 Laura J. Smith
Laura J. Smith Chiao-Yin Lee1,2
Chiao-Yin Lee1,2 Elisa Menozzi
Elisa Menozzi Anthony H. V. Schapira
Anthony H. V. Schapira- 1Department of Clinical and Movement Neurosciences, Queen Square Institute of Neurology, University College London (UCL), London, United Kingdom
- 2Aligning Science Across Parkinson's (ASAP) Collaborative Research Network, Chevy Chase, MD, United States
Variants in the GBA1 and LRRK2 genes are the most common genetic risk factors associated with Parkinson disease (PD). Both genes are associated with lysosomal and autophagic pathways, with the GBA1 gene encoding for the lysosomal enzyme, glucocerebrosidase (GCase) and the LRRK2 gene encoding for the leucine-rich repeat kinase 2 enzyme. GBA1-associated PD is characterized by earlier age at onset and more severe non-motor symptoms compared to sporadic PD. Mutations in the GBA1 gene can be stratified into severe, mild and risk variants depending on the clinical presentation of disease. Both a loss- and gain- of function hypothesis has been proposed for GBA1 variants and the functional consequences associated with each variant is often linked to mutation severity. On the other hand, LRRK2-associated PD is similar to sporadic PD, but with a more benign disease course. Mutations in the LRRK2 gene occur in several structural domains and affect phosphorylation of GTPases. Biochemical studies suggest a possible convergence of GBA1 and LRRK2 pathways, with double mutant carriers showing a milder phenotype compared to GBA1-associated PD. This review compares GBA1 and LRRK2-associated PD, and highlights possible genotype-phenotype associations for GBA1 and LRRK2 separately, based on biochemical consequences of single variants.
Introduction
Parkinson disease (PD) is the second most common neurodegenerative disorder. The disease is characterized by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the presence of intracellular proteinaceous inclusions, named Lewy bodies which are made up primarily of alpha-synuclein protein aggregates (1, 2). PD patients exhibit a classic triad of motor symptoms including bradykinesia, rigidity and resting tremor. A spectrum of non-motor symptoms has also been described, including cognitive decline, sleep disturbances, hyposmia and psychiatric symptoms (3).
Approximately 10–15% of all PD is caused by an identifiable genetic mutation (4), with large genome wide association studies (GWAS) having identified several additional genes and genetic loci important in familial and sporadic PD, many of which are associated with lysosomal and autophagic functions. Among these are the GBA1 gene (OMIM 606463), which encodes the lysosomal hydrolase enzyme glucocerebrosidase (GCase; EC 3.2.1.45), and LRRK2 (OMIM 609007) which encodes the leucine-rich repeat kinase 2 enzyme. Variants in these genes are widely recognized as the two most common genetic risk factors of PD worldwide (5–7).
In this review, we highlight the differences between GBA1 and LRRK2 variants, from both a clinical and biochemical perspective, and disentangle the complexity and heterogeneity of GBA1- and LRRK2-associated PD. We also summarize the recent findings on PD patients carrying both GBA1 and LRRK2 variants, and their particular clinical phenotype compared to single respective mutants, and possible pathomechanisms involved. Understanding the functional consequences of these variants and how they ultimately lead to specific PD phenotypes, is crucial to develop novel, gene-targeting therapies and direct patients to appropriate clinical trials.
The GBA1 gene to protein
The GBA1 gene and is located on chromosome 1 (1q21) and is made up of 11 exons and 10 introns spanning a sequence of 7.6 kb. It encodes a 60 kDa lysosomal hydrolase enzyme, glucocerebrosidase (GCase). The mature GCase peptide consists of 497 residues and is comprised of three non-continuous domains (as shown in Figure 1). The active site is located in Domain III, which is a (β/α)8 triosephosphate isomerase (TIM). Domain I consists of an antiparallel β-sheet, and Domain II resembles an immunoglobulin fold made up of 8 β-sheets (8–10). Within the mature GCase structure are three important flexible loops, which cap the active site. In an acidic environment, the conformation of loop 3 changes to allow substrates to access the active site (11, 12).
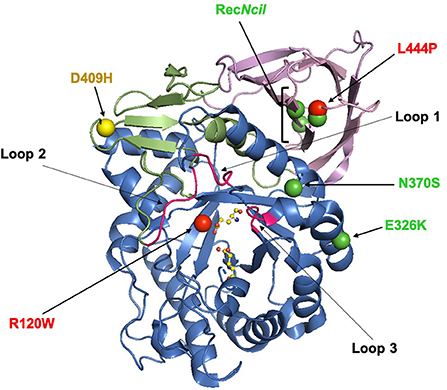
Figure 1. The X-ray structure of glucocerebrosidase (PDB code 3GXI). Domain I is shown in orange. Domain II is shown in pink. Domain III, the catalytic domain, is shown in blue and contains the active-site residues E253 and E340 which are shown as ball-and-stick models. The six significant glucocerebrosidase variants (R120W, L444P, E326K, N370S, D409H, and RecNcil) are shown with spheres. The color of the spheres corresponds with the odds ratio associated with the variant: green (<5); yellow (5–10) and red (>9) (13, 14). This figure was created using The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
GCase cleaves the sphingolipid glucosylceramide (GlcCer) into glucose and ceramide at the lysosome. Bi-allelic GBA1 mutations cause the lysosomal storage disorder Gaucher disease (GD), which presents as widespread accumulation of GlcCer and glucosylsphingosine (GlcSph) within the lysosomes of many cell types, particularly macrophages, across several tissues and organs. GCase is folded in the endoplasmic reticulum (ER) and binds to the lysosomal integral membrane protein type 2 (LIMP-2) to be trafficked to the lysosome, through the secretory pathway where it undergoes N-linked glycosylation (15–17). These post-translational modifications are thought to be imperative to the production of a fully active enzyme (18).
Common GBA1 variants
Almost 300 unique variants have been reported in the GBA1 gene, which span the entire protein (Figure 1). These include missense or non-sense mutations, insertions or deletions, complex alleles and splice junction mutations. The point mutations c.1226A>G (N370S) and c.1448T>C (L444P) are the most commonly associated with GD (19, 20). Generally the L444P variant causes a severe, neuronopathic type II or III GD, whereas the N370S variant is generally associated with non-neuronopathic type I GD (21). Some GBA1 mutations arise from recombination events between the functional GBA1 gene and a highly homologous pseudogene (GBA1P), an example of which is the complex allele RecNcil (19, 20).
Many mutations in the GBA1 gene, including the common R120W variant, occur in and around the active site, influencing its stability and affecting enzyme activity. Other common mutations including, D409H and L444P, occur far from the active site, suggesting important roles for Domains I and II (9). In the case of the L444P variant, the substitution of leucine to proline causes rigidity in the protein backbone, potentially disrupting the hydrophobicity of the domain (22) which may influence protein folding. This variant is also thought to be influenced by a lack of N-linked glycosylation and subsequent structural instability (23). To date, the crystal structure of N370S GBA1 is the only X-ray structure resolved. The N370S mutation occurs at the interface of domains II and III (9) and prevents stabilization of loop 3 at an acidic pH, impairing the ability of GCase to bind its substrate (12, 24).
GBA1 variants and Parkinson disease
Biallelic or monoallelic variants in the GBA1 gene are found in 10-15% of PD cases worldwide, and up to 30% of cases of Ashkenazi Jewish (AJ) ancestry (25). The penetrance of GBA1 variants in PD is variable. The probability of developing PD is ~5–7 and 9–12% among GD patients and 1.5–14 and 8–19% among GBA1 heterozygous carriers, by age 60 and 80, respectively (3, 26–29).
Within PD, GBA1 gene variants are stratified into complex, severe, mild and risk variants. The severity of a GBA1 mutation is based upon the phenotype it presents when homozygous in those with GD. Risk variants are referred to as such as they do not present any clinical features of GD when homozygous or compound heterozygous, but increase the risk of PD (30–32).
The type of variant differently influences the risk of PD, with higher odd ratios (OR) for complex or severe variants (e.g., L444P), followed by mild (e.g., N370S) and risk (e.g., E326K) variants (OR: 15, 4, and 2, respectively) (13, 14, 30, 33), as highlighted in Figure 1.
GBA1-Parkinson disease: Clinical picture and genotype-phenotype associations
From a pathological point of view, GBA1 associated PD (GBA1-PD) cases present with diffuse Lewy body pathology (34–39). From a clinical perspective, the most striking differences between GBA1-PD and sporadic PD cases are an earlier presentation and more severe non-motor phenotype, mainly within the cognitive, psychiatric, and olfactory domains (34, 40–44). However, this more severe phenotype is more clearly recognizable in patients carrying complex or severe variants, supporting a genotype-phenotype association (42, 45).
In terms of cognitive function, GBA1-PD patients with mild or risk variants showed slower occurrence of cognitive impairment compared to complex or severe variants (42, 45, 46), or to non-carriers (47). Psychiatric symptoms, hallucinations and hyposmia are also more common in GBA1-PD vs. non-carriers (40, 42, 44, 48), and these are more frequent in carriers of severe and complex variants compared to mild or risk variants (42, 49).
Controversy surrounds disease progression in GBA1-PD. In one study, GBA1-PD was characterized by a more aggressive progression and reduced survival rates compared to non-carriers (41), however, in another longitudinal study evaluating AJ patients, no significant effect on survival of either severe or mild variants was detected (50). When stratifying by variant type, risk variants were associated with similar mortality rates compared to non-carriers (51), with the greatest association with increased mortality in patients carrying severe variants (46).
Severe variants are generally associated with faster development of motor complications (42, 51). However, more recent longitudinal studies suggest that GBA1 status does not influence the risk of developing motor complications, even where different types of variants were considered separately (52–54).
Evaluating the biochemical consequences of GBA1 variants and their relationship with clinical features may aid in understanding of the complexity of GBA1-PD. Among markers of GBA1 dysfunction, GCase enzymatic activity is the most studied. GCase activity was found to be reduced in leucocytes (42), dried blood spots (55–57), and cerebrospinal fluid (CSF) (58) of patients with GBA1-PD compared to non-carriers. GCase activity presented a steeper decline among GBA1-PD patients according to variant severity (42). In a longitudinal analysis, increasing severity of GBA1 variants was associated with increasingly steeper decline in GCase activity, however the latter was not correlated overall with increasing severity of motor or cognitive features (56). Similarly, no genotype-phenotype correlation was found between GCase enzymatic activity and disease severity outcomes in a cross-sectional study (57), suggesting that GCase enzymatic activity might not be a reliable marker of disease severity or progression in GBA1-PD. Longitudinal studies evaluating other biochemical consequences of GBA1 dysfunction (e.g., sphingolipid metabolism), maybe in combination with GCase deficiency, and their ultimate impact on disease course, are needed.
GBA1 variants and Parkinson disease: Pathogenic mechanisms
Both loss- and gain- of function pathways are proposed to influence PD risk and onset (59, 60), and it is thought that these two hypotheses are not mutually exclusive. An overview of the pathogenic mechanisms associated with individual GBA1 mutations can be found in Table 1.
