 Frederik Teicher Kirk1*†
Frederik Teicher Kirk1*† Ditte Emilie Munk1†Jakob Ek2Lisbeth Birk Møller2†Mette Bendixen Thorup3Erik Hvid Danielsen4
Ditte Emilie Munk1†Jakob Ek2Lisbeth Birk Møller2†Mette Bendixen Thorup3Erik Hvid Danielsen4 Hendrik Vilstrup1
Hendrik Vilstrup1 Peter Ott1†Thomas Damgaard Sandahl1†
Peter Ott1†Thomas Damgaard Sandahl1†- 1Department of Hepatology and Gastroenterology, Aarhus University Hospital, ERN Rare Liver, Aarhus, Denmark
- 2Department of Genetics, Copenhagen University, Rigshospitalet, København, Denmark
- 3Department of Radiology and MR-center, Aarhus University Hospital, Aarhus, Denmark
- 4Department of Neurology, Aarhus University Hospital, Aarhus, Denmark
Background: Huppke–Brendel (HB) syndrome is an autosomal recessive disease caused by variants in the SLC33A1 gene. Since 2012, less than ten patients have been reported, none survived year six. With neurologic involvement and ceruloplasmin deficiency, it may mimic Wilson disease (WD).
Objectives and methods: We report the first adult patient with HB.
Results: The patient suffered from moderate intellectual disability, partial hearing loss, spastic ataxia, hypotonia, and unilateral tremor of parkinsonian type. At age 29, she was diagnosed with WD based on neurology, elevated 24H urinary copper, low ceruloplasmin, and pathological 65Cu test. Approximately 25 years later, genetic testing did not support WD or aceruloplasminemia. Full genome sequencing revealed two likely pathogenic variants in SLC33A1 which combined with re-evaluation of neurologic symptoms and MRI suggested the diagnosis of HB.
Conclusion: Adult patients with HB exist and may be confused with WD. Low ceruloplasmin and the absence of ATP7B variants should raise suspicion.
Introduction
Huppke–Brendel (HB) syndrome was first described in 2012 (1). Since less than ten patients have been reported, all pediatric (2). Clinical presentation is characterized by congenital cataracts, deafness, developmental delay, and death before the age of 6. MRI shows hypomyelination, cerebral atrophy with wide subarachnoid spaces, and cerebellar hypoplasia (1, 3).
Huppke–Brendel syndrome is caused by pathogenic biallelic variants in the SLC33A1 gene located on the long arm of chromosome 3. The protein product is acetyl-coenzyme A transporter 1 (AT-1), a highly conserved transmembrane protein located in the endoplasmic reticulum (ER) (4). AT-1 is involved in the acetylation of gangliosides and glycoproteins, by transporting acetyl-CoA from cytosol to the ER lumen (5). AT-1 dysfunction alters protein modification, delays Golgi-to-plasma protein trafficking, and increases in number of lysosomes (6). Thus AT-1 dysfunction may affect many proteins and processes due to its involvement in the secretory pathway (1, 7, 8).
One consequence of AT-1 dysfunction in HB is ceruloplasmin deficiency with low plasma copper and severe neurological phenotype. Biochemically, HB resembles other copper metabolism disorders such as Wilson disease (WD), Menkes disease, and aceruloplasminemia (1, 9).
Here, we present a Danish woman diagnosed in 1996 at age 29 with WD. In 2021, the diagnosis was revised, and HB was identified after the detection of two pathogenic variants in the SLC33A1 gene.
Case report
The patient was born in 1967 with developmental delays and partial hearing loss. Most of her schooling was spent in special education classes where she acquired rudimentary reading skills. In her late teenage years, she was hospitalized with anorexia nervosa and treated for depression.
1996–2017
In 1996, the patient, aged 29, was admitted to the hospital with unilateral Parkinsonian tremor, spastically ataxic gait disturbances, apraxia, hypotonia, and dysarthria. She was described as mentally and intellectually inferior. Dysphagia was not observed.
