 Ángel Aledo-Serrano1*
Ángel Aledo-Serrano1* Ana Mingorance2Vicente Villanueva3Juan José García-Peñas4Antonio Gil-Nagel1Susana Boronat5
Ana Mingorance2Vicente Villanueva3Juan José García-Peñas4Antonio Gil-Nagel1Susana Boronat5 JoséÁngel Aibar6Silvia Cámara4
JoséÁngel Aibar6Silvia Cámara4 María José Yániz7
María José Yániz7 Luis Miguel Aras8Bárbara Blanco9
Luis Miguel Aras8Bárbara Blanco9 Rocío Sánchez-Carpintero7 on behalf of the Charlotte Project Group
Rocío Sánchez-Carpintero7 on behalf of the Charlotte Project Group- 1Epilepsy Program, Neurology Department, Ruber Internacional Hospital, Madrid, Spain
- 2Dracaena Consulting SL, Loulou Foundation, London, United Kingdom
- 3Hospital Universitario y Politécnico La Fe, Valencia, Spain
- 4Hospital Infantil Universitario Niño Jesús, Madrid, Spain
- 5Hospital de la Santa Creu i Sant Pau, Barcelona, Spain
- 6Fundación Síndrome de Dravet, Madrid, Spain
- 7Clínica Universidad Navarra, Pamplona, Spain
- 8Apoyo Dravet, San Sebastian, Spain
- 9Hospital Universitario Virgen del Rocío, Seville, Spain
Objective: The appropriate management of patients with Dravet Syndrome (DS) is challenging, given the severity of symptoms and the burden of the disease for patients and caregivers. This study aimed to identify, through a qualitative methodology and a Delphi consensus-driven process, a set of recommendations for the management of DS to guide clinicians in the assessment of the clinical condition and quality of life (QoL) of DS patients, with a special focus on patient- and caregiver-reported outcomes (PROs).
Methods: This study was conducted in five phases, led by a multidisciplinary scientific committee (SC) including pediatric neurologists, epileptologists, a neuropsychologist, an epilepsy nurse, and members of DS patient advocates. In phases 1 and 2, a questionnaire related to patients' QoL was prepared and answered by caregivers and the SC. In phase 3, the SC generated, based on these answers and on a focus group discussion, a 70-item Delphi questionnaire, covering six topic categories on a nine-point Likert scale. In phase 4, 32 panelists, from different Spanish institutions and with a multidisciplinary background, answered the questionnaire. Consensus was obtained and defined as strong or moderate if ≥80% and 67–79% of panelists, respectively, rated the statement with ≥7. Phase 5 consisted of the preparation of the manuscript.
Results: The panelists agreed on a total of 69 items (98.6%), 54 (77.14%), and 15 (21.43%) with strong and moderate consensus, respectively. The experts' recommendations included the need for frequent assessment of patient and caregivers QoL parameters. The experts agreed that QoL should be assessed through specific questionnaires covering different domains. Likewise, the results showed consensus regarding the regular evaluation of several clinical parameters related to neurodevelopment, attention, behavior, other comorbidities, and sudden unexpected death in epilepsy (SUDEP). A consensus was also reached on the instruments, specific parameters, and caregivers' education in the routine clinical management of patients with DS.
Conclusions: This consensus resulted in a set of recommendations for the assessment of clinical and QoL parameters, including PROs, related to the general evaluation of QoL, neurodevelopment, attention, behavior, other comorbidities affecting QoL, SUDEP, and QoL of caregivers/relatives and patients with DS.
Introduction
Dravet syndrome (DS) is a life-threatening epilepsy syndrome that begins in infancy or early childhood and includes a wide spectrum of symptoms ranging from mild to severe (1). Additionally, DS is included in the group of developmental and epileptic encephalopathies (DEE). Patients with DS initially present with prolonged focal or generalized onset motor seizures, which are usually fever-induced, starting before 15 months of age (often during the first year) (2). As the disease progresses, DS patients develop other symptoms, including neurodevelopmental impairments (3). Up to 88% of DS patients have mutations in the SCN1A gene (4), which encodes a sodium channel involved in nervous system function. Pathogenic variants of the SCN1A gene result in a wide range of disease severities, from severe DS, on one end of the spectrum, to milder pathologies on the other end, such as genetic epilepsy with febrile seizures plus (GEFS+) and the genetic syndrome with febrile seizures (FS or FS+) (5–8).
Dravet syndrome, first described in 1978 (1), has an estimated incidence of 1/12,000 to 1/40,000 live births (9, 10), although there is still a diagnostic gap, especially in adult patients. Children with DS experience significant developmental delays associated with behavioral problems, which become particularly apparent in the second to fourth years of life (11, 12). Even though these symptoms become more stable after adolescence, they persist throughout adulthood, impairing patients' quality of life (QoL) (12). Furthermore, the severity of these cognitive and behavioral problems cause psychosocial sequelae in the short and long term, requiring extensive care from caregivers and relatives (1, 13–15). DS patients experience fine and gross motor skill impairments and other physical disorders, including ataxia and gait disturbances (1, 16). Due to the severity and the burden of the disease, the quantity and quality of the support required from caregivers and relatives can be emotionally and financially challenging, resulting in a significant financial impact for families (17, 18). Furthermore, mortality rates in DS are high and, in 59% of the cases, they are due to Sudden Unexpected Death in Epilepsy (SUDEP) (19). Accordingly, experts acknowledge the need to assess different aspects of DS beyond seizures in the clinical management of DS, including the impact of DS on caregivers' and relatives' QoL (14). In this regard, patient-reported outcome measures (PROMs) are an increasingly used tool that has proven useful in various pathologies (20), but there is still little evidence in the context of patients with DS (21).
Several guidelines and recommendations on DS are available, including a recently published European treatment guideline (22). However, owing to the complexity of DS involvement and the significant uncertainty associated with its clinical assessment (23–27), there is still a need for instruments assessing the relevant aspects of DS for patients follow-up in the routine clinical practice, such as PROMs. Similarly, there is limited literature summarizing recommendations regarding the comorbidities of DS from different perspectives, such as professionals from multiple disciplines and caregivers. This study aimed to identify, through a qualitative focus group including patients and caregivers and a Delphi consensus-driven process, a core set of recommendations for the management of DS to guide clinicians in the evaluation of both the clinical condition and QoL of DS patients, with a special focus on patient/caregiver reported outcomes. Recommendations regarding the assessment of the QoL of caregivers and relatives of patients with DS were a secondary objective.
Materials and methods
Study design
The Delphi technique is a structured method broadly used to collect relevant information on a specific issue and consists of a series of questionnaires or “rounds” targeted to experts (28). The key features of this method are participant anonymity and controlled feedback. The Charlotte Project was carried out in five phases. In phases 1 and 2, a questionnaire assessing different QoL aspects was prepared. In Phase 2, caregivers answered the questionnaire and the results were discussed using a qualitative methodology to collect caregivers' perspectives. Patient characteristics and relationship with participating caregivers are shown in Supplementary Table 1. The scientific committee also answered the questionnaire. During phase 3, the Delphi statements were developed based on the results of the previous phases. In phases 4 and 5, the Delphi questionnaire was answered by a panel of experts, results were analyzed, and the manuscript was prepared.
