 Dongcheng Li
Dongcheng Li Tengzhu Ren
Tengzhu Ren Hao Li
Hao Li Geng Liao
Geng Liao Xiong Zhang
Xiong Zhang- 1Department of Neurology, Affiliated Maoming People's Hospital, Southern Medical University, Maoming, China
- 2Department of Neurology, Guangdong Second Provincial General Hospital, Guangzhou, China
Cognitive impairment (CI) is a common complication of Parkinson's disease (PD). The major features of Parkinson's disease with cognitive impairment (PD-CI) include convergence of α-Synuclein (α-Syn) and Alzheimer's disease (AD)-like pathologies, neuroinflammation, and dysbiosis of gut microbiota. Porphyromonas gingivalis (P. gingivalis) is an important pathogen in periodontitis. Recent research has suggested a role of P. gingivalis and its virulence factor in the pathogenesis of PD and AD, in particular concerning neuroinflammation and deposition of α-Synuclein (α-Syn) and amyloid-β (Aβ). Furthermore, in animal models, oral P. gingivalis could cause neurodegeneration through regulating the gut-brain axis, suggesting an oral-gut-brain axis might exist. In this article, we discussed the pathological characteristics of PD-CI and the role of P. gingivalis in them.
Introduction
Parkinson's disease (PD) is a prevalent neurodegenerative disease caused by the death of dopaminergic neurons in the substantia nigra pars compacta (SNpc). Cognitive impairment (CI) is a common complication of PD non-motor symptoms, including PD with mild cognitive impairment (PD-MCI) and PD dementia (PDD). PD-MCI, as an independent risk factor for PDD (1), is characterized by the transitional state that fails to meet the diagnostic criteria for PDD. A 5-year follow-up study of patients with PD showed that the cumulative incidence of PD-MCI in patients aged ≥ 65 years is 41.3% after 5 years, and the conversion rate of patients with PD-MCI progressing to PDD within 5 years is about 39–50% (2).
The pathogenesis of PD-CI is unclear. Current studies have found that PD-CI-related pathologies include the deposition of α-Synuclein (α-Syn), Alzheimer's disease-type pathologies (amyloid-β, tau, and neurofibrillary tangles), and neuroinflammation (3–5). Recently, many studies have shown a correlation between Porphyromonas gingivalis (P. gingivalis) infection and PD (5–8). Interestingly, P. gingivalis has recently been shown to be associated with cognitive impairment and a potential microbial driver in Alzheimer s disease (AD) (9–15). Therefore, this review summarizes the literature on the potential role of P. gingivalis in PD-CI development and is aimed as a reference for further research and therapy.
Porphyromonas gingivalis
Porphyromonas gingivalis is a Gram-negative, anaerobic, and rod-shaped bacteria that colonizes the oral epithelium and is an important component of subgingival microbiomes. P. gingivalis is responsible for the chronic form of periodontitis through its capacity to remodel the commensal bacterial community, which promotes a state of dysbiosis (16). It can engineer its environment or modify the host's immune response to modulate the entire ecosystem. It can also persist in host tissues through unique and intricate mechanisms, such as the alteration of inflammatory signaling pathways, the complement system, the cell cycle, apoptosis, and the interaction with various host receptors (17). During common activities, such as brushing, flossing, chewing, and dental procedures (18), P. gingivalis invades the vasculature from the infected periodontal pocket. Furthermore, a study using an animal model showed that it can also increase the permeability of the blood-brain barrier (BBB) and facilitate access of bacteria into the brain (19).
The strategies and pathogenicity of P. gingivalis largely rely on its various virulence factors. Among these, the secretory components and gingipains are major virulence factors, consist of lysine-gingipain (Kgp) and arginine-gingipain (Rgp), which play essential roles in host colonization, host defense deactivation, tissue destruction, and nutrient acquisition (20). In addition, lipopolysaccharide (LPS), a structural component, is also a crucial pathogenic factor that can trigger the innate immune response via the activation of toll-like receptors (TLRs), leading to more lasting destruction of periodontal tissues (21). P. gingivalis LPS induces the expression of interleukin (IL)-6 and C-C motif chemokine ligand 2 (CCL2) in the brain microvascular endothelial cells, which may contribute to dysfunction of the BBB and subsequent neurological disorders (22, 23). Furthermore, P. gingivalis LPS can induce IL-8 elevation (24) and IL-10 decline (25) in the brain, which is consistent with cytokine changes in the cerebrospinal fluid (CSF) of patients with PD (26). Indeed, gingipains and LPS have attracted growing attention as virulence factors of P. gingivalis and recent studies have found that these two factors are present in the blood circulation and brain of PD and AD populations (5, 27). In addition to neurodegenerative diseases, studies have shown that P. gingivalis may be associated with other systemic inflammatory diseases, such as type 2 diabetes mellitus, rheumatoid arthritis, and cardiopulmonary disease (28).
Pathological characteristics of PD-CI
α-Syn and AD-type pathologies
Current neuropathological studies indicated that the convergence of α-Syn and AD-type pathologies is the major pathological features of PD-CI (4).
The misfolding and aggregation of aberrant α-Syn in the patients' brain are the major characteristics of PD (29) that result in neuron loss and the clinical syndrome of idiopathic PD (30). Moreover, it was found that pathogenic α-Syn can transfer between cells leading to neurodegeneration (30–32). The importance of α-Syn is further emphasized in PD-CI studies which showed that α-Syn pathology is more extensive and severe in PDD than in PD without dementia. PDD cases are almost exclusively of the predominant stage of α-Syn pathology in the limbic system or neocortex and α-Syn pathology level can distinguish between PD and PDD (3, 33, 34). The progression of α-Syn pathology stage or cortical α-Syn pathology burden is highly correlated with the cognitive level decline (35–37).
