
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 17 October 2022
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.951054
This article is part of the Research Topic Improving our Understanding of the Management and Pathogenesis of Rare and Neglected Tumors of the Central and Peripheral Nervous System View all 23 articles
 Yaheng Li1,2
Yaheng Li1,2 Xiaohong Xin3,4
Xiaohong Xin3,4 Wenzhu Song5Xuan Zhang6Shengli Chen6
Wenzhu Song5Xuan Zhang6Shengli Chen6 Qian Wang1,2Aizhong Li1,2
Qian Wang1,2Aizhong Li1,2 Yafeng Li1,2,3,4*
Yafeng Li1,2,3,4*Objective: To analyze the clinical manifestations and imaging features of a hospitalized patient with intermittent headache who was finally diagnosed with von Hippel–Lindau (VHL) syndrome and to perform whole-exon gene detection to improve the understanding of the diagnosis and treatment strategies of the disease.
Methods: A case of suspected VHL syndrome in Shanxi Provincial People's Hospital was analyzed. Proband DNA was also extracted for whole exome sequencing and screened for causative mutation sites, which were validated by Sanger sequencing. The literature about VHL gene mutations in Chinese patients in the past 10 years were also reviewed.
Results: There is a heterozygous mutation site c.499C > G on the VHL gene on the short arm of chromosome 3 of the patient, which is a missense mutation. The mutation results in the substitution of arginine with glycine at amino acid 167 of the encoded protein, which may be primarily responsible for the disease in the patient with VHL syndrome. However, the mutation did not occur in other family members.
Conclusion: Early recognition and treatment of VHL syndrome can be available with genetic testing technology. Strengthening the understanding of this complex genetic disease and improving the diagnostic rate of VHL syndrome are helpful for the precise treatment of patients with this disease, which may help prolong the survival time of patients to a certain extent and improve their quality of life.
Von Hippel–Lindau (VHL) syndrome is a rare clinical familial autosomal dominant tumor syndrome involving multiple body systems. Characteristic tumors in VHL syndrome include central nervous system hemangioblastomas (CNS HBs), renal cell carcinomas (RCCs), pheochromocytomas (PHEOs), and pancreatic cystadenomas. The most common tumors are retinal or CNS HBs (60–80%) and VHL syndrome-related renal lesions, which may range from simple cysts to multiple and renal cell carcinomas (RCC) (24–45%)(1). VHL syndrome-associated tumors frequently lose the function of the wild-type VHL allele in the process, which is known as loss of heterozygosity (LOH). According to statistics, about 1/36,000–53,000 people have VHL gene mutations (2). The majority of VHL syndrome affected have a positive family history, whereas sporadic cases of VHL syndrome with possible de novo mutations are rare (about 20%) and may occur during germ cell formation or early embryogenesis (3, 4). The patient with a mild clinical phenotype may be considered a sporadic case in this setting. Now, we present the genomic findings in a 50-year-old man with multiple CNS HBs, RCCs, and pancreatic neuroendocrine tumors (PNETs) diagnosed as suspected VHL syndrome without a clear positive family history. It was finally confirmed that the patient with VHL syndrome had a mutation site on the pathogenic gene VHL, which may be the cause of the disease.
A 50-year-old man presented with a 3-month history of frequent headaches associated with vomiting, nausea, and dizziness was admitted to the hospital for intermittent headache for more than half a month, aggravated with dizziness, nausea, and vomiting for 3 days. The patient had normal diet and sleep, normal urine and defecation, no significant weight loss, and no other discomforts.
The patient was generally in good condition. The systemic superficial lymph nodes were not palpable and enlarged, and the cardiopulmonary abdominal examination showed no abnormality. The patient had bilateral pupils of equal size and round, 2.5 cm in diameter, and sensitivity to light, and fundus examination showed no abnormality. Auxiliary examination: blood routine and urine routine were normal, fecal occult blood (-); serum amylase and urine amylase were normal; liver and kidney function, tumor markers, and coagulation showed no significant abnormality.
