
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 15 July 2022
Sec. Applied Neuroimaging
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.931006
This article is part of the Research TopicGenotype-Phenotype Correlations in Neurodegenerative Diseases: From Clinical Features to Neuroimaging SignaturesView all 6 articles
 Edoardo Gioele Spinelli1,2
Edoardo Gioele Spinelli1,2 Alma Ghirelli1,2,3
Alma Ghirelli1,2,3 Nilo Riva4,5
Nilo Riva4,5 Elisa Canu1
Elisa Canu1 Veronica Castelnovo1
Veronica Castelnovo1 Teuta Domi5
Teuta Domi5 Laura Pozzi5
Laura Pozzi5 Paola Carrera6
Paola Carrera6 Vincenzo Silani7,8
Vincenzo Silani7,8 Adriano Chiò9
Adriano Chiò9 Massimo Filippi1,2,3,4,10
Massimo Filippi1,2,3,4,10 Federica Agosta1,2,3*
Federica Agosta1,2,3*Objective: Mutations in the TARDBP gene are a rare cause of genetic motor neuron disease (MND). Morphologic MRI characteristics of MND patients carrying this mutation have been poorly described. Our objective was to investigate distinctive clinical and MRI features of a relatively large sample of MND patients carrying TARDBP mutations.
Methods: Eleven MND patients carrying a TARDBP mutation were enrolled. Eleven patients with sporadic MND (sMND) and no genetic mutations were also selected and individually matched by age, sex, clinical presentation and disease severity, along with 22 healthy controls. Patients underwent clinical and cognitive evaluations, as well as 3D T1-weighted and diffusion tensor (DT) MRI on a 3 Tesla scanner. Gray matter (GM) atrophy was first investigated at a whole-brain level using voxel-based morphometry (VBM). GM volumes and DT MRI metrics of the main white matter (WM) tracts were also obtained. Clinical, cognitive and MRI features were compared between groups.
Results: MND with TARDBP mutations was associated with all possible clinical phenotypes, including isolated upper/lower motor neuron involvement, with no predilection for bulbar or limb involvement at presentation. Greater impairment at naming tasks was found in TARDBP mutation carriers compared with sMND. VBM analysis showed significant atrophy of the right lateral parietal cortex in TARDBP patients, compared with controls. A distinctive reduction of GM volumes was found in the left precuneus and right angular gyrus of TARDBP patients compared to controls. WM microstructural damage of the corticospinal tract (CST) and inferior longitudinal fasciculi (ILF) was found in both sMND and TARDBP patients, compared with controls, although decreased fractional anisotropy of the right CST and increased axial diffusivity of the left ILF (p = 0.017) was detected only in TARDBP mutation carriers.
Conclusions: TARDBP patients showed a distinctive parietal pattern of cortical atrophy and greater damage of motor and extra-motor WM tracts compared with controls, which sMND patients matched for disease severity and clinical presentation were lacking. Our findings suggest that TDP-43 pathology due to TARDBP mutations may cause deeper morphologic alterations in both GM and WM.
Motor neuron disease (MND) refers to a group of adult-onset neurodegenerative conditions leading to the degeneration of upper and/or lower motor neurons. MND clinically manifests as a progressive loss of motor function, which ultimately leads to death due to the involvement of respiratory muscles (1). Amyotrophic lateral sclerosis (ALS) is the classic form of MND and is characterized by a combination of signs and symptoms of upper and lower motor neuron involvement. However, pure lower motor neuron [i.e., primary muscular atrophy (PMA)] or upper motor neuron [i.e., primary lateral sclerosis (PLS)] involvement can also be appreciated in a minority of cases (2).
One out of ten ALS patients has a familial form, the remaining cases being sporadic (3, 4). Mutations of more than 20 genes have been found to cause MND (5), the most common being chromosome 9 open reading frame 72 (C9Orf72) (33% of familial ALS) and superoxidase dismutase 1 (SOD1) (14% of familial ALS) (6). The third genetic cause of ALS in terms of frequency is the mutation in the transactive response (TAR) DNA binding protein 43 (TARDBP) gene (4.2% of familial cases) (6–9), which encodes the TDP-43 protein. In normal conditions, TDP-43 is ubiquitously expressed at the nuclear level, where it has a role in regulating transcription, messenger RNA (mRNA) spicing and transport, as well as in scaffolding nuclear bodies during interaction with the survival motor neuron protein (10, 11). In the central nervous system of MND patients, an abnormal accumulation of toxic aggregates of hyperphosphorylated TDP-43 is typically observed in the cytoplasm of motor neurons (12). TARDBP mutations have been described in a few cases of ALS, frontotemporal dementia (FTD), or combined presentations thereof (13–22), and are thought to facilitate or accelerate such neuropathological alterations. Consistent with this hypothesis, TARDBP mutations have been associated with increased TDP-43 intracellular aggregation, aberrant cytoplasmic localization, altered protein stability, resistance to protease action or modified interactions with other proteins (11, 13, 23–26). Furthermore, the level of TDP-43 that accumulates in granules, as well as the size or number of granules appear to be bigger in some forms of genetic ALS compared to sporadic cases (27, 28). Different mutations associated with familial ALS have been demonstrated to increase half-life and improve stability of the TDP-43 protein product compared to the wild-type form (29). This could be a potential mechanism for the accelerated disease onset of familial ALS (30, 31).
On magnetic resonance imaging (MRI), no typical pattern of atrophy has emerged in TARDBP patients, probably due to the limited number of cases described. Patterns of atrophy were mostly consistent with the individual clinical presentation, since cases of behavioral and linguistic variants of FTD associated with TARDBP mutations have shown variable combinations of temporal and/or frontal lobe involvement (14, 16, 18, 21), whereas cases of ALS associated with TARDBP mutation reported in literature presented either a normal MRI (15, 17, 32, 33) or mild bilateral temporal atrophy (33). One reported case of behavioral variant of FTD presented also with mesencephalic and caudate nuclei atrophy (19) in the presence of supranuclear gaze palsy and chorea. This far, no study has analyzed TARDBP mutated patients with advanced MRI techniques. The aim of this work was to investigate distinctive clinical and MRI features of a relatively large sample of 11 MND patients carrying TARDBP mutations.
A total of 379 patients with a confirmed diagnosis of MND were referred to IRCCS San Raffaele Scientific Institute in Milan between October 2007 and November 2021 to perform an MRI scan on a 3 Tesla scanner. Their diagnoses included ALS (34–36), PMA (34–36) and PLS (34–36). Patients were screened for known pathogenic mutations on the C9Orf72, GRN, MAPT, FUS, TARDBP, SOD1, TBK1, TREM2, OPTN, or VCP genes. As a result of the screening process, 11 TARDBP mutation carriers were identified, all presenting a pure MND phenotype (ALS, n = 7; PMA, n = 3; PLS, n = 1). As a control group, we included 11 MND patients [sporadic MND (sMND); ALS, n = 8; PMA, n = 3) who proved negative for known pathogenic mutations or variants of unknown significance on the evaluated MND-related genes, and were matched by age, sex, clinical presentation, and disease severity as measured by the ALS Functional Rating Scale Revised (ALSFRS-r) (37), as best as possible on a one-to-one basis. All patients underwent a thorough neurological examination and brain MRI at study entry, as well as a comprehensive, multi-domain clinical, cognitive and behavioral assessment. Twenty-two healthy controls matched for age, sex, and MRI scanner type were recruited among spouses of patients and by word of mouth. Healthy controls were included if the following criteria were satisfied: normal neurological assessment; MMSE score ≥28; no family history of neurodegenerative diseases. All included subjects (i.e., ALS patients and healthy controls) were right-handed, Caucasian, native Italian speakers. Exclusion criteria for all subjects were: significant medical illnesses or substance abuse that could interfere with cognitive functioning; any (other) major systemic, psychiatric, or neurological illnesses; and other causes of focal or diffuse brain damage, including lacunae and extensive cerebrovascular disorders at routine MRI.