Clinical evaluation revealed immeasurably low serum-ceruloplasmin (S-Cp) (<0.05 g/L; normal range 0.20–0.45 g/L) and low serum-copper (S-Cu) (3, 12–23 μmol/L). 24H urinary copper excretion (24H U-Cu) was moderately elevated (6.5, 0.1–1.3 μmol/24H). Liver function tests, iron, hemoglobin, and ferritin were normal. Liver histology was normal as were copper levels (7.9 mg/kg dry weight). Slit-lamp examination revealed no Kayser–Fleischer rings. Genetic testing was unavailable. According to the Sternlieb criteria valid in 1996, typical neurological symptoms and very low ceruloplasmin were sufficient to diagnose WD, elevated 24H U-Cu supported the diagnosis (10). However, normal hepatic copper raised uncertainty and it was decided to use the ultimate diagnostic tool at time, assessment of plasma 65Cu after oral administration (11). As typical for WD, the test was without the secondary peak in plasma of labeled copper at 72 h seen in healthy controls. Thus, the diagnosis of WD was accepted. Revised diagnostic “Leipzig” criteria were published in 2003 (12) and did not challenge the WD diagnosis. WD was still “highly likely” with 2 points each for neurology, elevated 24H U-Cu, low ceruloplasmin, and −1 for normal liver Cu, total 5 points. The AASLD guideline in 2008 or that of EASL in 2012 (13, 14) did not change that situation.
Zinc therapy was initiated for presumed WD. Neuropsychiatric symptoms were treated with antiepileptic and antipsychotic drugs. Until 2017, she remained stable albeit psychologically vulnerable and without progression in her neurological symptoms. She lived in her own home, with her husband, but received help from healthcare workers and family to assist in daily living.
2017
In 2017 at age 49, the patient developed severe dysphagia over several months. She lost weight, her mobility decreased, and she lost bladder and bowel control. She was hospitalized for aspiration pneumonia and admitted to an intensive care unit. Dysphagia was thought to be a progression of WD; therefore, she was transferred to our facility for treatment optimization. Penicillamine 600 mg × 2 daily was added to the treatment regimen. The 24H U-Cu excretion was normal (0.57 μmol/24H; normal range 0.61–1.62) and increased to 3.79 μmol/24H on penicillamine. Since the symptoms could also be caused by an overdose of her antipsychotic and/or antiepileptic medication, these were reduced and discontinued, respectively. Following the medication adjustments, the dysphagia and mobility improved over 2 months.
2021
Lack of genetic testing and less characteristic neurology led us to revisit the diagnosis. Liver function tests and standard blood chemistry were normal. Like in 1996, both S-Cu (1.5 μmol/L; normal range: 7.9–23.6 μmol/L) and ceruloplasmin (0.03, 0.15–0.45 g/L) were low. Exchangeable copper (CuEXC) (0.81, 0.61–1.61 μmol/L) was normal and relative exchangeable copper (REC) was elevated (54%, normal range: 3–9.7%) as expected in well-treated WD, apparently confirming the diagnosis (15, 16). However, the extensive genetic analysis did not detect potentially pathogenic variants of neither the ATP7B gene responsible for WD nor the CP gene responsible for aceruloplasminemia.
Therefore, whole exome sequencing was performed [Twist Human Core Exome Library Kit (TWIST Bioscience, USA) and sequencing on the NovaSeq platform (Illumina, US)] to identify variants in genes associated with WD or WD-like diseases. The patient proved heterozygote for two variants: c.817_819 del, p.(Thr273del) and c.1331T>C, p.(Ile444Thr) in the SLC33A1 gene (NM004733.4). None of these variants have been reported as pathogenic. In the gnomAD database containing more than 125,000 healthy individuals, only the c.817_819del variant is described in 3 alleles (in heterozygous form) (https://gnomad.broadinstitute.org/). The variant c.1331T>C, p.(Ile444Thr) has not been described previously. The variant, c.817_819del leads to a single amino acid deletion. Both amino acids are very evolutionary conserved and in silico both variants are predicted to be pathogenic (CADD scores 20.3 and 26.8, respectively). Following ACMG classification, we classify both variants as variants of uncertain significance. No clinical information is available on the patient's now deceased parents; the patient has no siblings or children.
Brain MRI scans from 2017 were revisited. No signal changes related to the basal ganglia and brainstem were found as would be expected in WD. Only small unspecific T2-hyperintensities were seen in the frontal lobes. Further, global atrophy including cerebellar atrophy was indicated by an ectatic ventricular system and sulcal widening (Figure 1).