Contrast-enhanced MRI of the head and cervicothoracic spine (Figure 1) showed nodular enhancement in the lesions of the right cerebellar hemisphere, about 1.0 × 0.6 cm, with clear contour; the fourth ventricle crushed and narrowed, enlargement of the lateral and third ventricles; multiple nodular and tortuous linear enhancement lesions at C5-T2 of the spinal cord, with blurred contour; and round abnormal enhancement shadows of the C6 vertebral body, with blurred contour, which presented suspected radiological diagnosis of HB. Contrast-enhanced abdominal CT scan showed multiple low-density lesions of different sizes in the pancreas and partially fused; pancreatic duct dilatation; calcification of the pancreatic head; and after enhancement, a significant round enhanced nodule of about 1.1 cm in diameter was observed on the pancreatic head, which was considered a neuroendocrine tumor. Multiple round soft tissue density lesions were observed in both kidneys, the larger one was about 3.7 cm in diameter, and the enhancement in the arterial phase was higher than that in the adjacent renal cortex and renal cancer was considered. Multiple low-density lesions were observed in both kidneys, some of which were accompanied by septum and calcification, and multiple cysts were considered.
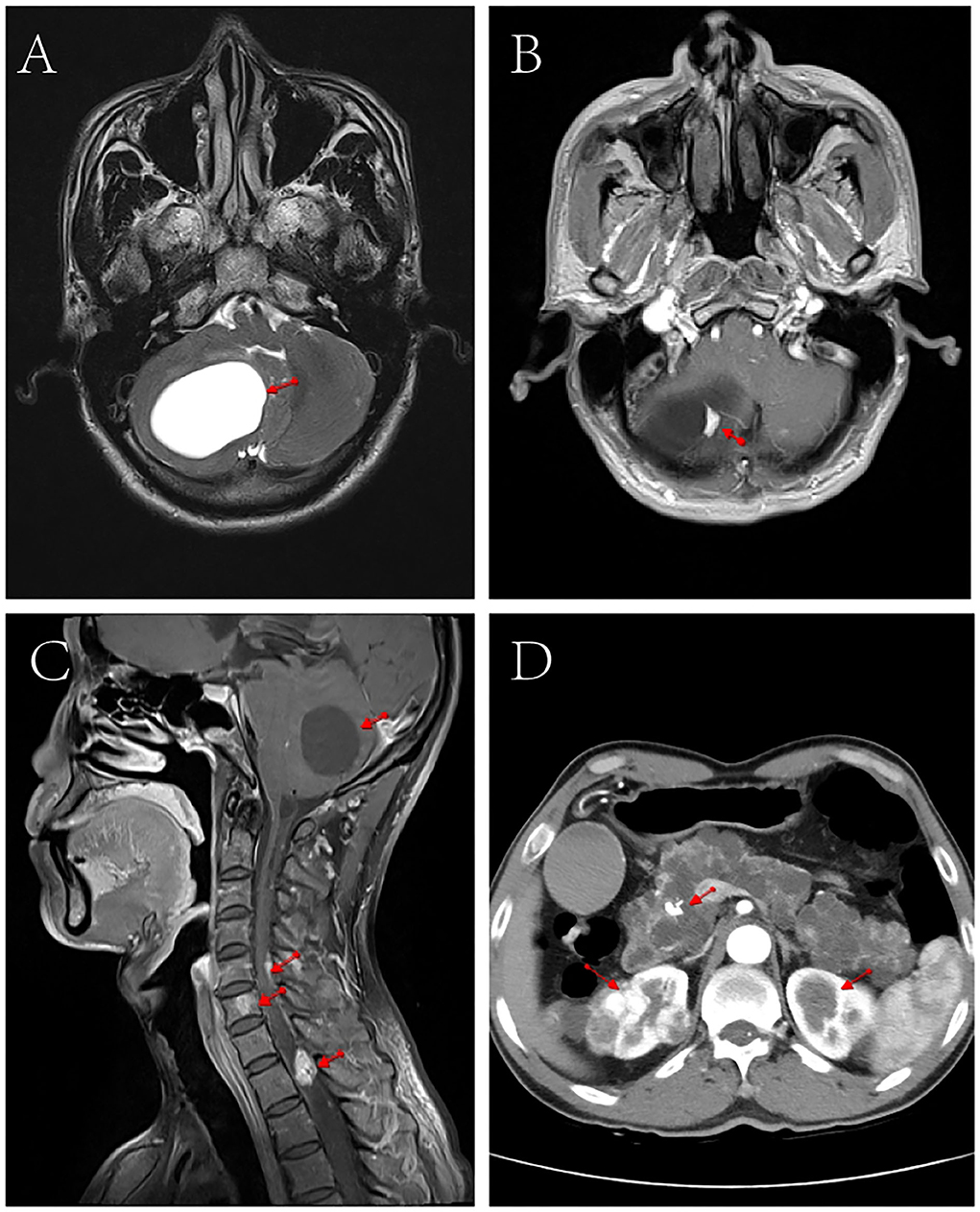
Figure 1. CT and MRI scan images of the patient. (A) Cranial MRI showed space- occupying lesions in the right cerebellar hemisphere. (B) Cranial enhanced MRI showed nodular enhancement in the right cerebellar hemisphere lesion (1.0 cm × 0.6 cm). (C) Contrast-enhanced MRI of the head and neck showed narrowing of the space in the fourth ventricle, multiple nodular and tortuous line-like enhancement in the spinal cord from C5 to T2, and round abnormal enhancement in the C6 vertebrae. (D) Abdominal enhanced CT showed multiple round soft tissue density lesions and multiple cystic low-density lesions in both kidneys, multiple low-density lesions of different sizes in the pancreas, and calcifications in the head of the pancreas.