Study phases
A diagram of the study phases is presented in Figure 1. In the initial phase, conducted between March and April 2021, the literature regarding the assessment of QoL, comorbidities, and patient-reported outcomes measures (PROMs) in DS and other related DEEs was reviewed. Keywords searched (in English language) in the literature databases PUBMED and EMBASE in March–April 2021 (last 10 years period) included “Dravet Syndrome,” “Patient-reported outcome,” “Quality of life,” “Comorbidity,” and “Caregiver”. Also, during this phase, an ad hoc questionnaire was designed for this project to assess the views of the patients and the scientific committee regarding aspects related to QoL, the impact of comorbidities on QoL, and caregivers' QoL. The coordinator of the project (Dr. Aledo) was in charge of designing the questionnaire. In the beginning, the QoL-related publications found by the literature search were examined. Relying on literature and clinical experience, the questions were developed (Supplementary Table 2). In the second phase, the qualitative phase completed between May and June 2021, representatives of the Dravet Syndrome Foundation (DSF) Spain contacted caregivers (i.e., patients' relatives) and invited them to participate in a meeting. During the meeting, the eight invited caregivers answered the questionnaire and discussed relevant issues in a focus group. Additionally, caregivers of DS patients shared their opinions and experiences related to the care and management of the disease from physical, social, and emotional perspectives. The 10 members of the scientific committee also answered the questionnaire individually. During the third phase, the scientific committee compared, analyzed, and discussed the answers provided both by patients and the members of the scientific committee. The conclusions obtained during the meeting were used to prepare the statements to be included in the Delphi questionnaire. The final version of the questionnaire included 70 items written as statements to be answered on a nine-point Likert scale, where 1 was totally disagree and 9 totally agree. Additionally, the scientific committee selected the expert panel to participate in the Delphi phase. The fourth phase was the Delphi phase, conducted between September and October 2021, in which the panelists answered the questionnaire in one round. A free text space was also included to allow participants to provide additional comments for each item. A second round was not considered necessary, due to the high degree of consensus reached. In the fifth and final phase (November 2021), the manuscript was prepared.
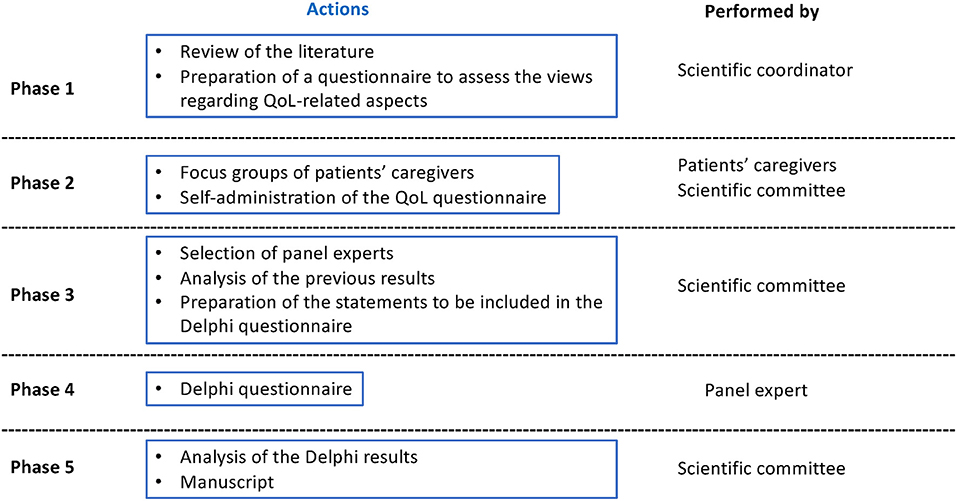
Figure 1. Study phase diagram.
Scientific committee
The project was led by a scientific committee that was comprised of a multidisciplinary team with seven experts in the field of DS, including three pediatric neurologists, three epileptologists (both pediatric and adult), one neuropsychologist, one epilepsy nurse, one neuroscientist specialized in DEEs and working with patient organizations, and two members of DS patient advocates.
Expert panel
The scientific committee selected a group of 32 epilepsy experts with a multidisciplinary background as participants (Supplementary Figure 1). The criteria for their selection included professional knowledge and experience in the field of DS. All participants were members of the Spanish Epilepsy Society, as well as the Epilepsy section of the Spanish Society of Pediatric Neurology or the Spanish Society of Neurology.
Consensus definition
An item or statement was considered consensual when there was “agreement” of opinion in the panel. This happens when the panelists' votes outside one of the three-point regions [(1–3), (4–6), (7–9)] containing the median were less than one-third of the answers (<33.3%). The median value determines the group consensus: majority “disagreement” if the median was within 1–3 and majority “agreement” if the median was within 7–9. Cases where the median was within the 4–6 region were considered “doubtful”. Conversely, “discordance” was considered when the scores of one-third or more of the panelists were in the (1–3) region and another third or more in the (7–9) region. The remaining statements for which no concordance or discordance was obtained were considered to have an “undetermined” level of consensus. A strong consensus was defined if 80% or more of panelists providing an opinion rated the statement with 7 or higher. A moderate consensus was considered if 67–79% of panelists rated the statement with 7 or higher (29). Statements that did not reach this level of agreement were interpreted as “undetermined”.
Data analysis
SPSS Statistics version 20 by IBM (Armonk, NY, USA) was used to create and analyze the database. The median and the percentage of responses in the 7–9 range were calculated, and their values were used to define consensus.
Ethical aspects
This study was performed following the Helsinki Declaration. Data from the Delphi questionnaire were anonymized for the analysis. All the personal data were dissociated from the results in compliance with the EU General Data Protection Regulation (GDPR).
Results
The 70 statements developed by the scientific committee covered a total of six categories: (1) general aspects of the QoL evaluation of patients with DS (15 items), (2) evaluation of neurodevelopment (24 items), (3) assessment of attention and behavior (9 items), (4) evaluation of other comorbidities affecting QoL (14 items), (5) Sudden unexpected death in epilepsy (SUDEP) (4 items), and (6) assessment of the QoL of caregivers/relatives (3 items). Of the 32 DS experts invited to participate in the Delphi process, a total of 28 (87.5%) answered the questionnaire (Supplementary Table 3). The panel experts reached a consensus on “agreement” in the first round on a total of 69 of the 70 items (98.57%). A strong consensus was obtained on 54 items (77.14%) and a moderate consensus was reached on 15 items (21.43%). Key results for each category are detailed below.
General aspects of the evaluation of the QoL of patients with DS
A 100% consensus was obtained across category 1 with an agreement on all the 15 statements, although 4 (26.7%) were categorized into the moderate consensus range (Table 1). The panelists agreed that the evaluation of the QoL of the patient at each visit with the specialist was important. Self-administered structured questionnaires for relatives or caregivers in an electronic format was the recommended method for evaluating QoL.
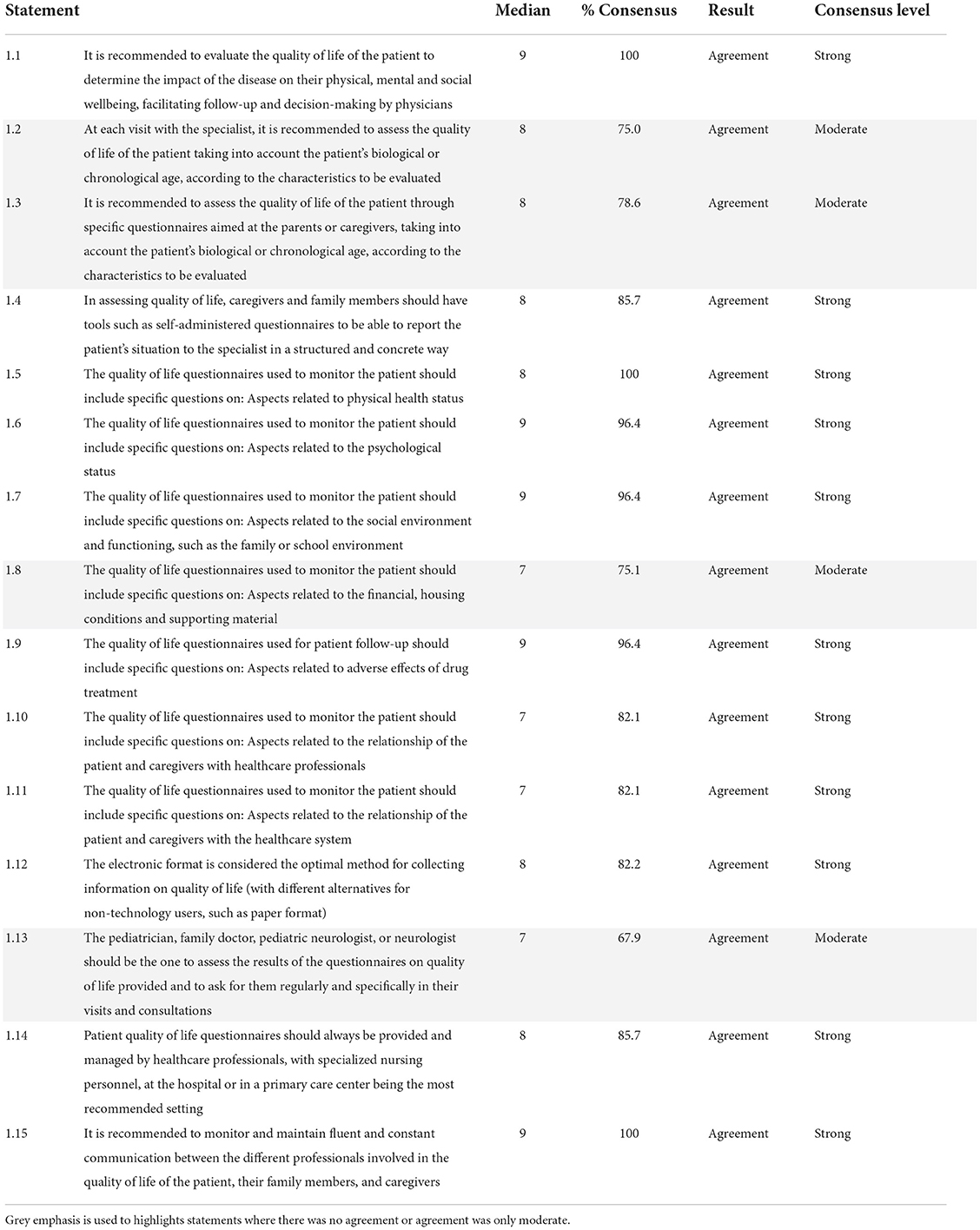
Table 1. General aspects on the assessment of the quality of life of patients with Dravet syndrome (block 1).
