 Mark B. Vestergaard
Mark B. Vestergaard Jette L. Frederiksen2,3
Jette L. Frederiksen2,3- 1Functional Imaging Unit, Department of Clinical Physiology and Nuclear Medicine, Copenhagen University Hospital Rigshospitalet, Glostrup, Denmark
- 2Danish Multiple Sclerosis Center, Department of Neurology, Copenhagen University Hospital Rigshospitalet, Glostrup, Denmark
- 3Department of Clinical Medicine, Faculty of Health and Medical Science, University of Copenhagen, Copenhagen, Denmark
The inflammatory processes observed in the central nervous system in multiple sclerosis (MS) could damage the endothelium of the cerebral vessels and lead to a dysfunctional regulation of vessel tonus and recruitment, potentially impairing cerebrovascular reactivity (CVR) and neurovascular coupling (NVC). Impaired CVR or NVC correlates with declining brain health and potentially plays a causal role in the development of neurodegenerative disease. Therefore, we examined studies on CVR or NVC in MS patients to evaluate the evidence for impaired cerebrovascular function as a contributing disease mechanism in MS. Twenty-three studies were included (12 examined CVR and 11 examined NVC). Six studies found no difference in CVR response between MS patients and healthy controls. Five studies observed reduced CVR in patients. This discrepancy can be because CVR is mainly affected after a long disease duration and therefore is not observed in all patients. All studies used CO2 as a vasodilating stimulus. The studies on NVC demonstrated diverse results; hence a conclusion that describes all the published observations is difficult to find. Future studies using quantitative techniques and larger study samples are needed to elucidate the discrepancies in the reported results.
Introduction
Multiple sclerosis (MS) is characterized by autoimmune inflammation in the central nervous system, which primarily targets myelin and oligodendrocytes, with secondary damage to the axons and potential degeneration of the neuron itself through Wallerian degeneration (1). Activated astrocytes and microglia contribute to the creation of a proinflammatory milieu by releasing cytokines and chemokines, attracting immune cells to the lesion area and allowing the migration of inflammatory cells into the brain parenchyma through a compromised blood–brain barrier (BBB) (2–7). The inflammatory processes also cause damage to the vessel endothelium in the brain, possibly through cytokine-mediated mechanisms or by the production of reactive oxygen species (ROS) (8–10). Endothelium damage can cause cerebrovascular dysfunction and impaired cerebral perfusion modulation, as the endothelium is involved in the regulation of vasomotor tone trough the release of vasodilating agents (11). Dysfunction in endothelially mediated dilation will reduce the responsiveness of the vessels to vasodilating stimuli.
Cerebrovascular dysfunction is observed in Alzheimer's disease (AD) and vascular dementia (VD), and increasing evidence suggests that cerebrovascular dysfunction can be a causal aspect in the development of age-related neurodegenerative diseases (12–14). Both reduced cerebrovascular reactivity (CVR) to vasodilating stimuli such as carbon dioxide (CO2) and impaired neurovascular coupling (NVC) are observed in patients with AD or VD (15–21). Reduced CVR also correlates with cognitive performance in elderly human subjects without neurodegenerative disease (22), whereas a healthy lifestyle can maintain a responsive cerebrovascular function with advancing age (23–25). In summary, these studies demonstrate that having healthy cerebrovascular function, which can readily increase cerebral blood flow (CBF) to a region with an increased metabolic demand, is likely an important factor in maintaining a healthy brain, especially considering age-related declines in brain health.
Studies on postmortem brain cells from MS patients have demonstrated hypoxia-like tissue damage in both gray and white matter (26–28), which could be an indication of vessel damage and cerebrovascular dysfunction. Experimental-autoimmune-encephalopathy (EAE) animal models of MS have also demonstrated that tissue hypoxia plays a vital role not only in the formation of demyelinating lesions but also in cortical atrophy and that hyperbaric treatment may dampen this process (29–31). Moreover, whether tissue hypoxia may be an important factor in explaining the highly transient neurological deficits observed in MS remains to be elucidated (31). Overall, these observations have led to speculations on whether tissue hypoxia and impaired cerebrovascular function make important contributions to the pathophysiology of MS, warranting further research from a treatment perspective. If patients with MS have impaired cerebrovascular function, then this could be part of a pathophysiological cascade aggravating brain atrophy. Therefore, examining and understanding how cerebrovascular function is affected in MS is of high clinical importance. Several studies have shown that cerebral hypometabolism and hypoperfusion are present in MS (32–34); however, these studies are not ideal to conclude on the causality between vasophysiology and brain atrophy as a reduced resting metabolism, especially in later stages of MS disease in itself can be a result of brain atrophy. Thus, the cerebrovascular response to a vasodilating stimulus may be a more suitable indicator of vascular dysfunction.
In the present systemic review, we examined the existing literature on CVR and NVC in patients with MS. We investigated whether evidence of impaired cerebrovascular function in MS could be found.
Examination of Cerebrovascular Reactivity
CBF is highly regulated to maintain adequate perfusion in the brain. Changes in metabolic demand rapidly leads to increased perfusion to the affected brain area. Similarly, changes in systemic factors, such as changes in blood pressure or arterial oxygen saturation, are rapidly counteracted to maintain a stable blood and oxygen supply to the brain (35). Cerebral perfusion is mainly regulated by changes in vascular resistance through the dilation and constriction of the cerebral arteries and capillaries. The dilation and constriction of arteries and arterioles are controlled by vascular smooth muscle cells (VSMCs), and the dilation and constriction of capillaries are controlled by pericytes (36).
CVR is the reactivity of the CBF response to an external isometabolic vasodilating stimulus. The preferred stimulus is exposure to CO2, a potent vasodilator of the cerebral arteries and arterioles (37), which is thought to cause minimal to no effect on cerebral metabolism (38, 39). The vasodilating effect of CO2 on VSMCs is mediated through the production of nitric oxide (NO) and bicarbonate () from a decrease in pH (40). Thus, using CO2 as a vasodilating agent provides a good measure of VSMC function and the vasomotor capabilities of the cerebral arteries. Therefore, damage to the endothelium from inflammatory processes in patients with MS is therefore hypothesized to cause reduced vasodilation from a CO2 stimulus. The typical procedure when examining CVR is to measure CBF at rest and to repeat this measurement during CO2 exposure. Exposure to CO2 is obtained either by the inhalation of air with added CO2 or by breath-hold challenges that increase tissue and blood CO2 partial pressure (41–43). CBF is measured by either neuroimaging techniques, such as positron emission tomography (PET) or arterial spin labeling (ASL) magnetic resonance imaging (MRI), or by measuring the blood flow or velocity in the feeding cerebral arteries using transcranial Doppler ultrasound (TCD) or phase contrast mapping (PCM) MRI (42, 44–48).
Examination of Neurovascular Coupling
The NVC describes the local increase in CBF evoked by neuronal activation. The NVC functions in a highly complex manner and involves multiple intermediate steps. The release of neurotransmitters from neuroactivation, most notably glutamate, leads to the release of vasoactive agents through a feedforward mechanism (49). A major pathway for vasodilation acts through synaptic glutamate by triggering N-methyl-d-aspartate (NMDA) receptors, which in turn mediate the influx of Ca2+ into the neurons, where the higher intracellular Ca2+ promotes NO synthase (nNOS) and the production of NO (50, 51). NO easily diffuses into the surrounding arteries, causing vasodilation through the generation of cyclic guanosine monophosphate (cGMP) in VSMCs (52). Astrocytes also play an important role in NVC and in the mediation of vasodilation (53–55). The influx of Ca2+ in astrocytes from glutamate activation promotes the release of arachidonic acid and prostaglandins, both of which have vasodilating effects. Additionally, the higher Ca2+ concentration in astrocyte end-feet will open K+ channels, releasing K+ to the intercellular space and causing direct vasodilation effects on the surrounding vessels (56). Reductions in the local oxygen pressure also have a marked regulatory effect on the accumulation of vasoactive agents. Reduced oxygen availability promotes astrocytic lactate production and release. Extracellular lactate attenuates the uptake of extracellular prostaglandin E2. Prostaglandin E2 accumulation leads to vasodilation (53). At the capillary level, the dilation of the vessel is controlled by pericytes through pathways similar to those of the VSMC control (36). Endothelial cells of the arterioles propagate the vasodilating signal upstream to the penetrating and pial arteries (57). Thus, the quantification of the NVC reflects the integrated sum of a complex cerebrovascular function with multiple steps aimed at increasing CBF. Reduced NVC in MS patients could arise from a malfunction in one or more of the multiple steps in the neurovascular cascade.