The patient underwent a large solid cerebellar cystic mass resection. Pathological diagnosis confirmed the lesion as HB.
After 2 years of follow-up, the condition of the patient was relieved and relatively stable.
To detect the VHL gene mutation in the patient and his family, we collected peripheral blood from the patient, his daughter, his son, and his only younger brother. Exons of VHL gene were amplified from genomic DNA by polymerase chain reaction. Primer pairs are shown in Table 1.

Table 1. Primer sequences of exons of VHL gene.
Sanger DNA sequencing was performed and each exon was identified and confirmed by forward and reverse analysis. A heterozygous mutation (c.499 C > G) in exon 3 of VHL gene was detected in genomic DNA from a peripheral blood sample collected from the patient. This mutation was not detected in the peripheral blood of any other family member (Figure 2). After investigating the family history of the patient, it was found that his parents, grandparents, and maternal grandparents had died. The proband's father died of chronic heart disease, and the mother was healthy before her death; however, she died unexpectedly due to a car accident. None of them had a clear history of VHL syndrome-related disease.
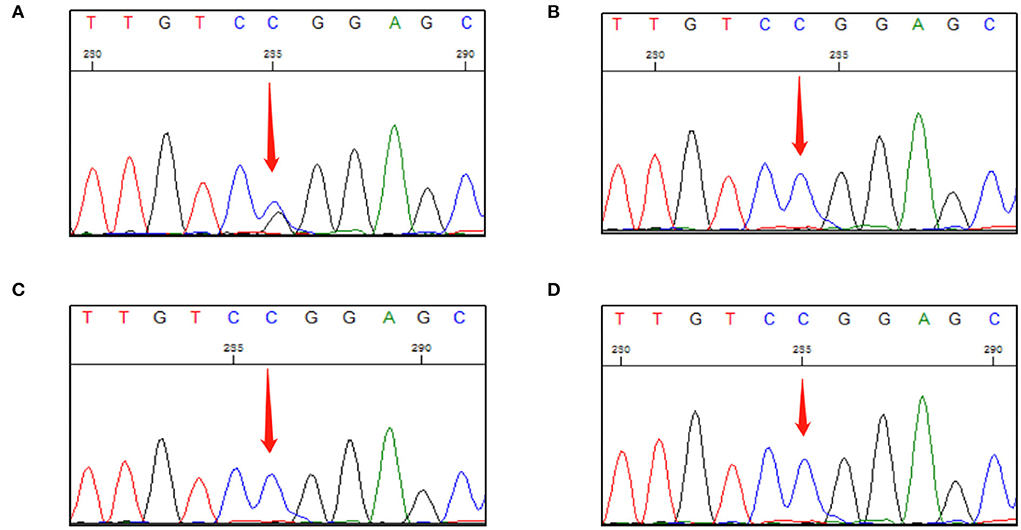
Figure 2. First-generation sequencing peak diagram. (A) The proband carried a heterozygous variant at the VHL c.499C>G site. (B) The proband's daughter did not carry this heterozygous mutation. (C) The proband's son did not carry this heterozygous mutation. (D) The proband's only younger brother did not carry this heterozygous mutation.
Von Hippel–Lindau syndrome is a progressive multisystem familial tumor syndrome characterized by phenotypically similar vascular tumors in the CNS and viscera. It's usually caused by germline mutations in the VHL tumor suppressor gene located on the short arm of chromosome 3 (3p25–26) (3).
At present, gene mutation types such as missense mutations, frameshift mutations, and in-frame deletions/insertional mutations have been detected in VHL syndrome patients. Depending on the presence or absence of PHEO, it can be divided into two clinical types. Patients with VHL type I have a low risk of developing PHEO (5). Most cases of VHL type I might be caused by missense mutations affecting the hydrophobic core of the protein, and the mutations lead to protein truncation, and partial gene deletions, which may be associated with VHL instability and high HIF activity, resulting in complete defects in protein function. In contrast, while type II is often subdivided into type IIA (with low risk of clear cell RCC (ccRCC)), type IIB (with increased risk of ccRCC), and IIC (pheochromocytoma only).VHL type II is often associated with missense mutations affecting the binding site of VHL protein (6).