Local ethical standards committee on human experimentation approved the study protocol and all participants provided written informed consent.
Blood samples were collected from all patients. The coding sequences and intron/exon boundaries of TARDBP gene were amplified by PCR using optimized protocols and analyzed using Sanger sequencing, looking for known pathogenic mutations (38). Furthermore, the presence of GGGGCC hexanucleotide expansion in the first intron of C9Orf72 was assessed using a repeat-primed polymerase chain reaction (PCR) assay (39). The coding sequences and intron/exon boundaries of GRN, MAPT, SOD1, FUS, TBK1, TREM2, OPTN, and VCP genes were also evaluated by Sanger sequencing (38).
Clinical evaluation was performed by experienced neurologists blinded to genetic status and MRI results, recording disease duration and site of disease onset. Disease severity was assessed using the ALSFRS-r (37). The rate of disease progression was defined according to the following formula: (48–ALSFRS-r score)/time from symptom onset. Muscular strength was assessed by manual muscle testing based on the Medical Research Council (MRC) scale.
Neuropsychological assessment was performed by an experienced neuropsychologist unaware of genetic status and MRI results. The following cognitive functions were evaluated: global cognitive functioning with the MMSE (40); long- and short-term verbal memory with the Rey Auditory Verbal Learning Test (41) and the digit span forward (42), respectively; attentive and executive functions with the digit span backward (43) and the Ravens colored progressive matrices (44); fluency with the phonemic and semantic fluency tests (45) and the relative fluency indices (controlling for individual motor disabilities) (46); visuospatial abilities with the Rey Figure copy (47); language with the Italian battery for the assessment of aphasic disorders (48) and the Token test (49); mood and the presence of behavioral disturbances with the Frontal Behavioral Inventory (50). Although most patients here included were enrolled prior to the revision of Strong criteria (51) and the validation of the Edinburgh Cognitive and Behavioral ALS Screen (ECAS) scale (52), for 11 patients (six mutation carriers and five sMND), sufficient neuropsychological data were available to make a diagnosis of cognitive and/or behavioral impairment according to the revised Strong criteria (51).
All patients and healthy controls underwent brain MRI on a 3.0 T scanner (Philips Medical Systems, Best, the Netherlands) at IRCSS San Raffaele Scientific Institute between 2007 and 2021. The original scanner (for brevity, Scanner 1) was substituted with a different device from the same manufacturer in 2016 (below defined as Scanner 2).
Using Scanner 1, the following brain MRI sequences were obtained: T2-weighted spin echo (SE) [repetition time (TR) = 3,500 ms; echo time (TE) = 85 ms; echo train length = 15; flip angle = 90; 22 contiguous, 5-mm-thick, axial slices; matrix size = 512 × 512; field of view (FOV) = 230 × 184 mm2]; fluid-attenuated inversion recovery (TR = 11 s; TE = 120 ms; flip angle = 90; 22 contiguous, 5-mm-thick, axial slices; matrix size = 512 × 512; FOV = 230 mm2); and 3D T1-weighted fast field echo (TR = 25 ms, TE = 4.6 ms, flip angle = 30, 220 contiguous axial slices with voxel size = 0.89 × 0.89 × 0.8 mm, matrix size = 256 × 256, FOV = 230 × 182 mm2) and pulsed-gradient SE echo planar with sensitivity encoding (acceleration factor = 2.5, TR = 8,986 ms, TE = 80 ms, 55 contiguous, 2.5 mm-thick axial slices, number of acquisitions = 2; acquisition matrix 96 × 96, with an in-plane pixel size of 1.87 × 1.87 mm and a FOV = 240 × 240 mm2) and diffusion gradients applied in 32 non-collinear directions using a gradient scheme which is standard on this system (gradient over-plus) and optimized to reduce echo time as much as possible. The b factor used was 1,000 s/mm. Fat saturation was performed to avoid chemical shift artifacts.
Using Scanner 2, the following brain MRI sequences were obtained: 3D T2-weighted [FOV = 256 × 256, pixel size = 1.21 × 1.21 mm, 192 slices, 1 mm thick, matrix = 256 × 256, TR = 5,500 ms, TE = 247 ms, inversion time (TI) 1–2 = 2,550- echo train length (ETL) = 173, acquisition time (TA) = 3.45 min]; sagittal 3D fluid-attenuation inversion recovery (FOV = 256 × 256, pixel size = 1 × 1 mm, 192 slices, 1 mm thick, matrix = 256 × 256, TR = 4,800 ms, TE = 270 ms, TI = 1,650 ms, ETL = 167, TA = 6.15 min); 3D high resolution T1-weighted turbo field echo. FOV = 256 × 256, pixel size = 1 × 1 mm, 204 slices, 1 mm thick, matrix = 256 × 256, TR = 7 ms, TE = 3.2 ms, TI = 1,000 ms, FA = 8, ETL = 240, TA = 8.53 min); and a diffusion-weighted sequence. FOV = 240 × 232 mm, pix = 2.14 × 2.69 mm, 56 slice, 2.3 mm thick, matrix = 112 × 85 TR = 5,900 ms, TE = 78 ms, 3 shells b-value = 700/1,000/2,855 s/mm2, along 6/30/60 non-collinear directions and 10 b = 0 volumes were acquired.
For acquisitions on both scanners, all slices were positioned to run parallel to a line that joins the most inferoanterior and inferoposterior parts of the corpus callosum.
Voxel-based morphometry (VBM) was performed using SPM12 (http://www.fil.ion.ucl.ac.uk/spm/) and Diffeomorphic Anatomical Registration Exponentiated Lie Algebra (DARTEL) registration method (53) to investigate gray matter (GM) volume alterations, as described previously (54).
GM maps of patients and healthy controls were parcellated into 90 Automated Anatomical Labeling (AAL) regions of interest to obtain regional GM volumes. Specifically, the AAL atlas was registered to the subjects' T1-weighted images using linear and non-linear registrations (FLIRT and FNIRT, respectively) (55, 56), as implemented in the FMRIB software library (FSL, http://www.fmrib.ox.ac.uk/fsl). Cortical GM maps were obtained from the segmentation step of VBM procedure (as described previously) (54), while maps of the basal ganglia (i.e., bilateral caudate, globus pallidus, putamen, and thalamus), hippocampus and amygdala were obtained using the FIRST tool in FSL (http://www.fmrib.ox.ac.uk/fsl/first/index.html). GM volumes were multiplied by the normalization factor derived from SIENAx (part of FSL; http://www.fmrib.ox.ac.uk/fsl/sienax/index.html) to correct for head size.
DT MRI analysis was performed using the FMRIB Diffusion Toolbox in FSL (http://www.fmrib.ox.ac.uk/fsl/fdt/index.html) and the JIM6 software (Version 6.0, Xinapse Systems, Northants, UK, http://www.xinapse.com), as described previously (57). Maps of mean diffusivity (MD), fractional anisotropy (FA), axial diffusivity (axD) and radial diffusivity (radD) were obtained. Seeds for tractography of the corpus callosum (CC), corticospinal tract (CST), cingulate, inferior and superior longitudinal, and uncinate fasciculi were defined in the Montreal Neurological Institute (MNI) space on the FA template provided by FSL, as previously described (57, 58). The CC was segmented into three portions to identify the callosal fibers linking the precentral (CC-precentral), lateral premotor (CC-premotor) and supplementary motor areas (CC-supplementary motor), as previously described (59). Fiber tracking was performed in native DT MRI space using a probabilistic tractography algorithm implemented in FSL (probtrackx) (60). For each tract, the average MD, FA, axD and radD were calculated in the native space.