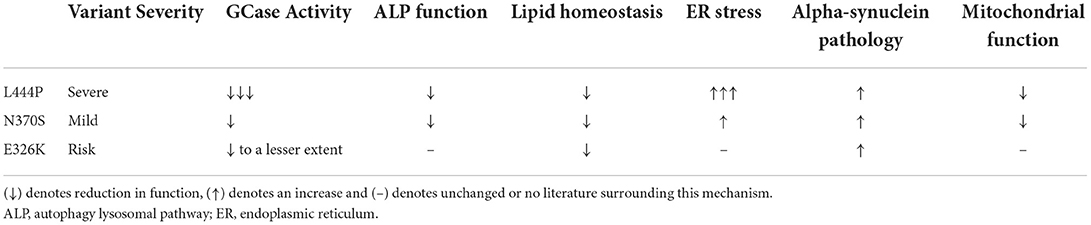
Table 1. Overview of the pathogenic mechanisms associated with the most common GBA1 variants associated with PD (L444P, N370S, and E326K).
Variants in the GBA1 gene often lead to a loss of GCase function. Analysis of GCase activity in the blood of PD patients has demonstrated that patients with severe GBA1 mutations exhibit a greater reduction in GCase activity when compared to those with mild GBA1 mutations and risk variants (56). This is supported by functional analysis of recombinant GCase protein, showing that risk variants reduce GCase activity to a lesser extent than GD-causing variants. The L444P and N370S variants reduce catalytic activity by 75–97 and 65–97%, respectively, whereas the E326K variant was associated with a 43–58% reduction (10, 61–63). The same pattern has been observed in fibroblast lines from patients harboring these mutations (64). However, this genotype-phenotype correlation is absent in one study in induced pluripotent stem cell (iPSC)-derived dopamine neurons where GCase activity was similarly reduced in L444P and N370S variants (65).
A loss of GCase activity may explain some of the downstream pathogenic mechanisms observed in models of GBA1 variants, as in human cells a GCase deficiency was associated with lysosomal dysfunction and alpha-synuclein pathology (66). In iPSC-derived midbrain dopamine neurons, the N370S variant has been associated with a significant reduction in GCase activity and protein, accompanied by impairment of the lysosome, altered distribution of GlcCer and increased extracellular release of alpha-synuclein (67). Similarly, in neural crest stem cell-derived midbrain dopamine neurons, heterozygous N370S mutations cause a loss of GCase function, impaired macroautophagy and alpha-synuclein pathology. This was rescued by the small molecular chaperone, ambroxol, suggesting these arose due to improper trafficking and activity of N370S GCase protein (68).
In cells harboring the L444P mutation, impaired lysosomal and autophagic function has been demonstrated, accompanied by a significant reduction in GCase activity and protein (69–71). However, contrary to the hypothesis that a loss of GCase function is imperative for cellular pathology, in iPSC-derived dopamine neurons from patients with homozygous and heterozygous L444P and N370S mutations, activity did not correlate with pathology. In homozygous lines, GCase activity was reduced to a greater extent than heterozygous lines, however no difference was observed in alpha-synuclein pathology and autophagic defects (65).
Improper function of the autophagy-lysosomal pathway (ALP) can lead to the aberrant metabolism of alpha-synuclein. Such has been shown in models of L444P and N370S variants (67, 68). In L444P heterozygous mice, a significant loss of GCase activity led to an abundance of alpha-synuclein inclusions in the brain and altered levels of GlcSph (72). This variant has also been associated with increased neuronal vulnerability to and accelerated spread of alpha-synuclein pathology in mice (73, 74).
It has also been proposed that there may be a genotype-phenotype correlation between severe and mild GBA1 variants and alpha-synuclein pathology. In SH-SY5Y cells, the L444P variant was associated with a greater increase in alpha-synuclein accumulation and stabilization, compared to N370S and wild-type (75). Another study in fibroblasts and SH-SY5Y cells demonstrated that both L444P and N370S fibroblasts exhibited an increase in the release of extracellular vesicles compared to control lines. However, alpha-synuclein pathology in SH-SY5Y cells was only promoted when incubated with vesicles isolated from L444P lines, and not N370S lines (76). In addition, a recent study showed that the E326K and L444P variants, despite different GCase activity, both presented comparable levels of alpha-synuclein aggregates suggesting that loss of GCase activity is not the only mechanism involved in alpha-synuclein pathology and that other mechanisms are involved in this process, especially for risk variants (64).
In addition to alpha-synuclein, the metabolism of lipids can be affected by impairment of the ALP or mitochondria, the latter of which has also been demonstrated in models of L444P (70, 71) and N370S (77) variants. Changes in the composition of glycosphingolipids has been demonstrated in models of GBA1 variants, likely due to a loss of GCase function and poor lysosomal and autophagic degradation. In mice with N370S and L444P variants, a reduction in GCase function was concurrent with an accumulation of GlcSph, which promoted alpha-synuclein aggregation (78). Similarly, in N370S iPSC-derived dopamine neurons an accumulation of GlcCer and alpha-synuclein was observed (79).
Accumulation of glycosphingolipids may be key to the pathology of L444P and N370S GBA1 variants as in dopamine neurons with these variants, reducing the levels of GlcCer/GlcSph rescued alpha-synuclein pathology (79, 80). Interestingly, in one study of L444P mice an accumulation of GlcSph alone was observed, which accelerated alpha-synuclein aggregation (72). In fibroblasts from L444P heterozygous patients, a significant increase in glycosphingolipids has been demonstrated, which correlated with decreased GCase activity. When these lipids were extracted and incubated with recombinant alpha-synuclein, an increase in the pathogenic aggregation of alpha-synuclein was observed, due to a higher content of short-chain lipids in the L444P cells (81). This may occur as lipid membrane dynamics are required for macroautophagy and chaperone mediated autophagy (CMA) (82).
In addition to glycosphingolipids, the level of fatty acids may be altered by GBA1 variants. In SH-SY5Y cells, expression of the E326K variant led to increased accumulation and formation of lipid droplets, which was accompanied by alpha-synuclein aggregation (64), suggesting alterations in the metabolism of several lipid types may be key to GBA1 pathology.
An additional pathogenic mechanism that has been proposed for GBA1-associated PD arises from the toxic gain-of-function hypothesis. As the majority of GBA1 variants are missense, a misfolded protein is often produced and retained in the endoplasmic reticulum (ER). This can activate ERAD, and lead to a deficiency in enzyme level through degradation and can activate pathway such as the unfolded protein response (UPR) and eventual ER stress. In some studies, in fibroblasts and Drosophila, activation of the UPR has been demonstrated in L444P and N370S variants (83, 84). Conversely, other studies have suggested a genotype-phenotype correlation between variant severity of UPR activation. In fibroblasts and SH-SY5Y cells, L444P has been associated with ER retention and ER stress, which was absent in N370S and E326K cells (64, 84). In another study, the severe L444P variant displayed extensive ERAD (85), suggesting that the extent of ER stress may correlate with disease severity, perhaps due to more pronounced conformational changes. However, another fibroblast study has demonstrated heterogeneity in ER retention and degradation across lines with the N370S genotype (86), weakening the genotype-phenotype correlation argument.
Overall, current evidence suggests that the mechanisms in which GBA1 variants predispose to PD are multifaceted. Different pathogenic mechanisms could explain the differences in risk and phenotypes of PD for single variants, and future studies will need to address these questions. The reasons why the majority of GD patients or heterozygous carriers do not develop PD, also remain unexplained.
GBA1-Parkinson disease: Current and future therapeutic strategies
The discovery of the GBA1 gene in PD has opened a new avenue to develop novel therapeutics for PD, with several GBA1-targeted strategies under development with the aim to enhance GCase activity [reviewed in smith et al. (87)].
Significant focus is on the development of molecular chaperones to penetrate the blood-brain-barrier (BBB) to bind and refold GCase in the ER, facilitating trafficking and rescuing enzyme activity (88). Within this class is the inhibitory, pH-dependant small molecular chaperone, ambroxol (89), which has been shown to increase GCase activity and reduce alpha-synuclein pathology in several cell and animal models (68, 90–95). Ambroxol has also demonstrated the ability to reduce UPR activation in Drosophila models of GCase deficiency (84, 96). In Type 1 GD patients, ambroxol has been shown to be safe and tolerable (ClinicalTrials.gov Identifier: NCT03950050) (97) and results from a phase II, single-centre trial, in PD patients with and without GBA1 mutations, demonstrate that ambroxol can cross the BBB and enter the CSF where it can alter GCase activity and protein level (ClinicalTrials.gov Identifier: NCT02941822) (98). Ambroxol also increased the alpha-synuclein concentration in the CSF and, importantly, improved motor function. A phase III clinical trial of ambroxol in treating PD is expected to commence in early 2023.
In addition to inhibitory chaperones, development of non-inhibitory chaperones for GCase is underway. Two compounds, NCGC758 and NCGC607, have been shown to improve GCase trafficking and rescue glycosphingolipid and alpha-synuclein accumulation in iPSC-derived dopamine neurons from GBA1-PD patients (99, 100).
Allosteric modulator small molecules, that can bind and enhance GCase activity, are also an area of interest. An example of which is LT1-291, which has been shown to cross the BBB (Trialregister.nl ID: NTR7299) (101). Pre-clinical studies have demonstrated that LT1-291 can reduce substrate accumulation (101), and this was also shown in a phase 1b placebo-controlled trial in GBA1-PD patients (NL6574). Further clinical trials are expected.
Small molecules are also being developed to modulate GCase activity through targeting other proteins. An example of this are histone deacetylase inhibitors (HDACis), which have been shown to increase GCase activity by preventing its ubiquitination and degradation (102, 103) or improving GCase folding and trafficking (104) in GD fibroblasts.
Enzyme replacement therapy (ERT) has shown great efficacy in improving the visceral symptoms of GD but fails to cross the BBB (105). Currently research is underway to improve the delivery of wild-type GCase enzyme and enhance its ability to cross the BBB. This involves ligating a peptide, usually a virus-associated protein, to the GCase enzyme (106). Denali Therapeutics have recently developed the transport-vehicle-modified recombinant GCase enzyme (ETV:GBA1) compound, using their transport vehicle platform technology which has the potential to actively transport enzymes across the BBB (107). Preclinical research is underway with this compound, but further studies are needed to investigate its efficacy in GBA1-PD patients.
Another avenue being explored to deliver wild-type GCase enzyme to the brain is gene therapy. Most commonly, the GBA1 gene is ligated into the adeno-associated virus (AAV) vector, and delivered to the brain. In mouse models of GD this method has been shown to rescue GCase activity and expression, reduce alpha-synuclein pathology and decrease glycosphingolipid accumulation (108–111). Prevail Therapeutics are currently testing their PR001A compound, which delivers the GBA1 gene using the AAV-9 vector, in phase I clinical trials (ClinicalTrials.gov Identifier: NCT04127578 and NCT04411654).
Strategies targeted to GCase to reduce the accumulation of glycosphingolipid substrates are also under development. Substrate reduction therapy (SRT), miglustat, has shown efficacy in reducing lipid accumulation in dopamine neurons from PD patients with GBA1 mutations, and can reduce alpha-synuclein pathology when coupled with GCase over-expression (79). However, miglustat cannot cross the BBB. Novel brain penetrant SRTs are therefore being developed. Sanofi's venglustat (GZ667161) had shown promise in GCase-deficient synucleinopathy mice models, able to reduce alpha-synuclein and glycosphingolipid accumulation and improve cognitive function (112). The phase I trials of venglustat demonstrated successful target engagement (ClinicalTrials.gov Identifier: NCT01674036 and NCT01710826), however the phase II trial failed to show a benefit, with patients with GBA1 mutations exhibiting a decline in motor function in PD (ClinicalTrials.gov Identifier: NCT02906020).
