 Ruihan Wang
Ruihan Wang Hui Gao
Hui Gao Hongsheng Xie2
Hongsheng Xie2 Zhiyun Jia
Zhiyun Jia Qin Chen
Qin Chen- 1Department of Neurology, West China Hospital of Sichuan University, Chengdu, China
- 2Department of Nuclear Medicine, West China Hospital of Sichuan University, Chengdu, China
Familial frontotemporal lobar degeneration (FTLD) is a pathologically heterogeneous group of neurodegenerative diseases with diverse genotypes and clinical phenotypes. Three major mutations were reported in patients with familial FTLD, namely, progranulin (GRN), microtubule-associated protein tau (MAPT), and the chromosome 9 open reading frame 72 (C9orf72) repeat expansion, which could cause neurodegenerative pathological changes years before symptom onset. Noninvasive quantitative molecular imaging with PET or single-photon emission CT (SPECT) allows for selective visualization of the molecular targets in vivo to investigate brain metabolism, perfusion, neuroinflammation, and pathophysiological changes. There was increasing evidence that several molecular imaging biomarkers tend to serve as biomarkers to reveal the early brain abnormalities in familial FTLD. Tau-PET with 18F-flortaucipir and 11C-PBB3 demonstrated the elevated tau position in patients with FTLD and also showed the ability to differentiate patterns among the different subtypes of the mutations in familial FTLD. Furthermore, dopamine transporter imaging with the 11C-DOPA and 11C-CFT in PET and the 123I-FP-CIT in SPECT revealed the loss of dopaminergic neurons in the asymptomatic and symptomatic patients of familial FTLD. In addition, PET imaging with the 11C-MP4A has demonstrated reduced acetylcholinesterase (AChE) activity in patients with FTLD, while PET with the 11C-DAA1106 and 11C-PK11195 revealed an increased level of microglial activation associated with neuroinflammation even before the onset of symptoms in familial FTLD. 18F-fluorodeoxyglucose (FDG)-PET indicated hypometabolism in FTLD with different mutations preceded the atrophy on MRI. Identifying molecular imaging biomarkers for familial FTLD is important for the in-vivo assessment of underlying pathophysiological changes with disease progression and future disease-modifying therapy. We review the recent progress of molecular imaging in familial FTLD with focused on the possible implication of these techniques and their prospects in specific mutation types.
Introduction
Frontotemporal lobar degeneration (FTLD) encompasses a set of clinical syndromes characterized by progressive abnormalities in behavior, executive function, language, or motor function. Patients with FTLD may present clinical syndromes with the behavioral variant of frontotemporal dementia (bvFTD), the nonfluent variant of a primary progressive aphasia (nfvPPA), a semantic variant of PPA (svPPA), and some patients also have amyotrophic lateral sclerosis (ALS), corticobasal syndrome (CBS) or progressive supranuclear palsy (PSP) (1). Approximately, 40% of patients with FTLD have a positive family history of autosomal dominant inheritance (2). Three major mutations were reported in patients with familial FTLD, namely, the microtubule-associated protein tau (MAPT), progranulin (GRN), and the repeat expansions in the chromosome 9 open reading frame 72 (C9orf72). These mutations could lead to neurodegenerative pathological changes years before symptom onset (2, 3).
Mutations in the MAPT gene located on chromosome 17q21 first reported in 1998 (4) were discovered in numerous pedigrees of familial FTLD. The majority of known mutations in the coding region occur in the repeats, causing the decreased ability of tau proteins to interact with microtubules, and resulting in hyperphosphorylated tau accumulation in neurons and glial cells (5). MAPT mutations of different subtypes have been linked to various tauopathies. Generally, the mutations inside exon 10 (i.e., N279K, S305N, P301L) and intron 10 (i.e., IVS 10 + 16) tend to form four tandem microtubule-binding domain repeat (4R-tau) pathology, while mutations outside exon 10 (i.e., V337M, R406W, Q351R) tend to form mixed 3R/4R tauopathy similar to the tauopathy in Alzheimer's disease (6).
Mutations in the GRN linked to chromosome 17q21 initially reported in 2006 (7, 8) could result in a lack of progranulin by haploinsufficiency and the accumulation of TAR DNA-binding protein (TDP)-43 protein (9). GRN mutation carriers have a wide range of clinical phenotypes and illness onset ages. The bvFTD, CBS, and PPA are the most common clinical syndromes in patients with GRN mutation (10, 11).
The hexanucleotide GGGGCC (G4C2) repeat expansions of the C9orf72 were identified as a common genetic cause of FTLD and ALS in 2011 (12, 13). TDP-43 aggregations were pathologically discovered in cases with C9orf72 expansions (14). The most prevalent clinical syndromes were bvFTD, ALS, or a mixture of both in C9orf72 mutation carriers (14, 15).
Currently, the visual inspection of MRI was demonstrated to easily increase the diagnostic confidence of underlying FTLD (16, 17). The cortical microstructure was found to be more sensitive than cerebral atrophy within patients with GRN mutations (18), suggesting the powerful value of MRI to correctly diagnose and capture the early abnormalities in familial FTLD. Noninvasive quantitative molecular imaging with PET or single-photon emission CT (SPECT) provided another perspective and allowed for selective visualization of the molecular targets in vivo to investigate the brain topographic and pathophysiological changes. The former included metabolism, perfusion, neuroinflammation, synaptic function, and neurotransmitters' activity, and the latter comprised Tau and Aβ aggregation. There was increasing evidence that several molecular imaging biomarkers tend to serve as biomarkers to reveal the early brain abnormalities in familial FTLD. Identifying molecular imaging biomarkers for familial FTLD is important for the in-vivo assessment of underlying pathophysiological changes with disease progression and future disease-modifying therapy. Thus, we review the recent progress of molecular imaging in familial FTLD with a focus on the possible implication of these techniques and their prospects in specific mutation types.
Methods
Search strategy
We performed electronic searches of Medline, PubMed, and Embase databases using the combination of a number of medical subject headings, Emtree subject headings, and free-text terms (“frontotemporal lobar degeneration,” and “frontotemporal dementia” for clinical categories; “microtubule-associated protein tau” or “MAPT,” “progranulin” or “GRN,” and “chromosome 9 open reading frame 72” or “C9orf72” for genes; “positron emission tomography” or “PET,” “single-photon emission CT” or “SPECT,” and “dopamine transporter imaging” for molecular imaging biomarkers). The retrieval deadline was 1 December 2021. All the relevant articles were retrieved, placing restrictions on #elds (free-text terms searched exclusively in the title or abstract of the articles) and publication type (original articles).
Discussion
Pathophysiological biomarkers
Tau studies
Tau-PET is currently being explored as a promising method to identify the tau protein in vivo (19). Several types of tracers have been applied to map the pattern of tau accumulation in familial FTLD, especially in individuals with MAPT mutations thought to be tauopathy. 18F-flortaucipir, the most commonly used tau tracer, has been proven to bind paired helical filaments composed of 3R/4R tau in Alzheimer's disease (AD) (20, 21). In recent years, other tracers, including 11C-PBB3 (22), 18F-MK6240 (23), and 18F-PM-PBB3 (24), started to be applied in MAPT mutation carriers. 11C-PBB3 could capture wide-ranging Tau pathologies, including 3R/4R tau and 4R tau (25, 26) compared to18F-flortaucipir (27). For 18F-MK6240 and 18F-PM-PBB3, no clear off-target binding was reported with the improved design (28–31). 18F-PM-PBB3 has shown higher binding affinities to 4R tau compared with the 3R/4R tracer 18F-MK6240 (24). The novel tau tracers might help show diverse tau pathologies in various mutation subtypes.
MAPT_Tau-PET
Frontotemporal lobar degeneration with MAPT mutations is regarded as tauopathy (32), and tau PET provides an effective way to explore biomarkers for multiform tau pathologies in a homogeneous patient group (33). Most individuals with MAPT mutations inside exon 10 (i.e., P301L, S305N, N279K) and intron 10 (i.e., IVS 10 + 16) had 4R tau pathology, while with MAPT mutations outside exon 10 (i.e., V337M, R406W, Q351R) had 3R/4R paired helical filament tau pathology (34). MAPT mutations have different types of underlying tauopathies, leading to different tracer binding patterns.
