampus after traumatic brain injury in mice. Epigenomics. (2019) 11:805–19. doi: 10.2217/epi-2019-0002
49. Shi H, Zhang X, Weng YL, Lu Z, Liu Y, Lu Z, et al. m(6)A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature. (2018) 563:249–53. doi: 10.1038/s41586-018-0666-1
50. Lai JQ, Shi YC, Lin S, Chen XR. Metabolic disorders on cognitive dysfunction after traumatic brain injury. Trends Endocrinol Metab. (2022) 33:451–62. doi: 10.1016/j.tem.2022.04.003
51. Yu J, Zhang Y, Ma H, Zeng R, Liu R, Wang P, et al. Epitranscriptomic profiling of n6-methyladenosine-related RNA methylation in rat cerebral cortex following traumatic brain injury. Mol Brain. (2020) 13:11. doi: 10.1186/s13041-020-0554-0
52. Kawabori M, Yenari MA. Inflammatory responses in brain ischemia. Curr Med Chem. (2015) 22:1258–77. doi: 10.2174/0929867322666150209154036
53. Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. (2007) 27:1129–38. doi: 10.1523/JNEUROSCI.4468-06.2007
54. Chokkalla AK, Mehta SL, Kim T, Chelluboina B, Kim J, Vemuganti R. Transient focal ischemia significantly alters the m6A epitranscriptomic tagging of RNAs in the brain. Stroke. (2019) 50:2912–21. doi: 10.1161/STROKEAHA.119.026433
55. Zheng Y, Nie P, Peng D, He Z, Liu M, Xie Y, et al. M6avar: a database of functional variants involved in M6a modification. Nucleic Acids Res. (2018) 46:D139–d45. doi: 10.1093/nar/gkx895
56. Mo XB, Lei SF, Zhang YH, Zhang H. Integrative analysis identified IRF6 and NDST1 as potential causal genes for ischemic stroke. Front Neurol. (2019) 10:517. doi: 10.3389/fneur.2019.00517
57. Zhu R, Tian D, Zhao Y, Zhang C, Liu X. Genome-wide detection of m(6)A-associated genetic polymorphisms associated with ischemic stroke. J Mol Neurosci. (2021) 71:2107–15. doi: 10.1007/s12031-021-01805-x
58. Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V, et al. Ros-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev. (2016) 2016:2183026. doi: 10.1155/2016/2183026
59. Diao MY, Zhu Y, Yang J, Xi SS, Wen X, Gu Q, et al. Hypothermia protects neurons against ischemia/reperfusion-induced pyroptosis via m6A-mediated activation of PTEN and the PI3K/AKT/GSK-3β signaling pathway. Brain Res Bull. (2020) 159:25–31. doi: 10.1016/j.brainresbull.2020.03.011
60. Xu K, Mo Y, Li D, Yu Q, Wang L, Lin F, et al. N(6)-methyladenosine demethylases Alkbh5/FTO regulate cerebral ischemia-reperfusion injury. Ther Adv Chronic Dis. (2020) 11:2040622320916024. doi: 10.1177/2040622320916024
61. Zheng L, Tang X, Lu M, Sun S, Xie S, Cai J, et al. MicroRNA-421-3p prevents inflammatory response in cerebral ischemia/reperfusion injury through targeting m6A reader YTHDF1 to inhibit P65 mRNA translation. Int Immunopharmacol. (2020) 88:106937. doi: 10.1016/j.intimp.2020.106937
62. Chen H, Li Y, Wu J, Li G, Tao X, Lai K, et al. RIPK3 collaborates with GSDMD to drive tissue injury in lethal polymicrobial sepsis. Cell Death Differ. (2020) 27:2568–85. doi: 10.1038/s41418-020-0524-1
63. Xu S, Li L, Wu J, An S, Fang H, Han Y, et al. Melatonin attenuates sepsis-induced small-intestine injury by upregulating SIRT3-mediated oxidative-stress inhibition, mitochondrial protection, and autophagy induction. Front Immunol. (2021) 12:625627. doi: 10.3389/fimmu.2021.625627
64. Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet (London, England). (2018) 392:75–87. doi: 10.1016/S0140-6736(18)30696-2
65. Xu XE, Liu L, Wang YC, Wang CT, Zheng Q, Liu QX, et al. Caspase-1 inhibitor exerts brain-protective effects against sepsis-associated encephalopathy and cognitive impairments in a mouse model of sepsis. Brain Behav Immun. (2019) 80:859–70. doi: 10.1016/j.bbi.2019.05.038