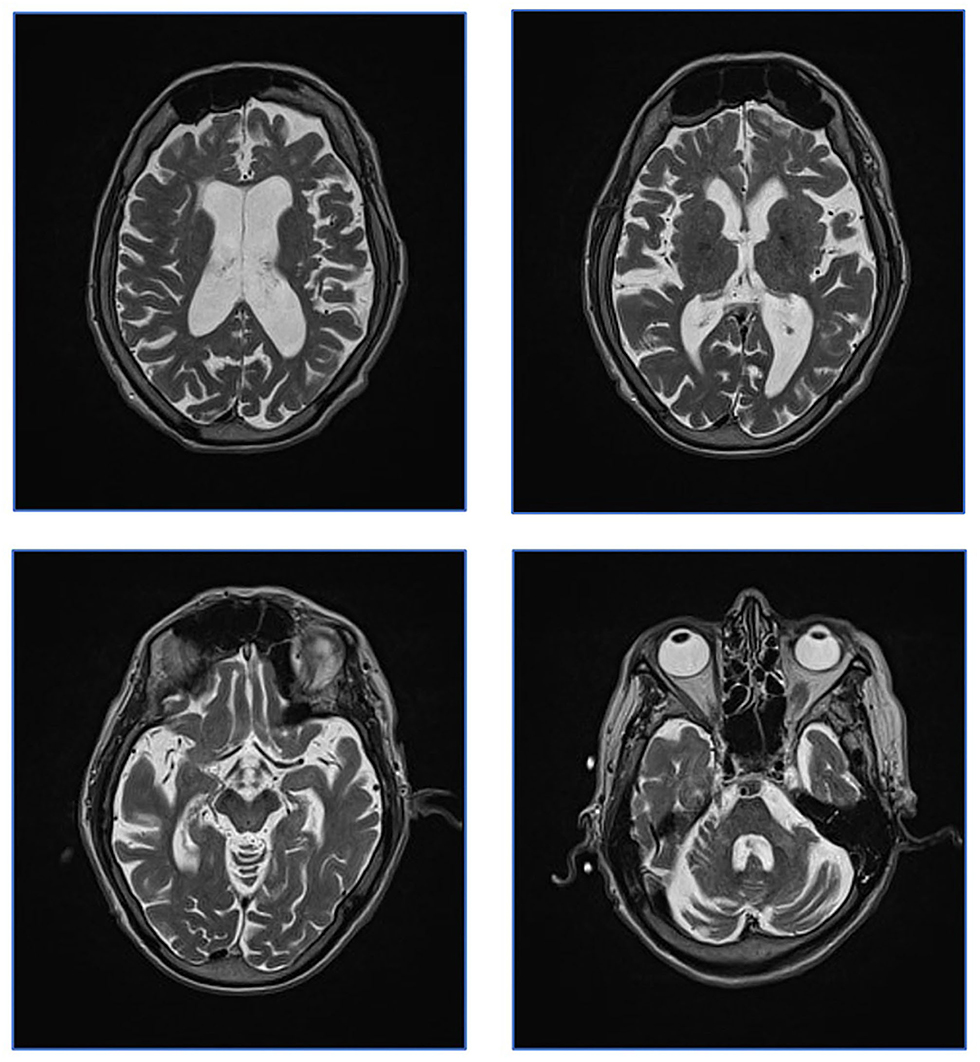
Figure 1. Patient MRI from 2017. Patient MRI. Axial T2 weighed images showing global atrophy and unspecific T2 hyperintensities. Characteristic WD changes to basal ganglia were not present.
Based on these findings, the most likely diagnosis was HB in a phenotypically less severe form compared to previous pediatric cases in the literature. Table 1 presents an overview of expected findings in HB, compared to other copper metabolic diseases. In support, the neurology included features such as intellectual disability and spastic ataxia that has been described in HB and has less characteristic of WD. Low S-Cp and S-Cu would be expected also in HB. So far, exchangeable copper assessment has not been examined in HB, but elevated REC could be explained by free cobber being normal and ceruloplasmin being extremely low. Without the ability to produce ceruloplasmin, the 65Cu test would miss the secondary 72H peak in both HB and WD. Therefore, the patient's neurological deterioration in 2017 was likely caused by large doses of neuroleptics rather than a WD manifestation.