Regarding the content of the QoL questionnaires used to monitor patients, the experts reached a consensus that these tools should be specific to the syndrome and should consider the biological or chronological age of the patients. The questionnaires should include aspects related to the patient's physical health, the psychological status, the social environment and functioning, the economic situation, housing conditions and supporting material, adverse effects of pharmacological treatment, and the relationships of the patient and caregivers with healthcare professionals and the healthcare system.
To conclude this category, the panelists reached a consensus agreement regarding the administration and assessment of the QoL questionnaire. The experts stated that an electronic format is an optimal method and that the questionnaire should be administered and evaluated by healthcare professionals, always in communication and coordination with each other and with the patient's family and caregivers.
Evaluation of neurodevelopment in patients with DS
The panelists reached a consensus agreement on all 24 statements (100%) of category 2, of which 6 (25.0%) had a moderate consensus (Table 2). In their opinion, a neurodevelopmental evaluation of DS patients is recommended at each visit with the specialist, at different frequencies depending on the age of the patient. The panelists agreed that the evaluation of various neurological parameters related to patient care and communication skills should be carried out at least every 3–6 months.
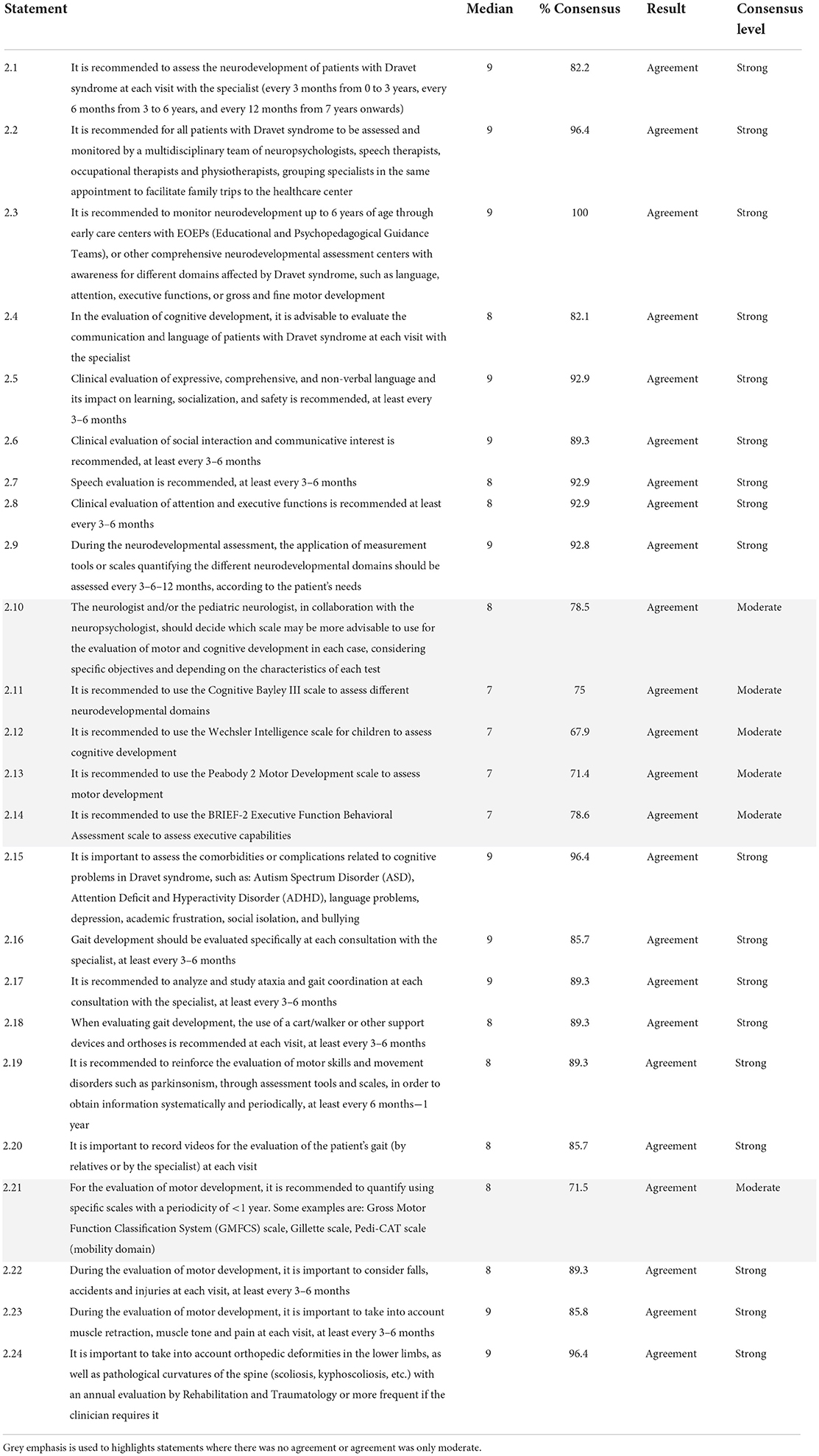
Table 2. Evaluation of neurodevelopment in patients with Dravet syndrome (block 2).
The panelists agreed on the importance of assessing several comorbidities or complications related to cognitive impairment. Likewise, experts agreed on recommendations related to the evaluation and study of gait and ataxia, including the frequency of evaluation.
Experts agreed on the likely suitability of a series of scales to measure neurological and motor development in patients with DS, including Bayley-III Cognitive Scale, Wechsler Intelligence Scale for Children-V, Peabody 2 Developmental Motor Scale-2, Behavior Rating Scale of Executive Function-2 (BRIEF-2), Gross Motor Function Classification System (GMFCS), Gillette Functional Assessment Questionnaire, and Pediatric Evaluation of Disability Inventory Computer Adaptive Test (PEDI-CAT). These scales should be used considering the specific objectives proposed by the adult/pediatric neurologist in collaboration with the neuropsychologist. Finally, the experts agreed on other parameters to be considered when assessing motor development, as well as on the frequency of these measurements.