However, despite α-Syn pathology being the major driving force of the development in cognitive impairment in patients with PD, AD pathology was also found to play an important role in PD-CI (4). Many clinical studies revealed that the tau concentration in the CSF is associated with cognitive impairment in Parkinson's disease (38–41). In some studies, AD neuropathology seemed to be even more associated with PDD than with α-Syn pathology, but most of these patients with PD were assigned a diagnosis of PDD + AD (3, 4, 33). In fact, α-Syn pathological levels in the cortex and limbic system of patients with PDD + AD appeared to be higher than those of patients with PDD without AD comorbidities. An increased severity of cortical senile plaques (SPs) and burden of neurofibrillary tangles (NFTs) were also related to an increased cortical α-Syn pathological density (3, 33, 36, 42). Recently, Bassi et al. demonstrated that amyloid-β (Aβ) deposits dramatically accelerate α-Syn pathogenesis and spread throughout the brain after injecting α-Syn preformed fibrils into mice with abundant Aβ plaques. Recent pathological studies in vitro showed that AD-related pathologies could exacerbates α-Syn seeding activity and neurotoxicity (43, 44), suggesting an interaction between α-Syn pathology and AD-type pathologies in PD-CI.
Neuroinflammation
In addition to α-Syn and AD pathology, neuroinflammation is also a crucial factor in PD-CI. Although it is not clear how inflammation contributes to the pathogenesis of PD-CI, it is universally acknowledged that both central and peripheral inflammations contribute to the progression of neurodegeneration in PD-CI. A study by Lindqvist et al. (45) indicated that PD non-motor features were associated with higher CSF levels of inflammatory markers. The C-reactive protein (CRP) level in the CSF of patients with PDD was significantly higher than that of patients with non-demented PD (p = 0.032) (45). In addition, a large cohort study of newly diagnosed patients with PD showed that higher levels of interferon gamma (IFN-γ), TNF-α, and CRP in blood are associated with a lower Mini-Mental State Examination (MMSE) score in patients with PD, and that IL-1β and IL-2 are related to a faster rate of cognitive decline (46). Recently, an animal study demonstrated that peripherally induced neuroinflammation potentiates the harmful effects of α-Syn. Furthermore, in genetic pathologic PD models, LPS-induced neuroinflammation aggravated cognitive deficits (47).
Neuroinflammation is mainly promoted by microglia and astrocytes. Under chronic peripheral inflammation, microglial and astroglial cells are overactivated by toll-like receptors (TLRs), resulting in the release of various inflammatory cytokines (e.g., TNF- a, IL-6, and CXCL1), leading to chronic neuroinflammation (48, 49). Neuroinflammation induces and exacerbates α-Syn and AD-type pathologies. Activated glial cells interact with α-Syn and AD-type pathologies (48, 50), and mediate their detrimental effects on both memory and neuroinflammation (47). These processes can promote a vicious circle and lead to PD-CI.
Dysbiosis of gut microbiota
Increasing evidence from studies of the gut–brain axis has suggested that the gut microbiome plays a critical role in neurodegenerative diseases, such as AD and PD (51). Previously, our research (52) has found that the gut microbiota of patients with PD-MCI was significantly altered compared with those healthy controls (HCs) and patients with PD with normal cognition (PD-NC). This is particularly manifesting in enriched genera from Porphyromonadaceae family, providing powerful evidence that the dysbiosis of gut microbiota may contribute to PD-CI (52).
Although the role of intestinal flora alteration in PD-CI is not completely known, we can still deduce hints from the results of recent studies. First, according to Braak's hypothesis (53), the accumulation of aberrant α-Syn is initiated in the gut and propagates via the vagus nerve to the brain. Furthermore, growing evidence showed that α-Syn may be transmitted in a prion-like manner (54–57). Recent studies also supported Braak's hypothesis in the etiology of PD and the prion-like theory (58), and indicated that the prion-like seeding activity of aberrant α-Syn may be related with its post-translational modifications (such as, carboxy-truncation) or oligomerization (59, 60). Several factors, such as antibiotics, diet, birth mode, or stress may trigger or promote the translocation of intestinal microorganisms and microbial products (such as, LPS), which would cause oxidative stress and mucosal inflammation, and promote the accumulation of α-Syn in the enteric nervous system (ENS) (61–63). Indeed, recent experiments have shown that the LPS of intestinal microorganisms can regulate the aggregation and toxicity of α-Syn and lead to cognitive decline (47, 64). Second, LPS from gut microbiota can disrupt the integrity of the BBB (62, 65, 66), which may promote neuroinflammation and SNpc damage. Finally, an experiment using a mouse model showed that microbiome disturbances have influences on microglia-mediated brain Aβ deposition (67). Collectively, these results suggest that alterations in the intestinal flora may facilitate the deposition of α-Syn and Aβ, and neuroinflammation, resulting in the induction induce PD-CI.
P. gingivalis and PD
In earlier studies, the view was that motor and cognitive disturbances that are caused by PD, could contribute to the progression of periodontal disease (68, 69). However, as periodontal disease is gradually found to be related to the onset and progression of AD (10), a reverse correlation between periodontal disease and PD is increasingly attracting attention (68, 70). The studies by Chen et al. (6, 71) showed that the risk of developing PD in patients with periodontitis is significantly higher than that in controls [adjusted hazard ratio (HR) = 1.431, p = 0.002] and patients without periodontitis who had a significantly lower risk of developing PD after dental scaling over 5 consecutive years [adjusted odds ratio (OR) = 0.204, p = 0.0399]. Another similar study by Jeong et al. (7) suggested a weak association between periodontitis and PD (log rank p < 0.001).