The LRRK2 gene to protein
The LRRK2 gene (also known as PARK8), first discovered in 2002 encodes for the leucine-rich repeat kinase 2 (LRRK2, OMIM 609007) (113). It is located on chromosome 12, consists of 51 exons and encodes a large, 288 kDa multi-domain protein containing seven domains (as illustrated in Figures 2A,B): armadillo repeat motif (ARM); ankyrin repeat (ANK); leucine-rich repeat (LRR); Ras of complex (ROC) GTPase domain; C-terminal of ROC (COR) domain; kinase (KIN) domain; WD40 domain (114). LRRK2 is thought to dimerize via the ROC-COR and WD40 domains, while the WD40 domain has also been implicated in LRRK2-mediated neurotoxicity (115–117).
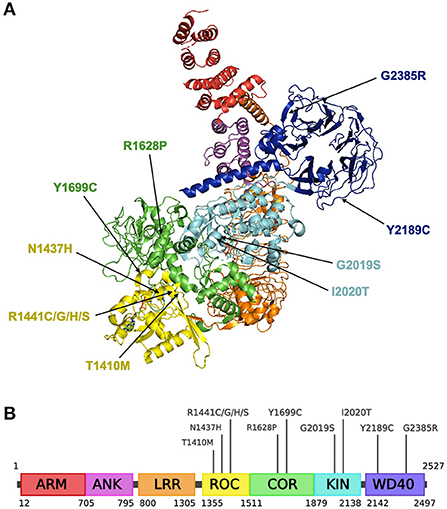
Figure 2. Structure of the LRRK2 protein and residing pathogenic variants. (A) Structural ribbon model of the LRRK2 monomer. PDB: 7LHW. This figure was created using The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. (B) Full-length LRRK2 protein. Created with BioRender.com.
LRRK2 is expressed ubiquitously in the brain, including neurons and glial cells, as well as in the kidneys, lungs, liver, heart and immune cells (118–121). The LRRK2 protein is thought to be primarily cytosolic but can also localize to a subset of organelles and inner cellular membranes, including mitochondria, ER, Golgi apparatus and microtubules (122, 123). However, the physiological roles of LRRK2 remain unclear, although it is suggested to be involved in many different processes such as adult neurogenesis, scaffolding, homeostasis of lysosome-related organelles, the innate immune response and neuroinflammation (124–126).
Common LRRK2 variants
There are several LRRK2 missense variants that have been confirmed to increase PD risk, including the most common variant G2019S, as well as N1437H, R1441C/G/H/S, Y1699C and I2020T (127, 128).
As seen in Figures 2A,B, G2019S resides in the activation loop of LRRK2's ATP binding site which regulates LRRK2 kinase activity (129). A computational prediction study suggests that G2019S may decrease the flexibility of the loop and improves the stability of the kinase domain, enabling it to remain in an active conformation for an extended period (130). This has been shown to increase phosphorylation of substrates by 2- to 3-fold (131). Another variant associated with increased PD risk, I2020T, is also located in the activation loop of the kinase domain and has been reported to significantly increase LRRK2 autophosphorylation by around 40% relative to the native enzyme (122).
Other variants that do not reside in the kinase domain may also modify LRRK2 kinase activity. The ROC domain contains motifs that are conserved amongst GTP-binding proteins, suggesting that LRRK2 is a functioning GTPase that can regulate LRRK2 kinase activity (132–134). An in vitro study showed that the R144C/G/H/S mutations located in the ROC domain, increases kinase activity while decreasing GTP hydrolysis and weakening LRRK2 dimerisation (132). N1437H in the ROC domain has been proposed to impair monomer-dimer conformational dynamics and hinder GTPase activity, permanently locking LRRK2 into a dimeric state (135). T1410M, found in the ROC domain, is a novel variant with unclear pathogenicity and may distort the tertiary structure of LRRK2 and disrupt GTP hydrolysis (136). Meanwhile, the Y1699C variant resides in the COR domain and is proposed to strengthen ROC-COR interactions, weaken ROC-COR dimerization and reduce GTPase activity (137).
Y2189C, identified in Arab-Berber populations (138), is located within the WD40 domain is presumed to have a deleterious effect for LRRK2 and induces high levels of cellular toxicity (139), however there is still controversy surrounding its pathogenicity for PD (128, 138). The G2385R and R1628P variants act as potential genetic risk factors in Chinese and Malaysian populations (140–142). G2385R is also located within the WD40 domain and causes dysfunctional synaptic vesicle trafficking (128, 143, 144), while R1628P is located in the COR domain.
LRRK2 gene variants and Parkinson disease
Worldwide, the frequency of LRRK2 G2019S is found in 1% of sporadic PD and 4% of familial PD cases (145). It is most frequently found in sporadic PD cases of north African Arabs and of AJ descent (30 and 10% of cases, respectively), whereas the variant is rarely found in Asians (only 0.1%) (145).
The penetrance of PD in subjects carrying a LRRK2 mutation is not fully elucidated and varies with age, which may explain both the high prevalence of mutations in sporadic PD cases and the detection of mutations in unaffected individuals (145). Although this finding has been repeatedly reported, the precise mutation penetrance rates vary across studies due to different populations considered and methodologies applied, and it is unclear whether distinct variants can differently impact on penetrance. Overall, cumulative risk has been estimated to be around 30–40% at age 80, with variable figures ranging from 7 and 80% (145–150). In one study considering effects of pathogenic LRRK2 mutations on penetrance, carriers of G2019S showed a lower penetrance compared to carriers of other pathogenic mutations combined, although the group of non-G2019S was relatively small (145).
LRRK2-Parkinson disease: Clinical picture and genotype-phenotype associations
LRRK2-PD patients are clinically very similar to sporadic PD. There are no differences in age at onset between LRRK2-PD patients carrying pathogenic variants vs. non-carriers (151, 152), as well as between carriers of different pathogenic mutations (G2019S vs. R1441C/G/H) (127), or carriers of risk variants vs. non-carriers (141) or vs. carriers of pathogenic variants (153). Interestingly, the male predominance seen in PD is less represented within LRRK2-PD patients (151, 152).
The motor phenotype of LRRK2-PD is that of levodopa-responsive parkinsonism, with sustained response over time, later onset of levodopa-induced dyskinesia (145, 151), and milder progression in motor symptoms over time (152) compared to non-carriers.
Although data comparing different genotypes is limited, there may be genotype-phenotype associations within LRRK2-PD, with risk variants showing a more rapid progression and G2019S a more benign course. Higher incidence of postural instability gait difficulty (PIGD) sub-type has been reported in PD patients of both AJ origin carrying G2019S (151, 152, 154), and Chinese origin carrying G2385R (155), when compared to non-carriers. Similar rates of PIGD sub-type were found in G2019S and G2385R when compared together (156). Within pathogenic variants, PD patients with G2019S showed more frequent PIGD when compared to patients carrying the R1441G variant (127). When analyzing disease course, carriers of pathogenic variants showed more sustained response to levodopa and lower motor scores when compared to carriers of risk variants (153, 156), and survival curves of AJ G2019S PD carriers were also not different from those of non-carriers (50, 157). Within pathogenic mutations, motor fluctuations were more frequently reported in carriers of p.R1441C/G/H mutation than in carriers of p.G2019S mutation (127).
From a non-motor perspective, the phenotype of all LRRK2-PD patients seems to be more benign compared to that of non-carriers. Slower cognitive decline has been observed in LRRK2-PD compared to sporadic PD or GBA1-PD (145, 158). Carriers of G2019S PD patients also showed better olfactory function, less severe mood disorders, and less frequent REM sleep behavior disorders (RBD) (159, 160) compared to non-carriers (156). In a cohort of Chinese patients, carriers of G2385R presented better cognitive performances and more severe RBD symptoms compared to non-carriers (155).
Overall, a genotype-phenotype relationship among LRRK2-PD patients might exist, with pathogenic variants showing a more benign motor disease course compared to risk variants. These observed clinical differences could reflect a lower pathogenicity for p.G2019S mutation, however additional genetic and environmental factors beyond mutational status might contribute to these different manifestations.
LRRK2 gene variants and Parkinson disease: Pathogenic mechanisms
As a major player in the ALP, pathogenic LRRK2 mutations have been shown to alter lysosomal activity, including late stage endocytosis, lysosome trafficking and synaptic vesicle endocytosis (161). In primary mouse astrocytes, G2019S, R1441C, and Y1699C reduce lysosomal capacity and increase lysosome size, and G2019S also reduces lysosomal pH, which is associated with dysfunctional lysosomal activity (162). Some reports also suggest a gain-of-function mechanism for G2019S involving ER stress and UPR, although the precise mechanisms and how they may underlie PD are poorly understood (163–165).
G2019S heterozygous and homozygous mice are reported to exhibit impaired extracellular release of dopamine and profound abnormalities of mitochondria in the striatum (166). More recent studies show that G2019S knock-in mice exhibit increased dopamine transporter levels, dopamine uptake and phosphorylation of α-synuclein from 9 months of age, while LRRK2-KO mice show slight elevation of total α-synuclein immunoreactivity at 23 months of age (167, 168). In addition, G2019S also alter glutaminergic synaptic transmission in midbrain dopaminergic neurons of 10–12 month old (middle-aged) mice which reflects aging before the onset of motor symptoms in PD (169). In astrocyte-dopaminergic neuron co-cultures from G2019S LRRK2-carrying PD patients, astrocytes accumulate α-synuclein and the neurons display shortened neurites and neurodegeneration which are not seen in co-cultures with control-patient-derived astrocytes (170). Collectively, this suggests that gain-of-function LRRK2 variants may increase the susceptibility of dopaminergic neurons to degeneration and implicates LRRK2 in α-synuclein clearance and homeostasis in PD pathology.
Although most studies focus on gain-of-function LRRK2 variants, large-scale genetic sequencing suggests that loss-of-function variants can also reduce LRRK2 protein levels in around 82% of heterozygous carriers. However, loss-of-function variants may not be strongly associated with a specific PD phenotype (171). This not only further emphasizes the link between increased kinase activity and familial PD, but also highlights the importance of additional research to elucidate both the physiological functions of LRRK2 as well as the precise mechanisms in which LRRK2 variants influence PD risk, onset and progression.
Rab proteins linked to LRRK2 in Parkinson disease
LRRK2 kinase has been shown to phosphorylate a subset of GTPases, called Rab GTPases (172). Rab proteins play important roles in vesicle trafficking, regulating the formation, transport, tethering and fusion of vesicles specific to each specific Rab, and dysfunction in Rab-mediated vesicle trafficking has been implicated in PD pathology (173). Although G2019S has been shown to increase phosphorylation of Rab proteins, in vivo assays show that other mutations such as R1441G also enhance Rab phosphorylation by up to 20-fold (172, 174). However, dysfunctional mutant T1348N LRRK2 demonstrates reduced kinase activity, suggesting the importance of GTP-binding in downstream signaling events (175).