Normal distribution assumption was checked by means of Q–Q plot and Shapiro-Wilks and Kolmogorov-Smirnov tests.
Sociodemographic and clinical features (i.e., age, sex, education, ALSFRS-r scores, disease duration, and progression rate) were compared between groups using ANOVA models or Pearson's chi square, as appropriate. Neuropsychological and MRI quantitative features (i.e., GM volumes and DT MRI metrics) were also compared between groups using separate ANOVA models, followed by post-hoc pairwise comparisons, Bonferroni-corrected for multiple comparisons and adjusted for age, sex, education, and—in the case of MRI variables—MR scanner. The threshold of statistical significance was set at p < 0.05. The SPSS Statistics 22.0 software was used.
VBM group comparisons were tested using ANCOVA model adjusting for total intracranial volume, age, sex, and MRI scanner type. Results were assessed at p < 0.05 Family-wise error (FWE)-corrected for multiple comparisons.
Table 1 summarizes the main sociodemographic and clinical variables of study groups. TARDBP and sMND patients were comparable in terms of sex, education, age at MRI and disease duration. Furthermore, patients were similar in terms of ALSFRS-r score and disease progression rate. Table 2 and Supplementary Table 1 report individual diagnoses and clinical features of included patients. The group of TARDBP patients included six men and five women with an age of onset ranging from 43 to 67 years old (59.8 ± 8.2). Their diagnoses included ALS (n = 7), PMA (n = 3), and PLS (n = 1). Nine out of 11 mutated patients presented with a limb onset of disease, with a tendency of having the right side involved first (6 out of 7 patients with a lateralized limb onset), although at the time of clinical evaluation muscle atrophy was mostly bilateral and symmetrical. Two patients with a limb onset were unable to date back a side of onset. Two patients had a bulbar onset (i.e., dysarthria and mild difficulties when swallowing). One of them (subject 5) had the fastest progression rate. At the time of MRI, ALSFRS-r was ranging from 20 to 44 (34.18 ± 8.83), considering that diagnosis was formulated from three up to 35 months after symptom onset. Disease progression rate was highly variable among mutated subjects (being faster in patients with bulbar presentation). No significant differences in disease progression velocity were recorded between sMND and mutated patients. Consistent with the fact that all MND phenotypes were represented, different combinations of upper and lower motor neuron signs were present at an individual basis (see Supplementary Table 1). No patients among TARDBP mutated and matched sMND fulfilled clinical criteria for behavioral or linguistic variants of FTD (57, 61, 62).
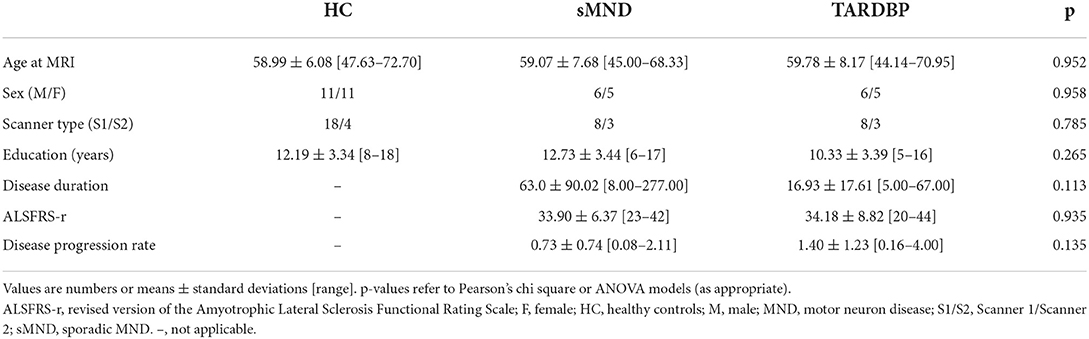
Table 1. Main clinical and demographic characteristics of subjects.
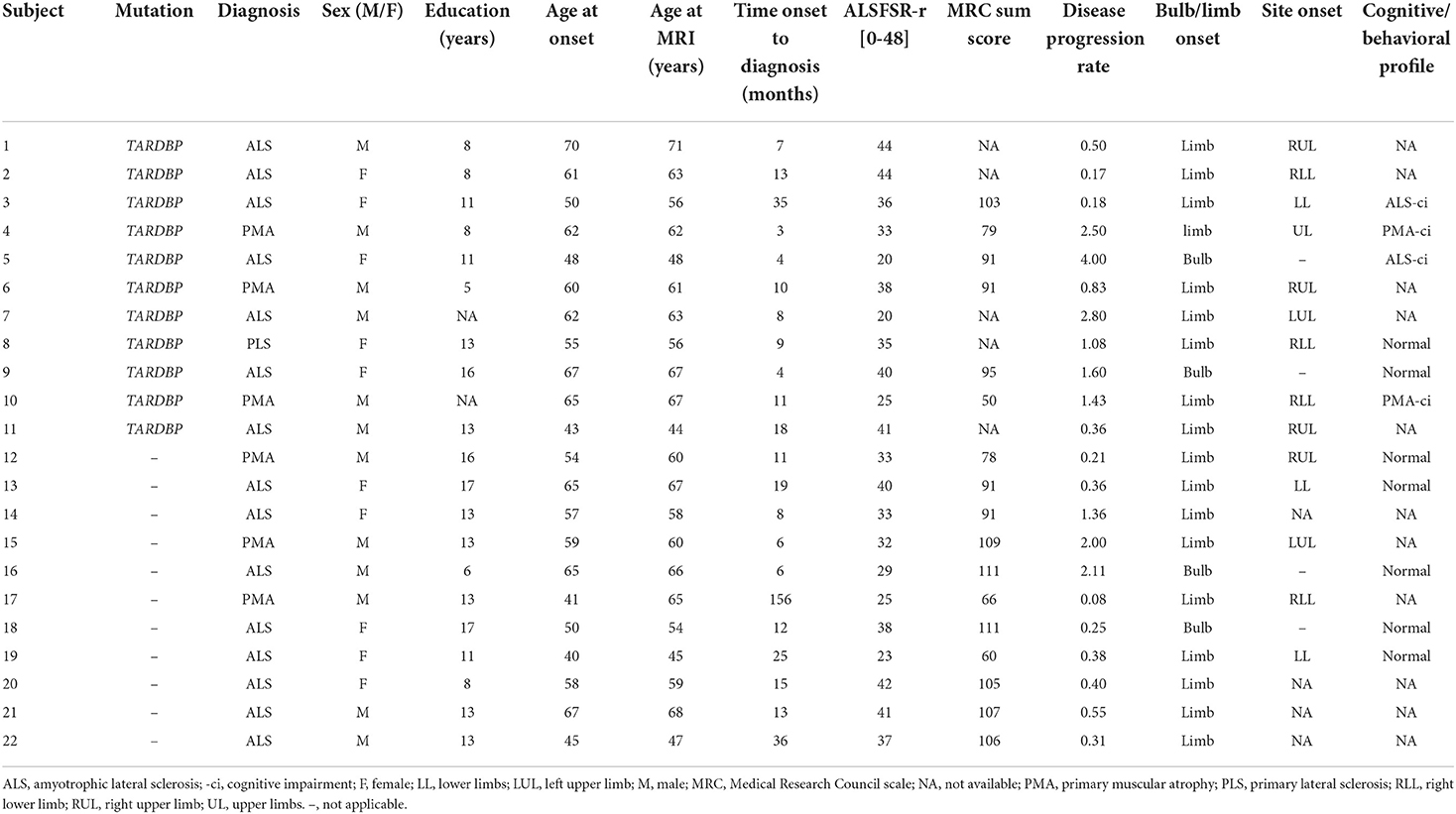
Table 2. Individual socio-demographic and clinical features of MND patients.