Recently, a study by Adams et al. (5) showed that gingipain R1 (RgpA), produced by P. gingivalis, is present in the blood circulation, highlighting the potential involvement of P. gingivalis in Parkinson's disease. In addition, Adams et al. found that the whole blood of patients with PD is hypercoagulable, due to the presence of hyperactivated platelets and fibrin(ogen) amyloid features. These results are consistent with previous finding (72) that clots are denser and hyperclottable in patients with PD. Moreover, preliminary data suggested a role of P. gingivalis LPS and gingipain in the systemic inflammatory and hypercoagulable pathology of PD.
The R1441G mutation in the leucine-rich repeat kinase 2 (LRRK2) gene results in late-onset PD (73). A recent animal study by Feng et al. found that (8), orally administrating live P. gingivalis to LRRK2 R1441G mice three times a week for 1 month, can induce a mutant LRRK2-dependent reduction of dopaminergic neurons in the substantia nigra, an increase in mutant LRRK2 expression, and the activation of microglia, leading to peripheral IL-17A secretion and IL-17 receptor A (IL-17RA) upregulation. These results provide further evidence on the correlation between P. gingivalis and PD.
P. gingivalis and cognitive impairment
Alzheimer's disease is the most common neurodegenerative disorder that causes cognitive impairment (49). Many studies have revealed that periodontitis increases the risk of AD (9–11). P. gingivalis, the main pathogen in periodontitis, was shown to play an important role in AD. In a cross-sectional study, patients with high P. gingivalis IgG had worse delayed verbal memory and impaired subtraction in a dose-response relationship (12). The study by Stein et al. indicated that after adjusting for confounders, such as age and smoking, serum antibody levels of P. gingivalis were higher in AD patients with cognitive impairment and positively correlated with the stage of AD development (13). Furthermore, P. gingivalis DNA, LPS (14), and gingipains (15) have been recently detected in the brains of patients with AD.
Amyloid-β plaques, neurofibrillary tangles, and neuroinflammation are the major hallmarks of AD. P. gingivalis may promote the progression of AD by contributing to these pathologies. The study by Wu et al. (74) showed that chronic exposure to P. gingivalis LPS for 5 consecutive weeks causes AD-like phenotypes, such as learning and memory deficits, microglia-mediated neuroinflammation and Aβ accumulation in neurons of middle-aged wild-type mice. In addition, cathepsin B (CatB) may be crucial for this process, as P. gingivalis LPS-induced AD-like phenotypes were found to be CatB-dependent. Interestingly, CatB plays a critical role in peripheral Aβ generation, as P. gingivalis infection induces the production of Aβ in inflammatory macrophages via activating the CatB/NF-κB signaling (75). Subsequently, a study by Zeng et al. (76) indicated that P. gingivalis infection can promote the CatB/NF-κB-dependent receptor for advanced glycation end (RAGE) expression in cerebral endothelial cells, which mediates the influx of peripheral Aβ into the brain across the BBB. These results revealed a potential pathogenesis of AD, associated with P. gingivalis induction of induced peripheral Aβ production and influx, resulting in AD-type pathologies in the brain, and in which CatB plays an important role.
To the best of our knowledge, no study has directly demonstrated the correlation between P. gingivalis and PD-CI. However, in a report by La Vitola et al. (47), it was shown that Escherichia coli (E. coli) LPS-induced neuroinflammation can aggravate the toxic effects of α-Syn and cognitive deficits. However, in the study by Zhang et al., although either P. gingivalis LPS or E. coli LPS was shown to impair spatial learning and memory in the MWM test, no significant differences were observed between the effects of the two LPS species (24). These results suggest that, despite their structural differences, P. gingivalis LPS may have similar mechanisms to those of E. coli LPS in exacerbating the α -Syn detrimental effects and cognitive impairment.
P. gingivalis and gut microbiota
According to previous reports, the density of P. gingivalis in saliva of patients with severe periodontitis can reach to 106/ml (77–79). Since humans produce 1–1.5 L of saliva a day, about 1012-1013 P. gingivalis bacteria might be daily swallowed by patients with severe periodontitis (80). Animal studies showed that the oral administration of P. gingivalis at simulated doses causes gut microbiota dysbiosis and gut permeability impairment, resulting in systemic inflammation in mice (80, 81). It was reported that P. gingivalis-induced dysbiosis is related to arthritis (65, 82), type 2 diabetes (83), and non-alcoholic fatty liver disease (NAFLD) (84).
As gut microbiota plays an important role in the gut-brain axis, whether P. gingivalis promotes neurodegenerative diseases through regulating the gut-brain axis, is still under research. Feng et al. (8) have found that the oral administration of P. gingivalis can lead to gut permeability impairment and an increase in the accumulation of α-Syn in the colon neurons of LRRK2 R1441G mice. Although the authors did not detect α-Syn in the brain or small intestine, this finding supports the previous point that the accumulation of aberrant α-Syn is initiated in the gut (53, 58). In addition, emerging evidence by Chi et al. (85) shows that oral P. gingivalis induces gut microbiota dysbiosis, exacerbates neuroinflammation, and ultimately lead to a decline in cognitive function. Furthermore, the number of neurons in the hippocampal and cortical regions was significantly decreased, and amyloid plaques appeared in brain. The results from these two studies are consistent with the characteristics of PD-CI mentioned above, indicating an oral-gut-brain axis may exist and contribute to PD-CI, which is worthy of further confirmation in future studies.
How is P. gingivalis involved in PD-CI?
Based on the evidence mentioned above, we hypothesize that P. gingivalis may be involved in PD-CI through two pathways (Figure 1).