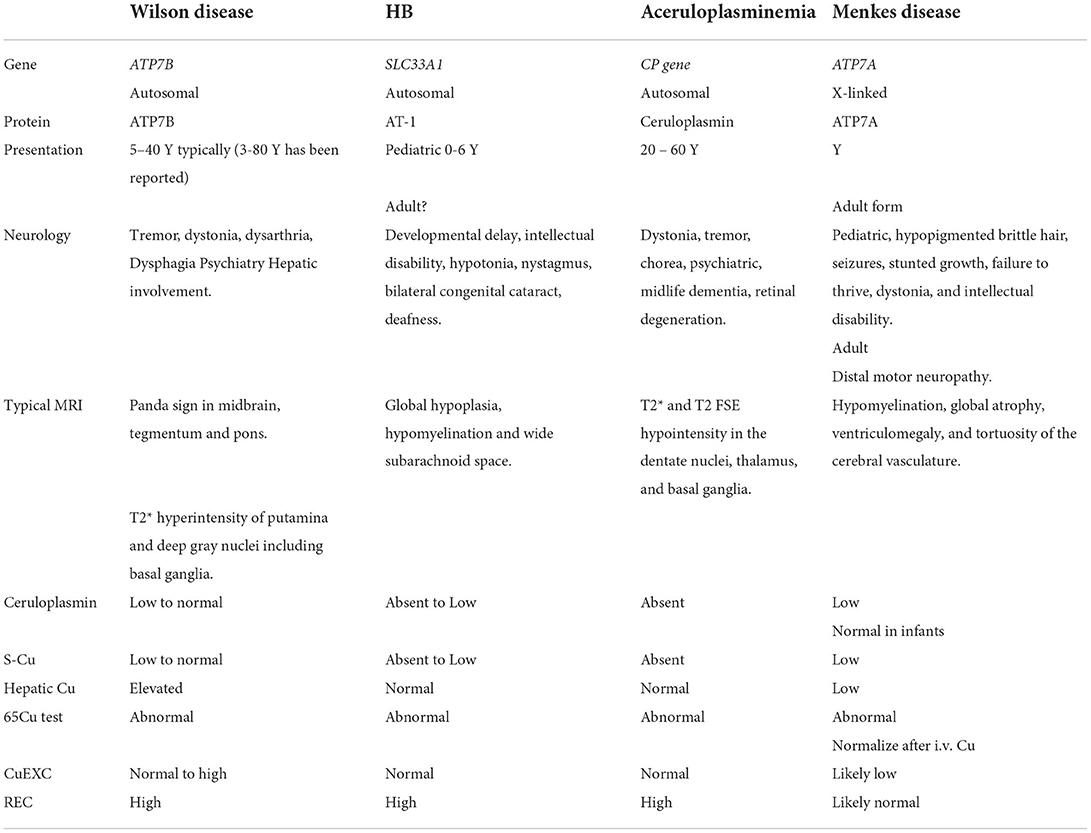
Table 1. Overview of Huppke–Brendel (HB) syndrome and other copper-related diseases.
No treatment of HB has been described. The patient has tolerated zinc therapy for 26 years without symptoms of copper deficiency and lived longer than any other patient with HB. On presentation in 1996, 24H U-Cu was moderately elevated. On this background, we decided to slowly taper the chelation therapy under close monitoring of symptoms and copper status.
Discussion
We present a 53-year-old woman with a phenotypically milder form of HB who was mistaken for WD for 25 years.
The suspicion of milder HB was raised when genetic analysis did not support the diagnosis of WD (ATP7B) or aceruloplasminemia (CP gene). Whole genome sequencing detected two variants of uncertain significance in the SLC33A1 gene indicative of HB. These variants were predicted to cause dysfunction of AT-1 potentially affecting the synthesis of a number of proteins, including the absence of ceruloplasmin.
The previous pediatric cases of HB also showed low ceruloplasmin, low P-Copper, and normal hepatic copper content (1, 3). Since nearly all copper is complexed within ceruloplasmin, low S-Cu is expected in HB because ceruloplasmin is low (2). Since ATP7B function is normal in HB, biliary excretion is maintained which explains normal hepatic copper (2). During the 65Cu test, 65Cu first disappears from plasma and then reappears after 72 h due to the formation of ceruloplasmin. The second peak is absent in WD but also in other conditions with low ceruloplasmin such as aceruloplasminemia and HB. Exchangeable or “free” copper, CuEXC, was normal, while the ratio of free vs. total copper, REC, was elevated because total copper was low (15). That would also be the case in WD.