18F-flortaucipir was most commonly used to track 3R/4R tau, so most participants with MAPT mutations outside exon 10 showed a higher-level tracer binding than mutations in exon 10 (34). Only a few 18F-flortaucipir studies included 4 asymptomatic MAPT mutation carriers. 3 of them (1 N279K, 1 R406W, and 1 IVS 10 + 16) had little to no uptake, but the other MAPT R406W mutation carrier had a signal in the AD range (34, 35). The heterogeneous results might be explained by quite a limited sample size. In symptomatic MAPT mutation carriers, temporal (36–42), insular (37, 41, 42), and frontal (38, 39, 41) regions were most commonly reported for increased 18F-flortaucipir uptake. Especially, in two MAPT R406W mutation carriers with short disease duration, the hippocampus and adjacent temporal lobe regions were mainly involved, whereas in another MAPT R406W mutation carrier with a long duration, tau aggregation spreads across the whole temporal, frontal lobes, and the basal ganglia (39). Moreover, a longitudinal study of a MAPT Q351R mutation carrier also demonstrated that tau aggregation expanded from the insula region cortically, and the medial temporal, putamen, and pallidum regions subcortically to the temporal region cortically and caudate and thalamus regions subcortically even just over 1 year (37). These findings suggested that 18F-flortaucipir might be a sensitive biomarker for disease progression in symptomatic MAPT mutation carriers. However, the majority of research was based on case reports or cross-sectional studies with small sample size. Longitudinal data with larger cohorts will be required for such investigations.
Two studies applied 11C/18F-PBB3 tracking both the 3R/4R tau and 4R tau in symptomatic MAPT mutation carriers (22, 24). In four patients with MAPT N279K mutation, the kindred with slow progression exhibited mild binding; in contrast, kindreds with rapid progression showed profoundly increased binding in widespread regions from an early disease stage (22). Recently, a study of 18F-MK-6240 in two asymptomatic MAPT P301L mutation carriers showed modest tau deposition about 5 years from estimated onset (23), indicating that 18F-MK-6240 uptake might be an early biomarker for MAPT P301L mutation carriers (Table 1).
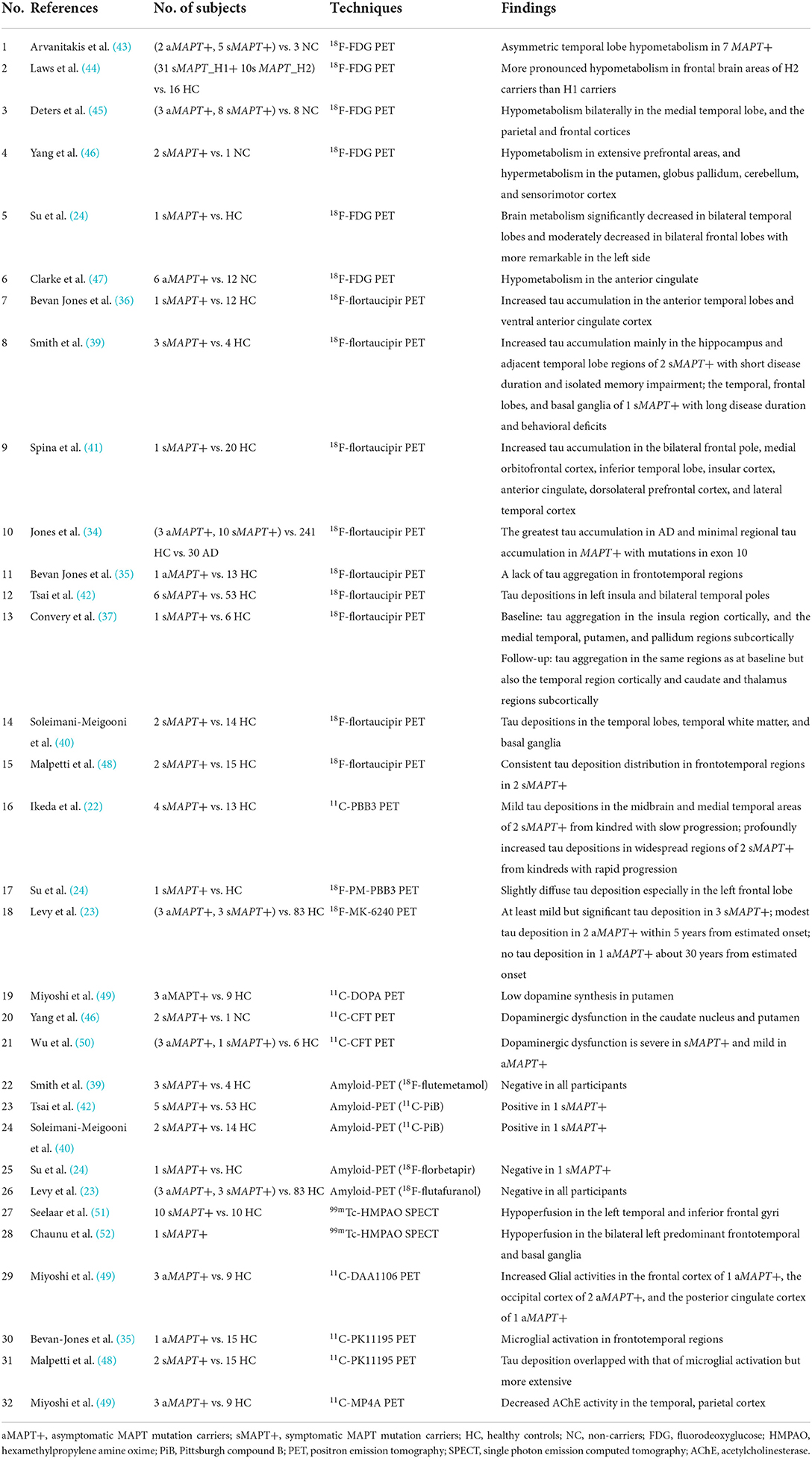
Table 1. Studies investigating MAPT mutation vs. controls.
In MAPT mutation carriers, the value of tau PET for capturing tau accumulation has been primarily proved, and the tau aggregation patterns were associated with the subtypes of mutations and tracers. Therefore, novel tracers for multiform tau pathologies need to be further explored in longitudinal studies with larger cohorts.
GRN/C9orf72_Tau-PET
Three studies reported 18F-flortaucipir binding in the frontotemporal region in five symptomatic GRN mutation carriers (38, 40, 42), whereas another research found no 18F-flortaucipir binding in a patient with GRN mutation (53) (Table 2). Similarly, findings among symptomatic C9orf72 mutation carriers were contradictory. Ten patients with C9orf72 mutation had increased 18F-flortaucipir binding in the frontal lobe (38, 40, 42, 64), while another study found no tau deposition in six patients with C9orf72 mutation (65) (Table 3). The contradictory results might be associated with the small number of participants and different clinical phenotypes. Moreover, a study showed that 18F-MK-6240 PET scan was negative for three individuals with GRN or C9orf72 mutations (23), implying that 18F-MK-6240 might not be an optimal method for tracking tau deposition in GRN or C9orf72 mutation carriers.
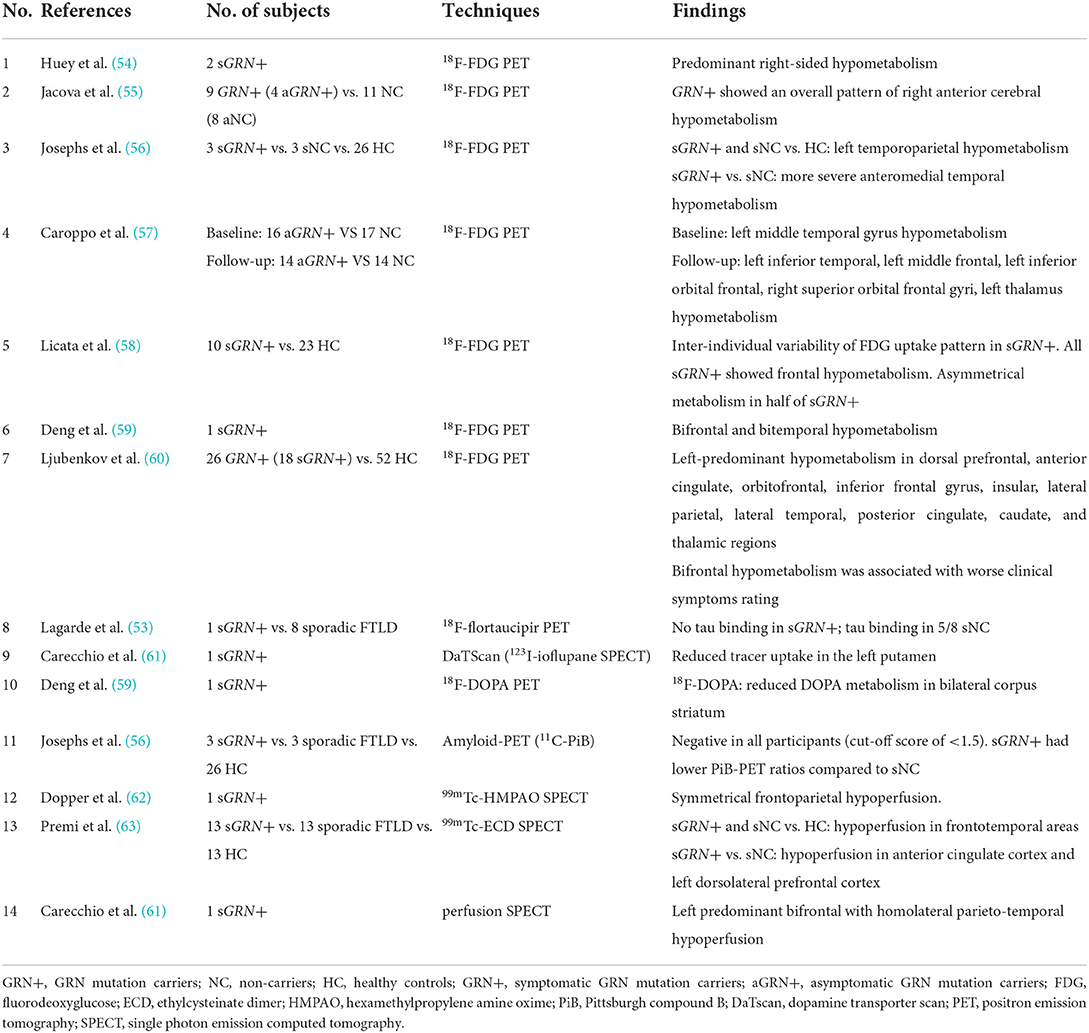
Table 2. Studies investigated asymptomatic/symptomatic GRN carriers.