Assessment of attention and behavior in patients with DS
All the initial nine statements proposed in category 3 reached consensus agreement status (Table 3), although one (11.11%) fell into the moderate consensus category. The panelists recommended evaluating the behavior of the patient at each consultation with the specialist or at least every 3–6 months through educational/stimulation centers. According to the experts' opinion, this evaluation should consider certain comorbidities and/or situations that might be associated with the behavior. The recommended parameters for behavior evaluation included aggressiveness, impulsivity, hyperactivity, and difficulty paying attention, low cognitive flexibility, frustration intolerance, temper tantrums, family stress, and the school situation. Finally, the experts recommended assessing the use of scales on a regular basis to systematize and formalize the behavior assessment, as well as to support a possible therapeutic decision.
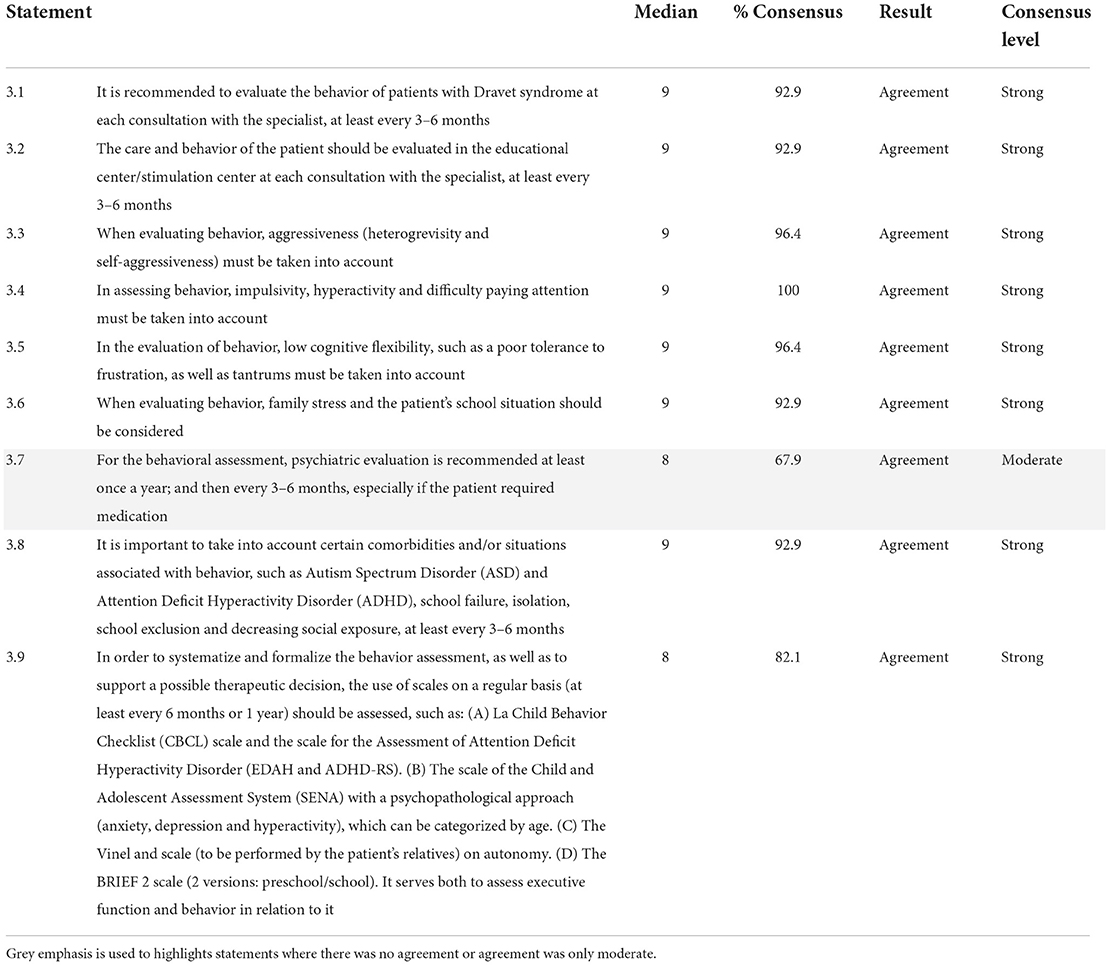
Table 3. Assessment of attention and behavior in patients with Dravet syndrome.
Assessment of other comorbidities that affect the QoL in patients with DS
The panelists overall agreed on category 4 items, obtaining consensus agreement on 13 of the 14 statements (92.86%), as shown in Table 4. Two of these items (14.28%) were considered as a moderate consensus according to the panelists' opinions. The experts recommended enquiring about the patient's sleep (quantity and quality) at each visit with the specialist, at least every 3–6 months, considering the need for sleep medication and its effectiveness as well as the frequency of nighttime seizures, in particular generalized tonic-clonic seizures.
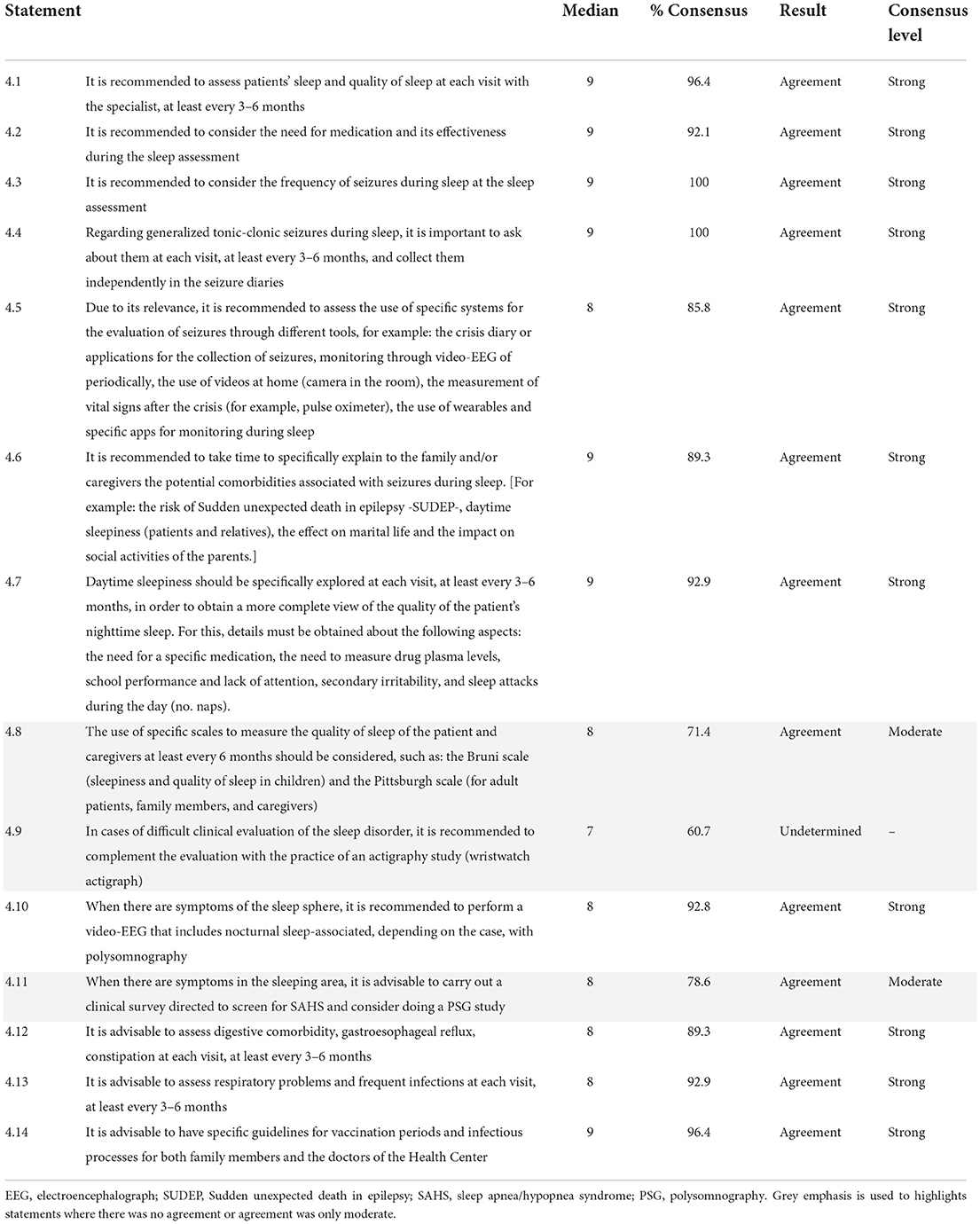
Table 4. Evaluation of other comorbidities that affect quality of life in patients with Dravet syndrome.