Measuring NVC in vivo in humans is challenging. To directly examine NVC, measurements of both the change in neuronal electrical activity and the corresponding change in CBF are needed and should preferably be examined in a quantitative manner. The measurements must also be acquired with a sufficiently fast time resolution to assess the rapid focal change in metabolism and electrical activity from neuroactivation. Ideally, the measurements of neuronal activity and CBF should also be acquired simultaneously. Brain electrical activity can be examined using electroencephalography (EEG) and magnetoencephalography (MEG) techniques (58). By default, these techniques are not quantitative; however, by calculating the spectral power of the measurements, the relative change in neuronal activity can be assessed (59, 60). Instead of measuring electrical activity, some studies instead acquire the change in oxygen usage [cerebral metabolic rate of oxygen (CMRO2)] from neuroactivation under the assumption of a strong correlation between electrical activity and energy consumption in neurons (61–63).
Given the difficulties in measuring neuronal activation, many studies only acquire the CBF response as a surrogate marker of NVC and then assume that neuronal electrical activation is constant across the studied population. Perfusion PET imaging can be used to measure the CBF response to neuroactivation by acquiring resting perfusion maps and perfusion maps during neuroactivation (64, 65). PET imaging is considered the gold standard for measuring quantitative CBF maps in vivo; however, due to the invasive procedure from the use of radioactive tracers and the need for arterial blood samples for an input function for kinetic modeling (46, 66, 67), PET imaging is rarely used to examine NVC in a research setting. Instead, the non-invasive arterial spin labeling (ASL) MRI technique can be applied to measure changes in CBF from neuronal activation (68). Additionally, by combining ASL with blood-oxygen-level-dependent (BOLD) imaging, a technique referred to as calibrated BOLD (cBOLD), the change in CMRO2 from neuroactivation can be calculated (69). Traditional BOLD imaging has also been used to examine the CBF response to activation as an indirect measure of NVC. By using the BOLD imaging, the temporal evolution of the hemodynamic response function (HRF), e.g., time to peak and peak amplitude, of the NVC can be characterized.
TCD has also been used to assess the CBF response to neuroactivation (70). This technique measures the blood velocity in one of the larger cerebral arteries supplying the activated brain region. Typically, the change in blood velocity in the posterior cerebral arteries (PCA) or middle cerebral artery (MCA) during visual activation is used as a marker of the CBF response to neuroactivation. The examination of CBF to neuroactivation by TCD serves as an indirect measurement because the regional change in perfusion in the activated region is not obtained, but only the upstream change in blood flow velocity (CBFv) in the large artery supplying the activated brain region is measured. Given that the artery supplies not only the activated region but also other areas of the brain, the measured change in blood velocity is a diluted average of a much larger brain area. When measuring CBF by TCD, the response to visual stimulation results in an increase in the blood velocity through the PCA by ~5–40% and through the MCA by ~0–10% (71–73), in contrast to the much larger focal increase in perfusion in the visual cortex, which is ~50-100% (64, 66, 68, 74). Thus, the examination of the CBF response by TCD is not fully quantifiable.
When assessing NVC, neuroactivation is typically obtained by visual stimulation of the participants, often in the form of a flickering light pattern eliciting a strong neuroactivation in the visual cortex (75).
Methods
We conducted the review according to the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) Statement (76).
Search Strategy
The search for articles to include in the systematic review was carried out via PubMed using a combination of free text words and MeSH terms. The initial search was performed on 31st of May 2021. The search was repeated on 30th of November 2021 to include papers published since the initial search.
The following PubMed search string was used:
[“multiple sclerosis”[MeSH Terms] OR “multiple sclerosis, relapsing remitting”[MeSH Terms] OR “progressive multiple sclerosis”[Text Word] OR “multiple sclerosis”[Text Word]) AND (“cerebrovascular reactivity”[Text Word] OR “Vasomotor”[Text Word] OR “vasodilation”[Text Word] OR “vasomotor reactivity”[Text Word] OR “cerebrovascular reserve capacity”[Text Word] OR “Cerebral autoregulation”[Text Word] OR “arterial stiffness“[Text Word] OR “endothelium dysfunction”[Text Word] OR “endothelium damage”[Text Word] OR “endothelium injury”[Text Word] OR “hemodynamic”[Text Word] OR “hemodynamic response function”[Text Word] OR “neurovascular coupling”[Text Word]].
Eligibility Criteria
Studies investigating CVR or NVC in a quantitative or semiquantitative fashion in MS patients were included in the review. Only original research studies were included. Articles in languages other than English were not included. All papers were initially screened by MBV based on the title and abstract. Papers that passed the initial screening were included in a full-text eligibility assessment. The reference lists of the included papers were reviewed for additional eligible articles.
Data Extraction
The CVR and NVC results were extracted as the primary output. The techniques used to measure CVR and NVC (MRI, TCD etc.) and the means of vasodilation and neuronal activation (CO2 inhalation, breath-holding, visual stimulation etc.) were additionally extracted. Finally, we identified data regarding the participants (i.e., type of MS, disease duration, medication status, current disease status, Expanded Disability Status Scale (EDSS) and additional disease parameters if provided).
The data extracted from the included studies are summarized in Tables 1, 2.

Table 1. Summary of studies examining cerebrovascular reactivity in MS.
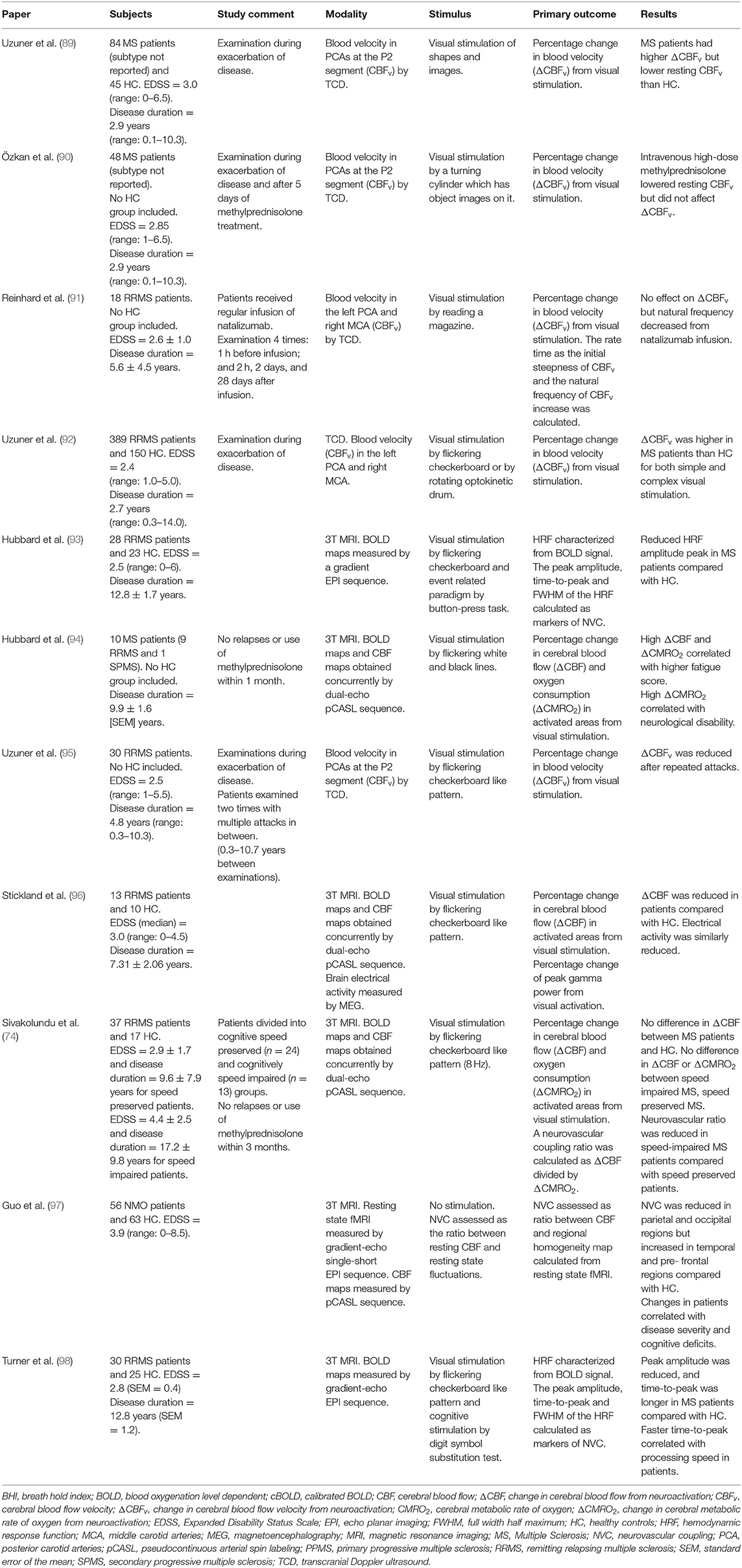
Table 2. Summary of studies examining neurovascular coupling in MS.