Patients with VHL disease are subjected to an increased risk of developing HBs, which may affect 60–80% of patients (7). These tumors are usually multiple and no more than 50% of HBs will enlarge when followed up for more than 5 years of follow-up (8). The most common HBs are cerebellar or retinal capillary HBs, which can also occur in the spinal cord or brainstem (1). Although the tumor is benign, HB may also greatly increase morbidity and mortality in patients with VHL syndrome due to its significant impact on central nervous system architecture, and retinal edema, hemorrhage, detachment, and visual loss due to tumor exudation (9). The results of enhanced MRI examination of the cranial and cervicothoracic spine as well as CT examination showed that HB was present in the cerebellar of our reported case and cerebellar mass resection was performed. A fundus examination of the patient was not performed at that time because the eye examination showed normal vision, and the fundus examination showed no abnormality at the reexamination of the patient 1 year later. Since retinal capillary HB is the most common manifestation of VHL disease, the patient should be advised to examine the fundus at each reexamination to prevent the progression of the disease and delay in treatment. Multiple renal cysts were frequently found to coexist with RCC in 60% of patients with VHL syndrome (10), and CT diagnostic results showed renal carcinoma with multiple cysts, as described in our report. The frequency of RCC increases with age, with up to 70% of VHL patients developing RCC at age 60 and ultimately leading to death (11). About 10–20% of patients with VHL develop PHEO which can cause systemic paragangliomas. It has been associated with secondary hypertension or stroke and ultimately with death in 5% of patients with VHL syndrome (12). Approximately 35–70% of VHL syndrome patients have pancreatic manifestations (13), and as in this patient, CT examination showed multiple cysts of different sizes in the pancreas, with calcifications in the pancreatic head, which are considered neuroendocrine tumors. Although pancreatic neuroendocrine tumors are rarely responsible for morbidity and mortality, tumor transformation to malignancy or metastasis may result in a poor prognosis.
VHL syndrome can be clinically diagnosed in patients with positive family history and with central nervous system HB, RCC, or PHEO. Those without a family history must have two or more HBs or one HB and one visceral tumor (RCC, PHEO, or pancreatic tumor). In this case report, a 50-year-old patient was found to have multiple CNS HBs, multiple pancreatic and renal cysts, and RCC. Although the patient we reported had no clear family history of VHL syndrome, he still met the diagnostic criteria for VHL syndrome.
The c.499 C > G mutation of our reported case is expected to result in a mutation of amino acid 167 of the encoded protein from arginine to glycine. The arginine residue is highly conserved, with a moderate physicochemical difference between arginine and glycine. Algorithms developed to predict the impact of missense changes on protein structure and function (SIFT, PolyPhen-2, Align-GVGD) suggest that this variant may be disruptive, but these predictions require further confirmation by functional studies. Most patients with VHL syndrome inherit a germline mutation from one copy of the VHL gene in affected parents and one normally functioning wild-type allele from unaffected parents. Two studies have reported that the c.499 C > G mutation in VHL gene was associated with VHL syndrome (14, 15). However, one patient had sporadic PHEO, and the other patient had a positive family history of VHL syndrome, but there was no PHEO and no clear family history of VHL syndrome in this patient. Due to the death of his parents and the absence of VHL syndrome-related conditions, first-generation sequencing verification showed that his asymptomatic younger brother, daughter, and son did not carry the heterozygous mutation. The heterozygous mutation may be de novo. In addition, we searched the relevant literature in PubMed, Web of Science, Embase, and other literature databases for the past 10 years (January 2012–August 2022) by using keywords such as “VHL syndrome”, “VHL disease”, “gene mutation”, and “case report”, and selected the literature with more complete clinical data for summary, and 26 cases met the conditions. We summarized the VHL gene mutation reports of the Chinese population in Table 2. Of the 26 Chinese patients we summarized, 16 cases belonged to VHL type I and 10 cases belonged to VHL type II, and the gene mutation types involved missense mutations, frameshift mutations, deletions and duplications of bases, and alternative splicing of introns. These mutations often occurred in the second half of exon 1 (14 cases), the first half of exon 2 (3 cases), and exon 3 (6 cases). The study has shown that VHL gene mutations often occurred at codons 75 and 82, cleavage sites between exon 2 and exon 3, and codons 157–189, and the hotspot of mutations was at codon 167 (CpG island region) (35). In addition to the common VHL syndrome-related neoplasms, our summary also showed that nonobstructive azoospermia, clear cell peritoneal epithelioid mesothelioma, and familial polycythemia type 2 were also associated with VHL syndrome, and the basic main treatment modalities were surgical resections or chemoradiotherapy. They are mainly based on point mutation and splice mutation. By detecting the characteristics of gene mutations and comprehensively considering family characteristics, clinical manifestations, and pathological diagnosis, it is helpful to deeply study the pathogenesis and pathogenic characteristics of VHL syndrome, thereby enriching the knowledge of this disease.