Even if data for a cognitive/behavioral diagnosis according to the revised Strong criteria (51) were available for only about half of our cohort, due to the fact that most patients here included were enrolled before 2017, 5/5 sMND patients presented with a normal cognitive profile, while 4/6 mutated patients showed signs of mild cognitive impairment (51). Table 3 reports the neuropsychological test scores of sMND and TARDBP mutated patients. In terms of global cognition, the two groups of patients scored similar results at MMSE. Furthermore, both groups were comparable in terms of memory, executive functions, visuospatial abilities and fluency. Of note, visuospatial abilities were only tested with Rey figure copy and data from the sMND population were available for only one subject. Language was evaluated with the Battery for aphasic deficit analysis (BADA). At the action naming subtest of BADA, TARBDP showed a lower performance compared to controls (p = 0.003) and sMND (p = 0.019). At the noun naming subtest of BADA, TARDBP mutation carriers performed poorer than controls and sMND, although pairwise comparisons did not reach statistical significance (p = 0.081 and p = 0.088, respectively).
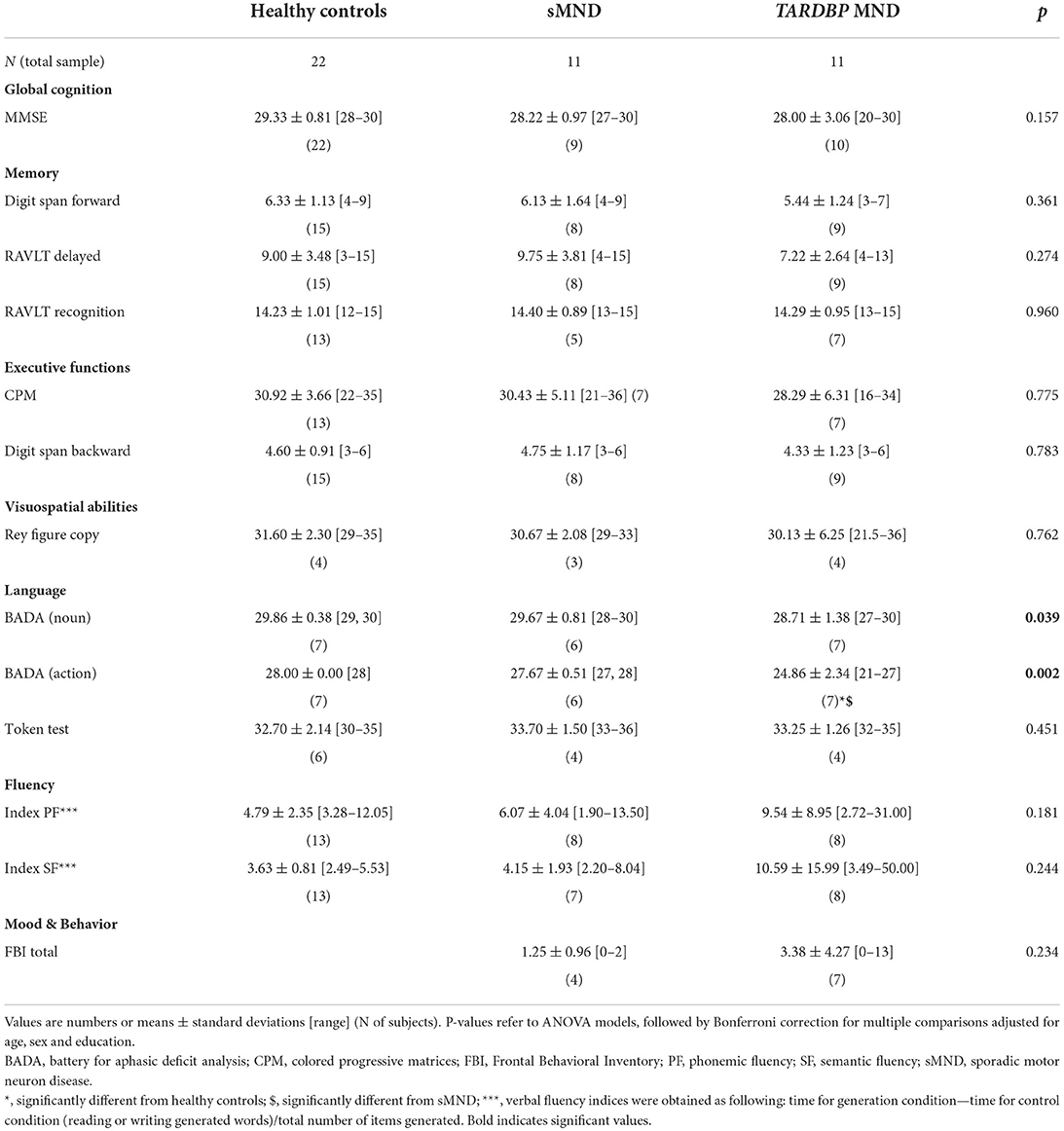
Table 3. Neuropsychological features of healthy controls and MND patients.
Figure 1 reports results of VBM for regions that survived a p < 0.05 FWE, corrected at cluster level. Compared with controls, TARDBP patients showed GM atrophy at the level of the right lateral parietal cortical regions, including the supramarginal and angular gyri (cluster size = 49; MNI coordinates of peak of significance: x = 60, y = −40, z = 45; T-value = 5.83).
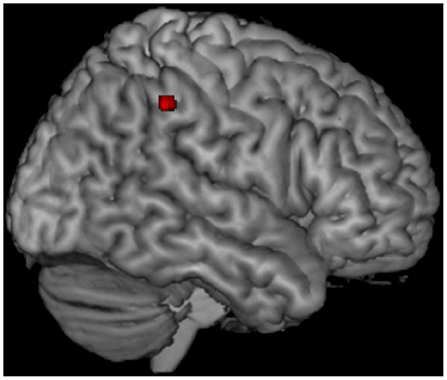
Figure 1. Voxel-based morphometry (VBM) results. Voxel-based analysis showing gray matter (GM) volume loss in TARDBP MND patients relative to healthy controls. Results are overlaid on a three-dimensional rendering of the Montreal Neurological Institute standard brain and displayed at p < 0.05 corrected for multiple comparisons. Only clusters comprising at least 20 contiguous voxels are shown. Analysis is corrected for intracranial volume, age, sex, and MR scanner type.
Figure 2 and Supplementary Table 2 summarize GM volume comparisons. A significant GM volume reduction was found in the left precuneus (p = 0.002) and right angular gyrus of TARDBP patients (p = 0.037), compared to controls. No other significant results emerged in our analysis, although a trend toward a greater atrophy of the left angular gyrus was also observed in TARDBP patients, compared with controls (p = 0.08).
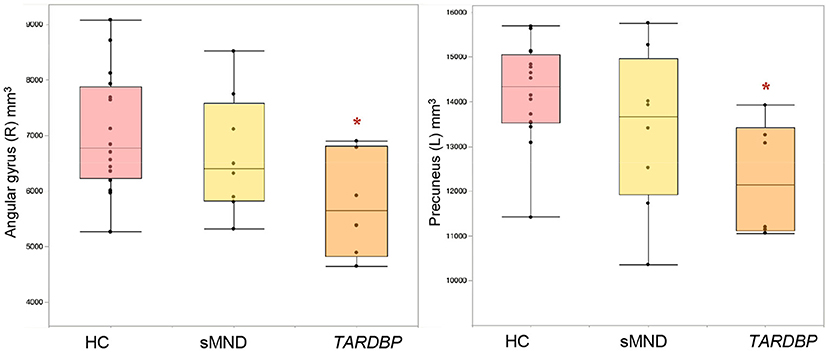
Figure 2. Gray matter (GM) cortical volumes showing significant differences in MND patients and controls. Values refer to mm3. Comparisons between groups were performed using age-, sex-, and MRI scanner-adjusted analysis of variance models, followed by post-hoc pairwise comparisons, Bonferroni-corrected for multiple comparisons. HC, healthy controls; L, left; R, right; sMND, sporadic motor neuron disease. *Significantly different from HC.