Results
Literature Search
The PubMed search resulted in 159 articles. Twelve articles examined CVR (Figure 1; Table 1). Six articles examined NVC and five additional studies were included from the reference lists, resulting in a total of 11 articles on NVC (Figure 1; Table 2).
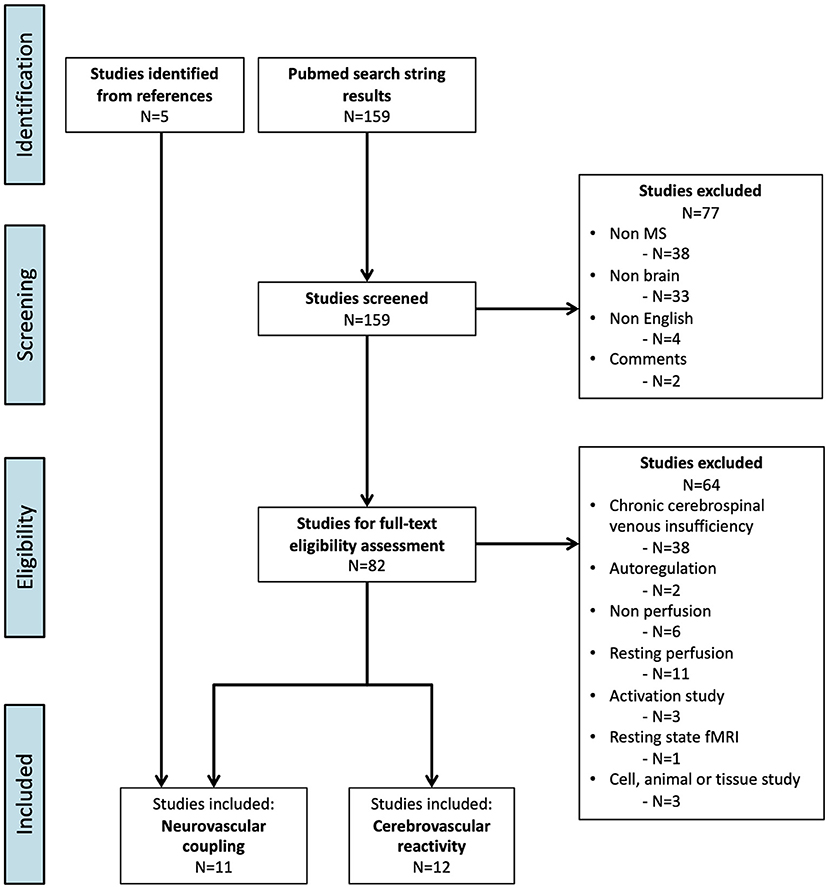
Figure 1. Flowchart of the inclusion process (PRISMA flow diagram) (76).
Cerebrovascular Reactivity
A summary of the 12 studies on CVR is presented in Table 1. All studies included a healthy control (HC) group for comparison with the MS patient group. The studies demonstrated mixed results. Six studies resulted in no differences between MS patients and HC, and 5 studies resulted in reduced CVR in patients compared with HC. In one study, the controls consisted of migraine patients, where MS patients demonstrated higher CVR than migraine patients. All studies used CO2 as the vasodilating agent, either by inhaling air with added CO2 or through breath-hold challenges. The studies used either the TCD methodology (n = 6), the BOLD imaging MRI technique (n = 2) or the ASL MRI technique (n = 4). The participating patients were primarily relapsing remitting MS (RRMS) patients; however, in some studies, patients with secondary progressive MS (SPMS) were also included.
Neurovascular Coupling
A summary of the 11 studies on NVC is presented in Table 2. In eight of the studies, HC were included for comparison. These studies showed mixed results: two studies demonstrated higher NVC in MS patients; two studies found no differences; three studies reported reduced NVC in patients; and one study observed increased NVC in some brain regions but decreased NVC in other brain regions compared with HC. Four of the studies used TCD Doppler to measure the increase in blood velocity in the PCA or MCA from visual stimulation as a marker of NVC. Three of the studies used calibrated BOLD and ASL MRI to measure the increase in CBF and CMRO2 in response to visual stimulation in the visual cortex. One of the studies used a combination of resting-state fMRI and ASL to acquire metrics describing NVC. Two studies used conventional BOLD imaging and calculated the characteristics of the HRF as markers of NVC.
All studies used visual stimulation for neuronal activation, except for Hubbard et al. (93), who used a combination of visual stimulation and a button press task, and Turner et al. (98), who used a combination of visual stimulation and a cognitive task.
The patients included in the studies consisted of RRMS patients, except for the work of Hubbard et al. (94), in which the studied population consisted of RRMS and one SPMS patient, and that of Guo et al. (97) who examined neuromyelitis optica (NMO) patients.
Discussion
Cerebrovascular Reactivity
In all studies, CO2 was used as the vasodilating agent, either by inhalation of air with added CO2 or through breath-hold challenges. The studies found either no difference or a reduced CVR between MS patients and HC.
Using the ASL MRI technique, Marshall et al. demonstrated in 2014 (78), and again in 2016 (79), that patients with MS have a reduced CVR in response to the inhalation of air with 5% CO2 compared with HC. The mean Expanded Disability Status Scale (EDSS) score was 2.9, and most subjects received second-line treatment. Recent relapse was not an exclusion criterion. A more recent study by Pelizzari et al. (83) that also used ASL MRI found no significant differences in CVR between MS patients and HC from inhalation of air with 5% CO2. This cohort had a mean EDSS of 1.5 and predominantly received no treatment or first-line treatment. Patients were excluded from the study if they had experienced a recent relapse. The authors speculate that the low degree of inflammation in their MS cohort may explain the lack of difference between patients and HC (83).
The role of severity and disease stage in the magnitude of possible CVR changes in MS was suggested in a recent study by Lattanzi et al. (84), who used TCD to measure the breath-hold-index (BHI) as a marker of CVR in a total of 80 RRMS patients outside of relapse and 80 HC, making it the largest current TCD study addressing CVR differences in MS. The authors report a groupwise progressively decreasing BHI from HC to RRMS and SPMS. The authors used bilateral simultaneous measurements of the MCA, repeating the breath hold experiment three times for each subject, and then calculating their mean value. This strategy reduced the variability of the reported BHI when compared with other TCD studies. Smoliński et al. (86), who found no difference in BHI between MS patients and HC, performed one unilateral measurement in each subject and reported a standard deviation in RRMS patients outside of relapse 2.5 times larger than the reported results of Lattanzi et al. (84) This finding indicates that for BHI, which is a method reported to have a good short-term but poor long-term reproducibility, a concern is methodological variability.