66. Tian M, Qingzhen L, Zhiyang Y, Chunlong C, Jiao D, Zhang L, et al. Attractylone attenuates sepsis-associated encephalopathy and cognitive dysfunction by inhibiting microglial activation and neuroinflammation. J Cell Biochem. (2019) 120:7101–8. doi: 10.1002/jcb.27983
67. Ren C, Yao RQ, Zhang H, Feng YW, Yao YM. Sepsis-associated encephalopathy: a vicious cycle of immunosuppression. J Neuroinflamm. (2020) 17:1–15. doi: 10.1186/s12974-020-1701-3
68. Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCs pathways. Nature. (2017) 548:338–42. doi: 10.1038/nature23450
69. Xing Y, Cheng D, Shi C, Shen Z. The protective role of YTHDF1-knock down macrophages on the immune paralysis of severe sepsis rats with ECMO. Microvasc Res. (2021) 137:104178. doi: 10.1016/j.mvr.2021.104178
70. Wang B, Liu Y, Jiang R, Liu Z, Gao H, Chen F, et al. Emodin relieves the inflammation and pyroptosis of lipopolysaccharide-treated 1321n1 cells by regulating methyltransferase-like 3-mediated NLR family pyrin domain containing 3 expression. Bioengineered. (2022) 13:6740–9. doi: 10.1080/21655979.2022.2045836
71. Yang SH, Lee DK, Shin J, Lee S, Baek S, Kim J, et al. NEC-1 Alleviates cognitive impairment with reduction of Aβ and tau abnormalities in APP/PS1 mice. EMBO Mol Med. (2017) 9:61–77. doi: 10.15252/emmm.201606566
72. Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, et al. Alzheimer disease. Nat Rev Disease Primers. (2021) 7:33. doi: 10.1038/s41572-021-00269-y
73. Han M, Liu Z, Xu Y, Liu X, Wang D, Li F, et al. Abnormality of m6A mRNA methylation is involved in Alzheimer's disease. Front Neurosci. (2020) 14:98. doi: 10.3389/fnins.2020.00098
74. Shu L, Huang X, Cheng X, Li X. Emerging roles of N6-methyladenosine modification in neurodevelopment and neurodegeneration. Cells. (2021) 10:2694. doi: 10.3390/cells10102694
75. Huang H, Camats-Perna J, Medeiros R, Anggono V, Widagdo J. Altered expression of the m6A methyltransferase METTL3 in Alzheimer's disease. eNeuro. (2020) 7:0125-20. doi: 10.1523/ENEURO.0125-20.2020
76. Zhao F, Xu Y, Gao S, Qin L, Austria Q, Siedlak SL, et al. METTL3-dependent RNA m(6)A dysregulation contributes to neurodegeneration in Alzheimer's disease through aberrant cell cycle events. Mol Neurodegener. (2021) 16:70. doi: 10.1186/s13024-021-00484-x
77. Shafik AM, Zhang F, Guo Z, Dai Q, Pajdzik K, Li Y, et al. N6-methyladenosine dynamics in neurodevelopment and aging, and its potential role in Alzheimer's disease. Genome Biol. (2021) 22:17. doi: 10.1186/s13059-020-02249-z
78. Gordon PH, Cheng B, Katz IB, Mitsumoto H, Rowland LP. Clinical features that distinguish PLS, upper motor neuron-dominant ALS, and typical ALS. Neurology. (2009) 72:1948–52. doi: 10.1212/WNL.0b013e3181a8269b
79. Johnson FO, Atchison WD. The role of environmental mercury, lead and pesticide exposure in development of amyotrophic lateral sclerosis. Neurotoxicology. (2009) 30:761–5. doi: 10.1016/j.neuro.2009.07.010
80. Vinceti M, Filippini T, Violi F, Rothman KJ, Costanzini S, Malagoli C, et al. Pesticide exposure assessed through agricultural crop proximity and risk of amyotrophic lateral sclerosis. Environ Health. (2017) 16:91. doi: 10.1186/s12940-017-0297-2
81. Andrew A, Zhou J, Gui J, Harrison A, Shi X, Li M, et al. Pesticides applied to crops and amyotrophic lateral sclerosis risk in the US. Neurotoxicology. (2021) 87:128–35. doi: 10.1016/j.neuro.2021.09.004
82. Diaz-Garcia S, Ko VI, Vazquez-Sanchez S, Chia R, Arogundade OA, Rodriguez MJ, et al. Nuclear depletion of RNA-binding protein ELAVL3 (HUC) in sporadic and familial amyotrophic lateral sclerosis. Acta Neuropathol. (2021) 142:985–1001. doi: 10.1007/s00401-021-02374-4