Overall, the panelists agreed on various recommendations related to the assessment of sleep-related problems (including the use of the Bruni and the Pittsburgh scales), with the exception of the actigraphy study, for which consensus was undetermined. Additionally, the experts recommended spending time specifically explaining to the family and/or caregivers the potential comorbidities associated with seizures during sleep.
Experts also recommended assessing other comorbidities at least every 3–6 months, including digestive and respiratory disorders, and available specific guidelines for vaccination periods and infectious processes for both family members and the physicians of the Healthcare Center.
Sudden unexpected death in epilepsy
Consensus was reached on all 4 items in category 5 (100%), with a strong consensus for all of them (Table 5). The panelists recommended providing information on the risk of SUDEP in DS through a specific conversation with the patient and family/caregivers. According to the experts, the pediatric/adult neurologist should lead the conversation and include prevention strategies and instructions for the management of “near-SUDEP” situations. The panelists agreed on the possibility of training caregivers on cardiopulmonary resuscitation techniques. Finally, the experts recommended carrying out an annual or biannual cardiological evaluation, based on patient risk.
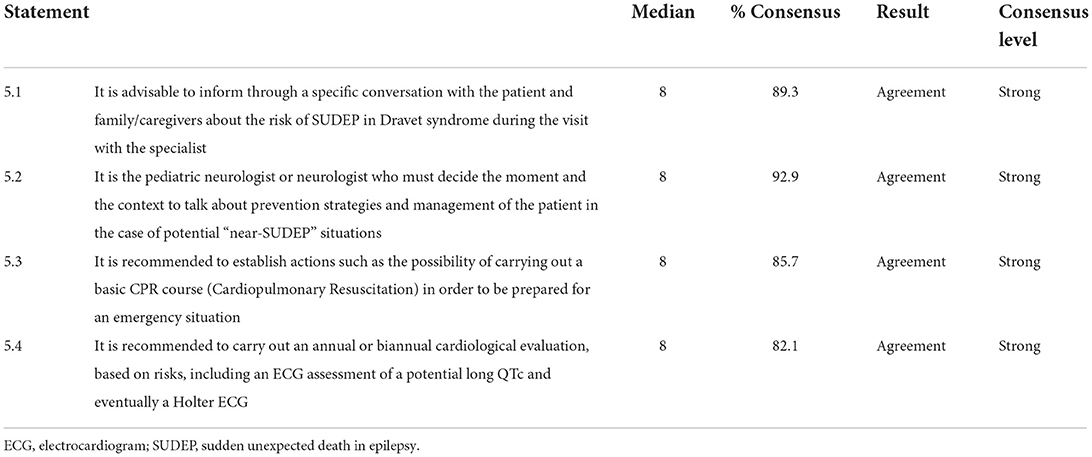
Table 5. Sudden unexpected death in epilepsy (SUDEP).
Assessment of the QoL of caregivers/relatives
The results of the category regarding the assessment of the QoL of caregivers/relatives are shown in Table 6. Although the three items reached an agreement, only one was classified as a strong consensus (33.33%), and the other two (66.66%) fell into the moderate consensus category. The experts reached a consensus agreement on the 3 statements included in the questionnaire. The panelists recommended collecting information on the impact of the disease on the QoL of caregivers and studying the dynamics between the patient, caregivers, and siblings at least every 3–6 months. The experts recommended using quantitative assessment scales for caregiver burden and QoL, such as the Zarit Burden Interview (ZBI).
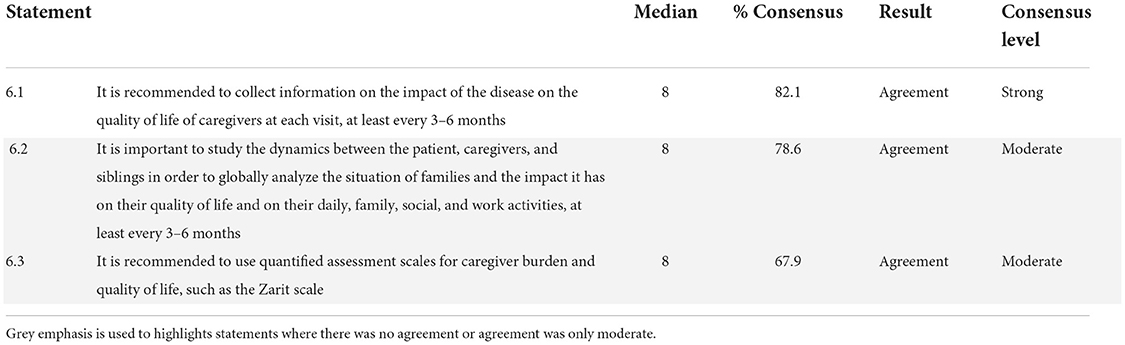
Table 6. Assessment of the quality of life of caregivers/relatives.
Discussion
To our knowledge, this is the first study to establish recommendations regarding QoL and PROs assessment and the main comorbidities in DS, using a Delphi technique and qualitative methodology, including caregivers and a multidisciplinary group of experts. Caregivers with a variety of familiar relationships and providing care to patients with a wide range of ages were included, and experts represented highly qualified professionals involved in the treatment and care of DS patients in Spain. The experts reached a consensus on recommendations for the management and follow-up of DS, focusing on the assessment of patients' and caregivers' QoL and other outcomes. The high degree of consensus reached by the panelists is noteworthy, with consensus agreement reached for 69 of the total 70 items proposed (Table 7), covering the six categories related to (1) general aspects of QoL evaluation of patients with DS, (2) neurodevelopmental evaluation in patients with DS, (3) assessment of attention and behavior in patients with DS, (4) evaluation of other comorbidities affecting QoL in patients with DS, (5) Sudden unexpected death in epilepsy (SUDEP), and (6) assessment of the QoL of caregivers/relatives. Full consensus agreement was obtained on the items included in all the categories, with the exception of category 4, where one item was considered undetermined. Fifteen out of the 70 items (21.43%) obtained a moderate consensus, indicating a certain level of controversy.

Table 7. Summary of recommendations.
Appropriate assessment of clinical and QoL parameters of patients with DS is crucial due to the importance of the symptoms and burden of the disease (14, 30, 31). In this regard, it is important to note that some treatments can negatively affect DS patients, as is the case of sodium channel blockers (SCBs) on cognition (32). Although a European clinical guideline was recently published (22), the publication focused on the treatment of DS, leaving gaps with respect to the QoL and comorbidity assessment in DS. Seizure severity, cognition, and motor and behavioral problems appear to be the primary drivers of QoL (32, 33). However, despite the available publications, clear guidelines for its routine assessment in both DS patients and caregivers are still missing (23–27). This Delphi consensus was developed to meet such needs and aimed to provide a set of recommendations for clinicians involved in the management of patients with DS.
The recommendations regarding general aspects of QoL evaluation of patients with DS indicate that experts overall consider the evaluation of the patient's QoL at each visit with the clinician to be of great importance. This opinion is supported by several publications indicating an association between clinical parameters and the QoL status of patients with DS (23, 30, 34). The experts also recommended including a wide range of parameters in the assessment of patients' QoL, considering its multidimensional nature. Although there was a consensus agreement on most of the contents of the instruments for assessing QoL, including pharmacological treatment, social, physical, and psychological aspects (35–37), a higher proportion of panelists disagreed on the assessment of the financial and home situation, support material, relationships with healthcare professionals and the healthcare system. Likewise, despite reaching a consensus agreement, several panelists disagreed on the need for questionnaires to be administered by physicians. In conclusion, the recommendations of this category are in favor of establishing the use of PROs in the clinical practice of DS. Given the infrequent use of PROs in this pathology (21), the publication of these recommendations may be a stimulus for their development and implementation.