RAB29, also known as RAB7L1, is contained within the PD-linked PARK16 locus (176, 177). RAB29 is thought to be the master regulator of LRRK2, recruiting LRRK2 to the trans-Golgi network and stimulating kinase activity. The R1441G/C and Y1699C pathogenic variants have been shown to enhance this recruitment (175), and GTP-binding is thought to be crucial for RAB29-mediated activation of LRRK2. This then triggers downstream phosphorylation of various Rab proteins, such as RAB8A/B and RAB10 (124, 178).
RAB8A/B and RAB10 have been shown to be involved in primary ciliogenesis, although direct links between LRRK2 and ciliogenesis in PD have yet to be established (124). RAB29, RAB8A, and RAB10 are all implicated in maintaining lysosome homeostasis, and Liu et al. reported that phosphorylated RAB10 may also play a role in phagocytic immune response (179), further supporting any links between LRRK2 and lysosomal dysfunction in PD (180).
LRRK2 gene variants and Parkinson disease: Current and future therapeutic strategies
There have been many recent developments in LRRK2-targeted strategies in PD, with a strong focus on small molecule LRRK2 kinase inhibitors which has been shown to trigger neuroprotective effects (181, 182).
The majority of LRRK2 kinase inhibitors are ATP-competitive, where the molecules compete with ATP for binding to the ATP-binding pocket in the kinase domain (183, 184). MLi-2 is a compound that exhibits exceptional potency and specificity both in vitro and in mouse models, where it has been shown to be well-tolerated with no adverse effects on body weight, food intake or behavior (185, 186). Although MLi-2 failed to slow or halt the progression of PD in mice and never reached clinical trials, it is an important compound for researchers to study LRRK2 function and pathobiology. PF-06685360, or PFE-360, also shows high potency, kinase selectivity and good brain permeability in rats (187). Two inhibitors [DNL-201 and DNL-151 (NCT03710707 and NCT04056689, respectively, https://clinicaltrials.gov)] are already in clinical trials (188, 189).
However, there are several challenges facing therapies targeting LRRK2 kinase. As LRRK2 protein expression is not limited to only the brain, it is crucial to assess any adverse effects on other systems in the body, such as in kidneys, lungs and immune cells. Preclinical toxicology studies show possible kidney and lung pathology as a results of various LRRK2 inhibitors (185, 187, 190, 191), and both activation and inhibition of LRRK2 kinase in immune cells have been associated with immune function (192).
Another challenge is the current lack of biomarkers and scalable assays that can measure LRRK2 activity in patients. To date, the most promising candidate biomarker is phosphorylated Rab protein (172, 193), as well as levels of auto-phosphorylated LRRK2 at Ser1292 (194). For example, phosphorylated RAB10 has been shown to be significantly increased in the brain of idiopathic PD patients (195). The development of reliable biomarkers is critical for early PD diagnosis (193), patient selection for the enrolment to clinical trials, to identify patients in which LRRK2 inhibition may be most effective and allow for personalized dose adjustments (196).
Finally, although increased LRRK2 kinase activity is present in other forms of genetic PD and especially sporadic PD (197), further research must be conducted to assess LRRK2 activity and function in these forms of PD order to assess the viability of LRRK2 inhibitors to treat all types of PD.
GBA1 and LRRK2 interactions
Although there are many clear differences between GBA1- and LRRK2- associated PD, highlighted in Table 2, there is increasing evidence suggesting a possible interaction between GBA1 and LRRK2 in PD (193). Clinical studies show that individuals carrying both the G2019S LRRK2 variant and a GBA1 variant exhibit symptoms that closely mimic G2019S-LRRK2 PD symptoms and are milder than patients carrying only a GBA1 variant. This includes slower rates of cognitive and motor decline and milder olfactory dysfunction (158). Compound variant carriers may have higher risk of developing PD, coupled with a tendency for a slightly earlier age at onset, compared with patients carrying just one variant and sporadic PD patients (158, 198–200). This suggests that the G2019S LRRK2 variant might be dominant over pathogenic GBA1 variants, although it could also depend on the varying penetrance of the two genes. In addition, it is also possible that the GBA1/LRRK2-PD patients in the study are exhibiting LRRK2-mediated PD and the GBA1 variants act as a bystander in pathology progression.
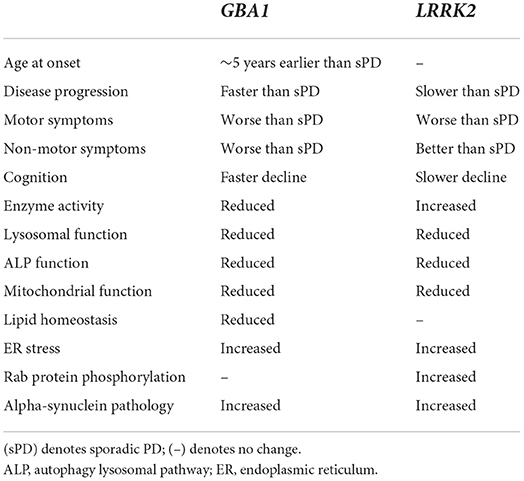
Table 2. Overview of the clinical presentation and pathological differences between GBA1- and LRRK2- associated PD.
Biochemical studies appear to support a link between LRRK2 kinase activity and GCase activity. For example, G2019S and R1441G/C variants reduce GCase activity (but not GCase protein levels) in dopaminergic neurons through increased RAB10 phosphorylation (200, 201). However, G2019S and the gain-of-function LRRK2 variant M1646T [association with PD risk is unclear (138, 202)] are reported to increase GCase activity in dried blood spots (203, 204). Therefore, the influence of LRRK2 variants on GCase activity appears to be inconsistent between the blood and dopaminergic neurons. However, there are currently a lack of studies focussing on GCase activity on LRRK2 which warrants further investigation. In addition, progression and onset are very difficult to study in cell models and compound mutant carriers are extremely rare, posing further difficulties in investigating the convergence of the two pathways.
Concluding remarks
The discovery of the GBA1 and LRRK2 mutations as the most important genetic risk factors for developing PD has led to enhanced understanding of the underlying causes of PD. Understanding the functional consequences associated with individual variants is imperative to develop highly efficacious gene-targeted therapies to halt or restore neurodegeneration. Further evaluation of GBA1 and LRRK2 variants and clinical presentation, as well as investigations into interactions between the two genes, is needed to develop biomarkers for early diagnosis and intervention and treatment of PD.
Author contributions
LJS, C-YL, EM, and AHVS contributed to drafting and editing the manuscript and have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by Aligning Science Across Parkinson's (Grant number: ASAP-000420) through the Michael J. Fox Foundation for Parkinson's Research (MJFF) and by the EU Joint Programme—Neurodegenerative Research (JPND) through the MRC grant code MR/T046007/1. For the purpose of open access, the author has applied a CC BY 4.0 public copyright license to all Author Accepted Manuscripts arising from this submission. AHVS was supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Braak H. Neuropathological staging of brain pathology in Sporadic Parkinson's disease: separating the wheat from the chaff. J Parkinsons Dis. (2017) 7(Suppl. 1):S71–85. doi: 10.3233/JPD-179001
2. Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. he wheat from the chaff. s. Nature. (1997) 388:839–40. doi: 10.1038/42166
3. Balestrino R, Schapira AHV. Parkinson disease. Eur J Neurol. (2020) 27:27–42. doi: 10.1111/ene.14108
4. Cherian A, Divya KP. Genetics of Parkinson's disease. Acta Neurol Belgica. (2020) 120:1297–305. doi: 10.1007/s13760-020-01473-5
5. Abe T, Kuwahara T. Targeting of lysosomal pathway genes for Parkinson's disease modification: insights from cellular and animal models. Front Neurol. (2021) 12:681369. doi: 10.3389/fneur.2021.681369
6. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet. (2014) 46:989–93. doi: 10.1038/ng.3043
7. Gan-Or Z, Dion PA, Rouleau GA. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy. (2015) 11:1443–57. doi: 10.1080/15548627.2015.1067364
8. Dvir H, Harel M, McCarthy AA, Toker L, Silman I, Futerman AH, et al. X-ray structure of human acid-beta-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. (2003) 4:704–9. doi: 10.1038/sj.embor.embor873
9. Smith L, Mullin S, Schapira AHV. Insights into the structural biology of Gaucher disease. Exp Neurol. (2017) 298(Pt. B):180–90. doi: 10.1016/j.expneurol.2017.09.010
10. Liou B, Kazimierczuk A, Zhang M, Scott CR, Hegde RS, Grabowski GA. Analyses of variant acid beta-glucosidases: effects of Gaucher disease mutations. J Biol Chem. (2006) 281:4242–53. doi: 10.1074/jbc.M511110200
11. Lieberman RL, D'Aquino J A, Ringe D, Petsko GA. Effects of pH and iminosugar pharmacological chaperones on lysosomal glycosidase structure and stability. Biochemistry. (2009) 48:4816–27. doi: 10.1021/bi9002265
12. Lieberman RL, Wustman BA, Huertas P, Powe AC Jr, Pine CW, et al. Structure of acid beta-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat Chem Biol. (2007) 3:101–7. doi: 10.1038/nchembio850
13. Gan-Or Z, Amshalom I, Kilarski LL, Bar-Shira A, Gana-Weisz M, Mirelman A, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology. (2015) 84:880–7. doi: 10.1212/WNL.0000000000001315
14. Zhang Y, Shu L, Sun Q, Zhou X, Pan H, Guo J, et al. Integrated genetic analysis of racial differences of common GBA variants in Parkinson's disease: a meta-analysis. Front Mol Neurosci. (2018) 11:43. doi: 10.3389/fnmol.2018.00043
15. Berg-Fussman A, Grace ME, Ioannou Y, Grabowski GA. Human acid beta-glucosidase. N-glycosylation site occupancy and the effect of glycosylation on enzymatic activity. J Biol Chem. (1993) 268:14861–6. doi: 10.1016/S0021-9258(18)82412-7
16. Bendikov-Bar I, Maor G, Horowitz M. Processing and maturation of human glucocerebrosidase. in Advances in Gaucher Disease: Basic and Clinical Perspectives. Elsevier: Amsterdam (1990). p. 140–57. doi: 10.2217/ebo.12.298
17. Reczek D, Schwake M, Schrake MC, Hughes H, Blanz J, Jin X, et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of vityntoure and stabil Cell. (2007) 131:770–83. doi: 10.1016/j.cell.2007.10.018
18. Grace ME, Grabowski GA. Human acid beta-glucosidase: glycosylation is required for catalytic activity. Biochem Biophys Res Commun. (1990) 168:771–7. doi: 10.1016/0006-291X(90)92388-G
19. Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. (2008) 29:567–83. doi: 10.1002/humu.20676
20. Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet. (2008) 372:1263–71. doi: 10.1016/S0140-6736(08)61522-6