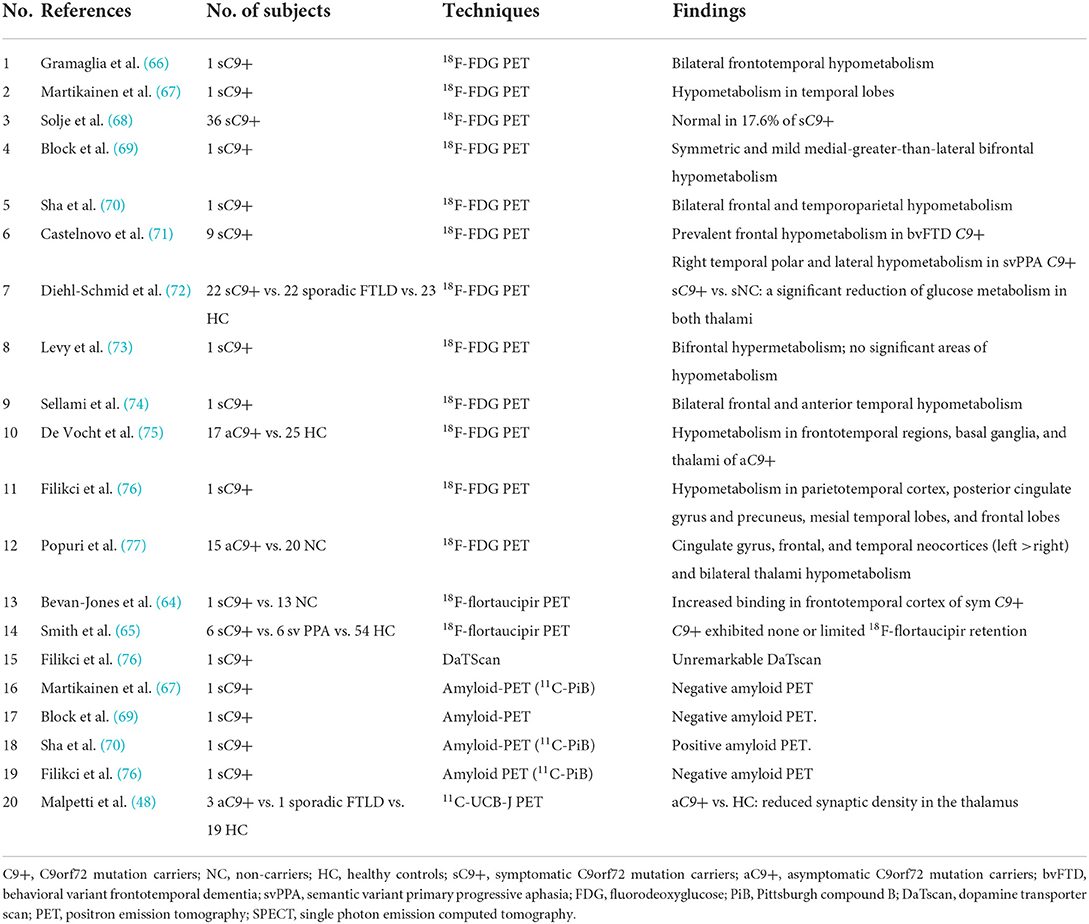
Table 3. Studies investigated asymptomatic/symptomatic C9 carriers.
Amyloid studies
To detect the underlying AD pathology, amyloid-PET with tracers, including 11C-Pittsburgh compound B (PiB) (42, 67, 70, 76), 18F-florbetapir (24), 18F-florbetaben (23), 18F-flutafuranol (78), 18F-flutemetamol (39, 79), etc., is applied in patients with familial FTLD.
Most patients with MAPT mutation indicated negative results with 11C-PiB or 18F-florbetapir PET (23, 24, 39, 42), while two patients with MAPT P301L mutation had a positive 11C-PiB scan (40, 42). However, one might imply an incidental rather than preclinical β-amyloid pathology since the SUVRs were well below those seen in AD (42); in contrast, the other regarded as combining with AD presented higher SUVRs close to AD (40). Negative results with 11C-PiB or 18F-flutafuranol were reported in patients with GRN and C9orf72 mutation carriers so far (23, 42, 56, 76). Thus, amyloid-PET may help discriminate true underlying AD co-pathology from incidental β-amyloid pathology (80) (Table 4).
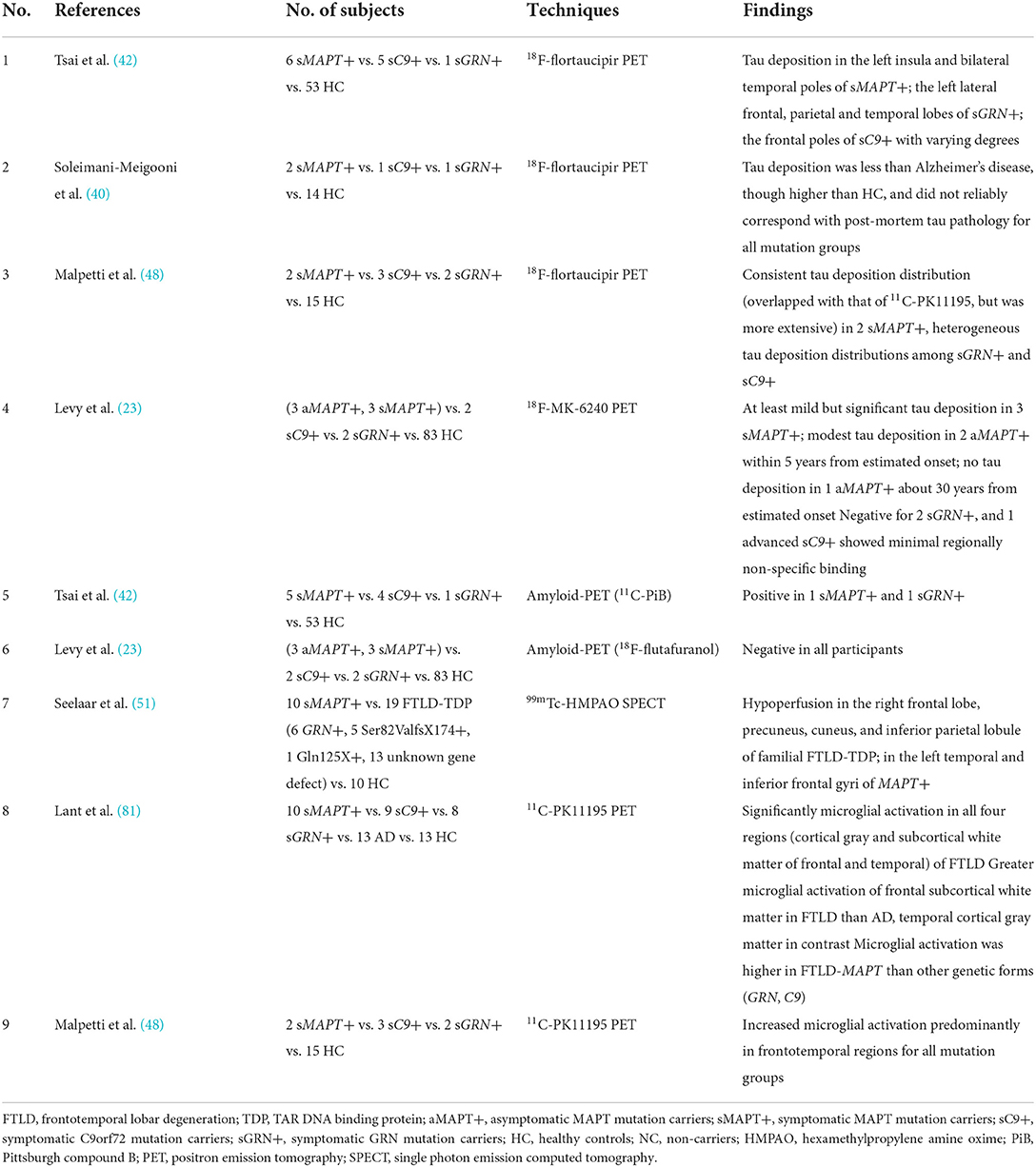
Table 4. Studies investigating multiple different mutations in FTLD.