As shown in Figure 3 and Supplementary Table 3, the right CST showed decreased fractional anisotropy (FA) only in TARDBP patients, compared to controls (p = 0.035). The left inferior longitudinal fasciculus (ILF) showed higher values of axial diffusivity (axD) in TARDBP compared to controls (0.017); the right ILF instead showed increased axD both in sMND compared to controls (p = 0.047) and in TARDBP cases compared to controls (p = 0.019).
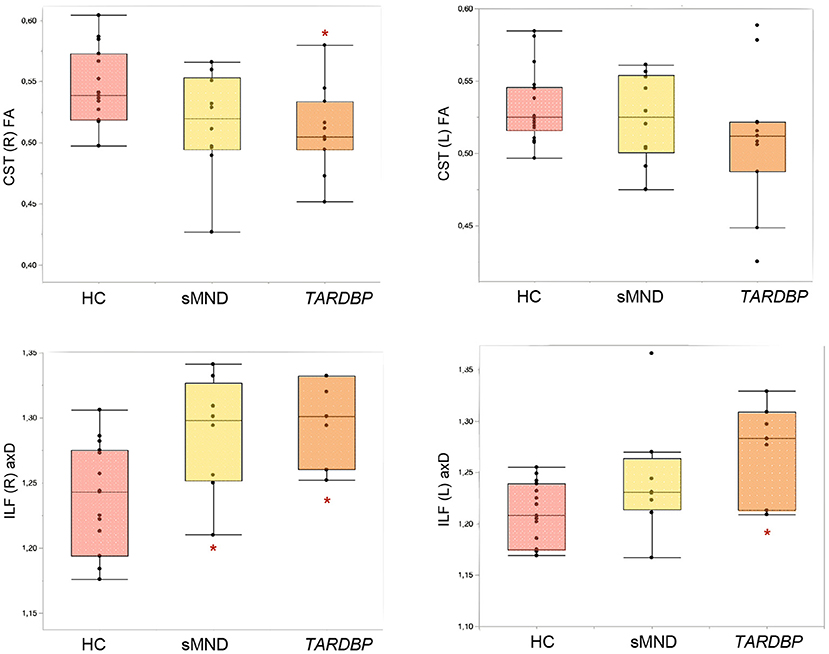
Figure 3. Diffusion tensor (DT) MRI metrics of white matter (WM) tracts showing significant differences in MND patients and controls. Comparisons between groups were performed using age-, sex-, and MRI scanner-adjusted analysis of variance models, followed by post hoc pairwise comparisons, Bonferroni-corrected for multiple comparisons. CST, corticospinal tract; HC, healthy controls; ILF, inferior longitudinal fasciculus; L, left; R, right; sMND, sporadic motor neuron disease. *Significantly different from HC.
To the best of our knowledge, this was the first study analyzing a relatively large cohort of TARDBP mutated MND patients with quantitative MRI advanced techniques. Previous studies have described single cases or small case series of families or unrelated subjects affected by TARDBP mutations causing a syndrome of the FTD-ALS spectrum (14–22, 33). However, none of these reports involved the use of advanced MRI techniques. Available data in the literature only included description of cortical atrophy by visual inspection of conventional MRI sequences, without displaying quantitative information on GM volumes or any information at all on WM alterations of patients affected by TARDBP mutations. We have previously described GM atrophy in a cohort of genetic FTLD (32), which included some of the TARDBP patients included here. However, in the present study, we expanded our cohort and performed a systematic analysis exploring WM features at DT MRI as well. Our study provides therefore a thorough, combined picture of GM and WM structural alterations in patients carrying this mutation, highlighting neuroanatomical differences compared to clinically matched sMND cases.
In the present cohort, all patients carrying mutations in the TARDBP gene presented with a pure MND phenotype, as no patient fulfilled established clinical criteria for a defined FTD syndrome (61, 62). The clinical presentation was heterogeneous, with patients presenting with variable combinations of upper and lower motor neuron signs, configuring all the main MND phenotypes, including ALS, PMA and PLS. Furthermore, mutated patients presented with either a limb or a bulbar onset (in one case, with a very fast disease progression). Therefore, in our cohort, no specific motor phenotype was associated with a TARDBP mutation. The right-sided onset of motor symptoms reported by most TARDBP patients with a limb onset might have been influenced by the fact that all patients were right-handed, as at time of neurological evaluation there was no clear consistent lateralization of clinical findings. Such heterogeneity diverges from previous reports, in which TARDBP mutations with MND have been mostly associated with an ALS syndrome, with the exception of one case of PMA (63), in a patient whose first symptom was camptocormia. All other cases reported an ALS presentation (14–22, 33), with variable associations of FTD phenotypes (of note, often consistent with temporal variant of FTD or semantic dementia) (16).
When examining neuropsychological data, sMND and TARDBP-mutated MND patients were comparable in terms of global cognitive measures, as well as performance in most cognitive domains and behavioral symptoms. However, TARDBP patients differed from controls and matched sMND patients in terms of linguistic performance. Indeed, at the action and noun naming subtests of a battery examining comprehension, denomination and repetition of nouns and verbs, mutated patients performed significantly worse than sMND. Even considering the limitation that sufficiently complete neuropsychological data for a formal cognitive diagnosis (51) was not available for all subjects, four mutated patients fulfilled Strong criteria for MND with cognitive impairment (51), suggesting a more globally distributed, although mild, damage to cognitive functions compared with the sMND sample, matched for all other clinical features. This is consistent with data reported in the literature, in which there are cases of TARDBP patients presenting with (or developing in the course of the disease) cognitive impairment, particularly in the executive and linguistic domains (15, 16). However, further data are warranted to confirm this as a characteristic feature of TARDBP mutated patients presenting with isolated MND.
Compared with controls, VBM showed a circumscribed, distinctive atrophy of the right lateral parietal cortex in MND patients with a TARDBP mutation, confirmed by GM volumetric reduction of the right angular gyrus. Moreover, GM volumetric analysis also demonstrated significant atrophy of the left precuneus, as well as an almost significant trend for the left angular gyrus in TARDBP mutation carriers. The lateral parietal cortex and the precuneus are known to be cross-modal hubs where multisensory information converging from frontal and temporal inputs is processed and integrated (64, 65). Among lateral parietal cortical regions involved, the key region showing atrophy only in mutation carriers was the angular gyrus. This area has been implied in higher language abilities, and its activation has been registered during semantic processing (66) and semantic tasks on auditory (67) and visual (68) stimuli. A recent report has specifically related hypoperfusion of the angular gyrus with language deficits in ALS (57, 69). With the limitations of the small number of patients included in our study, which could not allow us to properly run a correlation analysis, atrophy of these cortical regions could possibly justify, at least in part, the lower scores obtained at language assessments by mutated patients.
Previous studies have described cortical atrophy in more anterior regions in sMND patients, such as the primary motor and premotor cortices, as well as prefrontal and temporal regions (32, 70, 71). A more posterior pattern of atrophy has instead been associated with other genetic forms of MND—mostly, related to C9orf72 pathologic expansions (32, 72, 73)—, leading to the hypothesis that genetic mutations may promote neurodegeneration also in areas that are not typically involved in MND, possibly due to an accelerated neurodegenerative process (32, 72, 73).