21. Sidransky E. Gaucher disease: insights from a rare Mendelian disorder. Discov Med. (2012) 14:273–81.
22. Lieberman RL. A guided tour of the structural biology of gaucher disease: acid-beta-glucosidase and saposin C. Enzyme Res. (2011) 2011:973231. doi: 10.4061/2011/973231
23. Pol-Fachin L, Siebert M, Verli H, Saraiva-Pereira ML. Glycosylation is crucial for a proper catalytic site organization in human glucocerebrosidase. Glycoconj J. (2016) 33:237–44. doi: 10.1007/s10719-016-9661-7
24. Offman MN, Krol M, Rost B, Silman I, Sussman JL, Futerman AH. Comparison of a molecular dynamics model with the X-ray structure of the N370S acid-beta-glucosidase mutant that causes Gaucher disease. Prot Eng Design Select. (2011) 24:773–5. doi: 10.1093/protein/gzr032
25. Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med. (2004) 351:1972–7. doi: 10.1056/NEJMoa033277
26. Alcalay RN, Dinur T, Quinn T, Sakanaka K, Levy O, Waters C, et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. (2014) 71:752–7. doi: 10.1001/jamaneurol.2014.313
27. McNeill A, Duran R, Hughes DA, Mehta A, Schapira AH. A clinical and family history study of Parkinson's disease in heterozygous glucocerebrosidase mutation carriers. J Neurol Neurosurg Psychiatry. (2012) 83:853–4. doi: 10.1136/jnnp-2012-302402
28. Rana HQ, Balwani M, Bier L, Alcalay RN. Age-specific Parkinson disease risk in GBA mutation carriers: information for genetic counseling. Genet Med. (2013) 15:146–9. doi: 10.1038/gim.2012.107
29. Rosenbloom B, Balwani M, Bronstein JM, Kolodny E, Sathe S, Gwosdow AR, et al. The incidence of Parkinsonism in patients with type 1 Gaucher disease: data from the ICGG Gaucher registry. Blood Cells Mol Dis. (2011) 46:95–102. doi: 10.1016/j.bcmd.2010.10.006
30. Duran R, Mencacci NE, Angeli AV, Shoai M, Deas E, Houlden H, et al. The glucocerobrosidase E326K variant predisposes to Parkinson's disease, but does not cause Gaucher's disease. Mov Disord. (2013) 28:232–6. doi: 10.1002/mds.25248
31. Chabas A, Gort L, Diaz-Font A, Montfort M, Santamaria R, Cidras M, et al. Perinatal lethal phenotype with generalized ichthyosis in a type 2 Gaucher disease patient with the [L444P;E326K]/P182L genotype: effect of the E326K change in neonatal and classic forms of the disease. Blood Cells Mol Dis. (2005) 35:253–8. doi: 10.1016/j.bcmd.2005.04.007
32. Liou B, Grabowski GA. Is E326K glucocerebrosidase a polymorphic or pathological variant? Mol Genet Metab. (2012) 105:528–9. doi: 10.1016/j.ymgme.2011.12.002
33. Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology. (2008) 70:2277–83. doi: 10.1212/01.wnl.0000304039.11891.29
34. Neumann J, Bras J, Deas E, O'Sullivan SS, Parkkinen L, Lachmann RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain. (2009) 132(Pt. 7):1783–94. doi: 10.1093/brain/awp044
35. Westbroek W, Gustafson AM, Sidransky E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol Med. (2011) 17:485–93. doi: 10.1016/j.molmed.2011.05.003
36. Goker-Alpan O, Giasson BI, Eblan MJ, Nguyen J, Hurtig HI, Lee VMY, et al. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology. (2006) 67:908–10. doi: 10.1212/01.wnl.0000230215.41296.18
37. Choi JH, Stubblefield B, Cookson MR, Goldin E, Velayati A, Tayebi N, et al. Aggregation of α-synuclein in brain samples from subjects with glucocerebrosidase mutations. Mol Genet Metab. (2011) 104:185–8. doi: 10.1016/j.ymgme.2011.06.008
38. Tayebi N, Walker J, Stubblefield B, Orvisky E, LaMarca ME, Wong K, et al. Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab. (2003) 79:104–9. doi: 10.1016/S1096-7192(03)00071-4
39. Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. (2004) 82:192–207. doi: 10.1016/j.ymgme.2004.04.011
40. Brockmann K, Srulijes K, Hauser AK, Schulte C, Csoti I, Gasser T, et al. GBA-associated PD presents with nonmotor characteristics. Neurology. (2011) 77:276–80. doi: 10.1212/WNL.0b013e318225ab77
41. Brockmann K, Srulijes K, Pflederer S, Hauser AK, Schulte C, Maetzler W, et al. GBA-associated Parkinson's disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord. (2015) 30:407–11. doi: 10.1002/mds.26071
42. Petrucci S, Ginevrino M, Trezzi I, Monfrini E, Ricciardi L, Albanese A, et al. GBA-related Parkinson's disease: dissection of genotype-phenotype correlates in a large Italian cohort. Mov Disord. (2020) 35:2106–11. doi: 10.1002/mds.28195
43. Zhang Y, Shu L, Zhou X, Pan H, Xu Q, Guo J, et al. A meta-analysis of GBA-related clinical symptoms in Parkinson's disease. Parkinson's Dis. (2018) 2018:3136415. doi: 10.1155/2018/3136415
44. Wang C, Cai Y, Gu Z, Ma J, Zheng Z, Tang BS, et al. Clinical profiles of Parkinson's disease associated with common leucine-rich repeat kinase 2 and glucocerebrosidase genetic variants in Chinese individuals. Neurobiol Aging. (2014) 35:725 e1–6. doi: 10.1016/j.neurobiolaging.2013.08.012
45. Liu G, Boot B, Locascio JJ, Jansen IE, Winder-Rhodes S, Eberly S, et al. Specifically neuropathic Gaucher's mutations accelerate cognitive decline in Parkinson's. Ann Neurol. (2016) 80:674–85. doi: 10.1002/ana.24781
46. Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, et al. Survival and dementia in GBA-associated Parkinson's disease: the mutation matters. Ann Neurol. (2016) 80:662–73. doi: 10.1002/ana.24777
47. Simuni T, Brumm MC, Uribe L, Caspell-Garcia C, Coffey CS, Siderowf A, et al. Clinical and dopamine transporter imaging characteristics of leucine rich repeat kinase 2 (LRRK2) and glucosylceramidase beta (GBA) Parkinson's disease participants in the Parkinson's progression markers initiative: a cross-sectional study. Mov Disord. (2020) 35:833–44. doi: 10.1002/mds.27989
48. Malek N, Weil RS, Bresner C, Lawton MA, Grosset KA, Tan M, et al. Features of GBA-associated Parkinson's disease at presentation in the UK Tracking Parkinson's study. J Neurol Neurosurg Psychiatry. (2018) 89:702–9. doi: 10.1136/jnnp-2017-317348
49. Thaler A, Bregman N, Gurevich T, Shiner T, Dror Y, Zmira O, et al. Parkinson's disease phenotype is influenced by the severity of the mutations in the GBA gene. Parkinsonism Relat Disord. (2018) 55:45–9. doi: 10.1016/j.parkreldis.2018.05.009
50. Thaler A, Kozlovski T, Gurevich T, Bar-Shira A, Gana-Weisz M, Orr-Urtreger A, et al. Survival rates among Parkinson's disease patients who carry mutations in the LRRK2 and GBA genes. Mov Disord. (2018) 33:1656–60. doi: 10.1002/mds.27490
51. Stoker TB, Camacho M, Winder-Rhodes S, Liu G, Scherzer CR, Foltynie T, et al. Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson's disease. J Neurol Neurosurg Psychiatry. (2020) 91:695–702. doi: 10.1136/jnnp-2020-322857
52. Kim HJ, Mason S, Foltynie T, Winder-Rhodes S, Barker RA, Williams-Gray CH. Motor complications in Parkinson's disease: 13-year follow-up of the CamPaIGN cohort. Mov Disord. (2020) 35:185–90. doi: 10.1002/mds.27882
53. Oeda T, Umemura A, Mori Y, Tomita S, Kohsaka M, Park K, et al. Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson's disease. Neurobiol Aging. (2015) 36:3306–13. doi: 10.1016/j.neurobiolaging.2015.08.027
54. Maple-Grodem J, Paul KC, Dalen I, Ngo KJ, Wong D, Macleod AD, et al. Lack of association between GBA mutations and motor complications in European and American Parkinson's disease cohorts. J Parkinson's Dis. (2021) 11:1569–78. doi: 10.3233/JPD-212657
55. Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain. (2015) 138(Pt. 9):2648–58. doi: 10.1093/brain/awv179
56. Huh YE, Chiang MSR, Locascio JJ, Liao Z, Liu G, Choudhury K, et al. beta-Glucocerebrosidase activity in GBA-linked Parkinson disease: the type of mutation matters. Neurology. (2020) 95:e685–96. doi: 10.1212/WNL.0000000000009989
57. Omer N, Giladi N, Gurevich T, Bar-Shira A, Gana-Weisz M, Glinka T, et al. Glucocerebrosidase activity is not associated with parkinson's disease risk or severity. Mov Disord. (2022) 37:190–5. doi: 10.1002/mds.28792
58. Parnetti L, Paciotti S, Eusebi P, Dardis A, Zampieri S, Chiasserini D, et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in parkinson's disease patients. Mov Disord. (2017) 32:1423–31. doi: 10.1002/mds.27136
59. Clark LN, Ross BM, Wang Y, Mejia-Santana H, Harris J, Louis ED, et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology. (2007) 69:1270–7. doi: 10.1212/01.wnl.0000276989.17578.02
60. Nichols WC, Pankratz N, Marek DK, Pauciulo MW, Elsaesser VE, Halter CA, et al. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology. (2009) 72:310–6. doi: 10.1212/01.wnl.0000327823.81237.d1
61. Grace ME, Ashton-Prolla P, Pastores GM, Soni A, Desnick RJ. Non-pseudogene-derived complex acid beta-glucosidase mutations causing mild type 1 and severe type 2 gaucher disease. J Clin Invest. (1999) 103:817–23. doi: 10.1172/JCI5168
62. Grace ME, Newman KM, Scheinker V, Bergfussman A, Grabowski GA. Analysis of human acid beta-glucosidase by site-directed mutagenesis and heterologous expression. J Biol Chem. (1994) 269:2283–91. doi: 10.1016/S0021-9258(17)42166-1
63. Alfonso P, Rodriguez-Rey JC, Ganan A, Perez-Calvo JI, Giralt M, Giraldo P, et al. Expression and functional characterization of mutated glucocerebrosidase alleles causing Gaucher disease in Spanish patients. Blood Cells Mol Dis. (2004) 32:218–25. doi: 10.1016/j.bcmd.2003.10.010
64. Smith LJ, Bolsinger MM, Chau K-Y, Gegg ME, Schapira AHV. The GBA variant E326K is associated with alpha-synuclein aggregation and lipid droplet accumulation in human cell lines. bioRxiv [Preprint]. (2022). doi: 10.1101/2022.06.01.494130
65. Schöndorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B, et al. iPSC-derived neurons from GBA1-associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun. (2014) 5:4028. doi: 10.1038/ncomms5028
66. Bae EJ, Yang NY, Lee C, Lee HJ, Kim S, Sardi SP, et al. Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and alpha-synuclein aggregation. Exp Mol Med. (2015) 47:e153. doi: 10.1038/emm.2014.128