Table 2. VHL gene mutations in Chinese patients.
The protein pVHL encoded by the VHL gene is a master regulator of hypoxia-inducible factor-α (HIF-α). pVHL consists mainly of α domain and β domain. The α domain mainly forms a VCB-CUL2 complex with elongin B and elongin C, further constituting E3 ubiquitinase (36). The β domain mainly interacts with the hypoxia-inducible factor HIF-α (37). Under normoxic conditions, the VCB-CUL2 complex directly acts on HIF-α for polyubiquitination and degradation. Mutations in the VHL gene may reduce the degradation ability of E3 ubiquitinase, resulting in the accumulation of HIF-1α protein, which in turn activates key carcinogenic pathways such as angiogenesis, glycolysis, glucose transport, and erythropoiesis (38, 39). The upregulated cytokines such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), erythropoietin, and transforming growth factor alpha (TGF-α) (40, 41) may lead to abnormal proliferation of tumor cells, inhibit cell apoptosis, and promote the tumor occurrence. The c.499 C > G mutation we reported is located in the α domain and may affect the binding of VHL to elongin C and affect a series of downstream biological responses (42, 43), leading to the onset and progression of disease.
The treatment of VHL syndrome is mainly based on the surgical treatment of lesions in various organ systems to remove the lesion and relieve symptoms, but it cannot fundamentally improve the prognosis. With precision medicine and sequencing technology springing up, the diagnosis and treatment of VHL syndrome have gradually opened a new chapter. The use of drugs that block the pro-angiogenesis of VEGF signal transduction pathway can block the occurrence and development of the disease to a certain extent. Studies have shown that the use of anti-VEGF drug bevacizumab in patients with metastatic renal cancer can improve the survival rate of patients to some extent (44). However, bevacizumab may have a moderate impact on retinal HBs (45, 46). The use of the receptor tyrosine kinase inhibitor sunitinib can reduce the volume of PHEOs as well as renal and pancreatic tumors (47). In 2018, Zhang et al. discovered a new VHL target ZHX2 (zinc fingers and homeoboxes 2). ZHX2 may regulate nuclear factor B (NF-KB) signaling in VHL-deficient tissues to promote RCC tumorigenesis, potentially becoming a new target for the treatment of RCC (48). In 2021, the US FDA approved HIF-2α inhibitor Belzutifan (Welireg) for the treatment of CNS HBs, RCCs, or PNET associated with VHL syndrome that does not require immediate surgery (49). With the approval of Belzutifan, the evolving technology of next-generation sequencing (NGS) may power impetus for the precision medicine of VHL syndrome-related tumors (50). With the continuous deepening of the clinical treatment of VHL gene and VHL syndrome, surgical treatment, targeted drug therapy, gene therapy, and other methods will enable patients with VHL syndrome to obtain a wider treatment space and more effective symptomatic treatment options.