Several smaller TCD studies have also reported no difference in BHI between MS patients and HC; however, they applied variable methodologies, which ranged from using a short breath hold of 15 sec (77), which may not be long enough to achieve a sufficient CO2 increase (99), to using migraine patients as control subjects (80). It is noteworthy that most of these negative studies have utilized a single unilateral measurement and generally reported relatively high standard deviations of the measured BHI (86, 87). Finally, three of these negative studies showed a non-significant trend toward lower BHI in RRMS when compared with HC (77, 82, 87), and may have been underpowered and unable to detect potentially small differences. Two studies utilized the change in MRI BOLD response to CO2 inhalation as a marker of CVR and reported that CVR changes may be secondary to cognitive impairment in MS patients (81, 85). However, a similar relationship has been observed for neurodegenerative diseases (100), and thus, it remains to be seen how much of the CVR changes observed in MS are driven by early cognitive impairment, since this potential confounder has not been addressed in most studies. Other potential confounding factors may be recent relapses and steroid treatment, which has been found to significantly decrease CVR on a single subject level in one study (86).
Methodological Considerations
Burley et al. (101) investigated the correlations between four different methods for estimating CVR, namely TCD, BOLD MRI, ASL MRI and PCM MRI, in healthy subjects (101). The authors found no clear relationship between TCD and BOLD measures, highlighting that the different methodological approaches address different anatomical parts of the vasculature (TCD: arteries, BOLD: venules/veins, ASL: arterioles and capillaries). This finding calls for caution when comparing the CVR metrics derived from different imaging modalities. On the positive side, resting CBF was highly correlated between ASL, PCM and TCD, suggesting that vasodilation by CO2 cannot automatically be assumed to exert similar homogenous effects throughout the different anatomical parts of the cerebral vasculature.
The TCD methodology has an inherent problem when attempting to address changes in blood flow and thus CVR, because it does not directly measure flow but rather velocity. To use the blood velocity as a proxy for blood flow, the assumption that there is no change in the cross-sectional area of the artery following a stimulus must be valid. If such an effect differs significantly between the groups to be compared, for example between HC and patients, true physiological differences may be overlooked. Recently, it has been pointed out that blood vessel diameter is likely to be affected in the context of a gas challenge (102, 103), coinciding with an increase in blood velocity. This factor leads to an overestimation of CVR from a gas challenge in cases where blood vessels are less pliable.
Neurovascular Coupling
Studies examining NVC demonstrate mixed and contradictory results. Three studies, Stickland et al. (96), Sivakolundu et al. (74) and Hubbard et al. (94) measured the change in CBF from neuronal activation (ΔCBF) in the visual cortex using the ASL MRI technique. Furthermore, Hubbard et al. (94) and Sivakolundu et al. (74) additionally calculated the change in CMRO2 (ΔCMRO2) using a calibrated BOLD technique to examine the coupling between CBF and CMRO2 as a marker for NVC. Stickland et al. (96) acquired brain electrical activity using MEG to examine the NVC as the coupling between CBF and MEG gamma band power. Guo et al. (97) also examined NVC using the BOLD and ASL techniques. However, the study did not measure NVC in relation to external stimuli but examined the NVC from the resting fluctuation of brain activity and CBF in the resting state networks.
Sivakolundu et al. (74) found no differences in the ΔCBF response to visual stimulation between MS patients and HC. The study further examined the differences in ΔCBF response between MS patients with reduced cognitive processing speed and patients with normal processing speed but no significance was observed. However, when examining the ratio between ΔCBF and ΔCMRO2 as a marker of NVC, they found the ratio to be significantly reduced in patients with impaired processing speed, suggesting a neurovascular uncoupling. The authors hypothesize that the impaired NVC can, in part, explain the reduced processing speed and cognitive impairment observed in some MS patients. Stickland et al. (96) observed a significantly smaller ΔCBF in MS patients than in HC. However, the neuronal response measured by the MEG gamma band power was similarly reduced, which indicates that the NVC is not affected in MS. This finding is in contrast to the results of Sivakolundu et al. (74) who observed no differences in ΔCBF but found a reduction of NVC in MS. In contrast to both the studies of Stickland et al. (96) and Sivakolundu et al. (74), Hubbard et al. (94) found that a higher ΔCBF and ΔCMRO2 was correlated with fatigue and more severe MS disease, suggesting that NVC was increased in MS patients. The higher metabolic response was also correlated with white matter myelin abnormalities examined by the diffusion tensor imaging (DTI) MRI technique. The study did not include a control group and the group of patients was small (n = 10). Finally, Guo et al. (97) examined NVC in NMO patients based on the resting state fluctuation of the BOLD signal and CBF in the brain using fMRI and ASL MRI techniques. The study found that NVC was reduced in some brain regions but increased in other regions in NMO patients compared with HC. The different NVCs, both higher and reduced NVC, correlated with the disease severity of the NMO patients.
Two studies by Hubbard et al. (93) and Turner et al. (98) indirectly examined the CBF response to activation using traditional BOLD imaging, in which the peak of amplitude and the time to peak of the BOLD HRF were used to describe the NVC. Both studies found that the peak amplitude of the BOLD signal was reduced in MS patients compared with HC, suggesting a reduced cerebrovascular response in MS. Turner et al. (98) also demonstrated a longer time to peak in MS patients and that a longer time to peak correlated with poor cognitive processing speed performance, indicating a correlation between cognitive decline and an impaired CBF response to neuroactivation.
Two studies by Uzuner et al. (89, 92) indirectly examined the CBF response to neuroactivation by measuring the blood velocity in PCA (ΔCBFv) using TCD. Both studies found higher CBFv from visual stimulation in patients with MS during a period of exacerbation of the disease compared with HC. The studies also demonstrated a lower resting CBFv in MS patients than in HC. Overall, these results correspond with Hubbard et al. (93), who found higher ΔCBF in MS patients measured using the ASL MRI technique.
Two studies examined whether infusion of MS medication affected NVC. Özkan et al. (90) and Reinhard et al. (91) examined NVC before and after infusion of methylprednisolone or natalizumab, respectively. Both studies used TCD to examine the NVC. Methylprednisolone treatment lowered the resting CBFv; however, ΔCBFv from neuroactivation was unaffected. Treatment with natalizumab did not affect ΔCBFv but increased the very initial response in ΔCBFv from neuroactivation. Furthermore, the authors suggest that these findings are evidence for increased reactivity of the neurons or cerebral vessels after treatment.
The cause for these mixed observations is not obvious. Potential causes for differences in ΔCBF and ΔCMRO2 could be due to the heterogeneity of the examined patients with important differences in factors such as age, disease severity, MS subtype and treatment status. Variation in the degree of atrophy in MS patients could also affect the metabolic response to activation, as patients with MS demonstrate increased brain atrophy compared with age-matched control subjects (104–107). If the examined MS patients have fewer neurons due to atrophy, the metabolic response to activation would also be reduced. Ideally, studies should acquire some measure of atrophy or cortical thickness to address this potential confounder. However, brain volume estimation in MS patients must be interpreted with caution since MS patients can exhibit transient increases in brain volume due to inflammatory processes causing increased water and inflammatory cell volume. This phenomenon is often referred to as pseudoatrophy, as it is often observed in MS patients following disease-modifying anti-inflammatory treatment (104, 108). Therefore, differences in brain volume therefore do not necessarily directly reflect changes in neurons or glia cells. For example, Stickland et al. (96), who observed a reduced CBF response in MS patients, examined the gray matter volume in the visual cortex and did not observe a higher degree of atrophy in the MS patients than in the HC. This indicates that the reduced metabolic response is not a result of fewer viable neurons. However, the authors speculate that the lack of differences in atrophy between patients and HC could be due to the inflammatory processes in the MS brain causing swelling of the brain, resulting in higher brain volume estimates derived from anatomical images.