67. Fernandes HJR, Hartfield EM, Christian HC, Emmanoulidou E, Zheng Y, Booth H, et al. ER stress and autophagic per turbations lead to elevated extracellular alpha-synuclein in GBA-N370S LEParkinson's iPSC-derived dopamine neurons. Stem Cell Rep. (2016) 6:342–56. doi: 10.1016/j.stemcr.2016.01.013
68. Yang SY, Beavan M, Chau KY, Taanman JW, Schapira AH. A Human neural crest stem cell-derived dopaminergic neuronal model recapitulates biochemical abnormalities in GBA1 mutation carriers. Stem Cell Rep. (2017) 8:728–42. doi: 10.1016/j.stemcr.2017.01.011
69. Magalhaes J, Gegg ME, Migdalska-Richards A, Doherty MK, Whitfield PD, Schapira AHV. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum Mol Genet. (2016) 25:3432–45. doi: 10.1093/hmg/ddw185
70. Yun SP, Kim D, Kim S, Kim S, Karuppagounder SS, Kwon S-H, et al. α-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol Neurodegen. (2018) 13:1. doi: 10.1186/s13024-017-0233-5
71. Li H, Ham A, Ma TC, Kuo S-H, Kanter E, Kim D, et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy. (2019) 15:113–30. doi: 10.1080/15548627.2018.1509818
72. Mahoney-Crane CL, Viswanathan M, Russell D, Curtiss RAC, Freire J, Bobba SS, et al. Neuronopathic GBA1 L444P mutation accelerates Glucosylsphingosine levels and formation of hippocampal alpha-synuclein inclusions. bioRxiv [Preprint]. (2022). doi: 10.1101/2022.04.07.487391
73. Migdalska-Richards A, Wegrzynowicz M, Harrison IF, Verona G, Bellotti V, Spillantini MG, et al. L444P Gba1 mutation increases formation and spread of α-synuclein deposits in mice injected with mouse α-synuclein pre-formed fibrils. PLoS ONE. (2020) 15:e0238075. doi: 10.1371/journal.pone.0238075
74. Migdalska-Richards A, Wegrzynowicz M, Rusconi R, Deangeli G, Di Monte DA, Spillantini MG, et al. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain. (2017) 140:2706–21. doi: 10.1093/brain/awx221
75. Maor G, Rapaport D, Horowitz M. The effect of mutant GBA1 on accumulation and aggregation of alpha-synuclein. Hum Mol Genet. (2019) 28:1768–81. doi: 10.1093/hmg/ddz005
76. Cerri S, Ghezzi C, Ongari G, Croce S, Avenali M, Zangaglia R, et al. GBA Mutations influence the release and pathological effects of small extracellular vesicles from fibroblasts of patients with Parkinson's disease. Int J Mol Sci. (2021) 22:2215. doi: 10.3390/ijms22042215
77. Morén C, Juárez-Flores DL, Chau K-Y, Gegg M, Garrabou G, Gonzabou GSci release, et al. GBA mutation promotes early mitochondrial dysfunction in 3D neurosphere models. Aging. (2019) 11:10338–55. doi: 10.18632/aging.102460
78. Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, et al. Glucosylsphingosine promotes alpha-synuclein pathology in mutant GBA-associated Parkinson's disease. J Neurosci. (2017) 37:9617–31. doi: 10.1523/JNEUROSCI.1525-17.2017
79. Kim S, Yun SP, Lee S, Umanah GE, Bandaru VVR, Yin X, et al. GBA1 deficiency negatively affects physiological alpha-synuclein tetramers and related multimers. Proc Natl Acad Sci USA. (2018) 115:798–803. doi: 10.1073/pnas.1700465115
80. Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, et al. Reversible conformational conversion of alpha-synuclein into toxic assemblies by glucosylceramide. Neuron. (2018) 97:92–107.e10. doi: 10.1016/j.neuron.2017.12.012
81. Galvagnion C, Marlet FR, Cerri S, Schapira AHV, Blandini F, Di Monte DA. Sphingolipid changes in Parkinson L444P GBA mutation fibroblasts promote Schapira AHV, Blandini Brain. (2022) 145:1038–51. doi: 10.1093/brain/awab371
82. Gegg ME, Schapira AHV. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. (2018) 285:3591–603. doi: 10.1111/febs.14393
83. Maor G, Rencus-Lazar S, Filocamo M, Steller H, Segal D, Horowitz M. Unfolded protein response in Gaucher disease: from human to Drosophila. Orphanet J Rare Dis. (2013) 8:140. doi: 10.1186/1750-1172-8-140
84. Sanchez-Martinez A, Beavan M, Gegg ME, Chau KY, Whitworth AJ, Schapira AH. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci Rep. (2016) 6:31380. doi: 10.1038/srep31380
85. Bendikov-Bar I, Ron I, Filocamo M, Horowitz M. Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol Dis. (2011) 46:4–10. doi: 10.1016/j.bcmd.2010.10.012
86. Ron I, Horowitz M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet. (2005) 14:2387–98. doi: 10.1093/hmg/ddi240
87. Smith L, Schapira AHV. GBA variants and Parkinson disease: mechanisms and treatments. Cells. (2022) 11:1261. doi: 10.3390/cells11081261
88. Jung O, Patnaik S, Marugan J, Sidransky E, Westbroek W. Progress and potential of non-inhibitory small molecule chaperones for the treatment of Gaucher disease and its implications for Parkinson disease. Expert Rev Proteomics. (2016) 13:471–9. doi: 10.1080/14789450.2016.1174583
89. Maegawa GHB, Tropak MB, Buttner JD, Rigat BA, Fuller M, Pandit D, et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J Biol Chem. (2009) 284:23502–16. doi: 10.1074/jbc.M109.012393
90. Migdalska-Richards A, Daly L, Bezard E, Schapira AH. Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann Neurol. (2016) 80:766–75. doi: 10.1002/ana.24790
91. Migdalska-Richards A, Ko WK, Li Q, Bezard E, Schapira AH. Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse. (2017) 71:e21967. doi: 10.1002/syn.21967
92. Magalhaes J, Gegg ME, Migdalska-Richards A, Schapira AH. Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci Rep. (2018) 8:1385. doi: 10.1038/s41598-018-19479-8
93. McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain. (2014) 137(Pt. 5):1481–95. doi: 10.1093/brain/awu020
94. Ambrosi G, Ghezzi C, Zangaglia R, Levandis G, Pacchetti C, Blandini F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson's disease cells. Neurobiol Dis. (2015) 82:235–42. doi: 10.1016/j.nbd.2015.06.008
95. Kopytova AE, Rychkov GN, Nikolaev MA, Baydakova GV, Cheblokov AA, Senkevich KA, et al. Ambroxol increases glucocerebrosidase (GCase) activity and restores GCase translocation in primary patient-derived macrophages in Gaucher disease and Parkinsonism. Parkinsonism Relat Disord. (2021) 84:112–21. doi: 10.1016/j.parkreldis.2021.02.003
96. Suzuki T, Shimoda M, Ito K, Hanai S, Aizawa H, Kato T, et al. Expression of human Gaucher disease gene GBA generates neurodevelopmental defects and ER stress in Drosophila eye. PloS ONE. (2013) 8:e69147. doi: 10.1371/journal.pone.0069147
97. Zimran A, Altarescu G, Elstein D. Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells Mol Dis. (2013) 50:134–7. doi: 10.1016/j.bcmd.2012.09.006
98. Mullin S, Smith L, Lee K, D'Souza G, Woodgate P, Elflein J, et al. Ambroxol for the treatment of patients with parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol. (2020) 77:427–34. doi: 10.1001/jamaneurol.2019.4611
99. Aflaki E, Borger DK, Moaven N, Stubblefield BK, Rogers SA, Patnaik S, et al. A new glucocerebrosidase chaperone reduces alpha-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and Parkinsonism. J Neurosci. (2016) 36:7441–52. doi: 10.1523/JNEUROSCI.0636-16.2016
100. Patnaik S, Zheng W, Choi JH, Motabar O, Southall N, Westbroek W, et al. Discovery, structure-activity relationship, and biological evaluation of noninhibitory small molecule chaperones of glucocerebrosidase. J Med Chem. (2012) 55:5734–48. doi: 10.1021/jm300063b
101. den Heijer JM, Kruithof AC, van Amerongen G, de Kam ML, Thijssen E, Grievink HW, et al. A randomized single and multiple ascending dose study in healthy volunteers of LTI-291, a centrally penetrant glucocerebrosidase activator. Br J Clin Pharmacol. (2021) 87:3561–73. doi: 10.1111/bcp.14772
102. Lu J, Yang C, Chen M, Ye DY, Lonser RR, Brady RO, et al. Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease. Proc Natl Acad Sci USA. (2011) 108:21200–5. doi: 10.1073/pnas.1119181109
103. Yang C, Rahimpour S, Lu J, Pacak K, Ikejiri B, Brady RO, et al. Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc Natl Acad Sci USA. (2013) 110:966–71. doi: 10.1073/pnas.1221046110
104. Fog CK, Zago P, Malini E, Solanko LM, Peruzzo P, Bornaes C, et al. The heat shock protein amplifier arimoclomol improves refolding, maturation and lysosomal activity of glucocerebrosidase. EBioMedicine. (2018) 38:142–53. doi: 10.1016/j.ebiom.2018.11.037
105. Shemesh E, Deroma L, Bembi B, Deegan P, Hollak C, Weinreb NJ, et al. Enzyme replacement and substrate reduction therapy for Gaucher disease. Cochrane Database Syst Rev. (2015) 2015:Cd010324. doi: 10.1002/14651858.CD010324.pub2
106. Gramlich PA, Westbroek W, Feldman RA, Awad O, Mello N, Remington MP, et al. A peptide-linked recombinant glucocerebrosidase for targeted neuronal delivery: design, production, and assessment. J Biotechnol. (2016) 221:1–21. doi: 10.1016/j.jbiotec.2016.01.015
107. Ysselstein D, Young TJ, Nguyen M, Padmanabhan S, Hirst WD, Dzamko N, et al. Evaluation of strategies for measuring lysosomal glucocerebrosidase activity. Mov Disord. (2021) 36:2719–30. doi: 10.1002/mds.28815
108. Rocha EM, Smith GA, Park E, Cao H, Brown E, Hayes MA, et al. Glucocerebrosidase gene therapy prevents sidase for targeted neuronal deliveryy of gluco Neurobiol Dis. (2015) 82:495–503. doi: 10.1016/j.nbd.2015.09.009
109. Sardi SP, Clarke J, Kinnecom C, Tamsett TJ Li L, Stanek LM, et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci USA. (2011) 108:12101–6. doi: 10.1073/pnas.1108197108
110. Sardi SP, Clarke J, Viel C, Chan M, Tamsett TJ, Treleaven CM, et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci USA. (2013) 110:3537–42. doi: 10.1073/pnas.1220464110
111. Sucunza D, Rico AJ, Roda E, Collantes M, Gonzalez-Aseguinolaza G, Rodriguez-Perez AI, et al. Glucocerebrosidase gene therapy induces alpha-synuclein clearance and neuroprotection of midbrain dopaminergic neurons in mice and macaques. Int J Mol Sci. (2021) 22:4825. doi: 10.3390/ijms22094825