Topographic biomarkers
Brain metabolism
18F-fluorodeoxyglucose (FDG)-PET is a technique for measuring glucose metabolism in vivo (82). Studies of FDG-PET could capture the different patterns of brain hypometabolism and even precede brain atrophy in familial FTLD mutation carriers (43, 45, 47, 55, 57, 72, 83).
MAPT_FDG-PET
A few cross-sectional FDG-PET studies demonstrated brain hypometabolism in both the asymptomatic and symptomatic MAPT mutation carriers (24, 43, 45–47). Hypometabolism in the temporal lobe (43, 45) and anterior cingulate cortex (47) was reported in asymptomatic MAPT mutation carriers, while temporal lobe hypometabolism even preceded the brain atrophy on MRI in the asymptomatic stage (43). In symptomatic MAPT mutation carriers, hypometabolism regions spread extensively to the frontotemporal lobes (24, 43, 46), while hypermetabolism was also found in the putamen, globus pallidum, cerebellum, and sensorimotor cortex (46). These findings all pointed to early involvement of the temporal lobe in asymptomatic MAPT mutation carriers. Furthermore, only one study compared three asymptomatic MAPT mutation carriers and 8 symptomatic MAPT mutation carriers, but found no difference in FDG uptake (45), which was mainly due to the small sample size. However, most current studies were cross-sectional with a small cohort, and further studies are needed to characterize the trajectories of metabolism patterns from asymptomatic to symptomatic MAPT mutation carriers.
GRN_FDG-PET
Two studies indicated asymmetric temporal lobe hypometabolism with FDG-PET in asymptomatic GRN mutation carriers (55, 57). After 20 months of follow-up, hypometabolism spread to the frontal lobe and thalamus (57). The metabolic changes appeared before brain atrophy on MRI and approximately more than 10 years before clinical onset (57), suggesting that FDG-PET changes can be detected as early biomarkers in GRN mutation carriers. In symptomatic GRN mutation carriers, the asymmetrical hypometabolism of temporoparietal (56) and frontal (58) lobes was reported primarily based on a small number of cross-sectional studies or case reports. Hypometabolism patterns were observed to correlate with clinical manifestations (56), but another study failed to find clear metabolic change pattern in each clinical subtype (58).
C9orf72_FDG-PET
In asymptomatic C9orf72 mutation carriers, extensive hypometabolism was observed in frontotemporal and subcortical regions in two studies (75, 77). Thalami hypometabolism was found in both the asymptomatic (75, 77) and symptomatic (72) individuals with C9orf72 mutation, especially when compared to sporadic FTLD patients (72), suggesting that thalami could be a distinguishing early biomarker for C9orf72 mutation carriers. In symptomatic C9orf72 mutation carriers, some studies showed that the hypometabolism patterns were consistent with the clinical diagnosis and correlated well with the brain atrophy on MRI, for example, prevalent frontal hypometabolism in patients with bvFTD and temporal polar and lateral temporal hypometabolism in patient with svPPA (66, 69, 71, 74). However, the cross-sectional studies above with small sample sizes still need to be replicated in longitudinal studies with larger cohorts.
Most studies demonstrated the concordance between structural MRI and FDG-PET in MAPT (43, 45), GRN (84, 85), and C9orf72 (74, 77) mutation carriers. However, controversy still existed regarding the earlier or more sensitive biomarkers (43, 45, 77). Some studies showed that additional informative MRI modalities such as diffusion tensor imaging (DTI) and arterial spin labeling (ASL) had equivalent or even better diagnostic utility of FTLD compared with FDG-PET (86–89), but others found a gap in sensitivity or accuracy that still remained (90, 91). Further investigations of familial FTLD need to compare the clinical value of microstructural MRI and PET.
Dopaminergic system
Dopamine functional deficits can be measured in vivo via PET or SPECT with various types of tracers assessing dopamine synthesis and storage [18F-DOPA, 11C-DOPA, 11C-dihydrotetrabenazine (DTBZ), 18F-fluoropropyl-DTBZ, etc.], transporter density (123I-FP-CIT, 123I-ioflupane,11C-CFT, 99mTc-TRODAT, etc.), or postsynaptic terminals [11C-raclopride, 123I-iodobenzamide (IBZM), etc.] (92). Dopaminergic deficits were evaluated by the techniques mentioned above, especially in patients with familial FTLD with Parkinsonism.
Parkinsonism may present as the initial symptom in MAPT mutation carriers, particularly individuals with MAPT N279K mutation. Tracers such as 11C-DOPA and 2b-carbomethoxy-3b-(4-trmethylstannylphenyl) tropane (11C-CFT) were used to reveal dopaminergic function. The 11C-CFT uptake in the putamen was mildly low in asymptomatic MAPT N279K mutation carriers (49, 50). In symptomatic patients, both the caudate nucleus and putamen were involved more heavily (46, 50).
Individuals with GRN mutations and Parkinsonism could show reduced DOPA metabolism in bilateral corpus striatum by 18F-DOPA PET (59) or reduced tracer uptake in left putamen by 123I-ioflupane SPECT (61). Parkinsonism is not uncommon in GRN mutation carriers and sporadic patients with FTLD.
Brain perfusion
Perfusion SPECT is a well-established technique for measuring regional cerebral blood flow (rCBF) to assess brain function (93). The tracers utilized in brain perfusion SPECT are technetium-99m-hexamethylpropyleneamineoxime (99mTc-HMPAO) and technetium-99m-ethylcysteinate dimer (99mTc-ECD), both which are distributed proportionally to rCBF (93). Perfusion imaging has been widely used in the clinical evaluation of patients with neurological and psychiatric diseases (94), including FTLD.
In 11 MAPT mutation carriers, including eight in P301L, two in G272V, and one in G389R, significant hypoperfusion detected by 99mTc-HMPAO SPECT was found in the asymmetric frontotemporal lobes (51, 52). Several studies indicated that hypoperfusion occurred in frontal areas of GRN mutation carriers (61–63). Compared with MAPT mutation carriers, patients with GRN mutation exhibited relatively more posterior hypoperfusion, including the precuneus and inferior parietal lobule detected by 99mTc-HMPAO SPECT (51). Perfusion SPECT might be a potential biomarker to identify MAPT and GRN mutation carriers.
Neuroinflammation
Previous studies of genome-wide association (95) and animal (96) suggest that neuroinflammation might be an earlier process in FTLD, even preceding tau accumulation. The neuroinflammation is accompanied by the activation of microglia, and 18 kDa TSPO, previously known as peripheral benzodiazepine receptors, is highly expressed (97). Thus, radioligands (11C-PK11195, 11C-DAA1106) have been developed to target TSPO to visualize neuroinflammation in vivo (98, 99).
In asymptomatic MAPT mutation carriers, two studies with 11C-PK11195 PET (35) or 11C-DAA1106 PET (49) revealed increased levels of microglial activation, even despite a lack of significant atrophy or 18F-flortaucipir uptake (35). In symptomatic patients, 18F-flortaucipir binding overlapped with 11C-PK11195 binding and was more extensive across the brain (38). These findings suggest that neuroinflammation might facilitate tau aggregation initially, then tau-mediated neurodegeneration takes the dominant role. Combining different modalities in a relatively homogeneous group such as familial FTLD with a specific mutation subtype would better understand the underlying mechanism of disease progression.
Across different mutation subtypes, familial patients with FTLD with MAPT, GRN, and C9orf72 mutations all showed increased 11C-PK11195 binding predominantly in frontotemporal regions (38), and 11C-PK11195 binding was significantly higher in temporal subcortical white matter in MAPT mutation carriers than in other genetic (GRN, C9orf72) mutation carriers or sporadic FTLD (81). Future studies could add more details to the neuroinflammation patterns of subtypes of familial FTLD.