The VHL gene mutation c.499 C > G in the patient we reported had no clear family history of VHL syndrome. Additionally, this patient had only a mild clinical phenotype of VHL disease. Even if there is no evidence of other VHL-related lesions or a positive family history of VHL disease, clinicians should consider the possibility of sporadic VHL in such patients. Attention should be paid to differential diagnosis and regular review to try to avoid reducing misdiagnosis and missed diagnoses. Meanwhile, molecular genetic testing of such patients and family members should be considered to help identify at-risk family members to improve diagnostic certainty and advance appropriate treatment and prevent disease progression. To improve the treatment of tumors such as CNS HBs and RCCs associated with VHL syndrome, it is necessary to continuously study new pathogenesis, discover new molecular targets, and ultimately improve the quality of life of patients.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Ethics Committee of Shanxi Provincial People's Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
YahL and XX were responsible for the drafting of the manuscript. WS helped polish the manuscript. XZ and SC offered precious knowledge on medical imaging. QW and AL were responsible for acquisition of the relevant clinical data. YafL was responsible for the conception and design of the article. All authors contributed to the manuscript and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chittiboina P, Lonser RR. Von Hippel-Lindau disease. Handb Clin Neurol. (2015) 132:139–56. doi: 10.1016/B978-0-444-62702-5.00010-X
2. Chou A, Toon C, Pickett J, Gill AJ. Von Hippel-Lindau syndrome. Front Horm Res. (2013) 41:30–49. doi: 10.1159/000345668
3. Ben-Skowronek I, Kozaczuk S. Von Hippel-Lindau syndrome. Horm Res Paediatr. (2015) 84:145–52. doi: 10.1159/000431323
4. Murro V, Lippera M, Mucciolo DP, Canu L, Ercolino T, De Filpo G, et al. Outcome and genetic analysis of patients affected by retinal capillary hemangioblastoma in Von Hippel Lindau syndrome. Mol Vis. (2021) 27:542–54. Available online at: http://www.molvis.org/molvis/v27/542
5. Maher ER, Webster AR, Richards FM, Green JS, Crossey PA, Payne SJ, et al. Phenotypic expression in Von Hippel-Lindau disease: correlations with germline Vhl gene mutations. J Med Genet. (1996) 33:328–32. doi: 10.1136/jmg.33.4.328
6. Varshney N, Kebede AA, Owusu-Dapaah H, Lather J, Kaushik M, Bhullar JS, et al. Review of Von Hippel-Lindau syndrome. J Kidney Cancer VHL. (2017) 4:20–9. doi: 10.15586/jkcvhl.2017.88
7. Cassol C, Mete O. Endocrine manifestations of Von Hippel-Lindau disease. Arch Pathol Lab Med. (2015) 139:263–8. doi: 10.5858/arpa.2013-0520-RS
8. Shanbhogue KP, Hoch M, Fatterpaker G, Chandarana H. Von Hippel-Lindau Disease: review of genetics and imaging. Radiol Clin North Am. (2016) 54:409–22. doi: 10.1016/j.rcl.2015.12.004
9. Lonser RR, Butman JA, Huntoon K, Asthagiri AR, Wu T, Bakhtian KD, et al. Prospective natural history study of central nervous system hemangioblastomas in Von Hippel-Lindau disease. J Neurosurg. (2014) 120:1055–62. doi: 10.3171/2014.1.JNS131431
10. Ganeshan D, Menias CO, Pickhardt PJ, Sandrasegaran K, Lubner MG, Ramalingam P, et al. Tumors in von hippel-lindau syndrome: From head to toe -comprehensive state-of-the-art review. Radiographics. (2018) 38:849–66. doi: 10.1148/rg.2018170156
11. Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT, et al. Clinical features and natural history of Von Hippel-Lindau disease. Q J Med. (1990) 77:1151–63. doi: 10.1093/qjmed/77.2.1151
12. Opocher G, Conton P, Schiavi F, Macino B, Mantero F. Pheochromocytoma in Von Hippel-Lindau disease and neurofibromatosis type 1. Fam Cancer. (2005) 4:13–6. doi: 10.1007/s10689-004-6128-y
13. Maher ER, Neumann HP, Richard S. Von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet. (2011) 19:617–23. doi: 10.1038/ejhg.2010.175
14. Gergics P, Patocs A, Toth M, Igaz P, Szucs N, Liko I, et al. Germline Vhl gene mutations in hungarian families with Von Hippel-Lindau disease and patients with apparently sporadic unilateral pheochromocytomas. Eur J Endocrinol. (2009) 161:495–502. doi: 10.1530/EJE-09-0399