Some studies have demonstrated higher ΔCBF or NVC in MS patients. Hubbard et al. (94), who observed that higher ΔCBF and ΔCMRO2 were correlated with more severe MS disease, suggests that the reason for a higher metabolic response in MS could be due to a reduced resting metabolism. If the patients have reduced resting metabolism, they will demonstrate a higher percentage increase in CBF and CMRO2 in response to neuroactivation. The study did not include a control group to test this hypothesis. However, other studies have demonstrated reduced resting perfusion in MS patients that correlates with disease severity (32, 109, 110). A study by West et al. (34) have subsequently demonstrated that MS patients also have lower resting CMRO2 than HC. However, in this study, it was also observed that patients with high resting CMRO2 had worse fatigue and cognitive symptoms, which contrasts with the hypothesis of higher NVC due to lower resting metabolism in patients with more severe disease symptoms.
Uzuner et al. (89, 92) found that ΔCBFv was higher in MS patients who were examined during disease exacerbation. The authors suggest that patients with MS have more reactive neurons and cerebral vessels in response to neuroactivation during active disease. A subsequent study from Uzuner et al. (95), where patients were examined again after 26.7 months (range: 4–120 months) of disease progression, demonstrated that ΔCBFv was reduced after repeated relapses. The authors suggest that following disease progression, neuronal damage secondary to inflammatory activity inhibits the neurovascular unit and reduces NVC, which suggests that MS disease will progressively impair NVC over time.
Methodological Considerations
The use of ASL to measure increases in CBF from neuroactivation has good reproducibility (68) and has been validated against 15O-H2O PET imaging (65), which is considered the best available method for the quantification of cerebral perfusion. However, CBF measurements from different ASL MRI techniques differ significantly between MRI scanners and the applied data acquisition sequence. Thus, the heterogeneity of the ASL sequences and applied methodology between research groups could potentially bias the results (111, 112).
The examination of NVC by TCD is an indirect measurement because it measures the change in blood velocity in a large upstream artery supplying the activated region of the brain. Several TCD studies have investigated the magnitude of the increase in blood velocity from neuroactivation by a visual stimulation as well as a cognitive task. However, to our knowledge, whether the percentage increase in blood velocity measured by TCD is proportional to the actual NVC in the affected tissue has not yet been validated. The indirect measurement of the upstream effect will have confounding factors. For instance, most notably, the effect of increased perfusion in the activated region will be greatly diluted as the large artery also supplies other regions of the brain. Therefore, a clear confounding factor of this upstream measurement is therefore the size of the activated region, as larger activated areas will have stronger effects on the measurement compared with smaller activated areas. Thus, when examining the NVC, the primary interest is not to measure the size of the activated area but instead to measure how much CBF has increased in the activated region. This factor makes it challenging to draw a conclusion about the amplitude of NVC when measuring the change in upstream blood flow velocity by TCD. Therefore, direct comparisons between studies using TDCs and studies using other neuroimaging techniques to examine cerebrovascular responses to neuroactivation should be performed with a high degree of caution.
The larger ΔCBFv observed in MS patients can also be a result of a failure to fulfill the assumption that there is no significant difference exists in the cross-sectional area of the artery following stimulation between the studied groups. The MS patients may demonstrate a smaller change in the cross-sectional area, which increases blood flow velocities to ensure an increase in CBF from activation. Similar concerns are present when examining CVR from CO2 stimulation as discussed earlier. Senzaki et al. (87) have demonstrated that MS patients have reduced dilation of the brachial artery from reactive hyperemia, suggesting an inhibited vasodilation in MS patients, perhaps due to endothelial dysfunction.
Activation Pattern Studies
Several studies have examined neuroactivation in MS using BOLD imaging and have found alterations in activation patterns (113–120). Most studies have shown larger activation patterns in MS, which could be a result of a hyperreactivity of neurons and cerebral vessels, resulting in a higher NVC in MS patients. The BOLD signal is generated by the NVC, and changes in this coupling would likely cause alterations of the activation patterns seen in MS patients.
Conclusion
Due to increasing evidence for tissue hypoxia and endothelial vessel damage in MS, it seems plausible to speculate that MS patients exhibit impaired cerebrovascular reactivity and perfusion modulation. Reduced cerebrovascular function is associated with declining brain health, especially in the context of age-related neurodegeneration. Patients with MS demonstrate a progressive decline in brain health in the form of neurodegeneration with advancing disease stages. Whether these processes are, at least partially, related to cerebrovascular function warrants a thorough investigation.
Studies on CVR in MS patients demonstrate either no significant differences between patients and HC or reduced CVR in patients. This discrepancy can be because CVR is not affected in early MS disease but becomes progressively more affected after a certain disease period, with advancing disease stage.
The studies on NVC demonstrate diverse results, and a conclusion that describes all the published observations is difficult to establish. Some studies found indications of an increased CBF response to neuroactivation and increased NVC, especially during exacerbation of disease, perhaps due to an increase in reactive neurons. A higher NVC can also be a result of a lower resting metabolism, which then causes a larger percentage increase in CBF and CMRO2 in response to neuronal activation. Other studies found indications of a reduced CBF response to activation, which can be a result of impaired vasodilation and endothelial function in MS patients. More studies are needed to elaborate on these findings. Ideally, future studies should directly measure the increase in CBF and neuronal activity. A measure of brain atrophy should be included to address the role of atrophy in explaining the changes in NVC. Resting perfusion and metabolism should also be acquired to investigate whether reduced resting physiology can explain a relatively higher cerebrovascular and neuronal activity response in MS patients. Finally, the studies should have sufficient power to examine the effect of confounding factors such as age, MS subgroup, disease severity, treatment status and recent acute inflammatory events.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author.