For neurodevelopmental evaluation in patients with DS, the panelists recommended that assessments be conducted at each visit with the specialist, with varying frequencies depending on the patient's age. Given the neurodevelopmental delay in DS children (38–40), the experts considered that a frequent measurement of neurodevelopment was appropriate. Because of the multiple areas that may be affected by a neurodevelopment delay (38–40), the panelists recommended its assessment using several scales that have been previously validated in the pediatric setting (41–46), although in the case of Bayley III, Wechsler, Peabody 2, and BRIEF-2, there was less agreement on the consensus.
Regarding the assessment of attention and behavior in patients with DS, the panelists recommended evaluating the behavior of patients with DS in the educational/stimulation center at each consultation with the specialist, with a frequency of at least 3–6 months. For this statement, the percentage of consensus reached 67.9%, close to the limit of agreement, which may be due to the indicated follow-up time. Several panelists commented that a follow-up of 6 months is sufficient and more in line with the reality of clinical practice, with limited time for each patient visit. The motivation for this recommendation is that behavioral problems in DS are very common and have a tremendous impact on these patients' lives (47). Patients with DS present multiple behavioral problems and, therefore, experts recommend evaluating several parameters within this domain (47).
During the assessment of other comorbidities affecting QoL in patients with DS, a total of 13 statements of the 14 proposed obtained a consensus agreement. The only item not validated in the entire Delphi questionnaire was the one related to the use of actigraphy study for evaluating a sleep disorder. The reason for this may be the scarcity of studies on this subject conducted in the SD population (48). The remaining items on the importance of assessing sleep, as well as other comorbidities, reached consensus agreement by the experts, likely motivated by the high prevalence of other diseases in patients with DS (49–51). In this regard, the advice to assess respiratory problems may be due to the fact that seizures can increase the risk of pulmonary complications, such as aspiration pneumonia, which occurs when patients inhale food, stomach acid, or saliva into the lungs, potentially resulting in sepsis (50).
SUDEP is the leading reported cause of death reported in DS, accounting for nearly half of all premature deaths (52). The experts recommended assessing several aspects related to SUDEP in category 5. The panelists agreed that it is advisable to inform through a specific discussion with the patient and family/caregivers about the risk of SUDEP and to educate them on prevention and self-assessment strategies. Given the association between SUDEP and heart failure, periodic cardiological evaluations were recommended (52).
DS is a disease with a significant impact on patients' caregivers and relatives (53, 54). For this reason, in category 6, the experts reached a consensus to recommend collecting information regarding the impact of the disease on the QoL of caregivers and to study the dynamics between patients, caregivers, and siblings at least every 3–6 months. The ZBI was proposed as an assessment measure, presumably because it has been used in the field of epilepsy (53).
The Delphi technique is widely used in health studies as a method to obtain useful information from experts when the published body of evidence is incomplete (23, 24, 28, 55). However, the conclusions obtained with this methodology may have several limitations associated with the setting and design of this Delphi questionnaire. In the present study, the panelists were exclusively from Spain and, thus, their experience was focused on the Spanish healthcare system. Therefore, the recommendations of this study, such as those related to the frequency of assessments, may not be appropriate for other countries or regions. Likewise, this study has not assessed the impact that the implementation of these recommendations could have on the current Spanish healthcare system from the point of view of the potential need for more human and non-human resources. Clinicians with experience in other healthcare systems should assess whether each recommendation in this paper can be translated into their clinical practice, and adapt or discard those that they do not consider appropriate. Nevertheless, this study used a Delphi questionnaire developed by experts with the input from patient caregivers, which allowed for capturing controversial issues from the experts' and caregivers' perspectives. Another possible limitation of the study relates to the limited number of caregivers who participated in the focus group. Although the intention was to make it as representative and diverse as possible, it is possible that some bias could become apparent at some points, given that not all the perspectives of caregivers of DS patients may have been represented. In addition, further studies involving professionals from a wider range of disciplines involved in the care and management of patients with DS, such as social workers or therapists, may provide a more accurate view on the management of PROs in the daily life of DS patients and their families. Another limitation of the study associated to the Delphi technique is related to the consensus definition, which is not standard, and therefore, different criteria can be found in the literature (23, 29). For this reason, we deemed appropriate to define two categories of consensus, moderate and strong (29). We considered that although agreement was reached on most of the items consulted, those that fell into the moderate consensus category may have raised more controversy among the experts.
The high burden on QoL for DS patients and their caregivers, as well as the lack of concise guidelines on the evaluation of various clinical parameters, makes it necessary to issue expert recommendations to fill these gaps. The present study intended to meet these needs through an expert consensus, which may be a useful guide for clinicians involved in the routine follow-up and management of patients with DS. In particular, this study focused on PROs, which constitute a patient-centered alternative to incorporate into future clinical practice of patients with DS, as classical measures fail to cover all aspects of the disease (56). Furthermore, this study may serve as an example for the development of recommendations to assess PROs and other outcomes in other DEEs.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent from the patients/participants or patients/participants' legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
The Charlotte Project group
Adrián García-Ron, Alberto Vieco, Alfonso Amado Puentes, Helena Alarcón Martínez, Irene García Morales, Javier Aparicio Calvo, Jesús Eirís Puñal, Juan Jesús Rodriguez Uranga, Juan José Poza Aldea, Julián Lara Herguedas, Julio Ramos-Lizana, Mercè Falip Centelles, Mercedes Garcés Sánchez, Miquel Raspall Chaure, Patricia Smeyers Dura, Rafael Toledano Delgado, Rocío Calvo Medina, Salvador Ibáñez Micó, Sergio Aguilera Albesa, Víctor Soto Insuga, and Xiana Rodríguez Osorio.
Author contributions
ÁA-S has contributed to the design and conception of the study. All authors have contributed to the acquisition, analysis, interpretation of the data, have participated in the drafting and reviewing of the manuscript, and approving the submitted version.
Funding
This study was funded and promoted by Zogenix Spain, now a part of UCB.
Acknowledgments
The authors wish to thank the panel experts for their participation in the Delphi questionnaire (Annex I). We would also like to thank patients' caregivers for contributing with their perspective to this project by reviewing and commenting on the Delphi questionnaire statements (Annex II), the Dravet Syndrome Foundation (DSF) Spain for recruiting them for this study and participating in the focus group and Apoyo Dravet for participating in the focus group. The authors would like to thank Luzán 5 for their collaboration with meetings logistics and the technical assistance and i2e3 Biomedical Research Institute for providing medical writing support.