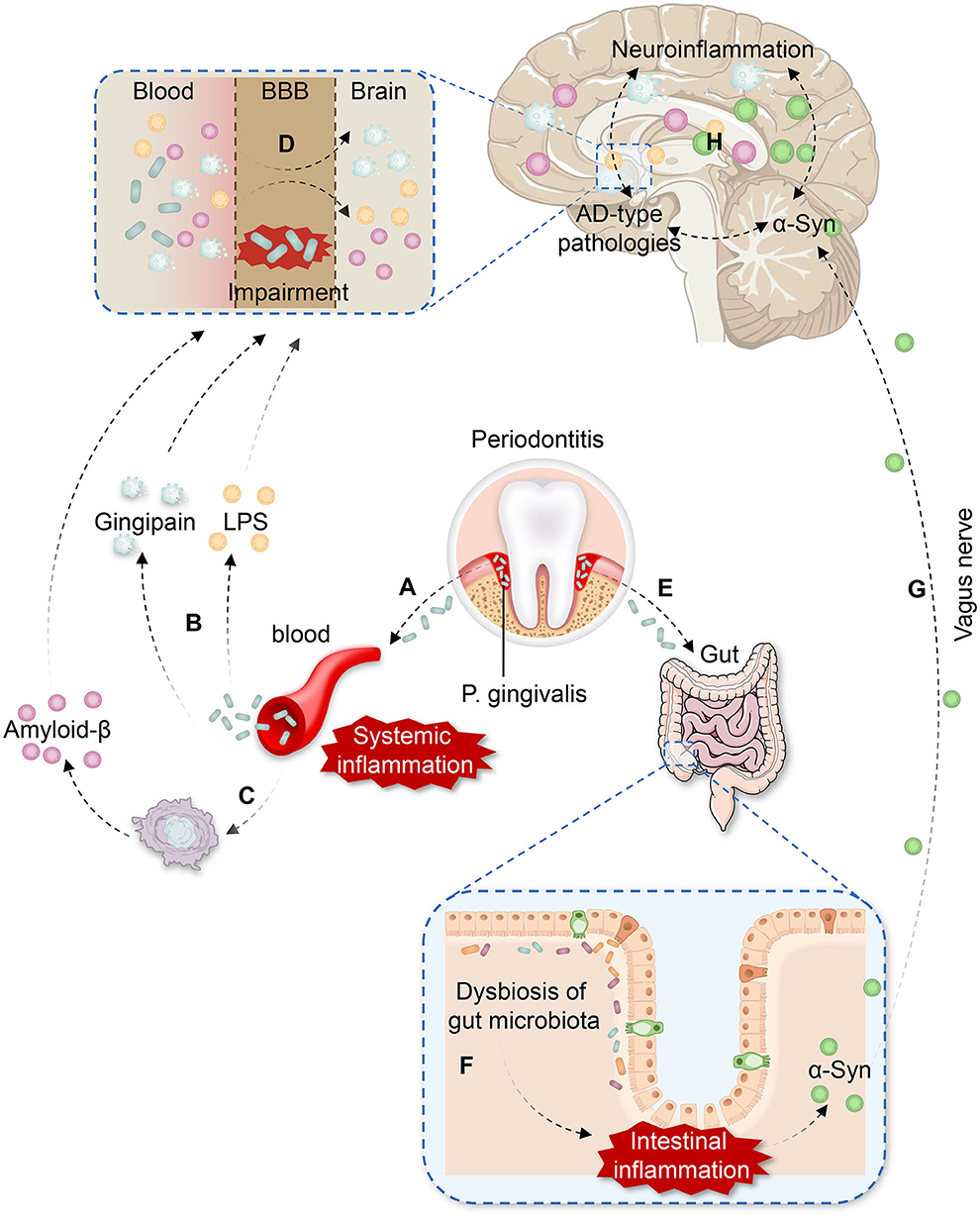
Figure 1. The potential pathways of P. gingivalis involvement in Parkinson's disease with cognitive impairment (PD-CI). (A) P. gingivalis enters the vasculature from the periodontal pocket, leading to bacteremia and systemic inflammation. (B) Gingipain and lipopolysaccharide (LPS) produced by P. gingivalis are present in the blood. (C) P. gingivalis infection induces the production of peripheral amyloid-β (Aβ) in inflammatory macrophages. (D) P. gingivalis impairs the function of the blood-brain barrier (BBB) by facilitating LPS, gingipain, and peripheral Aβ entry to the brain more easily, which causes neuroinflammation and Alzheimer's disease (AD)-type pathologies. (E) P. gingivalis is swallowed and enters the intestine. (F) P. gingivalis induces dysbiosis of the gut microbiota and intestinal inflammation, contributing to the accumulation of aberrant α-Synuclein (α-Syn) in the gut. (G) Aberrant α-Syn propagates from the gut to the brain via the vagus nerve. (H) Neuroinflammation, AD-type pathologies, and α-Syn promote each other effect in the brain, leading to a vicious cycle and resulting in PD-CI.
On the one hand, daily activities, such as tooth brushing and chewing, as well as dental procedures in patients with severe periodontitis can promote the entry of P. gingivalis into the vasculature from the periodontal pocket through ulcerated epithelium and lymphatic vessels (18, 28, 86). Additionally, P. gingivalis can destroy oral tissues to enter the bloodstream by gingipain (20). The entry of P. gingivalis into the bloodstream leads to bacteremia and the activation of the host immune response. Peripheral cytokines are subsequently increased (e.g., IL-1β, IL-2, IL-17A, and TNF-α), and systemic inflammation is induced, which facilitates the generation of peripheral Aβ. As P. gingivalis would impair the function of BBB, virulence factors, such as LPS, gingipain, and peripheral Aβ can easily enter the brain, resulting in the activation of microglia and astrocytes, causing neuroinflammation through TLRs. Ultimately neuroinflammation promotes aberrant α-Syn aggregation and AD-type pathologies in brain.