Author Contributions
MV and HL conceived and designed the study. MV and SC collected the data and wrote the paper. JF and HL edited and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
MV was funded by grant from the Lundbeck Foundation (R347-2020-217).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lassmann H, Brück W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol. (2007) 17:210–8. doi: 10.1111/j.1750-3639.2007.00064.x
2. Correale J, Villa A. The blood-brain-barrier in multiple sclerosis: functional roles and therapeutic targeting. Autoimmunity. (2007) 40:148–60. doi: 10.1080/08916930601183522
3. Pun PBL, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. (2009) 43:348–64. doi: 10.1080/10715760902751902
4. Cramer SP, Simonsen H, Frederiksen JL, Rostrup E, Larsson HBW. Abnormal blood-brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. NeuroImage Clin. (2014) 4:182–9. doi: 10.1016/j.nicl.2013.12.001
5. Larochelle C, Alvarez JI, Prat A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. (2011) 585:3770–80. doi: 10.1016/j.febslet.2011.04.066
6. Minagar A, Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Mult Scler. (2003) 9:540–9. doi: 10.1191/1352458503ms965oa
7. Lécuyer MA, Kebir H, Prat A. Glial influences on BBB functions and molecular players in immune cell trafficking. Biochim Biophys Acta. (2016) 1862:472–82. doi: 10.1016/j.bbadis.2015.10.004
8. Lassmann H. Hypoxia-like tissue injury as a component of multiple sclerosis lesions. J Neurol Sci. (2003) 206:187–91. doi: 10.1016/S0022-510X(02)00421-5
9. Alexander JS, Zivadinov R, Maghzi AH, Ganta VC, Harris MK, Minagar A. Multiple sclerosis and cerebral endothelial dysfunction: mechanisms. Pathophysiology. (2011) 18:3–12. doi: 10.1016/j.pathophys.2010.04.002
10. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxidants Redox Signal. (2014) 20:1126–67. doi: 10.1089/ars.2012.5149
11. Haacke EM, Ge Y, Sethi SK, Buch S, Zamboni P. An overview of venous abnormalities related to the development of lesions in multiple sclerosis. Front Neurol. (2021) 12:561458doi: 10.3389/fneur.2021.561458
12. Snyder HM, Corriveau RA, Craft S, Faber JE, Knopman D, Lamb BT, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer's disease. Alzheimer's Dement. (2015) 11:710–7. doi: 10.1016/j.jalz.2014.10.008
13. Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders berislav. Nat Rev Neurosci. (2011) 12:723–38. doi: 10.1038/nrn3114.Neurovascular
14. Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, et al. Vascular dysfunction — the disregarded partner of Alzheimer's disease. Alzheimer's Dement. (2019) 15:158–67. doi: 10.1016/j.jalz.2018.07.222
15. Glodzik L, Randall C, Rusinek H, de Leon MJ. Cerebrovascular reactivity to carbon dioxide in Alzheimer's disease. J Alzheimers Dis. (2013) 35:427–40. doi: 10.3233/JAD-122011
16. McKetton L, Sobczyk O, Duffin J, Poublanc J, Sam K, Crawley AP, et al. The aging brain and cerebrovascular reactivity. Neuroimage. (2018) 181:132–41. doi: 10.1016/j.neuroimage.2018.07.007
17. Silvestrini M, Pasqualetti P, Baruffaldi R, Bartolini M, Handouk Y, Matteis M, et al. Cerebrovascular reactivity and cognitive decline in patients with Alzheimer disease. Stroke. (2006) 37:1010–5. doi: 10.1161/01.STR.0000206439.62025.97
18. Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. (2006) 100:328–35. doi: 10.1152/japplphysiol.00966.2005
19. Toth P, Tarantini S, Csiszar A, Ungvari Z. Functional vascular contributions to cognitive impairment and dementia: Mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol - Hear Circ Physiol. (2017) 312:H1–H20. doi: 10.1152/ajpheart.00581.2016
20. Costantino Iadecola M. The pathobiology of vascular dementia. Neuron. (2013) 80:844–66. doi: 10.1016/j.neuron.2013.10.008
21. Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer's disease amy. Biochim Biophys Acta J. (2016) 1862:887–900. doi: 10.1016/j.bbadis.2015.12.016
22. Eskes GA, Longman S, Brown AD, McMorris CA, Langdon KD, Hogan DB, et al. Contribution of physical fitness, cerebrovascular reserve and cognitive stimulation to cognitive function in post-menopausal women. Front Aging Neurosci. (2010) 2:137. doi: 10.3389/fnagi.2010.00137
23. Guadagni V, Drogos LL, Tyndall AV, Davenport MH, Anderson TJ, Eskes GA, et al. Aerobic exercise improves cognition and cerebrovascular regulation in older adults. Neurology. (2020) 94:e2245–57. doi: 10.1212/WNL.0000000000009478
24. Davenport MH, Hogan DB, Eskes GA, Longman RS, Poulin MJ. Cerebrovascular reserve: the link between fitness and cognitive function? Exerc Sport Sci Rev. (2012) 40:153–8. doi: 10.1097/JES.0b013e3182553430
25. Vestergaard MB, Jensen MLF, Arngrim N, Lindberg U, Larsson HBW. Higher physiological vulnerability to hypoxic exposure with advancing age in the human brain. J Cereb Blood Flow Metab. (2020) 40:341–53. doi: 10.1177/0271678X18818291
26. Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. (2009) 8:280–91. doi: 10.1016/S1474-4422(09)70043-2
27. Juurlink BHJ. The evidence for hypoperfusion as a factor in multiple sclerosis lesion development. Mult Scler Int. (2013) 2013:1–6. doi: 10.1155/2013/598093
28. Aboul-Enein F, Lassmann H. Mitochondrial damage and histotoxic hypoxia: a pathway of tissue injury in inflammatory brain disease? Acta Neuropathol. (2005) 109:49–55. doi: 10.1007/s00401-004-0954-8
29. Davies AL, Desai RA, Bloomfield PS, McIntosh PR, Chapple KJ, Linington C, et al. Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Ann Neurol. (2013) 74:815–25. doi: 10.1002/ana.24006
30. Desai RA, Davies AL, Tachrount M, Kasti M, Laulund F, Golay X, et al. Cause and prevention of demyelination in a model multiple sclerosis lesion. Ann Neurol. (2016) 79:591–604. doi: 10.1002/ana.24607
31. Wekerle H. Tissue hypoxia in autoimmune brain disease: a Columbian discovery? Ann Neurol. (2013) 74:765–7. doi: 10.1002/ana.24054
32. Sun X, Tanaka M, Kondo S, Okamoto K, Hirai S. Clinical significance of reduced cerebral metabolism in multiple sclerosis: a combined PET and MRI study. Ann Nucl Med. (1998) 12:89–94. doi: 10.1007/bf03164835
33. Fan AP, Govindarajan ST, Kinkel RP, Madigan NK, Nielsen AS, Benner T, et al. Quantitative oxygen extraction fraction from 7-Tesla MRI phase: Reproducibility and application in multiple sclerosis. J Cereb Blood Flow Metab. (2015) 35:131–9. doi: 10.1038/jcbfm.2014.187
34. West K, Sivakolundu D, Maruthy G, Zuppichini M, Liu P, Thomas B, et al. Baseline cerebral metabolism predicts fatigue and cognition in Multiple Sclerosis patients. NeuroImage Clin. (2020) 27:102281. doi: 10.1016/j.nicl.2020.102281
35. Paulson OB, Strandgaard S, Edvinsson L. Cerebral autoregulation. Cerebrovasc Brain Metab Rev. (1990) 2:161–92.
36. Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland B a, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. (2014) 508:55–60. doi: 10.1038/nature13165
37. Willie CK, Tzeng Y-C, Fisher JA, Ainslie PN. Integrative regulation of human brain blood flow. J Physiol. (2014) 592:841–59. doi: 10.1113/jphysiol.2013.268953
38. Chen JJ, Pike GB. Global cerebral oxidative metabolism during hypercapnia and hypocapnia in humans: implications for BOLD fMRI. J Cereb Blood Flow Metab. (2010) 30:1094–9. doi: 10.1038/jcbfm.2010.42
39. Peiying L, De Vis J, Lu H. Cerebrovascular reactivity (CVR) MRI with CO2 challenge: a technical review. Neuroimage. (2019) 187:104–15. doi: 10.1016/j.neuroimage.2018.03.047
40. Yoon SH, Zuccarello M, Rapoport RM. pCO 2 and pH regulation of cerebral blood flow. Front Physiol. (2012) 3:1–8. doi: 10.3389/fphys.2012.00365
41. Vestergaard MB, Larsson HBW. Cerebral metabolism and vascular reactivity during breath-hold and hypoxic challenge in freedivers and healthy controls. J Cereb Blood Flow Metab. (2019) 39:834–48. doi: 10.1177/0271678X17737909
42. Ito H, Kanno I, Ibaraki M, Hatazawa J, Miura S. Changes in human cerebral blood flow and cerebral blood volume during hypercapnia and hypocapnia measured by positron emission tomography. J Cereb Blood Flow Metab. (2003) 23:665–70. doi: 10.1097/01.WCB.0000067721.64998.F5