Synaptic function and acetylcholinesterase activity
The synaptic vesicle glycoprotein 2A (SV2A) is a transmembrane protein ubiquitously expressed in secretory vesicles of synapsis in all the brain areas (100). It is critical for synaptic function (101), and it has been related to neurologic disorders such as AD and epilepsy (102–104). The density of SV2A could be quantified by the newly developed tracer 11C-UCB-J (105). Reduced synaptic density in the thalamus detected by 11C-UCB-J was found in three asymptomatic C9orf72 mutation carriers compared to healthy controls. It proved the role of the thalamus in C9orf72 mutation carriers again, especially before symptom onset (48). There is a lack of studies on synaptic density mapping in other early staged mutation carriers. Thus, its value and correspondence with other imaging techniques remain unknown.
11C-MP4A PET could reflect acetylcholinesterase (AChE) activity in vivo. A study showed reduced AchE activity in the temporoparietal cortex in one of three asymptomatic MAPT N279K mutation carriers (49). Therefore, more studies with larger sample sizes are needed to provide further evidence for 11C-MP4A PET in familial FTLD.
Challenges and limitations of molecular imaging
Even though more and more tracers were approved by the US Food and Drug Administration and by the European Medicines Agency for clinical usage (106), the higher cost and longer acquisition times compared to MRI might limit the wide applications in clinical practice (107). Changes in the levels of human fluid components could reflect underlying pathophysiological processes, and several fluid biomarkers were available or showed potential values such as Aβ, tau, NfL, and progranulin. A lack of multicenter standardization of procedures and quality control would compromise the stability and reliability of outcomes (108). By contrast, molecular imaging could provide more robust and comprehensive (quantitative and spatial distribution) information. However, the unspecific binding was still a challenge. Off-target binding of first-generation tau tracers such as 18F-flortaucipir might interfere with the quantification in several brain regions (109). Further development of 4R tau and TDP-43 specific tracers was needed to move toward precise diagnoses in FTLD. Several studies demonstrated that some molecular imaging biomarkers of FTLD with mutations could be different from sporadic individuals (72, 81), suggesting findings in genetic FTLD that may not translate to sporadic FTLD.
Conclusion
This review summarized recent molecular imaging findings in familial frontotemporal lobar degeneration regarding common genetic mutations. The application of advanced neuroimaging techniques in monogenetic familial FTLD provides a unique opportunity to study specific proteinopathies and their clinical phenotypes. Although various study designs and data analysis methods generated heterogeneous nonspecific results, some key biomarkers could still be identified, pointing to specific brain regions worth further exploring. The combination of multimodal neuroimaging would also help identify the underlying mechanism of these biomarkers. To date, this research topic has been limited by a large multicenter longitudinal cohort study and a comparison between asymptomatic/symptomatic mutation carriers and sporadic patients with FTLD. Thus, the changes in different time points of these biomarkers between FTLD mutation carriers and sporadic ones are largely unknown, and the prognostic value of these biomarkers is still unclear. Future studies could focus on these issues and provide more insight into the significance of these molecular imaging methods and their findings.
Author contributions
RW contributed to data collection, analysis and interpretation of the data, and drafting of the manuscript. HG contributed to analysis and interpretation of the data and drafting of the manuscript. HX and ZJ revised the manuscript. QC contributed to design the study, interpretation of the data, and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by National Natural Science Foundation of China (82071203), Science and Technology Innovation 2030 Major Projects (2022ZD0213600), and Natural Science Foundation of Sichuan (2022NSFSC1325).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Boeve BF, Boxer AL, Kumfor F, Pijnenburg Y, Rohrer JD. Advances and controversies in frontotemporal dementia: diagnosis, biomarkers, and therapeutic considerations. Lancet Neurol. (2022) 21:258–72. doi: 10.1016/S1474-4422(21)00341-0
2. Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia. Arch Neurol. (1999) 56:817–22. doi: 10.1001/archneur.56.7.817
3. Rohrer JD, Warren JD. Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol. (2011) 24:542–9. doi: 10.1097/WCO.0b013e32834cd442
4. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. (1998) 393:702–5. doi: 10.1038/31508
5. Bunker JM, Kamath K, Wilson L, Jordan MA, Feinstein SC. FTDP-17 mutations compromise the ability of tau to regulate microtubule dynamics in cells. J Biol Chem. (2006) 281:11856–63. doi: 10.1074/jbc.M509420200
6. McCarthy A, Lonergan R, Olszewska DA, O'Dowd S, Cummins G, Magennis B, et al. Closing the tau loop: the missing tau mutation. Brain. (2015) 138(Pt 10):3100–9. doi: 10.1093/brain/awv234
7. Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. (2006) 442:916–9. doi: 10.1038/nature05016
8. Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. (2006) 442:920–4. doi: 10.1038/nature05017
9. Pottier C, Ravenscroft TA, Sanchez-Contreras M, Rademakers R. Genetics of Ftld: overview and what else we can expect from genetic studies. J Neurochem. (2016) 138:32–53. doi: 10.1111/jnc.13622
10. Beck J, Rohrer JD, Campbell T, Isaacs A, Morrison KE, Goodall EF, et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain. (2008) 131(Pt 3):706–20. doi: 10.1093/brain/awm320
11. Kelley BJ, Haidar W, Boeve BF, Baker M, Graff-Radford NR, Krefft T, et al. Prominent phenotypic variability associated with mutations in progranulin. Neurobiol Aging. (2009) 30:739–51. doi: 10.1016/j.neurobiolaging.2007.08.022
12. Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9orf72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. (2011) 72:257–68.
13. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded Ggggcc hexanucleotide repeat in noncoding region of C9orf72 causes chromosome 9p-linked FTD and Als. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
14. Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the Ggggcc repeat expansion in C9orf72. Brain. (2012) 135(Pt 3):765–83. doi: 10.1016/j.jalz.2012.05.2129
15. Hsiung GY, DeJesus-Hernandez M, Feldman HH, Sengdy P, Bouchard-Kerr P, Dwosh E, et al. Clinical and pathological features of familial frontotemporal dementia caused by C9orf72 mutation on chromosome 9p. Brain. (2012) 135(Pt 3):709–22. doi: 10.1093/brain/awr354