On the other hand, the oral-gut-brain axis may be another crucial mechanism. Patients with severe periodontitis can swallow large amounts of P. gingivalis each day. When entering the intestine, P. gingivalis may induce dysbiosis of gut microbiota, which simultaneously activate TLRs in the intestine, leading to intestinal inflammation and increased intestinal permeability. In addition to exacerbating systemic inflammation, the gut dysbiosis also contribute to the accumulation of aberrant α-Syn in the gut. Subsequently, α-Syn could propagate from the gut to the brain via the vagus nerve in a prion-like manner, aggravating α-Syn deposition and misfolding in the brain.
Discussion
Collectively, the pathology of PD-CI has three major characteristics: convergence of α-Syn and Aβ pathologies, neuroinflammation, and dysbiosis of the gut microbiota. The currently available studies showed that P. gingivalis infection correlates with these features of PD-CI, underlining the association between P. gingivalis and PD, and between P. gingivalis and cognitive impairment, respectively. However, these studies have indirectly shown that P. gingivalis is associated with PD-CI, while no study has provided direct evidence of the association between P. gingivalis and PD-CI. Thus, the correlation between P. gingivalis and PD-CI needs further confirmation in subsequent studies.
Author contributions
XZ conceived the concept of the review. DL did initial systemic search to find articles and drafted the manuscript with TR. XZ, HL, and GL modified the article. All authors contributed to the final manuscript and approved the submitted version.
Funding
This study was supported by the Special Fund Project for Science and Technology Innovation Strategy of Guangdong Province and the High-level Hospital Construction Research Project of Maoming People's Hospital. XZ was supported by the Doctoral Research Foundation of Maoming People's Hospital.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hoogland J, Boel JA, de Bie RMA, Geskus RB, Schmand BA, Dalrymple-Alford JC, et al. Mild cognitive impairment as a risk factor for Parkinson's disease dementia. Movement Disord. (2017) 32:1056–65. doi: 10.1002/mds.27002
2. Pedersen KF, Larsen JP, Tysnes O, Alves G. Natural course of mild cognitive impairment in Parkinson disease. Neurology. (2017) 88:767–74. doi: 10.1212/WNL.0000000000003634
3. Compta Y, Parkkinen L, O'Sullivan SS, Vandrovcova J, Holton JL, Collins C, et al. Lewy- and alzheimer-type pathologies in Parkinson's disease dementia: which is more important? Brain. (2011) 134:1493–505. doi: 10.1093/brain/awr031
4. Irwin DJ, Lee VMY, Trojanowski JQ. Parkinson's disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci. (2013) 14:626–36. doi: 10.1038/nrn3549
5. Adams B, Nunes JM, Page MJ, Roberts T, Carr J, Nell TA, et al. Parkinson's disease: a systemic inflammatory disease accompanied by bacterial inflammagens. Front Aging Neurosci. (2019) 11:210. doi: 10.3389/fnagi.2019.00210
6. Chen C, Wu Y, Chang Y. Periodontal inflammatory disease is associated with the risk of Parkinson's disease: a population-based retrospective matched-cohort study. PeerJ. (2017) 5:e3647. doi: 10.7717/peerj.3647
7. Jeong E, Park J, Park Y. Evaluation of the association between periodontitis and risk of Parkinson's disease: a nationwide retrospective cohort study. Sci Rep. (2021) 11:16594. doi: 10.1038/s41598-021-96147-4
8. Feng Y, Wu Q, Peng Y, Liang F, You H, Feng Y, et al. Oral P. gingivalis impairs gut permeability and mediates immune responses associated with neurodegeneration in lrrk2 r1441g mice. J Neuroinflamm. (2020) 17:347. doi: 10.1186/s12974-020-02027-5
9. Ide M, Harris M, Stevens A, Sussams R, Hopkins V, Culliford D, et al. Periodontitis and cognitive decline in Alzheimer's disease. PLoS One. (2016) 11:e151081. doi: 10.1371/journal.pone.0151081
10. Kamer AR, Craig RG, Dasanayake AP, Brys M, Glodzik Sobanska L, Leon MJ. Inflammation and Alzheimer's disease: possible role of periodontal diseases. Alzheimer's Dement. (2008) 4:242–50. doi: 10.1016/j.jalz.2007.08.004
11. Chen C, Wu Y, Chang Y. Association between chronic periodontitis and the risk of Alzheimer's disease: a retrospective, population-based, matched-cohort study. Alzheimer's Res Ther. (2017) 9:56. doi: 10.1186/s13195-017-0282-6
12. Noble JM, Borrell LN, Papapanou PN, Elkind MSV, Scarmeas N, Wright CB. Periodontitis is associated with cognitive impairment among older adults: analysis of NHANES-III. J Neurol Neurosurg Psychiatry. (2009) 80:1206–11. doi: 10.1136/jnnp.2009.174029
13. Sparks Stein P, Steffen MJ, Smith C, Jicha G, Ebersole JL, Abner E, et al. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer's disease. Alzheimer's Dement. (2012) 8:196–203. doi: 10.1016/j.jalz.2011.04.006
14. Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer's disease brain tissue. J Alzheimer's Dis. (2013) 36:665–77. doi: 10.3233/JAD-121918
15. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer's disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. (2019) 5:u3333. doi: 10.1126/sciadv.aau3333