15. Crossey PA, Richards FM, Foster K, Green JS, Prowse A, Latif F, et al. Identification of intragenic mutations in the Von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum Mol Genet. (1994) 3:1303–8. doi: 10.1093/hmg/3.8.1303
16. Ma X, Tan Z, Zhang Q, Ma K, Xiao J, Wang X, et al. Vhl Ser65 mutations enhance Hif2α signaling and promote epithelial-mesenchymal transition of renal cancer cells. Cell Biosci. (2022) 12:52. doi: 10.1186/s13578-022-00790-x
17. Heo SJ, Lee CK, Hahn KY, Kim G, Hur H, Choi SH, et al. A case of Von Hippel-Lindau disease with colorectal adenocarcinoma, renal cell carcinoma and hemangioblastomas. Cancer Res Treat. (2016) 48:409–14. doi: 10.4143/crt.2014.299
18. Jia D, Tang B, Shi Y, Wang J, Sun Z, Chen Z, et al. A deletion mutation of the Vhl gene associated with a patient with sporadic Von Hippel-Lindau disease. J Clin Neurosci. (2013) 20:842–7. doi: 10.1016/j.jocn.2012.06.013
19. Liu Q, Yuan G, Tong D, Liu G, Yi Y, Zhang J, et al. Novel genotype-phenotype correlations in five Chinese families with Von Hippel-Lindau disease. Endocr Connect. (2018) 7:870–8. doi: 10.1530/EC-18-0167
20. Lin G, Zhao Y, Zhang Z, Zhang H. Clinical diagnosis, treatment and screening of the Vhl gene in three Von Hippel-Lindau disease pedigrees. Exp Ther Med. (2020) 20:1237–44. doi: 10.3892/etm.2020.8829
21. Du XM, Wei YP, Gao Y, Li Z, Zhang JM, Chang H, et al. Clinicopathological characteristics of primary peritoneal epithelioid mesothelioma of clear cell type: a case report. Medicine. (2021) 100:e25264. doi: 10.1097/MD.0000000000025264
22. Yuan P, Sun Q, Liang H, Wang W, Li L, Wang Y, et al. Germline mutations in the Vhl gene associated with 3 different renal lesions in a Chinese Von Hippel-Lindau disease family. Cancer Biol Ther. (2016) 17:599–603. doi: 10.1080/15384047.2016.1167293
23. Yang B, Li Z, Wang Y, Zhang C, Zhang Z, Zhang X. Central nervous system hemangioblastoma in a pediatric patient associated with Von Hippel-Lindau disease: a case report and literature review. Front Oncol. (2021) 11:683021. doi: 10.3389/fonc.2021.683021
24. Man Y, Shang X, Liu C, Zhang W, Huang Q, Ma S, et al. Whole-exome sequencing identifies the Vhl mutation (C262t ≫ C, PTry88arg) in non-obstructive azoospermia-associated cystic renal cell carcinoma. Curr Oncol. (2022) 29:2376–84. doi: 10.3390/curroncol29040192
25. Zhang M, Wang J, Jiang J, Zhan X, Ling Y, Lu Z, et al. Von Hippel-Lindau Disease Type 2 in a Chinese Family with a Vhl PW88x truncation. Endocrine. (2015) 48:83–8. doi: 10.1007/s12020-014-0368-x
26. Fu XM, Zhao SL, Gui JC, Jiang YQ, Shen MN, Su DL, et al. A novel mutation links to Von Hippel-Lindau syndrome in a Chinese family with hemangioblastoma. Genet Mol Res. (2015) 14:4513–20. doi: 10.4238/2015.May.4.9
27. Wu X, Chen L, Zhang Y, Xie H, Xue M, Wang Y, et al. A novel mutation in the Vhl gene in a Chinese family with Von Hippel-Lindau Disease. BMC Med Genet. (2018) 19:204. doi: 10.1186/s12881-018-0716-4
28. Dagdeviren Çakir A, Turan H, Aykut A, Durmaz A, Ercan O, Evliyaoglu O. Two childhood pheochromocytoma cases due to Von Hippel-Lindau Disease, one associated with pancreatic neuroendocrine tumor: a very rare manifestation. J Clin Res Pediatr Endocrinol. (2018) 10:179–82. doi: 10.4274/jcrpe.5078
29. Ma X, Jing Y, Liu Y, Yu L. Effect of clarithromycin in Von Hippel-Lindau syndrome: a case report. J Int Med Res. (2019) 47:973–81. doi: 10.1177/0300060518792368
30. Lu Y, Lu J, Liu Q, Niu J, Zhang SM, Wu QY, et al. A C464t>a mutation in Vhl gene in a Chinese family with Vhl syndrome. J Neurooncol. (2013) 111:313–8. doi: 10.1007/s11060-012-1015-0
31. Liu Z, Zhou J, Li L, Yi Z, Lu R, Li C, et al. Intronic mutation of the Vhl gene associated with central nervous system hemangioblastomas in two Chinese families with Von Hippel-Lindau disease: case report. BMC Med Genet. (2020) 21:191. doi: 10.1186/s12881-020-01126-7
32. Zhang J, Ma J, Du X, Wu D, Ai H, Bai J, et al. Clinical and genetic investigation of a multi-generational chinese family afflicted with Von Hippel-Lindau disease. Chin Med J. (2015) 128:32–8. doi: 10.4103/0366-6999.147802
33. Wang J, Cao W, Wang Z, Zhu H. Novel gene mutation in Von Hippel-Lindau disease - a report of two cases. BMC Med Genet. (2019) 20:194. doi: 10.1186/s12881-019-0930-8
34. Zhang W, Bao S, Jiang LJ, Ma YP. A case of familial erythrocytosis type 2 caused by Vhl gene mutation. Zhonghua Xue Ye Xue Za Zhi. (2020) 41:1047–9.