Conflict of interest
Author ÁA-S has received funding for educational and research activities from Angelini, Zogenix, UCB, PTC pharma, Blueprint genetics, GW, Eisai. Author AG-N has received advisory or research funds by Arvelle/Angelini, Bial, Biocodex, EISAI, Esteve, GW Pharma, Jazz Pharmaceuticals, PTC Therapeutics, Stoke, UCB Pharma and Zogenix. Author JA is president of the Dravet Syndrome Foundation Spain (DSF). He and/or the DSF have received grants and/or financial support from GW Pharma, Zogenix, Ovid Therapeutics, Encoded Therapeutics, Biocodex, Praxis, Health in Code, Takeda, UCB, Epygenix, Jazz Pharmaceuticals and StrideBio to help carry out some of the DSF's foundational activities or provide consulting services. Author JA honoraria have always been donated directly or indirectly to the DSF. Author RS-C has been a trial investigator for GW Pharmaceuticals, Takeda Pharmaceutical Company Ltd. Author and Zogenix and has served on advisory boards for Novartis, GW Pharmaceuticals, Biocodex and Zogenix. Author JG-P has received advisory or lecture funds by BIAL, EISAI, GW, NUTRICIA, SANOFI, UCB and ZOGENIX. VV has participated in advisory boards and symposia organized by Angellini, Bial, Eisai Inc., GW pharma, Novartis, Takeda, UCB, Zogenix. Author AM was employed by Dracaena Consulting SL. Author LA was employed by Apoyo Dravet.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.975034/full#supplementary-material
References
1. Dravet C. The core Dravet syndrome phenotype. Epilepsia. (2011) 52(Suppl. 2):3–9. doi: 10.1111/j.1528-1167.2011.02994.x
2. Villeneuve N, Laguitton V, Viellard M, Lépine A, Chabrol B, Dravet C, et al. Cognitive and adaptive evaluation of 21 consecutive patients with Dravet syndrome. Epilepsy Behav. (2014) 31:143–8. doi: 10.1016/j.yebeh.2013.11.021
3. Dravet C, Oguni H. Dravet syndrome (severe myoclonic epilepsy in infancy). Handb Clin Neurol. (2013) 111:627–33. doi: 10.1016/B978-0-444-52891-9.00065-8
4. Rosander C, Hallböök T. Dravet syndrome in Sweden: a population-based study. Dev Med Child Neurol. (2015) 57:628–33. doi: 10.1111/dmcn.12709
5. Aledo-Serrano A, Sánchez-Alcudia R, Toledano R, García-Morales I, Beltrán-Corbellini Á, del Pino I, et al. Developmental and epileptic encephalopathies after negative or inconclusive genetic testing: what is next? J Transl Genet Genomics. (2021) 5:443–55. doi: 10.20517/jtgg.2021.40
6. Sadleir LG, Mountier EI, Gill D, Davis S, Joshi C, DeVile C, et al. Not all SCN1A epileptic encephalopathies are Dravet syndrome: early profound Thr226Met phenotype. Neurology. (2017) 89:1035–42. doi: 10.1212/WNL.0000000000004331
7. Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA. SCN1A mutations and epilepsy. Hum Mutat. (2005) 25:535–42. doi: 10.1002/humu.20178
8. Claes L, Ceulemans B, Audenaert D, Smets K, Löfgren A, Del-Favero J, et al. De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum Mutat. (2003) 21:615–21. doi: 10.1002/humu.10217
9. Symonds JD, Zuberi SM, Stewart K, McLellan A, O'Regan M, MacLeod S, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. (2019) 142:2303–18. doi: 10.1093/brain/awz195
10. Martin P, Rautenstrauβ B, Abicht A, Fahrbach J, Koster S. Severe myoclonic epilepsy in infancy - adult phenotype with bradykinesia, hypomimia, and perseverative behavior: report of five cases. Mol Syndromol. (2010) 1:231–8. doi: 10.1159/000326746
11. Aledo-Serrano A, García-Morales I, Toledano R, Jiménez-Huete A, Parejo B, Anciones C, et al. Diagnostic gap in genetic epilepsies: a matter of age. Epilepsy Behav. (2020) 111:107266. doi: 10.1016/j.yebeh.2020.107266
12. Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia. (2011) 52(Suppl. 2):44–9. doi: 10.1111/j.1528-1167.2011.03001.x
13. Guzzetta F. Cognitive and behavioral characteristics of children with Dravet syndrome: an overview. Epilepsia. (2011) 52(Suppl. 2):35–8. doi: 10.1111/j.1528-1167.2011.02999.x
14. Brunklaus A. Dravet syndrome - time to consider the burden beyond the disease. Eur J Paediatr Neurol. (2019) 23:344. doi: 10.1016/j.ejpn.2019.05.006
15. Nabbout R, Auvin S, Chiron C, Irwin J, Mistry A, Bonner N, et al. Development and content validation of a preliminary core set of patient- and caregiver-relevant outcomes for inclusion in a potential composite endpoint for Dravet Syndrome. Epilepsy Behav. (2018) 78:232–42. doi: 10.1016/j.yebeh.2017.08.029
16. Gitiaux C, Chemaly N, Quijano-Roy S, Barnerias C, Desguerre I, Hully M, et al. Motor neuropathy contributes to crouching in patients with Dravet syndrome. Neurology. (2016) 87:277–81. doi: 10.1212/WNL.0000000000002859
17. Nolan K, Camfield CS, Camfield PR. Coping with a child with Dravet syndrome: insights from families. J Child Neurol. (2008) 23:690–4. doi: 10.1177/0883073808314162
18. Whittington MD, Knupp KG, Vanderveen G, Kim C, Gammaitoni A, Campbell JD. The direct and indirect costs of Dravet Syndrome. Epilepsy Behav. (2018) 80:109–13. doi: 10.1016/j.yebeh.2017.12.034
19. Cooper MS, Mcintosh A, Crompton DE, McMahon JM, Schneider A, Farrell K, et al. Mortality in Dravet syndrome. Epilepsy Res. (2016) 128:43–7. doi: 10.1016/j.eplepsyres.2016.10.006
20. Rivera SC, Kyte DG, Aiyegbusi OL, Slade AL, McMullan C, Calvert MJ. The impact of patient-reported outcome (PRO) data from clinical trials: a systematic review and critical analysis. Health Qual Life Outcomes. (2019) 17:156. doi: 10.1186/s12955-019-1220-z
21. Teneishvili M, Khachatryan A, Chandak A, Toward T. PND94 quality of life of dravet syndrome patients: a cross-sectional study in France. Value Heal. (2020) 23:S639. doi: 10.1016/j.jval.2020.08.1419
22. Cardenal-Muñoz E, Auvin S, Villanueva V, Cross JH, Zuberi SM, Lagae L, et al. Guidance on Dravet syndrome from infant to adult care: road map for treatment planning in Europe. Epilepsia Open. (2022) 7:11–26. doi: 10.1002/epi4.12569
23. Wirrell EC, Laux L, Donner E, Jette N, Knupp K, Meskis MA, et al. Optimizing the diagnosis and management of dravet syndrome: recommendations From a North American Consensus Panel. Pediatr Neurol. (2017) 68:18–34.e3. doi: 10.1016/j.pediatrneurol.2017.01.025
24. Crudgington H, Rogers M, Bray L, Carter B, Currier J, Dunkley C, et al. Core health outcomes in childhood epilepsy (CHOICE): development of a core outcome set using systematic review methods and a Delphi survey consensus. Epilepsia. (2019) 60:857–71. doi: 10.1111/epi.14735
25. Chan EKH, Edwards TC, Haywood K, Mikles SP, Newton L. Implementing patient-reported outcome measures in clinical practice: a companion guide to the ISOQOL user's guide. Qual life Res an Int J Qual life Asp Treat care Rehabil. (2019) 28:621–7. doi: 10.1007/s11136-018-2048-4
26. Berg AT, Baca CB, Loddenkemper T, Vickrey BG, Dlugos D. Priorities in pediatric epilepsy research: improving children's futures today. Neurology. (2013) 81:1166–75. doi: 10.1212/WNL.0b013e3182a55fb9
27. Aledo-Serrano A, Mingorance A. Analysis of the family impact and needs of Dravet's syndrome in Spain. Rev Neurol. (2020) 70:75–83. doi: 10.33588/rn.7003.2019310
28. Niederberger M, Spranger J. Delphi technique in health sciences: a map. Front Public Heal. (2020) 8:457. doi: 10.3389/fpubh.2020.00457
29. Wirrell EC, Hood V, Knupp KG, Meskis MA, Nabbout R, Scheffer I, et al. The international consensus on diagnosis and management of dravet syndrome. Epilepsia. (2022) 63:1761–77. doi: 10.1111/epi.17274
30. Lagae L, Brambilla I, Mingorance A, Gibson E, Battersby A. Quality of life and comorbidities associated with Dravet syndrome severity: a multinational cohort survey. Dev Med Child Neurol. (2018) 60:63–72. doi: 10.1111/dmcn.13591
31. De Liso P, Pironi V, Mastrangelo M, Battaglia D, Craiu D, Trivisano M, et al. Fatal status epilepticus in dravet syndrome. Brain Sci. (2020) 10:889. doi: 10.3390/brainsci10110889
32. de Lange IM, Gunning B, Sonsma ACM, van Gemert L, van Kempen M, Verbeek NE, et al. Influence of contraindicated medication use on cognitive outcome in Dravet syndrome and age at first afebrile seizure as a clinical predictor in SCN1A-related seizure phenotypes. Epilepsia. (2018) 59:1154–65. doi: 10.1111/epi.14191
33. Auvin S, Damera V, Martin M, Holland R, Simontacchi K, Saich A. The impact of seizure frequency on quality of life in patients with Lennox-Gastaut syndrome or Dravet syndrome. Epilepsy Behav. (2021) 123:108239. doi: 10.1016/j.yebeh.2021.108239
34. Brunklaus A, Dorris L, Zuberi SM. Comorbidities and predictors of health-related quality of life in Dravet syndrome. Epilepsia. (2011) 52:1476–82. doi: 10.1111/j.1528-1167.2011.03129.x
35. Hsiao C-J, Dymek C, Kim B, Russell B. Advancing the use of patient-reported outcomes in practice: understanding challenges, opportunities, and the potential of health information technology. Qual life Res an Int J Qual life Asp Treat care Rehabil. (2019) 28:1575–83. doi: 10.1007/s11136-019-02112-0