16. Olsen I, Lambris JD, Hajishengallis G. Porphyromonas gingivalis disturbs host-commensal homeostasis by changing complement function. J Oral Microbiol. (2017) 9:1340011–85. doi: 10.1080/20002297.2017.1340085
17. Chopra A, Bhat SG, Sivaraman K. Porphyromonas gingivalis adopts intricate and unique molecular mechanisms to survive and persist within the host: a critical update. J Oral Microbiol. (2020) 12:1801090. doi: 10.1080/20002297.2020.1801090
18. Forner L, Larsen T, Kilian M, Holmstrup P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J Clin Periodontol. (2006) 33:401–7. doi: 10.1111/j.1600-051X.2006.00924.x
19. Poole S, Singhrao SK, Chukkapalli S, Rivera M, Velsko I, Kesavalu L, et al. Active invasion of Porphyromonas gingivalis and infection-induced complement activation in apoe-/- mice brains. J Alzheimer's Dis. (2014) 43:67–80. doi: 10.3233/JAD-140315
20. Guo Y, Nguyen KA, Potempa J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon's knife to a meat chopper-like brutal degradation of proteins. Periodontol 2000. (2010) 54:15–44. doi: 10.1111/j.1600-0757.2010.00377.x
21. Jia L, Han N, Du J, Guo L, Luo Z, Liu Y. Pathogenesis of important virulence factors of Porphyromonas gingivalis via toll-like receptors. Front Cell Infect Microbiol. (2019) 9:262. doi: 10.3389/fcimb.2019.00262
22. Sato N, Matsumoto T, Kawaguchi S, Seya K, Matsumiya T, Ding J, et al. Porphyromonas gingivalis lipopolysaccharide induces interleukin-6 and c-c motif chemokine ligand 2 expression in cultured hcmec/d3 human brain microvascular endothelial cells. Gerodontology. (2022) 39:139–47. doi: 10.1111/ger.12545
23. Kimura A, Yoshikura N, Hayashi Y, Inuzuka T. Cerebrospinal fluid c-c motif chemokine ligand 2 correlates with brain atrophy and cognitive impairment in Alzheimer's disease. J Alzheimer's Dis. (2018) 61:581–8. doi: 10.3233/JAD-170519
24. Zhang J, Yu C, Zhang X, Chen H, Dong J, Lu W, et al. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the tlr4 signaling pathway in c57bl/6 mice. J Neuroinflamm. (2018) 15:37. doi: 10.1186/s12974-017-1052-x
25. Zhang X, Zhang X, Qiu C, Shen H, Zhang H, He Z, et al. The imbalance of th17/treg via stat3 activation modulates cognitive impairment in p. Gingivalis lps-induced periodontitis mice. J Leukocyte Biol. (2021) 110:511–24. doi: 10.1002/JLB.3MA0521-742RRR
26. Wijeyekoon RS, Moore SF, Farrell K, Breen DP, Barker RA, Williams Gray CH. Cerebrospinal fluid cytokines and neurodegeneration-associated proteins in Parkinson's disease. Movement Disord. (2020) 35:1062–6. doi: 10.1002/mds.28015
27. Dioguardi M, Crincoli V, Laino L, Alovisi M, Sovereto D, Mastrangelo F, et al. The role of periodontitis and periodontal bacteria in the onset and progression of Alzheimer's disease: a systematic review. J Clin Med. (2020) 9:495. doi: 10.3390/jcm9020495
28. Mei F, Xie M, Huang X, Long Y, Lu X, Wang X, et al. Porphyromonas gingivalis and its systemic impact: current status. Pathogens. (2020) 9:944. doi: 10.3390/pathogens9110944
29. Braak H, Tredici KD, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003) 24:197–211. doi: 10.1016/S0197-4580(02)00065-9
30. Luk KC, Kehm VM, Zhang B, O Brien P, Trojanowski JQ, Lee VMY. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med. (2012) 209:975–86. doi: 10.1084/jem.20112457
31. Li J, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. (2008) 14:501–3. doi: 10.1038/nm1746
32. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body–like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. (2008) 14:504–6. doi: 10.1038/nm1747
33. Irwin DJ, White MT, Toledo JB, Xie SX, Robinson JL, Van Deerlin V, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. (2012) 72:587–98. doi: 10.1002/ana.23659
34. Braak H, Rub U, Jansen SE, Del TK, de Vos RA. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology. (2005) 64:1404–10. doi: 10.1212/01.WNL.0000158422.41380.82
35. Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, et al. Unified staging system for lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. (2009) 117:613–34. doi: 10.1007/s00401-009-0538-8
36. Mattila PM, Rinne JO, Helenius H, Dickson DW, Roytta M. Alpha-synuclein-immunoreactive cortical lewy bodies are associated with cognitive impairment in Parkinson's disease. Acta Neuropathol. (2000) 100:285–90. doi: 10.1007/s004019900168
37. Kövari E, Gold G, Herrmann FR, Canuto A, Hof PR, Bouras C, et al. Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson's disease. Acta Neuropathol. (2003) 106:83–8. doi: 10.1007/s00401-003-0705-2
38. Fereshtehnejad S, Zeighami Y, Dagher A, Postuma RB. Clinical criteria for subtyping Parkinson's disease: biomarkers and longitudinal progression. Brain. (2017) 140:1959–76. doi: 10.1093/brain/awx118
39. Liu C, Cholerton B, Shi M, Ginghina C, Cain KC, Auinger P, et al. CSF tau and tau/aβ42 predict cognitive decline in Parkinson's disease. Parkinsonism Relat D. (2015) 21:271–6. doi: 10.1016/j.parkreldis.2014.12.027
40. Parnetti L, Farotti L, Eusebi P, Chiasserini D, De Carlo C, Giannandrea D, et al. Differential role of csf alpha-synuclein species, tau, and Aî242 in Parkinson's disease. Front Aging Neurosci. (2014) 6:53. doi: 10.3389/fnagi.2014.00053