112. Sardi SP, Viel C, Clarke J, Treleaven CM, Richards AM, Park H, et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci USA. (2017) 114:2699–704. doi: 10.1073/pnas.1616152114
113. Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p112-q131. Ann Neurol. (2002) 51:296–301. doi: 10.1002/ana.10113
114. Myasnikov A, Zhu H, Hixson P, Xie B, Yu K, Pitre A, et al. Structural analysis of the full-length human LRRK2. Cell. (2021) 184:3519–27 e10. doi: 10.1016/j.cell.2021.05.004
115. Deniston CK, Salogiannis J, Mathea S, Snead DM, Lahiri I, Matyszewski M, et al. Structure of LRRK2 in Parkinson's disease and model for microtubule interaction. Nature. (2020) 588:344–9. doi: 10.1038/s41586-020-2673-2
116. Guaitoli G, Raimondi F, Gilsbach BK, G lsbach BK dise, Deyaert E, Renzi F, et al. Structural model of the dimeric Parkinson's protein LRRK2 reveals a compact architecture involving distant interdomain contacts. Proc Natl Acad Sci USA. (2016) 113:E4357–66. doi: 10.1073/pnas.1523708113
117. Jorgensen ND, Peng Y, Ho CC, Rideout HJ, Petrey D, Liu P, et al. The WD40 domain is required for LRRK2 neurotoxicity. PLoS ONE. (2009) 4:e8463. doi: 10.1371/journal.pone.0008463
118. Biskup S, Moore DJ, Rea A, Lorenz-Deperieux B, Coombes CE, Dawson VL, et al. Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC Neurosci. (2007) 8:102. doi: 10.1186/1471-2202-8-102
119. Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. (2010) 185:5577–85. doi: 10.4049/jimmunol.1000548
120. Maekawa T, Kubo M, Yokoyama I, Ohta E, Obata F. Age-dependent and cell-population-restricted LRRK2 expression in normal mouse spleen. Biochem Biophys Res Commun. (2010) 392:431–5. doi: 10.1016/j.bbrc.2010.01.041
121. Miklossy J, Arai T, Guo JP, Klegeris A, Yu S, McGeer EG, et al. LRRK2 expression in normal and pathologic human brain and in human cell lines. J Neuropathol Exp Neurol. (2006) 65:953–63. doi: 10.1097/01.jnen.0000235121.98052.54
122. Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, et al. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. (2006) 15:223–32. doi: 10.1093/hmg/ddi439
123. West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, et al. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. (2005) 102:16842–7. doi: 10.1073/pnas.0507360102
124. Araki M, Ito G, Tomita T. Physiological and pathological functions of LRRK2: implications from substrate proteins. Neuronal Signal. (2018) 2:NS20180005. doi: 10.1042/NS20180005
125. Harvey K, Outeiro TF. The role of LRRK2 in cell signalling. Biochem Soc Trans. (2019) 47:197–207. doi: 10.1042/BST20180464
126. Rui Q, Ni H, Li D, Gao R, Chen G. The role of LRRK2 in neurodegeneration of parkinson disease. Curr Neuropharmacol. (2018) 16:1348–57. doi: 10.2174/1570159X16666180222165418
127. Trinh J, Zeldenrust FMJ, Huang J, Kasten M, Schaake S, Petkovic S, et al. Genotype-phenotype relations for the Parkinson's disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord. (2018) 33:1857–70. doi: 10.1002/mds.27527
128. Abdalla-Carvalho CB, Santos-Reboucas CB, Guimaraes BC, Campos M, Pereira JS, de Rosso AL, et al. Genetic analysis of LRRK2 functional domains in Brazilian patients with Parkinson's disease. Eur J Neurol. (2010) 17:1479–81. doi: 10.1111/j.1468-1331.2010.03039.x
129. Liu M, Bender SA, Cuny GD, Sherman W, Glicksman M, Ray SS. Type II kinase inhibitors show an unexpected inhibition mode against Parkinson's disease-linked LRRK2 mutant G2019S. Biochemistry. (2013) 52:1725–36. doi: 10.1021/bi3012077
130. Agrahari AK, Doss GPC, Siva R, Magesh R, Zayed H. Molecular insights of the G2019S substitution in LRRK2 kinase domain associated with Parkinson's disease: a molecular dynamics simulation approach. J Theor Biol. (2019) 469:163–71. doi: 10.1016/j.jtbi.2019.03.003
131. Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, et al. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. (2007) 405:307–17. doi: 10.1042/BJ20070209
132. Guo L, Gandhi PN, Wang W, Petersen RB, Wilson-Delfosse AL, Chen SG. The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res. (2007) 313:3658–70. doi: 10.1016/j.yexcr.2007.07.007
133. Ito G, Okai T, Fujino G, Takeda K, Ichijo H, Katada T, et al. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. (2007) 46:1380–8. doi: 10.1021/bi061960m
134. West AB, Moore DJ, Choi C, Andrabi SA Li X, Dikeman D, et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. (2007) 16:223–32. doi: 10.1093/hmg/ddl471
135. Huang X, Wu C, Park Y, Long X, Hoang QQ, Liao J. The Parkinson's disease-associated mutation N1437H impairs conformational dynamics in the G domain of LRRK2. FASEB J. (2019) 33:4814–23. doi: 10.1096/fj.201802031R
136. Deng J, Lewis PA, Greggio E, Sluch E, Beilina A, Cookson MR. Structure of the ROC domain from the Parkinson's disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc Natl Acad Sci USA. (2008) 105:1499–504. doi: 10.1073/pnas.0709098105
137. Daniels V, Vancraenenbroeck R, Law BM, Greggio E, Lobbestael E, Gao F, et al. Insight into the mode of action of the LRRK2 Y1699C pathogenic mutant. J Neurochem. (2011) 116:304–15. doi: 10.1111/j.1471-4159.2010.07105.x
138. Ross OA, Soto-Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G, et al. Association of LRRK2 exonic variants with susceptibility to Parkinson's disease: a case-control study. Lancet Neurol. (2011) 10:898–908. doi: 10.1016/S1474-4422(11)70175-2
139. Refai FS, Ng SH, Tan EK. Evaluating LRRK2 genetic variants with unclear pathogenicity. Biomed Res Int. (2015) 2015:678701. doi: 10.1155/2015/678701
140. Di Fonzo A, Wu-Chou YH, Lu CS, van Doeselaar M, Simons EJ, Rohe CF, et al. A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson's disease risk in Taiwan. Neurogenetics. (2006) 7:133–8. doi: 10.1007/s10048-006-0041-5
141. Li C, Ting Z, Qin X, Ying W, Li B, Guo Qiang L, et al. The prevalence of LRRK2 Gly2385Arg variant in Chinese Han population with Parkinson's disease. Mov Disord. (2007) 22:2439–43. doi: 10.1002/mds.21763
142. Tan EK, Zhao Y, Skipper L, Tan MG, Di Fonzo A, Sun L, et al. The LRRK2 Gly2385Arg variant is associated with Parkinson's disease: genetic and functional evidence. Hum Genet. (2007) 120:857–63.
143. Carrion MDP, Marsicano S, Daniele F, Marte A, Pischedda F, Di Cairano E, et al. The LRRK2 G2385R variant is a partial loss-of-function mutation that affects synaptic vesicle trafficking through altered protein interactions. Sci Rep. (2017) 7:5377. doi: 10.1038/s41598-017-05760-9
144. Gopalai AA, Lim SY, Chua JY, Tey S, Lim TT, Mohamed Ibrahim N, et al. LRRK2 G2385R and R1628P mutations are associated with an increased risk of Parkinson's disease in the Malaysian population. Biomed Res Int. (2014) 2014:867321. doi: 10.1155/2014/867321
145. Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol. (2008) 7:583–90. doi: 10.1016/S1474-4422(08)70117-0
146. Clark LN, Wang Y, Karlins E, Saito L, Mejia-Santana H, Harris J, et al. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology. (2006) 67:1786–91. doi: 10.1212/01.wnl.0000244345.49809.36
147. Troiano AR, Elbaz A, Lohmann E, Belarbi S, Vidailhet M, Bonnet AM, et al. Low disease risk in relatives of north african lrrk2 Parkinson disease patients. Neurology. (2010) 75:1118–9. doi: 10.1212/WNL.0b013e3181f39a2e
148. Goldwurm S, Zini M, Mariani L, Tesei S, Miceli R, Sironi F, et al. Evaluation of LRRK2 G2019S penetrance: relevance for genetic counseling in Parkinson disease. Neurology. (2007) 68:1141–3. doi: 10.1212/01.wnl.0000254483.19854.ef
149. Lesage S, Ibanez P, Lohmann E, Pollak P, Tison F, Tazir M, et al. G2019S LRRK2 mutation in French and North African families with Parkinson's disease. Ann Neurol. (2005) 58:784–7. doi: 10.1002/ana.20636
150. Lee AJ, Wang Y, Alcalay RN, Mejia-Santana H, Saunders-Pullman R, Bressman S, et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov Disord. (2017) 32:1432–8. doi: 10.1002/mds.27059
151. Alcalay RN, Mirelman A, Saunders-Pullman R, Tang MX, Mejia Santana H, Raymond D, et al. Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov Disord. (2013) 28:1966–71. doi: 10.1002/mds.25647
152. Saunders-Pullman R, Mirelman A, Alcalay RN, Wang C, Ortega RA, Raymond D, et al. Progression in the LRRK2-asssociated Parkinson disease population. JAMA Neurol. (2018) 75:312–9. doi: 10.1001/jamaneurol.2017.4019
153. San Luciano M, Tanner CM, Meng C, Marras C, Goldman SM, Lang AE, et al. Nonsteroidal anti-inflammatory use and LRRK2 Parkinson's disease penetrance. Mov Disord. (2020) 35:1755–64. doi: 10.1002/mds.28189
154. Mirelman A, Heman T, Yasinovsky K, Thaler A, Gurevich T, Marder K, et al. Fall risk and gait in Parkinson's disease: the role of the LRRK2 G2019S mutation. Mov Disord. (2013) 28:1683–90. doi: 10.1002/mds.25587
155. Sun Q, Wang T, Jiang TF, Huang P, Li DH, Wang Y, et al. Effect of a leucine-rich repeat kinase 2 variant on motor and non-motor symptoms in Chinese Parkinson's disease patients. Aging Dis. (2016) 7:230–6. doi: 10.14336/AD.2015.1026
156. Marras C, Alcalay RN, Caspell-Garcia C, Coffey C, Chan P, Duda JE, et al. Motor and nonmotor heterogeneity of LRRK2-related and idiopathic Parkinson's disease. Mov Disord. (2016) 31:1192–202. doi: 10.1002/mds.26614
157. Yahalom G, Kaplan N, Vituri A, Cohen OS, Inzelberg R, Kozlova E, et al. Dyskinesias in patients with Parkinson's disease: effect of the leucine-rich repeat kinase 2 (LRRK2) G2019S mutation. Parkinsonism Relat Disord. (2012) 18:1039–41. doi: 10.1016/j.parkreldis.2012.05.014
158. Ortega RA, Wang C, Raymond D, Bryant N, Scherzer CR, Thaler A, et al. Association of Dual LRRK2 G2019S and GBA variations with Parkinson disease progression. JAMA Netw Open. (2021) 4:e215845. doi: 10.1001/jamanetworkopen.2021.5845
159. Saunders-Pullman R, Alcalay RN, Mirelman A, Wang C, Luciano MS, Ortega RA, et al. REM sleep behavior disorder, as assessed by questionnaire, in G2019S LRRK2 mutation PD and carriers. Mov Disord. (2015) 30:1834–9. doi: 10.1002/mds.26413