16. Falgàs N, Balasa M, Bargalló N, Borrego-Écija S, Ramos-Campoy O, Fernández-Villullas G, et al. Diagnostic accuracy of MRI visual rating scales in the diagnosis of early onset cognitive impairment. J Alzheimers Dis. (2020) 73:1575–83. doi: 10.3233/JAD-191167
17. Illán-Gala I, Falgàs N, Friedberg A, Castro-Suárez S, Keret O, Rogers N, et al. Diagnostic utility of measuring cerebral atrophy in the behavioral variant of frontotemporal dementia and association with clinical deterioration. JAMA Netw Open. (2021) 4:e211290. doi: 10.1001/jamanetworkopen.2021.1290
18. Illán-Gala I, Montal V, Borrego-Écija S, Mandelli ML, Falgàs N, Welch AE, et al. Cortical microstructure in primary progressive aphasia: a multicenter study. Alzheimers Res Ther. (2022) 14:27. doi: 10.1186/s13195-022-00974-0
19. Leuzy A, Chiotis K, Lemoine L, Gillberg PG, Almkvist O, Rodriguez-Vieitez E, et al. Tau pet imaging in neurodegenerative tauopathies-still a challenge. Mol Psychiatry. (2019) 24:1112–34. doi: 10.1038/s41380-018-0342-8
20. Marquié M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG, et al. Validating novel tau positron emission tomography tracer [F-18]-Av-1451 (T807) on postmortem brain tissue. Ann Neurol. (2015) 78:787–800. doi: 10.1002/ana.24517
21. Xia CF, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D, et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer's disease. Alzheimers Dement. (2013) 9:666–76. doi: 10.1016/j.jalz.2012.11.008
22. Ikeda A, Shimada H, Nishioka K, Takanashi M, Hayashida A, Li Y, et al. Clinical heterogeneity of frontotemporal dementia and Parkinsonism linked to chromosome 17 caused by Mapt N279k mutation in relation to tau positron emission tomography features. Mov Disord. (2019) 34:568–74. doi: 10.1002/mds.27623
23. Levy JP, Bezgin G, Savard M, Pascoal TA, Finger E, Laforce R, et al. 18f-Mk-6240 tau-pet in genetic frontotemporal dementia. Brain. (2021) 145:1763–72. doi: 10.1093/brain/awab392
24. Su Y, Fu J, Yu J, Zhao Q, Guan Y, Zuo C, et al. Tau pet imaging with [18f]Pm-Pbb3 in frontotemporal dementia with mapt mutation. J Alzheimers Dis. (2020) 76:149–57. doi: 10.3233/JAD-200287
25. Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, et al. Imaging of tau pathology in a tauopathy mouse model and in alzheimer patients compared to normal controls. Neuron. (2013) 79:1094–108. doi: 10.1016/j.neuron.2013.07.037
26. Perez-Soriano A, Arena JE, Dinelle K, Miao Q, McKenzie J, Neilson N, et al. Pbb3 Imaging in parkinsonian disorders: evidence for binding to tau and other proteins. Mov Disord. (2017) 32:1016–24. doi: 10.1002/mds.27029
27. Ono M, Sahara N, Kumata K, Ji B, Ni R, Koga S, et al. Distinct binding of pet ligands Pbb3 and Av-1451 to tau fibril strains in neurodegenerative tauopathies. Brain. (2017) 140:764–80. doi: 10.1093/brain/aww339
28. Pascoal TA, Therriault J, Benedet AL, Savard M, Lussier FZ, Chamoun M, et al. 18f-Mk-6240 pet for early and late detection of neurofibrillary tangles. Brain. (2020) 143:2818–30. doi: 10.1093/brain/awaa180
29. Tagai K, Ono M, Kubota M, Kitamura S, Takahata K, Seki C, et al. High-contrast in vivo imaging of tau pathologies in Alzheimer's and non-Alzheimer's disease tauopathies. Neuron. (2021) 109:42–58.e8. doi: 10.1016/j.neuron.2020.09.042
30. Yap SY, Frias B, Wren MC, Schöll M, Fox NC, Årstad E, et al. Discriminatory ability of next-generation tau pet tracers for Alzheimer's disease. Brain. (2021) 144:2284–90. doi: 10.1093/brain/awab120
31. Leuzy A, Pascoal TA, Strandberg O, Insel P, Smith R, Mattsson-Carlgren N, et al. A multicenter comparison of [(18)F]Flortaucipir, [(18)F]Ro948, and [(18)F]Mk6240 tau pet tracers to detect a common target roi for differential diagnosis. Eur J Nucl Med Mol Imaging. (2021) 48:2295–305. doi: 10.1007/s00259-021-05401-4
32. Strang KH, Golde TE, Giasson BI. Mapt mutations, tauopathy, and mechanisms of neurodegeneration. Lab Invest. (2019) 99:912–28. doi: 10.1038/s41374-019-0197-x
33. Ricci M, Cimini A, Camedda R, Chiaravalloti A, Schillaci O. Tau biomarkers in dementia: positron emission tomography radiopharmaceuticals in tauopathy assessment and future perspective. Int J Mol Sci. (2021) 22:13002. doi: 10.3390/ijms222313002
34. Jones DT, Knopman DS, Graff-Radford J, Syrjanen JA, Senjem ML, Schwarz CG, et al. In vivo (18)F-Av-1451 tau pet signal in mapt mutation carriers varies by expected tau isoforms. Neurology. (2018) 90:e947–54. doi: 10.1212/WNL.0000000000005117
35. Bevan-Jones WR, Cope TE, Jones PS, Passamonti L, Hong YT, Fryer T, et al. In vivo evidence for pre-symptomatic neuroinflammation in a mapt mutation carrier. Ann Clin Transl Neurol. (2019) 6:373–8. doi: 10.1002/acn3.683
36. Bevan Jones WR, Cope TE, Passamonti L, Fryer TD, Hong YT, Aigbirhio F, et al. [(18)F]Av-1451 pet in behavioral variant frontotemporal dementia due to mapt mutation. Ann Clin Transl Neurol. (2016) 3:940–7. doi: 10.1002/acn3.366
37. Convery RS, Jiao J, Clarke MTM, Moore KM, Koriath CAM, Woollacott IOC, et al. Longitudinal ((18)F)Av-1451 pet imaging in a patient with frontotemporal dementia due to a Q351r Mapt mutation. J Neurol Neurosurg Psychiatry. (2020) 91:106–8. doi: 10.1136/jnnp-2019-320904
38. Malpetti M, Rittman T, Jones PS, Cope TE, Passamonti L, Bevan-Jones WR, et al. In vivo pet imaging of neuroinflammation in familial frontotemporal dementia. J Neurol Neurosurg Psychiatry. (2021) 92:319–22. doi: 10.1136/jnnp-2020-323698
39. Smith R, Puschmann A, Schöll M, Ohlsson T, van Swieten J, Honer M, et al. 18f-Av-1451 tau pet imaging correlates strongly with tau neuropathology in mapt mutation carriers. Brain. (2016) 139(Pt 9):2372–9. doi: 10.1093/brain/aww163
40. Soleimani-Meigooni DN, Iaccarino L, La Joie R, Baker S, Bourakova V, Boxer AL, et al. 18f-flortaucipir pet to autopsy comparisons in alzheimer's disease and other neurodegenerative diseases. Brain. (2020) 143:3477–94. doi: 10.1093/brain/awaa276
41. Spina S, Schonhaut DR, Boeve BF, Seeley WW, Ossenkoppele R, O'Neil JP, et al. Frontotemporal dementia with the V337m mapt mutation: tau-pet and pathology correlations. Neurology. (2017) 88:758–66. doi: 10.1212/WNL.0000000000003636
42. Tsai RM, Bejanin A, Lesman-Segev O, LaJoie R, Visani A, Bourakova V, et al. (18)F-Flortaucipir (Av-1451) tau pet in frontotemporal dementia syndromes. Alzheimers Res Ther. (2019) 11:13. doi: 10.1186/s13195-019-0470-7
43. Arvanitakis Z, Witte RJ, Dickson DW, Tsuboi Y, Uitti RJ, Slowinski J, et al. Clinical-pathologic study of biomarkers in FTDP-17 (Ppnd family with N279k tau mutation). Parkinsonism Relat Disord. (2007) 13:230–9. doi: 10.1016/j.parkreldis.2006.10.007
44. Laws SM, Perneczky R, Drzezga A, Diehl-Schmid J, Ibach B, Bäuml J, et al. Association of the tau haplotype h2 with age at onset and functional alterations of glucose utilization in frontotemporal dementia. Am J Psychiatry. (2007) 164:1577–84. doi: 10.1176/appi.ajp.2007.06091456
45. Deters KD, Risacher SL, Farlow MR, Unverzagt FW, Kareken DA, Hutchins GD, et al. Cerebral hypometabolism and grey matter density in mapt intron 10 +3 mutation carriers. Am J Neurodegener Dis. (2014) 3:103–14.