41. Schrag A, Siddiqui UF, Anastasiou Z, Weintraub D, Schott JM. Clinical variables and biomarkers in prediction of cognitive impairment in patients with newly diagnosed Parkinson's disease: a cohort study. Lancet Neurol. (2017) 16:66–75. doi: 10.1016/S1474-4422(16)30328-3
42. Harding AJ, Halliday GM. Cortical lewy body pathology in the diagnosis of dementia. Acta Neuropathol. (2001) 102:355–63. doi: 10.1007/s004010100390
43. Pan L, Li C, Meng L, Tian Y, He M, Yuan X, et al. Tau accelerates alpha-synuclein aggregation and spreading in Parkinson's disease. Brain. (2022). doi: 10.1093/brain/awac171
44. Jin Y, Li F, Sonoustoun B, Kondru NC, Martens YA, Qiao W, et al. Apoe4 exacerbates α-synuclein seeding activity and contributes to neurotoxicity in Alzheimer's disease with lewy body pathology. Acta Neuropathol. (2022) 143:641–62. doi: 10.1007/s00401-022-02421-8
45. Lindqvist D, Hall S, Surova Y, Nielsen HM, Janelidze S, Brundin L, et al. Cerebrospinal fluid inflammatory markers in Parkinson's disease – associations with depression, fatigue, and cognitive impairment. Brain Behav Immun. (2013) 33:183–9. doi: 10.1016/j.bbi.2013.07.007
46. Williams Gray CH, Wijeyekoon R, Yarnall AJ, Lawson RA, Breen DP, Evans JR, et al. Serum immune markers and disease progression in an incident Parkinson's disease cohort (icicle-pd). Movement Disord. (2016) 31:995–1003. doi: 10.1002/mds.26563
47. La Vitola P, Balducci C, Baroni M, Artioli L, Santamaria G, Castiglioni M, et al. Peripheral inflammation exacerbates α-synuclein toxicity and neuropathology in Parkinson's models. Neuropath Appl Neuro. (2021) 47:43–60. doi: 10.1111/nan.12644
48. Fellner L, Stefanova N. The role of glia in alpha-synucleinopathies. Mol Neurobiol. (2013) 47:575–86. doi: 10.1007/s12035-012-8340-3
49. Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, et al. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. (2013) 61:349–60. doi: 10.1002/glia.22437
50. Navarro V, Sanchez-Mejias E, Jimenez S, Muñoz-Castro C, Sanchez-Varo R, Davila JC, et al. Microglia in Alzheimer's disease: activated, dysfunctional or degenerative. Front Aging Neurosci. (2018) 10:140. doi: 10.3389/fnagi.2018.00140
51. Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The central nervous system and the gut microbiome. Cell. (2016) 167:915–32. doi: 10.1016/j.cell.2016.10.027
52. Ren T, Gao Y, Qiu Y, Jiang S, Zhang Q, Zhang J, et al. Gut microbiota altered in mild cognitive impairment compared with normal cognition in sporadic Parkinson's disease. Front Neurol. (2020) 11:137. doi: 10.3389/fneur.2020.00137
53. Braak H, Sastre M, Bohl JRE, de Vos RAI, Del Tredici K. Parkinson's disease: lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol. (2007) 113:421–9. doi: 10.1007/s00401-007-0193-x
54. Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, et al. Exogenous α-synuclein fibrils induce lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. (2011) 72:57–71. doi: 10.1016/j.neuron.2011.08.033
55. Pan-Montojo F, Schwarz M, Winkler C, Arnhold M, O'Sullivan GA, Pal A, et al. Environmental toxins trigger pd-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep. (2012) 2:898. doi: 10.1038/srep00898
56. Luk KC, Kehm V, Carroll J, Zhang B, O Brien P, Trojanowski JQ, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. (2012) 338:949–53. doi: 10.1126/science.1227157
57. Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol. (2010) 6:702–6. doi: 10.1038/nrneurol.2010.145
58. Kim S, Kwon S, Kam T, Panicker N, Karuppagounder SS, Lee S, et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson's disease. Neuron. (2019) 103:627–41. doi: 10.1016/j.neuron.2019.05.035
59. Sorrentino ZA, Xia Y, Gorion KM, Hass E, Giasson BI. Carboxy-terminal truncations of mouse alpha-synuclein alter aggregation and prion-like seeding. FEBS Lett. (2020) 594:1271–83. doi: 10.1002/1873-3468.13728
60. Du X, Xie X, Liu R. The role of α-synuclein oligomers in Parkinson's disease. Int J Mol Sci. (2020) 21:8645. doi: 10.3390/ijms21228645
61. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell. (2016) 167:1469–80. doi: 10.1016/j.cell.2016.11.018
62. Bhattarai Y. Microbiota-gut-brain axis: interaction of gut microbes and their metabolites with host epithelial barriers. Neurogastroenterol Motil. (2018) 30:e13366. doi: 10.1111/nmo.13366
63. Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson's disease. PLoS One. (2011) 6:e28032. doi: 10.1371/journal.pone.0028032
64. Bhattacharyya D, Mohite GM, Krishnamoorthy J, Gayen N, Mehra S, Navalkar A, et al. Lipopolysaccharide from gut microbiota modulates α-synuclein aggregation and alters its biological function. Acs Chem Neurosci. (2019) 10:2229–36. doi: 10.1021/acschemneuro.8b00733
65. Banks WA, Gray AM, Erickson MA, Salameh TS, Damodarasamy M, Sheibani N, et al. Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J Neuroinflamm. (2015) 12:223. doi: 10.1186/s12974-015-0434-1
66. Peng X, Luo Z, He S, Zhang L, Li Y. Blood-brain barrier disruption by lipopolysaccharide and sepsis-associated encephalopathy. Front Cell Infect Microbiol. (2021) 11:768108. doi: 10.3389/fcimb.2021.768108