35. Kolomeyer AM, Eller AW, Martel JN. Spontaneous resolution of macular epiretinal membranes after fluorescein potentiated argon laser treatment of Von Hippel-Lindau associated retinal hemangiomas: case report and review of literature. Retin Cases Brief Rep. (2016) 10:145–50. doi: 10.1097/ICB.0000000000000206
36. Hudler P, Urbancic M. The role of Vhl in the development of Von Hippel-Lindau disease and erythrocytosis. Genes. (2022) 13:362. doi: 10.3390/genes13020362
37. Kaelin WG Jr. Molecular basis of the Vhl hereditary cancer syndrome. Nat Rev Cancer. (2002) 2:673–82. doi: 10.1038/nrc885
38. Gossage L, Eisen T, Maher ER. Vhl, the story of a tumour suppressor gene. Nat Rev Cancer. (2015) 15:55–64. doi: 10.1038/nrc3844
39. He W, Batty-Stuart S, Lee JE, Ohh M. Hif-1α hydroxyprolines modulate oxygen-dependent protein stability via single Vhl interface with comparable effect on ubiquitination rate. J Mol Biol. (2021) 433:167244. doi: 10.1016/j.jmb.2021.167244
40. Maher ER. Von Hippel-Lindau disease. Curr Mol Med. (2004) 4:833–42. doi: 10.2174/1566524043359827
41. Gläsker S, Vergauwen E, Koch CA, Kutikov A, Vortmeyer AO. Von Hippel-Lindau disease: current challenges and future prospects. Onco Targets Ther. (2020) 13:5669–90. doi: 10.2147/OTT.S190753
42. Hacker KE, Lee CM, Rathmell WK. Vhl type 2b mutations retain Vbc complex form and function. PLoS ONE. (2008) 3:e3801. doi: 10.1371/journal.pone.0003801
43. Jung YS, Lee SJ, Yoon MH, Ha NC, Park BJ. Estrogen receptor ? is a novel target of the Von Hippel-Lindau protein and is responsible for the proliferation of Vhl-deficient cells under hypoxic conditions. Cell Cycle. (2012) 11:4462–73. doi: 10.4161/cc.22794
44. Semenza GL. Targeting Hif-1 for cancer therapy. Nat Rev Cancer. (2003) 3:721–32. doi: 10.1038/nrc1187
45. Tano R, Kakurai K, Sakurai T, Fujiwara R, Mano T, Maeno T. Intravitreal bevacizumab (avastin) combined with vitrectomy for recurrences of proliferative vitreoretinopathy in Von Hippel-Lindau disease. Acta Ophthalmol. (2012) 90:e157–8. doi: 10.1111/j.1755-3768.2011.02108.x
46. Hrisomalos FN, Maturi RK, Pata V. Long-term use of intravitreal bevacizumab (avastin) for the treatment of Von Hippel-Lindau associated retinal hemangioblastomas. Open Ophthalmol J. (2010) 4:66–9. doi: 10.2174/1874364101004010066
47. Jimenez C, Cabanillas ME, Santarpia L, Jonasch E, Kyle KL, Lano EA, et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with Von Hippel-Lindau disease: targeting angiogenic factors in pheochromocytoma and other Von Hippel-Lindau disease-related tumors. J Clin Endocrinol Metab. (2009) 94:386–91. doi: 10.1210/jc.2008-1972
48. Sanchez DJ, Simon MC. transcriptional control of kidney cancer. Science. (2018) 361:226–7. doi: 10.1126/science.aau4385
Keywords: Von Hippel–Lindaut syndrome, whole exome sequencing, gene mutation, literature, genetic testing
Citation: Li Y, Xin X, Song W, Zhang X, Chen S, Wang Q, Li A and Li Y (2022) VHL syndrome without clear family history: A rare case report and literature review of Chinese patients. Front. Neurol. 13:951054. doi: 10.3389/fneur.2022.951054
Received: 23 May 2022; Accepted: 05 September 2022;
Published: 17 October 2022.
Edited by:
Ignazio Gaspare Vetrano, IRCCS Carlo Besta Neurological Institute Foundation, ItalyCopyright © 2022 Li, Xin, Song, Zhang, Chen, Wang, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yafeng Li, ZHIueWFmZW5nbGlAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.