46. Yang Y, Tang BS, Weng L, Li N, Shen L, Wang J, et al. Genetic identification is critical for the diagnosis of parkinsonism: a Chinese pedigree with early onset of parkinsonism. PLoS ONE. (2015) 10:e0136245. doi: 10.1371/journal.pone.0136245
47. Clarke MTM, St-Onge F, Beauregard JM, Bocchetta M, Todd E, Cash DM, et al. Early anterior cingulate involvement is seen in presymptomatic Mapt P301l mutation carriers. Alzheimers Res Ther. (2021) 13:42. doi: 10.1186/s13195-021-00777-9
48. Malpetti M, Holland N, Jones PS, Ye R, Cope TE, Fryer TD, et al. Synaptic density in carriers of C9orf72 mutations: a [(11) C]Ucb-J pet study. Ann Clin Transl Neurol. (2021) 8:1515–23. doi: 10.1002/acn3.51407
49. Miyoshi M, Shinotoh H, Wszolek ZK, Strongosky AJ, Shimada H, Arakawa R, et al. In vivo detection of neuropathologic changes in presymptomatic mapt mutation carriers: a pet and MRI study. Parkinsonism Relat Disord. (2010) 16:404–8. doi: 10.1016/j.parkreldis.2010.04.004
50. Wu L, Liu J, Feng X, Dong J, Qin W, Liu Y, et al. 11c-Cft-pet in presymptomatic FTDP-17: a potential biomarker predicting onset. J Alzheimers Dis. (2018) 61:613–8. doi: 10.3233/JAD-170561
51. Seelaar H, Papma JM, Garraux G, de Koning I, Reijs AE, Valkema R, et al. Brain perfusion patterns in familial frontotemporal lobar degeneration. Neurology. (2011) 77:384–92. doi: 10.1212/WNL.0b013e3182270456
52. Chaunu MP, Deramecourt V, Buée-Scherrer V, Le Ber I, Brice A, Ehrle N, et al. Juvenile frontotemporal dementia with parkinsonism associated with tau mutation G389r. J Alzheimers Dis. (2013) 37:769–76. doi: 10.3233/JAD-130413
53. Lagarde J, Olivieri P, Caillé F, Gervais P, Baron JC, Bottlaender M, et al. [(18)F]-Av-1451 tau pet imaging in alzheimer's disease and suspected non-ad tauopathies using a late acquisition time window. J Neurol. (2019) 266:3087–97. doi: 10.1007/s00415-019-09530-7
54. Huey ED, Grafman J, Wassermann EM, Pietrini P, Tierney MC, Ghetti B, et al. Characteristics of frontotemporal dementia patients with a progranulin mutation. Ann Neurol. (2006) 60:374–80. doi: 10.1002/ana.20969
55. Jacova C, Hsiung GY, Tawankanjanachot I, Dinelle K, McCormick S, Gonzalez M, et al. Anterior brain glucose hypometabolism predates dementia in progranulin mutation carriers. Neurology. (2013) 81:1322–31. doi: 10.1212/WNL.0b013e3182a8237e
56. Josephs KA, Duffy JR, Strand EA, Machulda MM, Vemuri P, Senjem ML, et al. Progranulin-associated pib-negative logopenic primary progressive aphasia. J Neurol. (2014) 261:604–14. doi: 10.1007/s00415-014-7243-9
57. Caroppo P, Habert MO, Durrleman S, Funkiewiez A, Perlbarg V, Hahn V, et al. Lateral temporal lobe: an early imaging marker of the presymptomatic Grn disease? J Alzheimers Dis. (2015) 47:751–9. doi: 10.3233/JAD-150270
58. Licata A, Grimmer T, Winkelmann J, Wagner M, Goldhardt O, Riedl L, et al. Variability of clinical syndromes and cerebral glucose metabolism in symptomatic frontotemporal lobar degeneration associated with progranulin mutations. Amyotroph Lateral Scler Frontotemporal Degener. (2020) 21:389–95. doi: 10.1080/21678421.2020.1779302
59. Deng B, Zheng Z, Zheng J, Yang W, Huang Y, Luo Y, et al. FTD-PSP is an unusual clinical phenotype in a frontotemporal dementia patient with a novel progranulin mutation. Aging Dis. (2021) 12:1741–52. doi: 10.14336/AD.2021.0309
60. Ljubenkov PA, Edwards L, Iaccarino L, La Joie R, Rojas JC, Koestler M, et al. Effect of the histone deacetylase inhibitor frm-0334 on progranulin levels in patients with progranulin gene haploinsufficiency: A randomized clinical trial. JAMA Netw Open. (2021) 4:e2125584. doi: 10.1001/jamanetworkopen.2021.25584
61. Carecchio M, Galimberti D, Fenoglio C, Serpente M, Scarpini E, Comi C, et al. Evidence of pre-synaptic dopaminergic deficit in a patient with a novel progranulin mutation presenting with atypical parkinsonism. J Alzheimers Dis. (2014) 38:747–52. doi: 10.3233/JAD-131151
62. Dopper EG, Seelaar H, Chiu WZ, de Koning I, van Minkelen R, Baker MC, et al. Symmetrical corticobasal syndrome caused by a novel C314dup progranulin mutation. J Mol Neurosci. (2011) 45:354–8. doi: 10.1007/s12031-011-9626-z
63. Premi E, Grassi M, Gazzina S, Paghera B, Pepe D, Archetti S, et al. The neuroimaging signature of frontotemporal lobar degeneration associated with granulin mutations: an effective connectivity study. J Nucl Med. (2013) 54:1066–71. doi: 10.2967/jnumed.112.111773
64. Bevan-Jones RW, Cope TE, Jones SP, Passamonti L, Hong YT, Fryer T, et al. [(18)F]Av-1451 binding is increased in frontotemporal dementia due to C9orf72 expansion. Ann Clin Transl Neurol. (2018) 5:1292–6. doi: 10.1002/acn3.631
65. Smith R, Santillo AF, Waldö ML, Strandberg O, Berron D, Vestberg S, et al. (18)F-flortaucipir in Tdp-43 associated frontotemporal dementia. Sci Rep. (2019) 9:6082. doi: 10.1038/s41598-019-42625-9
66. Gramaglia C, Cantello R, Terazzi E, Carecchio M, D'Alfonso S, Chieppa N, et al. Early onset frontotemporal dementia with psychiatric presentation due to the C9orf72 hexanucleotide repeat expansion: a case report. BMC Neurol. (2014) 14:228. doi: 10.1186/s12883-014-0228-6
67. Martikainen MH, Gardberg M, Jansson L, Röyttä M, Rinne JO, Kaasinen V. Brain 18f-Fdg and 11c-Pib pet findings in two siblings with FTD/ALS associated with the C9orf72 repeat expansion. Neurocase. (2014) 20:150–7. doi: 10.1080/13554794.2012.741252
68. Solje E, Aaltokallio H, Koivumaa-Honkanen H, Suhonen NM, Moilanen V, Kiviharju A, et al. The phenotype of the c9orf72 expansion carriers according to revised criteria for bvftd. PLoS ONE. (2015) 10:e0131817. doi: 10.1371/journal.pone.0131817
69. Block NR, Sha SJ, Karydas AM, Fong JC, De May MG, Miller BL, et al. Frontotemporal dementia and psychiatric illness: emerging clinical and biological links in gene carriers. Am J Geriatr Psychiatry. (2016) 24:107–16. doi: 10.1016/j.jagp.2015.04.007
70. Sha SJ, Khazenzon AM, Ghosh PM, Rankin KP, Pribadi M, Coppola G, et al. Early-onset Alzheimer's disease versus frontotemporal dementia: resolution with genetic diagnoses? Neurocase. (2016) 22:161–7. doi: 10.1080/13554794.2015.1080283
71. Castelnovo V, Caminiti SP, Riva N, Magnani G, Silani V, Perani D. Heterogeneous brain FDG-PET metabolic patterns in patients with C9orf72 mutation. Neurol Sci. (2019) 40:515–21. doi: 10.1007/s10072-018-3685-7
72. Diehl-Schmid J, Licata A, Goldhardt O, Förstl H, Yakushew I, Otto M, et al. FDG-PET underscores the key role of the thalamus in frontotemporal lobar degeneration caused by C9orf72 mutations. Transl Psychiatry. (2019) 9:54. doi: 10.1038/s41398-019-0381-1
73. Levy JP, Bocti C, Elie D, Paquet N, Soucy JP, Ducharme S. Bifrontal hypermetabolism on brain fdg-pet in a case of c9orf72-related behavioral variant of frontotemporal dementia. J Neuropsychiatry Clin Neurosci. (2019) 31:92–4. doi: 10.1176/appi.neuropsych.18050114
74. Sellami L, St-Onge F, Poulin S, Laforce R Jr. Schizophrenia phenotype preceding behavioral variant frontotemporal dementia related to C9orf72 repeat expansion. Cogn Behav Neurol. (2019) 32:120–3. doi: 10.1097/WNN.0000000000000189
75. De Vocht J, Blommaert J, Devrome M, Radwan A, Van Weehaeghe D, De Schaepdryver M, et al. Use of multimodal imaging and clinical biomarkers in presymptomatic carriers of C9orf72 repeat expansion. JAMA Neurol. (2020) 77:1008–17. doi: 10.1001/jamaneurol.2020.1087
76. Filikci Z, Gustafsson MAK, Henriksen OM, Marner L, Høgh P. C9orf72 hexanucleotide repeat expansion with Alzheimer's disease-like clinical phenotype: a case report with results from neuropsychology, CSF, FDG-PET, and PiB-PET. Clin Case Rep. (2020) 8:3416–20. doi: 10.1002/ccr3.3417
77. Popuri K, Beg MF, Lee H, Balachandar R, Wang L, Sossi V, et al. FDG-PET in presymptomatic C9orf72 mutation carriers. Neuroimage Clin. (2021) 31:102687. doi: 10.1016/j.nicl.2021.102687