In contrast to our patient, normal 24H U-Cu was reported in pediatric cases, but U-Cu quantification is uncertain in children (1, 3).
Disturbed copper metabolism may not be of pathophysiologic importance in HB. The AT-1 transporter is expressed in many tissues including brain and spinal cord and affects the function of a number of proteins (17). This may explain the neuropsychiatric symptoms in our patient who showed similarities with previously reported childhood cases (e.g., developmental delay, hearing loss, and ataxia). This case does not fully meet the previous reports of HB as she has no congenital cataract and survived into adulthood. MRI findings were inconclusive. Without pediatric MRI, it is impossible to determine whether the global atrophy seen in this case was acquired in adulthood; however, it may have been congenital in which case it would be described as hypoplasia. The wide subarachnoid spacing is a result of either hypoplasia or atrophy. Hypomyelination is primarily a term used in pediatrics and may refer to late myelination. While this subject did not show congenital hypomyelination nor acquired demyelination, late myelination would not be expected to be visible in the adult brain.
The previously reported homozygous and compound homozygous variants leading to exon skipping and/or premature termination of translation or alternatively mislocalization of the SLC33A1 protein variant might be very destructive for the protein product (1, 18). While these were associated with neurologic disease in childhood, a publication reported that missense mutation in SLC33A1 (p. S113R) leads to dominantly inherited spastic paraplegia (19). The presently identified variants: an in-frame deletion of a single amino acid combined with and a missense variant in the SLC33A1 gene, might be less destructive for the protein and explain this milder clinical presentation of the HB. Thus, the present case serves to expand the phenotypic spectrum for the sparse number of patients with SLC33A1 variants (1, 3, 19).
Because of the original WD diagnosis, the patient received zinc therapy for many years. Whether this affected her disease course is unclear, but she developed no signs of copper deficiency. Her life-threatening deterioration in 2017 was likely caused by an overdose of antipsychotics.
This case serves to expand the clinical picture of HB and highlights how adult HB may easily be mistaken for WD as well as the need for more specific diagnostic tools. Furthermore, this case serves as a reminder that patients may fulfill the Leipzig criteria without having WD. Thus, these patients may be found in the populations treated for WD. The newly described 64Cu PET/CT method may be useful for secure diagnosis but remains experimental and was not performed on this patient (20).
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the patient for the publication of any potentially identifiable images or data included in this article.
Author contributions
FK wrote the manuscript and acquired patient data. JE and LB aquired genetic data. MB analyzed patient MRI. EH analyzed patient neurology. DM, JE, LB, MB, EH, and TD assisted in writing the manuscript. HV, PO, and TD helped form the project and gave inputs to the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Huppke P, Brendel C, Kalscheuer V, Korenke GC, Marquardt I, Freisinger P, et al. Mutations in SLC33A1 cause a lethal autosomal-recessive disorder with congenital cataracts, hearing loss, and low serum copper and ceruloplasmin. Am J Hum Genet. (2012) 90:61–8. doi: 10.1016/j.ajhg.2011.11.030
2. Bindu PS, Chiplunkar S, Vandana VP, Nagappa M, Govindaraj P, Taly A. Huppke-Brendel Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al, editors. GeneReviews(®). Seattle (WA): University of Washington, Seattle (1993).