43. Kety SS, Schmidt CF. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest. (1948) 27:484–92.
44. Vestergaard MB, Lindberg U, Aachmann-Andersen NJ, Lisbjerg K, Christensen SJ, Rasmussen P, et al. Comparison of global cerebral blood flow measured by phase-contrast mapping MRI with 15O-H2O positron emission tomography. J Magn Reson Imaging. (2017) 45:692–9. doi: 10.1002/jmri.25442
45. Bakker CJG, Hartkamp MJ, Mali WPTM. Measuring blood flow by nontriggered 2D phase-contrast MR angiography. Magn Reson Imaging. (1996) 14:609–14. doi: 10.1016/0730-725X(96)00092-6
46. Vestergaard MB, Calvo OP, Hansen AE, Rosenbaum S, Larsson HBW, Henriksen OM, et al. Validation of kinetic modeling of [15O]H2O PET using an image derived input function on hybrid PET/MRI. Neuroimage. (2021) 233:117950. doi: 10.1016/j.neuroimage.2021.117950
47. Scheel P, Ruge C, Schöning M. Flow velocity and flow volume measurements in the extracranial carotid and vertebral arteries in healthy adults: reference data and the effects of age. Ultrasound Med Biol. (2000) 26:1261–6. doi: 10.1016/S0301-5629(00)00293-3
48. Battisti-Charbonney A, Fisher J, Duffin J. The cerebrovascular response to carbon dioxide in humans. J Physiol. (2011) 589:3039–48. doi: 10.1113/jphysiol.2011.206052
49. Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar B a, Newman E a. Glial and neuronal control of brain blood flow. Nature. (2010) 468:232–43. doi: 10.1038/nature09613
50. Meng W, Tobin JR, Busija DW. Glutamate-induced cerebral vasodilation is mediated by nitric oxide through n-methyl-d-aspartate receptors. Stroke. (1995) 26:857–63. doi: 10.1161/01.STR.26.5.857
51. Busija DW, Bari F, Domoki F, Louis T. Mechanisms involved in the cerebrovascular dilator effects of N- methyl-D-aspartate in cerebral cortex. Brain Res Rev. (2007) 56:89–100. doi: 10.1016/j.brainresrev.2007.05.011
52. Carvajal JA, Germain AM, Huidobro-Toro JP, Weiner CP. Molecular mechanism of cGMP-mediated smooth muscle relaxation. J Cell Physiol. (2000) 184:409–20. doi: 10.1002/1097-4652(200009)184:3<409::AID-JCP16>3.0.CO;2-K
53. Gordon GRJ, Choi HB, Rungta RL, Ellis-Davies GCR, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. (2008) 456:745–9. doi: 10.1038/nature07525
54. Filosa JA, Iddings JA. Astrocyte regulation of cerebral vascular tone. Am J Physiol - Hear Circ Physiol. (2013) 305:H609–19. doi: 10.1152/ajpheart.00359.2013
55. Lauritzen M, Mathiesen C, Schaefer K, Thomsen KJ. Neuronal inhibition and excitation, and the dichotomic control of brain hemodynamic and oxygen responses. Neuroimage. (2012) 62:1040–50. doi: 10.1016/j.neuroimage.2012.01.040
56. Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, et al. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. (2006) 9:1397–403. doi: 10.1038/nn1779
57. Chen BR, Kozberg MG, Bouchard MB, Shaik MA, Hillman EMC. A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J Am Heart Assoc. (2014) 3:1–14. doi: 10.1161/JAHA.114.000787
58. Lopes da Silva F. EEG and MEG: relevance to neuroscience. Neuron. (2013) 80:1112–28. doi: 10.1016/j.neuron.2013.10.017
59. Oakes TR, Pizzagalli DA, Hendrick AM, Horras KA, Larson CL, Abercrombie HC, et al. Functional coupling of simultaneous electrical and metabolic activity in the human brain. Hum Brain Mapp. (2004) 21:257–70. doi: 10.1002/hbm.20004
60. Oostenveld R, Fries P, Maris E, Schoffelen JM. FieldTrip: Open source software for advanced analysis of MEG, EEG, and invasive electrophysiological data. Comput Intell Neurosci. (2011) 2011:156869. doi: 10.1155/2011/156869
61. Shetty PK, Galeffi F, Turner DA. Cellular links between neuronal activity and energy homeostasis. Front Pharmacol. (2012) 3:43. doi: 10.3389/fphar.2012.00043
62. Alkire MT. Quantitative EEG correlations with brain glucose metabolic rate during anesthesia in volunteers. Anesthesiology. (1998) 89:323–33. doi: 10.1097/00000542-199808000-00007
63. Nagata K, Tagawa K, Hiroi S, Shishido F, Uemura K. Electroencephalographic correlates of blood flow and oxygen metabolism provided by positron emission tomography in patients with cerebral infarction. Electroencephalogr Clin Neurophysiol. (1989) 72:16–30. doi: 10.1016/0013-4694(89)90027-8
64. Fox PT, Raichle ME, Mintun MA, Carmen D. Nonoxidative glucose consumption during focal physiologic neural activity. Science. (1988) 241:462–4. doi: 10.1126/science.3260686
65. Feng CM, Narayana S, Lancaster JL, Jerabek PA, Arnow TL, Zhu F, et al. changes during brain activation: fMRI vs. PET Neuroimage. (2004) 22:443–6. doi: 10.1016/j.neuroimage.2004.01.017
66. Ito H, Takahashi K, Hatazawa J, Kim S-G, Kanno I. Changes in human regional cerebral blood flow and cerebral blood volume during visual stimulation measured by positron emission tomography. J Cereb Blood Flow Metab. (2001) 12:608–612. doi: 10.1097/00004647-200105000-00015
67. Zanotti-Fregonara P, Chen K, Liow JS, Fujita M, Innis RB. Image-derived input function for brain PET studies: many challenges and few opportunities. J Cereb Blood Flow Metab. (2011) 31:1986–98. doi: 10.1038/jcbfm.2011.107
68. Raoult H, Petr J, Bannier E, Stamm A, Gauvrit JY, Barillot C, et al. Arterial spin labeling for motor activation mapping at 3T with a 32-channel coil: reproducibility and spatial accuracy in comparison with BOLD fMRI. Neuroimage. (2011) 58:157–67. doi: 10.1016/j.neuroimage.2011.06.011
69. Davis TL, Kwong KK, Weisskoff RM, Rosen BR. Calibrated functional MRI: mapping the dynamics of oxidative metabolism. Proc Natl Acad Sci U S A. (1998) 95:1834–9.
70. Sushmita Purkayastha, Farzaneh P.. Transcranial doppler ultrasound: technique and application. Semin Neurol. (2014) 32:411–420. doi: 10.1055/s-0032-1331812
71. Lisak M, Trkanjec Z, Mikula I, Demarin V, Aleksić-Shibabi A, Šulentić V. Analysis of mean blood flow velocities in posterior cerebral arteries by transcranial Doppler during visual stimulation. Acta Clin Croat. (2006) 45:309–13.