67. Dodiya HB, Lutz HL, Weigle IQ, Patel P, Michalkiewicz J, Roman-Santiago CJ, et al. Gut microbiota–driven brain aβ amyloidosis in mice requires microglia. J Exp Med. (2022). doi: 10.1084/jem.20200895
68. Kaur T, Uppoor A, Naik D. Parkinson's disease and periodontitis - the missing link? A review. Gerodontology. (2016) 33:434–8. doi: 10.1111/ger.12188
69. Hanaoka A, Kashihara K. Increased frequencies of caries, periodontal disease and tooth loss in patients with Parkinson's disease. J Clin Neurosci. (2009) 16:1279–82. doi: 10.1016/j.jocn.2008.12.027
70. Olsen I, Kell DB, Pretorius E. Is Porphyromonas gingivalis involved in Parkinson's disease? Eur J Clin Microbiol. (2020) 39:2013–8. doi: 10.1007/s10096-020-03944-2
71. Chen C, Huang J, Wu Y, Chang Y. Dental scaling decreases the risk of Parkinson's disease: a nationwide population-based nested case-control study. Int J Env Res Pub Health. (2018) 15:1587. doi: 10.3390/ijerph15081587
72. Pretorius E, Bester J, Page MJ, Kell DB. The potential of LPS-binding protein to reverse amyloid formation in plasma fibrin of individuals with Alzheimer-type dementia. Front Aging Neurosci. (2018) 10:257. doi: 10.3389/fnagi.2018.00257
73. Gaig C, Ezquerra M, Marti MJ, Munoz E, Valldeoriola F, Tolosa E. Lrrk2 mutations in spanish patients with Parkinson disease: frequency, clinical features, and incomplete penetrance. Arch Neurol. (2006) 63:377–82. doi: 10.1001/archneur.63.3.377
74. Wu Z, Ni J, Liu Y, Teeling JL, Takayama F, Collcutt A, et al. Cathepsin B plays a critical role in inducing Alzheimer's disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun. (2017) 65:350–61. doi: 10.1016/j.bbi.2017.06.002
75. Nie R, Wu Z, Ni J, Zeng F, Yu W, Zhang Y, et al. Porphyromonas gingivalis infection induces amyloid-β accumulation in monocytes/macrophages. J Alzheimer's Dis. (2019) 72:479–94. doi: 10.3233/JAD-190298
76. Zeng F, Liu Y, Huang W, Qing H, Kadowaki T, Kashiwazaki H, et al. Receptor for advanced glycation end products up-regulation in cerebral endothelial cells mediates cerebrovascular-related amyloid β accumulation after Porphyromonas gingivalis infection. J Neurochem. (2021) 158:724–36. doi: 10.1111/jnc.15096
77. von Troil-Lindén B, Torkko H, Alaluusua S, Jousimies-Somer H, Asikainen S. Salivary levels of suspected periodontal pathogens in relation to periodontal status and treatment. J Dent Res. (1995) 74:1789–95. doi: 10.1177/00220345950740111201
78. Saygun I, Nizam N, Keskiner I, Bal V, Kubar A, Acikel C, et al. Salivary infectious agents and periodontal disease status. J Periodontal Res. (2011) 46:235–9. doi: 10.1111/j.1600-0765.2010.01335.x
79. Boutaga K, Savelkoul PHM, Winkel EG, van Winkelhoff AJ. Comparison of subgingival bacterial sampling with oral lavage for detection and quantification of periodontal pathogens by real-time polymerase chain reaction. J Periodontol. (2007) 78:79–86. doi: 10.1902/jop.2007.060078
80. Nakajima M, Arimatsu K, Kato T, Matsuda Y, Minagawa T, Takahashi N, et al. Oral administration of P. gingivalis induces dysbiosis of gut microbiota and impaired barrier function leading to dissemination of enterobacteria to the liver. PLoS ONE. (2015) 10:e134234. doi: 10.1371/journal.pone.0134234
81. Arimatsu K, Yamada H, Miyazawa H, Minagawa T, Nakajima M, Ryder MI, et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci Rep. (2015) 11:768108. doi: 10.1038/srep04828
82. Sato K, Takahashi N, Kato T, Matsuda Y, Yokoji M, Yamada M, et al. Aggravation of collagen-induced arthritis by orally administered Porphyromonas gingivalis through modulation of the gut microbiota and gut immune system. Sci Rep. (2017) 7:6955. doi: 10.1038/s41598-017-07196-7
83. Kashiwagi Y, Aburaya S, Sugiyama N, Narukawa Y, Sakamoto Y, Takahashi M, et al. Porphyromonas gingivalis induces entero-hepatic metabolic derangements with alteration of gut microbiota in a type 2 diabetes mouse model. Sci Rep. (2021) 11:18398. doi: 10.1038/s41598-021-97868-2
84. Wang T, Ishikawa T, Sasaki M, Chiba T. Oral and gut microbial dysbiosis and non-alcoholic fatty liver disease: the central role of Porphyromonas gingivalis. Front Med. (2022). doi: 10.3389/fmed.2022.822190
85. Chi L, Cheng X, Lin L, Yang T, Sun J, Feng Y, et al. Porphyromonas gingivalis-induced cognitive impairment is associated with gut dysbiosis, neuroinflammation, and glymphatic dysfunction. Front Cell Infect Microbiol. (2021) 11:755925. doi: 10.3389/fcimb.2021.755925
Keywords: Parkinson's disease, cognitive impairment, Porphyromonas gingivalis, neuroinflammation, α-Synuclein, amyloid-β, gut microbiota
Citation: Li D, Ren T, Li H, Liao G and Zhang X (2022) Porphyromonas gingivalis: A key role in Parkinson's disease with cognitive impairment? Front. Neurol. 13:945523. doi: 10.3389/fneur.2022.945523
Received: 16 May 2022; Accepted: 04 July 2022;
Published: 26 July 2022.
Edited by:
Jinyoung Youn, Sungkyunkwan University School of Medicine, South KoreaReviewed by:
Greg Sutherland, The University of Sydney, AustraliaTejaswani Kaur, CMC Ludhiana, India
Copyright © 2022 Li, Ren, Li, Liao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiong Zhang, eGlvbmc3MTVAMTI2LmNvbQ==