160. Trinh J, Amouri R, Duda JE, Morley JF, Read M, Donald A, et al. Comparative study of Parkinson's disease and leucine-rich repeat kinase 2 p.G2019S parkinsonism. Neurobiol Aging. (2014) 35:1125–31. doi: 10.1016/j.neurobiolaging.2013.11.015
161. Madureira M, Connor-Robson N, Wade-Martins R. LRRK2: autophagy and lysosomal activity. Front Neurosci. (2020) 14:498. doi: 10.3389/fnins.2020.00498
162. Henry AG, Aghamohammadzadeh S, Samaroo H, Chen Y, Mou K, Needle E, et al. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum Mol Genet. (2015) 24:6013–28. doi: 10.1093/hmg/ddv314
163. Costa CAD, Manaa WE, Duplan E, Checler F. The endoplasmic reticulum stress/unfolded protein response and their contributions to parkinson's disease physiopathology. Cells. (2020) 9:2495. doi: 10.3390/cells9112495
164. Lee JH, Han JH, Kim H, Park SM, Joe EH, Jou I. Parkinson's disease-associated LRRK2-G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes. Acta Neuropathol Commun. (2019) 7:68. doi: 10.1186/s40478-019-0716-4
165. Toyofuku T, Okamoto Y, Ishikawa T, Sasawatari S, Kumanogoh A. LRRK2 regulates endoplasmic reticulum-mitochondrial tethering through the PERK-mediated ubiquitination pathway. EMBO J. (2020) 39:e105826. doi: 10.15252/embj.2018100875
166. Yue M, Hinkle KM, Davies P, Trushina E, Fiesel FC, Christenson TA, et al. Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol Dis. (2015) 78:172–95. doi: 10.1016/j.nbd.2015.02.031
167. Domenicale C, Mercatelli D, Albanese F, Novello S, Vincenzi F, Varani K, et al. Dopamine transporter, phosphoSerine129 alpha-synuclein and alpha-synuclein levels in aged LRRK2 G2019S knock-in and knock-out mice. Biomedicines. (2022) 10:881. doi: 10.3390/biomedicines10040881
168. Longo F, Mercatelli D, Novello S, Arcuri L, Brugnoli A, Vincenzi F, et al. Age-dependent dopamine transporter dysfunction and Serine129 phospho-alpha-synuclein overload in G2019S LRRK2 mice. Acta Neuropathol Commun. (2017) 5:22. doi: 10.1186/s40478-017-0426-8
169. Skiteva O, Yao N, Sitzia G, Chergui K. LRRK2-G2019S mice display alterations in glutamatergic synaptic transmission in midbrain dopamine neurons. J Neurochem. (2022) 161:158–72. doi: 10.1111/jnc.15588
170. di Domenico A, Carola G, Calatayud C, Pons-Espinal M, Munoz JP, Richaud-Patin Y, et al. Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson's disease. Stem Cell Rep. (2019) 12:213–29. doi: 10.1016/j.stemcr.2018.12.011
171. Whiffin N, Armean IM, Kleinman A, Marshall JL, Minikel EV, Goodrich JK, et al. The effect of LRRK2 loss-of-function variants in humans. Nat Med. (2020) 26:869–77. doi: 10.1038/s41591-020-0893-5
172. Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, et al. Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. (2016) 5:e12813. doi: 10.7554/eLife.12813
173. Gao Y, Wilson GR, Stephenson SEM, Bozaoglu K, Farrer MJ, Lockhart PJ. The emerging role of Rab GTPases in the pathogenesis of Parkinson's disease. Mov Disord. (2018) 33:196–207. doi: 10.1002/mds.27270
174. Ito G, Katsemonova K, Tonelli F, Lis P, Baptista MA, Shpiro N, et al. Phos-tag analysis of Rab10 phosphorylation by LRRK2: a powerful assay for assessing kinase function and inhibitors. Biochem J. (2016) 473:2671–85. doi: 10.1042/BCJ20160557
175. Purlyte E, Dhekne HS, Sarhan AR, Gomez R, Lis P, Wightman M, et al. Rab29 activation of the Parkinson's disease-associated LRRK2 kinase. EMBO J. (2019) 38:1–18. doi: 10.15252/embj.201798099
176. Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. (2009) 41:1308–12. doi: 10.1038/ng.487
177. Tucci A, Nalls MA, Houlden H, Revesz T, Singleton AB, Wood NW, et al. Genetic variability at the PARK16 locus. Eur J Hum Genet. (2010) 18:1356–9. doi: 10.1038/ejhg.2010.125
178. Thirstrup K, Dachsel JC, Oppermann FS, Williamson DS, Smith GP, Fog K, et al. Selective LRRK2 kinase inhibition reduces phosphorylation of endogenous Rab10 and Rab12 in human peripheral mononuclear blood cells. Sci Rep. (2017) 7:10300. doi: 10.1038/s41598-017-10501-z
179. Liu Z, Xu E, Zhao HT, Cole T, West AB. LRRK2 and Rab10 coordinate macropinocytosis to mediate immunological responses in phagocytes. EMBO J. (2020) 39:e104862. doi: 10.15252/embj.2020104862
180. Eguchi T, Kuwahara T, Sakurai M, Komori T, Fujimoto T, Ito G, et al. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci USA. (2018) 115:E9115–24. doi: 10.1073/pnas.1812196115
181. West AB. Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp Neurol. (2017) 298(Pt. B):236–45. doi: 10.1016/j.expneurol.2017.07.019
182. Ho DH, Nam D, Seo M, Park SW, Seol W, Son I. LRRK2 inhibition mitigates the neuroinflammation caused by TLR2-specific alpha-synuclein and alleviates neuroinflammation-derived dopaminergic neuronal loss. Cells. (2022) 11:861. doi: 10.3390/cells11050861
183. Lu X, Smaill JB, Ding K. New promise and opportunities for allosteric kinase inhibitors. Angew Chem Int Ed Engl. (2020) 59:13764–76. doi: 10.1002/anie.201914525
184. Hatcher JM, Choi HG, Alessi DR, Gray NS. Small-molecule inhibitors of LRRK2. Adv Neurobiol. (2017) 14:241–64. doi: 10.1007/978-3-319-49969-7_13
185. Fell MJ, Mirescu C, Basu K, Cheewatrakoolpong B, DeMong DE, Ellis JM, et al. MLi-2, a potent, selective, and centrally active compound for exploring the therapeutic potential and safety of LRRK2 kinase inhibition. J Pharmacol Exp Ther. (2015) 355:397–409. doi: 10.1124/jpet.115.227587
186. Scott JD, DeMong DE, Greshock TJ, Basu K, Dai X, Harris J, et al. Discovery of a 3-(4-pyrimidinyl) indazole (MLi-2), an orally available and selective leucine-rich repeat kinase 2 (LRRK2) inhibitor that reduces brain kinase activity. J Med Chem. (2017) 60:2983–92. doi: 10.1021/acs.jmedchem.7b00045
187. Andersen MA, Wegener KM, Larsen S, Badolo L, Smith GP, Jeggo R, et al. PFE-360-induced LRRK2 inhibition induces reversible, non-adverse renal changes in rats. Toxicology. (2018) 395:15–22. doi: 10.1016/j.tox.2018.01.003
188. Ding X, Ren F. Leucine-rich repeat kinase 2 inhibitors: a patent review (2014-present). Expert Opin Ther Pat. (2020) 30:275–86. doi: 10.1080/13543776.2020.1729354
189. Jennings D, Huntwork-Rodriguez S, Henry AG, Sasaki JC, Meisner R, Diaz D, et al. Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson's disease. Sci Transl Med. (2022) 14:eabj2658. doi: 10.1126/scitranslmed.abj2658
190. Baptista MAS, Merchant K, Barrett T, Bhargava S, Bryce DK, Ellis JM, et al. LRRK2 inhibitors induce reversible changes in nonhuman primate lungs without measurable pulmonary deficits. Sci Transl Med. (2020) 12:eaav0820. doi: 10.1126/scitranslmed.aav0820
191. Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet. (2011) 20:4209–23. doi: 10.1093/hmg/ddr348
192. Zhao Y, Dzamko N. Recent developments in LRRK2-targeted therapy for Parkinson's disease. Drugs. (2019) 79:1037–51. doi: 10.1007/s40265-019-01139-4
193. Lee CY, Menozzi E, Chau KY, Schapira AHV. Glucocerebrosidase 1 and leucine-rich repeat kinase 2 in Parkinson disease and interplay between the two genes. J Neurochem. (2021) 159:826–39. doi: 10.1111/jnc.15524
194. Sheng Z, Zhang S, Bustos D, Kleinheinz T, Le Pichon CE, Dominguez SL, et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med. (2012) 4:164ra1. doi: 10.1126/scitranslmed.3004485
195. Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, et al. LRRK2 activation in idiopathic Parkinson's disease. Sci Transl Med. (2018) 10:eaar5429. doi: 10.1126/scitranslmed.aar5429
196. Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol. (2020) 16:97–107. doi: 10.1038/s41582-019-0301-2
197. Kluss JH, Mamais A, Cookson MR. LRRK2 links genetic and sporadic Parkinson's disease. Biochem Soc Trans. (2019) 47:651–61. doi: 10.1042/BST20180462
198. Omer N, Giladi N, Gurevich T, Bar-Shira A, Gana-Weisz M, Goldstein O, et al. A Possible modifying effect of the G2019S mutation in the LRRK2 gene on GBA Parkinson's disease. Mov Disord. (2020) 35:1249–53. doi: 10.1002/mds.28066
199. Yahalom G, Greenbaum L, Israeli-Korn S, Fay-Karmon T, Livneh V, Ruskey JA, et al. Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson's disease: risk estimates and genotype-phenotype correlations. Parkinsonism Relat Disord. (2019) 62:179–84. doi: 10.1016/j.parkreldis.2018.12.014
200. Ysselstein D, Nguyen M, Young TJ, Severino A, Schwake M, Merchant K, et al. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson's disease patients. Nat Commun. (2019) 10:5570. doi: 10.1038/s41467-019-13413-w
201. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. (2002) 4:160–4. doi: 10.1038/ncb748
202. Heckman MG, Soto-Ortolaza AI, Aasly JO, Abahuni N, Annesi G, Bacon JA, et al. Population-specific frequencies for LRRK2 susceptibility variants in the Genetic Epidemiology of Parkinson's Disease (GEO-PD) Consortium. Mov Disord. (2013) 28:1740–4. doi: 10.1002/mds.25600
203. Sanyal A, DeAndrade MP, Novis HS, Lin S, Chang J, Lengacher N, et al. Lysosome and inflammatory defects in GBA1-mutant astrocytes are normalized by LRRK2 inhibition. Mov Disord. (2020) 35:760–73. doi: 10.1002/mds.27994
Keywords: Parkinson's disease, glucocerebrosidase, LRRK2, GBA1, lysosome
Citation: Smith LJ, Lee C-Y, Menozzi E and Schapira AHV (2022) Genetic variations in GBA1 and LRRK2 genes: Biochemical and clinical consequences in Parkinson disease. Front. Neurol. 13:971252. doi: 10.3389/fneur.2022.971252
Received: 16 June 2022; Accepted: 25 July 2022;
Published: 12 August 2022.
Edited by:
Nikhil Panicker, Johns Hopkins Medicine, United StatesReviewed by:
Suzanne Lesage, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceMukesh Gautam, Northwestern University, United States
Copyright © 2022 Smith, Lee, Menozzi and Schapira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony H. V. Schapira, YS5zY2hhcGlyYUB1Y2wuYWMudWs=