As previously highlighted, the present study was the first to assess WM damage using DT MRI in TARDBP mutated patients in a systematic way. Based on our results, TARDBP patients and sMND shared a significant microstructural damage of the CST and ILF, although mutation carriers showed a slightly more extensive involvement, in comparison with the matched sMND sample. DT MRI of the CST is a well-known quantitative measure of upper motor neuron damage in MND (74, 75), whereas the ILF (connecting occipital and temporal lobes) is known to be involved not only in spatial processing but also in language processes (76–78). It has been suggested that the ILF acts in the interplay of the semantic-ventral stream (79–84), as supported by consistent evidence obtained from patients with semantic dementia, in which the ILF is highly disrupted (58, 85). Of note, a significant proportion of FTD patients with a TARDBP mutation were found to fulfill criteria for semantic dementia by other reports (16). So far, previous studies on ALS patients have demonstrated a correlation between alterations of the ILF and emotional processing disorders (86, 87), but future investigation will be needed to perfect our knowledge on the role of this WM tract for linguistic impairment in pure MND.
This study is not without limitations. First, to reach the greatest number of TARDBP patients, we had to include subjects who performed MRI on two different scanners. This limitation was partially overcome by including scanner type as a covariate in our statistical analyses. Secondly, accurate cognitive/behavioral phenotyping was not available for the whole cohort of patients, thus not allowing a proper correlation analysis between neuroanatomical and cognitive data in a sufficiently powered sample. Moreover, we did not have systematic information regarding mutations or variants of unknown significance on less common MND-related genes, other than those analyzed with Sanger sequencing (e.g., those related with hereditary spastic paraplegia, etc.). Although we could not completely exclude the presence of these alterations in the sMND sample, we still could describe the distinctive neuroanatomical alterations of the TARDBP mutated subjects, as per declared objective of the present study. One last pitfall of this study is its cross-sectional design, as a longitudinal approach would have allowed to keep track of cortical and subcortical damage as the disease unfolds and different symptoms come at play. Longitudinal studies are warranted to better understand the clinical relevance of these findings in mutated subjects, in order to identify useful outcome measures in future gene-targeting clinical trials.
In conclusion, our findings suggest that MND patients carrying a TARDBP may present with a heterogeneous clinical phenotype. However, we suggest that a distinctive, mild impairment of the linguistic domains, together with a prominent damage to parietal GM structures might be the hallmark of this uncommon, but significant cause of genetically-determined MND.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by IRCCS Ospedale San Raffaele. The patients/participants provided their written informed consent to participate in this study.
ES, AG, MF, and FA conceived and designed the study. ES, AG, NR, EC, VC, VS, and AC acquired data. ES, AG, EC, VC, TD, and LP analyzed data. All authors contributed to the article, drafted the manuscript, and approved the submitted version.
This work was supported by the Italian Ministry of Health (GR-2011-02351217, GR-2013-02357415, and RF-2011-02351193) and AriSLA (ConnectALS), European Research Council (StG-2016_714388_NeuroTRACK).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.931006/full#supplementary-material
1. Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. (2016) 539:197–206. doi: 10.1038/nature20413
2. Devenney E, Vucic S, Hodges JR, Kiernan MC. Motor neuron disease-frontotemporal dementia: a clinical continuum. Expert Rev Neurother. (2015) 15:509–22. doi: 10.1586/14737175.2015.1034108
3. Gros-Louis F, Gaspar C, Rouleau GA. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta. (2006) 1762:956–72. doi: 10.1016/j.bbadis.2006.01.004
4. Valdmanis PN, Rouleau GA. Genetics of familial amyotrophic lateral sclerosis. Neurology. (2008) 70:144–52. doi: 10.1212/01.wnl.0000296811.19811.db
5. Chia R, Chiò A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. (2018) 17:94–102. doi: 10.1016/S1474-4422(17)30401-5
6. Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. (2017) 88:540–9. doi: 10.1136/jnnp-2016-315018
7. Cady J, Allred P, Bali T, Pestronk A, Goate A, Miller TM, et al. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol. (2015) 77:100–13. doi: 10.1002/ana.24306
8. Chiò A, Calvo A, Mazzini L, Cantello R, Mora G, Moglia C, et al. Extensive genetics of ALS: a population-based study in Italy. Neurology. (2012) 79:1983–9. doi: 10.1212/WNL.0b013e3182735d36
9. Kenna KP, McLaughlin RL, Byrne S, Elamin M, Heverin M, Kenny EM, et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet. (2013) 50:776–83. doi: 10.1136/jmedgenet-2013-101795
10. Kwong LK, Uryu K, Trojanowski JQ, Lee VM. TDP-43 proteinopathies: neurodegenerative protein misfolding diseases without amyloidosis. Neurosignals. (2008) 16:41–51. doi: 10.1159/000109758
11. Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. (2008) 13:867–78. doi: 10.2741/2727
12. Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. (2010) 9:995–1007. doi: 10.1016/S1474-4422(10)70195-2
13. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. (2008) 319:1668–72. doi: 10.1126/science.1154584
14. Borroni B, Bonvicini C, Alberici A, Buratti E, Agosti C, Archetti S, et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum Mutat. (2009) 30:E974–83. doi: 10.1002/humu.21100
15. Chiò A, Calvo A, Moglia C, Restagno G, Ossola I, Brunetti M, et al. Amyotrophic lateral sclerosis-frontotemporal lobar dementia in 3 families with p.Ala382Thr TARDBP mutations. Arch Neurol. (2010) 67:1002–9. doi: 10.1001/archneurol.2010.173
16. Floris G, Borghero G, Cannas A, Di Stefano F, Murru MR, Corongiu D, et al. Clinical phenotypes and radiological findings in frontotemporal dementia related to TARDBP mutations. J Neurol. (2015) 262:375–84. doi: 10.1007/s00415-014-7575-5
17. Cheng YW, Lee MJ, Chen TF, Cheng TW, Lai YM, Hua MS, et al. A single nucleotide TDP-43 mutation within a Taiwanese family: a multifaceted demon. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:292–4. doi: 10.3109/21678421.2015.1111905
18. Gelpi E, van der Zee J, Turon Estrada A, Van Broeckhoven C, Sanchez-Valle R. TARDBP mutation p.Ile383Val associated with semantic dementia and complex proteinopathy. Neuropathol Appl Neurobiol. (2014) 40:225–30. doi: 10.1111/nan.12063
19. Kovacs GG, Murrell JR, Horvath S, Haraszti L, Majtenyi K, Molnar MJ, et al. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord. (2009) 24:1843–7. doi: 10.1002/mds.22697
20. Quadri M, Cossu G, Saddi V, Simons EJ, Murgia D, Melis M, et al. Broadening the phenotype of TARDBP mutations: the TARDBP Ala382Thr mutation and Parkinson's disease in sardinia. Neurogenetics. (2011) 12:203–9. doi: 10.1007/s10048-011-0288-3