3. Chiplunkar S, Bindu PS, Nagappa M, Bineesh C, Govindaraj P, Gayathri N, et al. Huppke-Brendel syndrome in a seven months old boy with a novel 2-bp deletion in SLC33A1. Metab Brain Dis. (2016) 31:1195–8. doi: 10.1007/s11011-016-9854-6
4. Hirabayashi Y, Nomura KH, Nomura K. The acetyl-CoA transporter family SLC33. Mol Aspects Med. (2013) 34:586–9. doi: 10.1016/j.mam.2012.05.009
5. Farrugia MA, Puglielli L. Nε-lysine acetylation in the endoplasmic reticulum—a novel cellular mechanism that regulates proteostasis and autophagy. J Cell Sci. (2018) 131:221747. doi: 10.1242/jcs.221747
6. Dieterich IA, Cui Y, Braun MM, Lawton AJ, Robinson NH, Peotter JL, et al. Acetyl-CoA flux from the cytosol to the ER regulates engagement and quality of the secretory pathway. Sci Rep. (2021) 11:2013. doi: 10.1038/s41598-021-81447-6
7. Mao F, Li Z, Zhao B, Lin P, Liu P, Zhai M, et al. Identification and functional analysis of a SLC33A1: c. 339T>G (pSer113Arg) variant in the original SPG42 family. Hum Mutat. (2015) 36:240–9. doi: 10.1002/humu.22732
8. Liu P, Jiang B, Ma J, Lin P, Zhang Y, Shao C, et al. S113R mutation in SLC33A1 leads to neurodegeneration and augmented BMP signaling in a mouse model. Dis Model Mech. (2017) 10:53–62. doi: 10.1242/dmm.026880
9. Bandmann O, Weiss KH, Kaler SG. Wilson's disease and other neurological copper disorders. Lancet Neurol. (2015) 14:103–13. doi: 10.1016/S1474-4422(14)70190-5
10. Sternlieb I. Perspectives on Wilson's disease. Hepatology. (1990) 12:1234–9. doi: 10.1002/hep.1840120526
11. Lyon TD, Fell GS, Gaffney D, McGaw BA, Russell RI, Park RH, et al. Use of a stable copper isotope (65Cu) in the differential diagnosis of Wilson's disease. Clin Sci (Lond). (1995) 88:727–32. doi: 10.1042/cs0880727
12. Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. (2003) 23:139–42. doi: 10.1034/j.1600-0676.2003.00824.x
13. Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology. (2008) 47:2089–111. doi: 10.1002/hep.22261
14. European Association for the Study of the L. EASL Clinical Practice Guidelines: Wilson's disease. J Hepatol. (2012) 56:671–85. doi: 10.1016/j.jhep.2011.11.007
15. Woimant F, Djebrani-Oussedik N, Poujois A. New tools for Wilson's disease diagnosis: exchangeable copper fraction. Ann Transl Med. (2019) 7:S70. doi: 10.21037/atm.2019.03.02
16. Guillaud O, Brunet AS, Mallet I, Dumortier J, Pelosse M, Heissat S, et al. Relative exchangeable copper: a valuable tool for the diagnosis of Wilson disease. Liver Int. (2018) 38:350–7. doi: 10.1111/liv.13520
17. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. (2009) 325:834–40. doi: 10.1126/science.1175371
18. Horváth R, Freisinger P, Rubio R, Merl T, Bax R, Mayr JA, et al. Congenital cataract, muscular hypotonia, developmental delay and sensorineural hearing loss associated with a defect in copper metabolism. J Inherit Metab Dis. (2005) 28:479–92. doi: 10.1007/s10545-005-0479-x
19. Lin P, Li J, Liu Q, Mao F, Li J, Qiu R, et al. A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42). Am J Hum Genet. (2008) 83:752–9. doi: 10.1016/j.ajhg.2008.11.003
Keywords: Wilson disease, copper, neurology, SLC33A1, rare disease, Huppke-Brendel syndrome, case report
Citation: Kirk FT, Munk DE, Ek J, Birk Møller L, Bendixen Thorup M, Hvid Danielsen E, Vilstrup H, Ott P and Damgaard Sandahl T (2022) Case report: Huppke–Brendel syndrome in an adult, mistaken for and treated as Wilson disease for 25 years. Front. Neurol. 13:957794. doi: 10.3389/fneur.2022.957794
Received: 31 May 2022; Accepted: 20 July 2022;
Published: 01 September 2022.
Edited by:
Félix Javier Jiménez-Jiménez, Hospital Universitario del Sureste, SpainReviewed by:
Oliver Bandmann, The University of Sheffield, United KingdomPingting Liu, Stanford University, United States
Copyright © 2022 Kirk, Munk, Ek, Birk Møller, Bendixen Thorup, Hvid Danielsen, Vilstrup, Ott and Damgaard Sandahl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frederik Teicher Kirk, ZnJraXJrJiN4MDAwNDA7cm0uZGs=
†ORCID: Frederik Teicher Kirk orcid.org/0000-0002-3058-9042
Ditte Emilie Munk orcid.org/0000-0002-8077-2806
Lisbeth Birk Møller orcid.org/0000-0002-9524-4301
Peter Ott orcid.org/0000-0002-3088-1983
Thomas Damgaard Sandahl orcid.org/0000-0001-9807-6852