36. Aaronson NK. Quality of life: what is it? How should it be measured? Oncology. (1988) 2:64, 69–76.
37. Olsen JA, Misajon R. A conceptual map of health-related quality of life dimensions: key lessons for a new instrument. Qual life Res an Int J Qual life Asp Treat care Rehabil. (2020) 29:733–43. doi: 10.1007/s11136-019-02341-3
38. Battaglia D, Chieffo D, Lucibello S, Marini C, Sibilia V, Mei D, et al. Multicenter prospective longitudinal study in 34 patients with Dravet syndrome: neuropsychological development in the first six years of life. Brain Dev. (2021) 43:419–30. doi: 10.1016/j.braindev.2020.10.004
39. Chieffo D, Battaglia D, Lettori D, Del Re M, Brogna C, Dravet C, et al. Neuropsychological development in children with Dravet syndrome. Epilepsy Res. (2011) 95:86–93. doi: 10.1016/j.eplepsyres.2011.03.005
40. Acha J, Pérez A, Davidson DJ, Carreiras M. Cognitive characterization of children with Dravet syndrome: a neurodevelopmental perspective. Child Neuropsychol J Norm Abnorm Dev Child Adolesc. (2015) 21:693–715. doi: 10.1080/09297049.2014.959480
41. Palisano RJ, Rosenbaum P, Bartlett D, Livingston MH. Content validity of the expanded and revised Gross Motor Function Classification System. Dev Med Child Neurol. (2008) 50:744–50. doi: 10.1111/j.1469-8749.2008.03089.x
42. Cumming MM, Poling DV, Qiu Y, Pham AV, Daunic AP, Corbett N, et al. A validation study of the BRIEF-2 among kindergarteners and first graders at-risk for behavior problems. Assessment. (2021) 22:10731911211032288. doi: 10.1177/10731911211032289
43. Towns M, Rosenbaum P, Palisano R, Wright FV. Should the Gross Motor Function Classification System be used for children who do not have cerebral palsy? Dev Med Child Neurol. (2018) 60:147–54. doi: 10.1111/dmcn.13602
44. Na SD, Burns TG. Wechsler intelligence scale for children-V: test review. Appl Neuropsychol Child. (2016) 5:156–60. doi: 10.1080/21622965.2015.1015337
45. Spencer-Smith MM, Spittle AJ, Lee KJ, Doyle LW, Anderson PJ. Bayley-III cognitive and language scales in preterm children. Pediatrics. (2015) 135:e1258–65. doi: 10.1542/peds.2014-3039
46. Scott K, Lewis J, Pan X, Heathcock J. Parent-reported PEDI-CAT mobility and gross motor function in infants with cerebral palsy. Pediatr Phys Ther Off Publ Sect Pediatr Am Phys Ther Assoc. (2021) 33:156–61. doi: 10.1097/PEP.0000000000000801
47. Sinoo C, de Lange IM-L, Westers P, Gunning WB, Jongmans MJ, Brilstra EH. Behavior problems and health-related quality of life in Dravet syndrome. Epilepsy Behav. (2019) 90:217–27. doi: 10.1016/j.yebeh.2018.11.029
48. Myers KA, Davey MJ, Ching M, Ellis C, Grinton BE, Roten A, et al. Randomized controlled trial of melatonin for sleep disturbance in Dravet syndrome: the DREAMS study. J Clin Sleep Med. (2018) 14:1697–704. doi: 10.5664/jcsm.7376
49. Huang C-H, Hung P-L, Fan P-C, Lin K-L, Hsu T-R, Chou I-J, et al. Clinical spectrum and the comorbidities of Dravet syndrome in Taiwan and the possible molecular mechanisms. Sci Rep. (2021) 11:20242. doi: 10.1038/s41598-021-98517-4
50. Villas N, Meskis MA, Goodliffe S. Dravet syndrome: characteristics, comorbidities, and caregiver concerns. Epilepsy Behav. (2017) 74:81–6. doi: 10.1016/j.yebeh.2017.06.031
51. Licheni SH, Mcmahon JM, Schneider AL, Davey MJ, Scheffer IE. Sleep problems in Dravet syndrome: a modifiable comorbidity. Dev Med Child Neurol. (2018) 60:192–8. doi: 10.1111/dmcn.13601
52. Shmuely S, Sisodiya SM, Gunning WB, Sander JW, Thijs RD. Mortality in Dravet syndrome: a review. Epilepsy Behav. (2016) 64:69–74. doi: 10.1016/j.yebeh.2016.09.007
53. Gallop K, Lloyd AJ, Olt J, Marshall J. Impact of developmental and epileptic encephalopathies on caregivers: a literature review. Epilepsy Behav. (2021) 124:108324. doi: 10.1016/j.yebeh.2021.108324
54. Jensen MP, Brunklaus A, Dorris L, Zuberi SM, Knupp KG, Galer BS, et al. The humanistic and economic burden of Dravet syndrome on caregivers and families: implications for future research. Epilepsy Behav. (2017) 70:104–9. doi: 10.1016/j.yebeh.2017.02.003
55. Gil-Nagel A, Sanchez-Carpintero R, San Antonio V, Mistry A, Barker G, Shepherd J, et al. Ascertaining the epidemiology, patient flow and disease management for Dravet syndrome in Spain. Rev Neurol. (2019) 68:75–81. doi: 10.33588/rn.6802.2018155
Keywords: neurodevelopment, developmental and epileptic encephalopathies, epilepsy, genetic epilepsy, caregivers, patient-reported outcomes, SCN1A
Citation: Aledo-Serrano Á, Mingorance A, Villanueva V, García-Peñas JJ, Gil-Nagel A, Boronat S, Aibar J, Cámara S, Yániz MJ, Aras LM, Blanco B and Sánchez-Carpintero R (2022) The Charlotte Project: Recommendations for patient-reported outcomes and clinical parameters in Dravet syndrome through a qualitative and Delphi consensus study. Front. Neurol. 13:975034. doi: 10.3389/fneur.2022.975034
Received: 21 June 2022; Accepted: 15 August 2022;
Published: 01 September 2022.
Edited by:
Marina Trivisano, Bambino Gesù Children's Hospital (IRCCS), ItalyReviewed by:
Ganna Balagura, Giannina Gaslini Institute (IRCCS), ItalyMaurizio Elia, IRCCS Oasi Maria SS, Italy
Giorgia Giussani, Mario Negri Pharmacological Research Institute (IRCCS), Italy
Copyright © 2022 Aledo-Serrano, Mingorance, Villanueva, García-Peñas, Gil-Nagel, Boronat, Aibar, Cámara, Yániz, Aras, Blanco and Sánchez-Carpintero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ángel Aledo-Serrano, YWFsZWRvQG5ldXJvbG9naWFjbGluaWNhLmVz