21. Synofzik M, Born C, Rominger A, Lummel N, Schöls L, Biskup S, et al. Targeted high-throughput sequencing identifies a TARDBP mutation as a cause of early-onset FTD without motor neuron disease. Neurobiol Aging. (2014) 35:1212.e1–5. doi: 10.1016/j.neurobiolaging.2013.10.092
22. Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. (2009) 65:470–3. doi: 10.1002/ana.21612
23. Johnson JO, Glynn SM, Gibbs JR, Nalls MA, Sabatelli M, Restagno G, et al. Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain. (2014) 137 (Pt. 12):e311. doi: 10.1093/brain/awu265
24. Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. (2010) 30:639–49. doi: 10.1523/JNEUROSCI.4988-09.2010
25. Mutihac R, Alegre-Abarrategui J, Gordon D, Farrimond L, Yamasaki-Mann M, Talbot K, et al. TARDBP pathogenic mutations increase cytoplasmic translocation of TDP-43 and cause reduction of endoplasmic reticulum Ca2+ signaling in motor neurons. Neurobiol Dis. (2015) 75:64–77. doi: 10.1016/j.nbd.2014.12.010
26. Mitsuzawa S, Akiyama T, Nishiyama A, Suzuki N, Kato M, Warita H, et al. TARDBP p.G376D mutation, found in rapid progressive familial ALS, induces mislocalization of TDP-43. eNeurologicalSci. (2018) 11:20–2. doi: 10.1016/j.ensci.2018.04.001
27. McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, et al. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet. (2011) 20:1400–10. doi: 10.1093/hmg/ddr021
28. Dewey CM, Cenik B, Sephton CF, Dries DR, Mayer P III, Good SK, et al. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol. (2011) 31:1098–108. doi: 10.1128/MCB.01279-10
29. Ling SC, Albuquerque CP, Han JS, Lagier-Tourenne C, Tokunaga S, Zhou H, et al. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci USA. (2010) 107:13318–23. doi: 10.1073/pnas.1008227107
30. Watanabe S, Kaneko K, Yamanaka K. Accelerated disease onset with stabilized familial amyotrophic lateral sclerosis (ALS)-linked mutant TDP-43 proteins. J Biol Chem. (2013) 288:3641–54. doi: 10.1074/jbc.M112.433615
31. Austin JA, Wright GS, Watanabe S, Grossmann JG, Antonyuk SV, Yamanaka K, et al. Disease causing mutants of TDP-43 nucleic acid binding domains are resistant to aggregation and have increased stability and half-life. Proc Natl Acad Sci USA. (2014) 111:4309–14. doi: 10.1073/pnas.1317317111
32. Spinelli EG, Ghirelli A, Basaia S, Cividini C, Riva N, Canu E, et al. Structural MRI signatures in genetic presentations of the frontotemporal dementia/motor neuron disease spectrum. Neurology. (2021) 97:e1594–607. doi: 10.1212/WNL.0000000000012702
33. Del Bo R, Ghezzi S, Corti S, Pandolfo M, Ranieri M, Santoro D, et al. TARDBP (TDP-43) sequence analysis in patients with familial and sporadic ALS: identification of two novel mutations. Eur J Neurol. (2009) 16:727–32. doi: 10.1111/j.1468-1331.2009.02574.x
34. Brooks BR, Miller RG, Swash M, Munsat TL. El escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1:293–9. doi: 10.1080/146608200300079536
35. Pringle CE, Hudson AJ, Munoz DG, Kiernan JA, Brown WF, Ebers GC. Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain. (1992) 115 (Pt. 2):495–520. doi: 10.1093/brain/115.2.495
36. Van Den Berg-Vos RM, Van Den Berg LH, Visser J, de Visser M, Franssen H, Wokke JH. The spectrum of lower motor neuron syndromes. J Neurol. (2003) 250:1279–92. doi: 10.1007/s00415-003-0235-9
37. Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS study group (p hase III). J Neurol Sci. (1999) 169:13–21. doi: 10.1016/S0022-510X(99)00210-5
38. Pozzi L, Valenza F, Mosca L, Dal Mas A, Domi T, Romano A, et al. TBK1 mutations in Italian patients with amyotrophic lateral sclerosis: genetic and functional characterisation. J Neurol Neurosurg Psychiatry. (2017) 88:869–75. doi: 10.1136/jnnp-2017-316174
39. Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
40. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. (1975) 12:189–98. doi: 10.1016/0022-3956(75)90026-6
41. Carlesimo GA, Caltagirone C, Gainotti G. The mental deterioration battery: normative data, diagnostic reliability and qualitative analyses of cognitive impairment. The group for the standardization of the mental deterioration battery. Eur Neurol. (1996) 36:378–84. doi: 10.1159/000117297
42. Orsini A, Fragassi NA, Chiacchio L, Falanga AM, Cocchiaro C, Grossi D. Verbal and spatial memory span in patients with extrapyramidal diseases. Percept Mot Skills. (1987) 65:555–8. doi: 10.2466/pms.1987.65.2.555
43. Monaco M, Costa A, Caltagirone C, Carlesimo GA. Forward and backward span for verbal and visuo-spatial data: standardization and normative data from an Italian adult population. Neurol Sci. (2013) 34:749–54. doi: 10.1007/s10072-012-1130-x
44. Basso A, Capitani E, Laiacona M. Raven's coloured progressive matrices: normative values on 305 adult normal controls. Funct Neurol. (1987) 2:189–94.
45. Novelli G, Papagno C, Capitani E, Laiacona M. Tre test clinici di ricerca e produzione les-sicale. Taratura su soggetti normali. Arch Psicol Neurol Psichiatria. (1986) 47:278–96.
46. Abrahams S, Leigh PN, Harvey A, Vythelingum GN, Grisé D, Goldstein LH. Verbal fluency and executive dysfunction in amyotrophic lateral sclerosis (ALS). Neuropsychologia. (2000) 38:734–47. doi: 10.1016/S0028-3932(99)00146-3
47. Caffarra P, Vezzadini G, Dieci F, Zonato F, Venneri A. Rey-Osterrieth complex figure: normative values in an Italian population sample. Neurol Sci. (2002) 22:443–7. doi: 10.1007/s100720200003
48. Miceli G, Capasso R, Caramazza A. The interaction of lexical and sublexical processes in reading, writing and repetition. Neuropsychologia. (1994) 32:317–33. doi: 10.1016/0028-3932(94)90134-1
49. De Renzi E, Vignolo LA. The token test: a sensitive test to detect receptive disturbances in aphasics. Brain. (1962) 85:665–78. doi: 10.1093/brain/85.4.665
50. Alberici A, Geroldi C, Cotelli M, Adorni A, Calabria M, Rossi G, et al. The frontal behavioural inventory (Italian version) differentiates frontotemporal lobar degeneration variants from Alzheimer's disease. Neurol Sci. (2007) 28:80–6. doi: 10.1007/s10072-007-0791-3
51. Strong MJ, Abrahams S, Goldstein LH, Woolley S, McLaughlin P, Snowden J, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:153–74. doi: 10.1080/21678421.2016.1267768
52. Abrahams S, Newton J, Niven E, Foley J, Bak TH. Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2014) 15:9–14. doi: 10.3109/21678421.2013.805784
53. Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage. (2007) 38:95–113. doi: 10.1016/j.neuroimage.2007.07.007
54. Filippi M, Basaia S, Canu E, Imperiale F, Magnani G, Falautano M, et al. Changes in functional and structural brain connectome along the Alzheimer's disease continuum. Mol Psychiatry. (2020) 25:230–9. doi: 10.1038/s41380-018-0067-8
55. Jenkinson M, Bannister P, Brady M, Smith S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage. (2002) 17:825–41. doi: 10.1006/nimg.2002.1132
56. Andersson JLR. Non-Linear Registration, Aka Spatial Normalisation. FMRIB technical report TR07JA2 (2007). Available online at: https://www.fmrib.ox.ac.uk/datasets/techrep/tr07ja2/tr07ja2.pdf
57. Agosta F, Galantucci S, Svetel M, Lukić MJ, Copetti M, Davidovic K, et al. Clinical, cognitive, and behavioural correlates of white matter damage in progressive supranuclear palsy. J Neurol. (2014) 261:913–24. doi: 10.1007/s00415-014-7301-3