78. Cselényi Z, Jönhagen ME, Forsberg A, Halldin C, Julin P, Schou M, et al. Clinical validation of 18f-Azd4694, an amyloid-?-specific pet radioligand. J Nucl Med. (2012) 53:415–24. doi: 10.2967/jnumed.111.094029
79. Gallucci M, Dell'Acqua C, Bergamelli C, Fenoglio C, Serpente M, Galimberti D, et al. A case with early onset Alzheimer's disease, frontotemporal hypometabolism, apoe genotype ε4/ε4 and c9orf72 intermediate expansion: a Treviso Dementia (TREDEM) registry case report. J Alzheimers Dis. (2019) 67:985–93. doi: 10.3233/JAD-180715
80. Ali F, Whitwell JL, Martin PR, Senjem ML, Knopman DS, Jack CR, et al. [(18)F] Av-1451 uptake in corticobasal syndrome: the influence of beta-amyloid and clinical presentation. J Neurol. (2018) 265:1079–88. doi: 10.1007/s00415-018-8815-x
81. Lant SB, Robinson AC, Thompson JC, Rollinson S, Pickering-Brown S, Snowden JS, et al. Patterns of microglial cell activation in frontotemporal lobar degeneration. Neuropathol Appl Neurobiol. (2014) 40:686–96. doi: 10.1111/nan.12092
82. Boecker H, Drzezga A. A perspective on the future role of brain pet imaging in exercise science. Neuroimage. (2016) 131:73–80. doi: 10.1016/j.neuroimage.2015.10.021
83. Cistaro A, Pagani M, Montuschi A, Calvo A, Moglia C, Canosa A, et al. The metabolic signature of C9orf72-related ALS: FDG pet comparison with nonmutated patients. Eur J Nucl Med Mol Imaging. (2014) 41:844–52. doi: 10.1007/s00259-013-2667-5
84. Dominguez J, Ng A, Guevarra AC, Daroy ML, Alfon A, Catindig JA, et al. Autosomal dominant frontotemporal lobar degeneration in a filipino family with progranulin mutation. Dement Geriatr Cogn Disord. (2021). doi: 10.1159/000510106
85. McDade E, Boeve BF, Burrus TM, Boot BP, Kantarci K, Fields J, et al. Similar clinical and neuroimaging features in monozygotic twin pair with mutation in progranulin. Neurology. (2012) 78:1245–9. doi: 10.1212/WNL.0b013e318251594c
86. Fällmar D, Haller S, Lilja J, Danfors T, Kilander L, Tolboom N, et al. Arterial spin labeling-based Z-maps have high specificity and positive predictive value for neurodegenerative dementia compared to FDG-PET. Eur Radiol. (2017) 27:4237–46. doi: 10.1007/s00330-017-4784-1
87. He S, Chen S, Xia MR, Sun ZK, Huang Y, Zhang JW. The role of mapt gene in chinese dementia patients: a P301l pedigree study and brief literature review. Neuropsychiatr Dis Treat. (2018) 14:1627–33. doi: 10.2147/NDT.S155521
88. Tosun D, Schuff N, Rabinovici GD, Ayakta N, Miller BL, Jagust W, et al. Diagnostic utility of Asl-MRI and FDG-PET in the behavioral variant of FTD and AD. Ann Clin Transl Neurol. (2016) 3:740–51. doi: 10.1002/acn3.330
89. Verfaillie SC, Adriaanse SM, Binnewijzend MA, Benedictus MR, Ossenkoppele R, Wattjes MP, et al. Cerebral perfusion and glucose metabolism in Alzheimer's disease and frontotemporal dementia: two sides of the same coin? Eur Radiol. (2015) 25:3050–9. doi: 10.1007/s00330-015-3696-1
90. Anazodo UC, Finger E, Kwan BYM, Pavlosky W, Warrington JC, Günther M, et al. Using simultaneous pet/Mri to compare the accuracy of diagnosing frontotemporal dementia by arterial spin labelling Mri and FDG-PET. Neuroimage Clin. (2018) 17:405–14. doi: 10.1016/j.nicl.2017.10.033
91. Krämer J, Lueg G, Schiffler P, Vrachimis A, Weckesser M, Wenning C, et al. Diagnostic value of diffusion tensor imaging and positron emission tomography in early stages of frontotemporal dementia. J Alzheimers Dis. (2018) 63:239–53. doi: 10.3233/JAD-170224
92. Nicastro N, Nencha U, Burkhard PR, Garibotto V. Dopaminergic imaging in degenerative Parkinsonisms, an established clinical diagnostic tool. J Neurochem. (2021). doi: 10.1111/jnc.15561
93. Santra A, Kumar R. Brain perfusion single photon emission computed tomography in major psychiatric disorders: from basics to clinical practice. Indian J Nucl Med. (2014) 29:210–21. doi: 10.4103/0972-3919.142622
94. Holman BL, Devous MD Sr. Functional brain spect: the emergence of a powerful clinical method. J Nucl Med. (1992) 33:1888–904.
95. Broce I, Karch CM, Wen N, Fan CC, Wang Y, Tan CH, et al. Immune-related genetic enrichment in frontotemporal dementia: an analysis of genome-wide association studies. PLoS Med. (2018) 15:e1002487. doi: 10.1371/journal.pmed.1002487
96. Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a p301s tauopathy mouse model. Neuron. (2007) 53:337–51. doi: 10.1016/j.neuron.2007.01.010
97. Doorduin J, de Vries EF, Dierckx RA, Klein HC. Pet imaging of the peripheral benzodiazepine receptor: monitoring disease progression and therapy response in neurodegenerative disorders. Curr Pharm Des. (2008) 14:3297–315. doi: 10.2174/138161208786549443
98. Kreisl WC, Kim MJ, Coughlin JM, Henter ID, Owen DR, Innis RB. Pet imaging of neuroinflammation in neurological disorders. Lancet Neurol. (2020) 19:940–50. doi: 10.1016/S1474-4422(20)30346-X
99. Zhang L, Hu K, Shao T, Hou L, Zhang S, Ye W, et al. Recent developments on pet radiotracers for Tspo and their applications in neuroimaging. Acta Pharm Sin B. (2021) 11:373–93. doi: 10.1016/j.apsb.2020.08.006
100. Bajjalieh SM, Peterson K, Shinghal R, Scheller RH. Sv2, a Brain synaptic vesicle protein homologous to bacterial transporters. Science. (1992) 257:1271–3. doi: 10.1126/science.1519064
101. Vogl C, Tanifuji S, Danis B, Daniels V, Foerch P, Wolff C, et al. Synaptic vesicle glycoprotein 2a modulates vesicular release and calcium channel function at peripheral sympathetic synapses. Eur J Neurosci. (2015) 41:398–409. doi: 10.1111/ejn.12799
102. Gillard M, Chatelain P, Fuks B. Binding characteristics of levetiracetam to synaptic vesicle protein 2a (Sv2a) in human brain and in cho cells expressing the human recombinant protein. Eur J Pharmacol. (2006) 536:102–8. doi: 10.1016/j.ejphar.2006.02.022
103. Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev MA, Jeng AT, et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol. (2015) 77:953–71. doi: 10.1002/ana.24394
104. Robinson JL, Molina-Porcel L, Corrada MM, Raible K, Lee EB, Lee VM, et al. Perforant path synaptic loss correlates with cognitive impairment and alzheimer's disease in the oldest-old. Brain. (2014) 137(Pt 9):2578–87. doi: 10.1093/brain/awu190
105. Nabulsi NB, Mercier J, Holden D, Carré S, Najafzadeh S, Vandergeten MC, et al. Synthesis and preclinical evaluation of 11c-Ucb-J as a pet tracer for imaging the synaptic vesicle glycoprotein 2a in the brain. J Nucl Med. (2016) 57:777–84. doi: 10.2967/jnumed.115.168179
106. Ni R, Nitsch RM. Recent developments in positron emission tomography tracers for proteinopathies imaging in dementia. Front Aging Neurosci. (2021) 13:751897. doi: 10.3389/fnagi.2021.751897
107. Dev SI, Dickerson BC, Touroutoglou A. Neuroimaging in frontotemporal lobar degeneration: research and clinical utility. Adv Exp Med Biol. (2021) 1281:93–112. doi: 10.1007/978-3-030-51140-1_7
108. Meeter LH, Kaat LD, Rohrer JD, van Swieten JC. Imaging and fluid biomarkers in frontotemporal dementia. Nat Rev Neurol. (2017) 13:406–19. doi: 10.1038/nrneurol.2017.75
Keywords: familial frontotemporal lobar degeneration, molecular imaging, biomarkers, MAPT, GRN, C9orf72
Citation: Wang R, Gao H, Xie H, Jia Z and Chen Q (2022) Molecular imaging biomarkers in familial frontotemporal lobar degeneration: Progress and prospects. Front. Neurol. 13:933217. doi: 10.3389/fneur.2022.933217
Received: 30 April 2022; Accepted: 25 July 2022;
Published: 16 August 2022.
Edited by:
Jennifer S. Yokoyama, University of San Francisco, United StatesReviewed by:
Ignacio Illán-Gala, Hospital de la Santa Creu i Sant Pau, SpainCatherine Pennington, University of Edinburgh, United Kingdom
Copyright © 2022 Wang, Gao, Xie, Jia and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qin Chen, Y2hlbi5xaW5Ac2N1LmVkdS5jbg==
†These authors have contributed equally to this work