83. Mann JR, Donnelly CJ. RNA modulates physiological and neuropathological protein phase transitions. Neuron. (2021) 109:2663–81. doi: 10.1016/j.neuron.2021.06.023
84. Sato S, Idogawa M, Honda K, Fujii G, Kawashima H, Takekuma K, et al. Beta-catenin interacts with the FUS proto-oncogene product and regulates pre-mRNA splicing. Gastroenterology. (2005) 129:1225–36. doi: 10.1053/j.gastro.2005.07.025
85. Reber S, Jutzi D, Lindsay H, Devoy A, Mechtersheimer J, Levone BR, et al. The phase separation-dependent FUS interactome reveals nuclear and cytoplasmic function of liquid-liquid phase separation. Nucleic Acids Res. (2021) 49:7713–31. doi: 10.1093/nar/gkab582
86. Yoneda R, Ueda N, Kurokawa R. M(6)A modified short RNA fragments inhibit cytoplasmic TLS/FUS aggregation induced by hyperosmotic stress. Int J Mol Sci. (2021) 22:11014. doi: 10.3390/ijms222011014
87. Balestrino R, Schapira AHV. Parkinson disease. Eur J Neurol. (2020) 27:27–42. doi: 10.1111/ene.14108
88. Tanudjojo B, Shaikh SS, Fenyi A, Bousset L, Agarwal D, Marsh J, et al. Phenotypic manifestation of α-synuclein strains derived from Parkinson's disease and multiple system atrophy in human dopaminergic neurons. Nat Commun. (2021) 12:3817. doi: 10.1038/s41467-021-23682-z
89. Batelli S, Invernizzi RW, Negro A, Calcagno E, Rodilossi S, Forloni G, et al. The Parkinson's disease-related protein DJ-1 protects dopaminergic neurons in vivo and cultured cells from alpha-synuclein and 6-hydroxydopamine toxicity. Neurodegener Dis. (2015) 15:13–23. doi: 10.1159/000367993
90. Hess ME, Hess S, Meyer KD, Verhagen LA, Koch L, Brönneke HS, et al. The fat mass and obesity associated gene (FTO) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci. (2013) 16:1042–8. doi: 10.1038/nn.3449
91. Bai L, Tang Q, Zou Z, Meng P, Tu B, Xia Y, et al. M6A demethylase FTO regulates dopaminergic neurotransmission deficits caused by arsenite. Toxicol Sci. (2018) 165:431–46. doi: 10.1093/toxsci/kfy172
92. Qiu X, He H, Huang Y, Wang J, Xiao Y. Genome-wide identification of M(6)a-associated single-nucleotide polymorphisms in Parkinson's disease. Neurosci Lett. (2020) 737:135315. doi: 10.1016/j.neulet.2020.135315
93. Dumitriu A, Pacheco CD, Wilk JB, Strathearn KE, Latourelle JC, Goldwurm S, et al. Cyclin-G-associated kinase modifies α-synuclein expression levels and toxicity in Parkinson's disease: results from the GENEPD study. Hum Mol Genet. (2011) 20:1478–87. doi: 10.1093/hmg/ddr026
94. Chen X, Yu C, Guo M, Zheng X, Ali S, Huang H, et al. Down-regulation of m6A mRNA methylation is involved in dopaminergic neuronal death. ACS Chem Neurosci. (2019) 10:2355–63. doi: 10.1021/acschemneuro.8b00657
95. Qi Z, Wang S, Li J, Wen Y, Cui R, Zhang K, et al. Protective Role of mRNA demethylase FTO on axon guidance molecules of Nigro-striatal projection system in manganese-induced Parkinsonism. J Hazard Mater. (2022) 426:128099. doi: 10.1016/j.jhazmat.2021.128099
Keywords: N6-methyladenosine, post-transcriptional RNA modification, RNA, brain injury, neurodegenerative disease
Citation: Deng J, Chen X, Chen A and Zheng X (2022) m6A RNA methylation in brain injury and neurodegenerative disease. Front. Neurol. 13:995747. doi: 10.3389/fneur.2022.995747
Received: 16 July 2022; Accepted: 17 August 2022;
Published: 08 September 2022.
Edited by:
Xiangrong Liu, Capital Medical University, ChinaReviewed by:
Xian-jian Huang, Shenzhen Second People's Hospital, ChinaCopyright © 2022 Deng, Chen, Chen and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaochun Zheng, emhlbmd4aWFvY2h1bjc3NjZAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.