58. Agosta F, Galantucci S, Canu E, Cappa SF, Magnani G, Franceschi M, et al. Disruption of structural connectivity along the dorsal and ventral language pathways in patients with nonfluent and semantic variant primary progressive aphasia: a DT MRI study and a literature review. Brain Lang. (2013) 127:157–66. doi: 10.1016/j.bandl.2013.06.003
59. Agosta F, Galantucci S, Riva N, Chio A, Messina S, Iannaccone S, et al. Intrahemispheric and interhemispheric structural network abnormalities in PLS and ALS. Hum Brain Mapp. (2014) 35:1710–22. doi: 10.1002/hbm.22286
60. Behrens TE, Berg HJ, Jbabdi S, Rushworth MF, Woolrich MW. Probabilistic diffusion tractography with multiple fibre orientations: what can we gain? Neuroimage. (2007) 34:144–55. doi: 10.1016/j.neuroimage.2006.09.018
61. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. (2011) 134 (Pt. 9):2456–77. doi: 10.1093/brain/awr179
62. Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. (2011) 76:1006–14. doi: 10.1212/WNL.0b013e31821103e6
63. Filippelli E, Valentino P, Annesi G, Nisticò R, Quattrone A. Camptocormia as presenting in lower motor neuron disease with TARDBP mutation: case report. Neurol Sci. (2017) 38:1885–7. doi: 10.1007/s10072-017-3005-7
64. Obleser J, Kotz SA. Expectancy constraints in degraded speech modulate the language comprehension network. Cereb Cortex. (2010) 20:633–40. doi: 10.1093/cercor/bhp128
65. Xu J, Kemeny S, Park G, Frattali C, Braun A. Language in context: emergent features of word, sentence, and narrative comprehension. Neuroimage. (2005) 25:1002–15. doi: 10.1016/j.neuroimage.2004.12.013
66. Van Ettinger-Veenstra H, McAllister A, Lundberg P, Karlsson T, Engström M. Higher language ability is related to angular gyrus activation increase during semantic processing, independent of sentence incongruency. Front Hum Neurosci. (2016) 10:110. doi: 10.3389/fnhum.2016.00110
67. Démonet JF, Chollet F, Ramsay S, Cardebat D, Nespoulous JL, Wise R, et al. The anatomy of phonological and semantic processing in normal subjects. Brain. (1992) 115 (Pt. 6):1753–68. doi: 10.1093/brain/115.6.1753
68. Vandenberghe R, Price C, Wise R, Josephs O, Frackowiak RS. Functional anatomy of a common semantic system for words and pictures. Nature. (1996) 383:254–6. doi: 10.1038/383254a0
69. Sakurai T, Hirano S, Abe M, Uji Y, Shimizu K, Suzuki M, et al. Dysfunction of the left angular gyrus may be associated with writing errors in ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2021) 22:267–75. doi: 10.1080/21678421.2020.1861021
70. Chang JL, Lomen-Hoerth C, Murphy J, Henry RG, Kramer JH, Miller BL, et al. A voxel-based morphometry study of patterns of brain atrophy in ALS and ALS/FTLD. Neurology. (2005) 65:75–80. doi: 10.1212/01.wnl.0000167602.38643.29
71. Walhout R, Westeneng HJ, Verstraete E, Hendrikse J, Veldink JH, van den Heuvel MP, et al. Cortical thickness in ALS: towards a marker for upper motor neuron involvement. J Neurol Neurosurg Psychiatry. (2015) 86:288–94. doi: 10.1136/jnnp-2013-306839
72. Schönecker S, Neuhofer C, Otto M, Ludolph A, Kassubek J, Landwehrmeyer B, et al. Atrophy in the thalamus but not cerebellum is specific for C9orf72 FTD and ALS patients - an atlas-based volumetric MRI study. Front Aging Neurosci. (2018) 10:45. doi: 10.3389/fnagi.2018.00045
73. Agosta F, Ferraro PM, Riva N, Spinelli EG, Domi T, Carrera P, et al. Structural and functional brain signatures of C9orf72 in motor neuron disease. Neurobiol Aging. (2017) 57:206–19. doi: 10.1016/j.neurobiolaging.2017.05.024
74. Agosta F, Pagani E, Petrolini M, Sormani MP, Caputo D, Perini M, et al. MRI predictors of long-term evolution in amyotrophic lateral sclerosis. Eur J Neurosci. (2010) 32:1490–6. doi: 10.1111/j.1460-9568.2010.07445.x
75. Spinelli EG, Riva N, Rancoita PMV, Schito P, Doretti A, Poletti B, et al. Structural MRI outcomes and predictors of disease progression in amyotrophic lateral sclerosis. Neuroimage Clin. (2020) 27:102315. doi: 10.1016/j.nicl.2020.102315
76. Shin J, Rowley J, Chowdhury R, Jolicoeur P, Klein D, Grova C, et al. Inferior longitudinal fasciculus' role in visual processing and language comprehension: a combined MEG-DTI study. Front Neurosci. (2019) 13:875. doi: 10.3389/fnins.2019.00875
77. Agosta F, Henry RG, Migliaccio R, Neuhaus J, Miller BL, Dronkers NF, et al. Language networks in semantic dementia. Brain. (2010) 133 (Pt. 1):286–99. doi: 10.1093/brain/awp233
78. Wong FC, Chandrasekaran B, Garibaldi K, Wong PC. White matter anisotropy in the ventral language pathway predicts sound-to-word learning success. J Neurosci. (2011) 31:8780–5. doi: 10.1523/JNEUROSCI.0999-11.2011
79. Saur D, Kreher BW, Schnell S, Kümmerer D, Kellmeyer P, Vry MS, et al. Ventral and dorsal pathways for language. Proc Natl Acad Sci USA. (2008) 105:18035–40. doi: 10.1073/pnas.0805234105
80. Turken AU, Dronkers NF. The neural architecture of the language comprehension network: converging evidence from lesion and connectivity analyses. Front Syst Neurosci. (2011) 5:1. doi: 10.3389/fnsys.2011.00001
81. Duffau H, Herbet G, Moritz-Gasser S. Toward a pluri-component, multimodal, and dynamic organization of the ventral semantic stream in humans: lessons from stimulation mapping in awake patients. Front Syst Neurosci. (2013) 7:44. doi: 10.3389/fnsys.2013.00044
82. Moritz-Gasser S, Herbet G, Duffau H. Mapping the connectivity underlying multimodal (verbal and non-verbal) semantic processing: a brain electrostimulation study. Neuropsychologia. (2013) 51:1814–22. doi: 10.1016/j.neuropsychologia.2013.06.007
83. Dick AS, Bernal B, Tremblay P. The language connectome: new pathways, new concepts. Neuroscientist. (2014) 20:453–67. doi: 10.1177/1073858413513502
84. Almairac F, Herbet G, Moritz-Gasser S, de Champfleur NM, Duffau H. The left inferior fronto-occipital fasciculus subserves language semantics: a multilevel lesion study. Brain Struct Funct. (2015) 220:1983–95. doi: 10.1007/s00429-014-0773-1
85. Acosta-Cabronero J, Patterson K, Fryer TD, Hodges JR, Pengas G, Williams GB, et al. Atrophy, hypometabolism and white matter abnormalities in semantic dementia tell a coherent story. Brain. (2011) 134 (Pt. 7):2025–35 doi: 10.1093/brain/awr119
86. Sedda A. Disorders of emotional processing in amyotrophic lateral sclerosis. Curr Opin Neurol. (2014) 27:659–65. doi: 10.1097/WCO.0000000000000147
Keywords: motor neuron disease (MND), amyotrophic lateral sclerosis (ALS), transactive response (TAR) DNA binding protein 43 (TARDBP), magnetic resonance imaging (MRI), voxel-based morphometry (VBM)
Citation: Spinelli EG, Ghirelli A, Riva N, Canu E, Castelnovo V, Domi T, Pozzi L, Carrera P, Silani V, Chiò A, Filippi M and Agosta F (2022) Profiling morphologic MRI features of motor neuron disease caused by TARDBP mutations. Front. Neurol. 13:931006. doi: 10.3389/fneur.2022.931006
Received: 28 April 2022; Accepted: 27 June 2022;
Published: 15 July 2022.
Edited by:
Eoin Finegan, Trinity College Dublin, IrelandCopyright © 2022 Spinelli, Ghirelli, Riva, Canu, Castelnovo, Domi, Pozzi, Carrera, Silani, Chiò, Filippi and Agosta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Federica Agosta, YWdvc3RhLmZlZGVyaWNhQGhzci5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.