72. Conrad B, Klingelhöfer J. Dynamics of regional cerebral blood flow for various visual stimuli. Exp Brain Res. (1989) 77:437–41. doi: 10.1007/BF00275003
73. Sturzenegger M, Newell DW, Aaslid R. Visually evoked blood flow response assessed by simultaneous two-channel transcranial doppler using flow velocity averaging. Stroke. (1996) 27:2256–61. doi: 10.1161/01.STR.27.12.2256
74. Sivakolundu DK, West KL, Zuppichini M, Turner MP, Abdelkarim D, Zhao Y, et al. The neurovascular basis of processing speed differences in humans: a model-systems approach using multiple sclerosis. Neuroimage. (2020) 215:116812. doi: 10.1016/j.neuroimage.2020.116812
75. Lin AL, Fox PT, Hardies J, Duong TQ, Gao JH. Nonlinear coupling between cerebral blood flow, oxygen consumption, and ATP production in human visual cortex. Proc Natl Acad Sci U S A. (2010) 107:8446–51. doi: 10.1073/pnas.0909711107
76. Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ. (2021) 372:2020–1. doi: 10.1136/bmj.n71
77. Uzuner N, Ozkan S, Cinar N. Cerebrovascular reactivity in multiple sclerosis patients. Mult Scler. (2007) 13:737–41. doi: 10.1177/1352458506074645
78. Marshall O, Lu H, Brisset J-C, Xu F, Liu P, Herbert J, et al. Impaired cerebrovascular reactivity in multiple sclerosis. JAMA Neurol. (2014) 71:1275–81. doi: 10.1001/jamaneurol.2014.1668.Impaired
79. Marshall O, Chawla S, Lu H, Pape L, Ge Y. Cerebral blood flow modulation insufficiency in brain networks in multiple sclerosis: a hypercapnia MRI study. J Cereb Blood Flow Metab. (2016) 36:2087–95. doi: 10.1177/0271678X16654922
80. Khorvash F, Masaeli A, Shaygannejad V, Saadatnia M. Vasomotor reactivity comparison in multiple sclerosis patients with white matter lesions and nonmultiple sclerosis subjects with white matter lesions in brain magnetic resonance imaging. Adv Biomed Res. (2016) 8:5:23. doi: 10.4103/2277-9175.175916
81. Metzger A, Le Bars E, Deverdun J, Molino F, Maréchal B, Picot MC, et al. Is impaired cerebral vasoreactivity an early marker of cognitive decline in multiple sclerosis patients? Eur Radiol. (2018) 28:1204–14. doi: 10.1007/s00330-017-5068-5
82. Krogias C, Christou I, Tsivgoulis G, Koutroulou I, Schroeder C, Lantinioti C, et al. Functional neurosonology reveals impaired cerebrovascular reactivity in multiple Sclerosis. J Neuroimaging. (2019) 29:589–91. doi: 10.1111/jon.12617
83. Pelizzari L, Laganà MM, Baglio F, Bergsland N, Cecconi P, Viotti S, et al. Cerebrovascular reactivity and its correlation with age in patients with multiple sclerosis. Brain Imaging Behav. (2020) 14:1889–98. doi: 10.1007/s11682-019-00132-5
84. Lattanzi S, Acciarri MC, Danni M, Taffi R, Cerqua R, Rocchi C, et al. Cerebral hemodynamics in patients with multiple sclerosis. Mult Scler Relat Disord. (2020) 44:102309. doi: 10.1016/j.msard.2020.102309
85. Sivakolundu DK, West KL, Maruthy GB, Zuppichini M, Turner MP, Abdelkarim D, et al. Reduced arterial compliance along the cerebrovascular tree predicts cognitive slowing in multiple sclerosis: evidence for a neurovascular uncoupling hypothesis. Mult Scler J. (2020) 26:1486–96. doi: 10.1177/1352458519866605
86. Smoliński Ł, Litwin T, Kruk K, Skowrońska M, Kurkowska-Jastrzebska I, Członkowska A. Cerebrovascular reactivity and disease activity in relapsing-remitting multiple sclerosis. Adv Clin Exp Med. (2020) 29:183–8. doi: 10.17219/acem/114762
87. Senzaki K, Okada Y, Ochi H, Ochi M, Takei SI, Miura S, et al. Vascular endothelial dysfunction associated with severity in multiple sclerosis. Mult Scler Relat Disord. (2021) 54:103135. doi: 10.1016/j.msard.2021.103135
88. Deverdun J, Coget A, Ayrignac X, Carra-Dalliere C, Krainik A, Metzger A, et al.. Cerebral vasoreactivity as an indirect MRI marker of white matter tracts alterations in multiple sclerosis. Brain Topogr. (2021) 34:245–55. doi: 10.1007/s10548-021-00819-3
89. Uzuner N, Özkan S, Gücüyener D, Özdemir G. Cerebral blood flow velocity changes to visual stimuli in patients with multiple sclerosis. Mult Scler. (2002) 8:217–21. doi: 10.1191/1352458502ms798oa
90. Özkan S, Uzuner N, Kutlu C, Özbabalik D, Özdemir G. The effect of methylprednisolone treatment on cerebral reactivity in patients with multiple sclerosis. J Clin Neurosci. (2006) 13:214–7. doi: 10.1016/j.jocn.2005.03.023
91. Reinhard M, Rosengarten B, Kirchhoff L, Hetzel A, Rauer S. Natalizumab and regulation of cerebral blood flow: results from an observational study. Eur Neurol. (2010) 64:124–8. doi: 10.1159/000316765
92. Tekgöl Uzuner G, Uzuner N. Neurovascular coupling in patients with relapsing-remitting multiple sclerosis. Clin Neurol Neurosurg. (2016) 146:24–8. doi: 10.1016/j.clineuro.2016.04.020
93. Hubbard NA, Turner M, Hutchison JL, Ouyang A, Strain J, Oasay L, et al. Multiple sclerosis-related white matter microstructural change alters the BOLD hemodynamic response. J Cereb Blood Flow Metab. (2016) 36:1872–84. doi: 10.1177/0271678X15615133
94. Hubbard NA, Turner MP, Ouyang M, Himes L, Thomas BP, Hutchison JL, et al. Calibrated imaging reveals altered grey matter metabolism related to white matter microstructure and symptom severity in multiple sclerosis. Hum Brain Mapp. (2017) 38:5375–90. doi: 10.1002/hbm.23727
95. Uzuner N, Uzuner GT. Neurovascular reactivity after repeated attacks in patients with multiple sclerosis. Int J Mult Scler Relat Disord. (2017) 1:1–5.
96. Stickland R, Allen M, Magazzini L, Singh KD, Wise RG, Tomassini V. Neurovascular coupling during visual stimulation in multiple sclerosis: a MEG-fMRI study. Neuroscience. (2019) 403:54–69. doi: 10.1016/j.neuroscience.2018.03.018
97. Guo X, Zhu J, Zhang N, Zhang L, Qi Y, Cai H, et al. Altered neurovascular coupling in neuromyelitis optica. Hum Brain Mapp. (2019) 40:976–86. doi: 10.1002/hbm.24426
98. Turner MP, Hubbard NA, Sivakolundu DK, Himes LM, Hutchison JL, Jr JH, et al. Preserved canonicality of the BOLD hemodynamic response reflects healthy cognition: insights into the healthy brain through the window of multiple sclerosis. Neuroimage. (2019) 190:46–55. doi: 10.1016/j.neuroimage.2017.12.081
99. Vernieri F, Pasqualetti P, Matteis M, Passarelli F, Troisi E, Rossini PM, et al. Effect of collateral blood flow and cerebral vasomotor reactivity on the outcome of carotid artery occlusion. Stroke. (2001) 32:1552–8. doi: 10.1161/01.STR.32.7.1552
100. Catchlove SJ, Pipingas A, Hughes ME, Macpherson H. Magnetic resonance imaging for assessment of cerebrovascular reactivity and its relationship to cognition: a systematic review. BMC Neurosci. (2018) 19:1–15. doi: 10.1186/s12868-018-0421-4
101. Burley CV, Francis ST, Thomas KN, Whittaker AC, Lucas SJE, Mullinger KJ. Contrasting measures of cerebrovascular reactivity between MRI and Doppler: a cross-sectional study of younger and older healthy individuals. Front Physiol. (2021) 12:1–17. doi: 10.3389/fphys.2021.656746
102. Verbree J, Bronzwaer ASGT, Ghariq E, Versluis MJ, Daemen MJAP, Van Buchem MA, et al. Assessment of middle cerebral artery diameter during hypocapnia and hypercapnia in humans using ultra-high-field MRI. J Appl Physiol. (2014) 117:1084–9. doi: 10.1152/japplphysiol.00651.2014
103. Coverdale NS, Badrov MB, Kevin Shoemaker J. Impact of age on cerebrovascular dilation versus reactivity to hypercapnia. J Cereb Blood Flow Metab. (2017) 37:344–55. doi: 10.1177/0271678X15626156
104. Bermel RA, Bakshi R. The measurement and clinical relevance of brain atrophy in multiple sclerosis. Lancet Neurol. (2006) 5:158–70. doi: 10.1016/S1474-4422(06)70349-0
105. De Stefano N, Airas L, Grigoriadis N, Mattle HP, O'Riordan J, Oreja-Guevara C, et al. Clinical relevance of brain volume measures in multiple sclerosis. CNS Drugs. (2014) 28:147–56. doi: 10.1007/s40263-014-0140-z
106. Giovannoni G, Butzkueven H, Dhib-Jalbut S, Hobart J, Kobelt G, Pepper G, et al. Brain health: time matters in multiple sclerosis. Mult Scler Relat Disord. (2016) 9:S5–S48. doi: 10.1016/j.msard.2016.07.003
107. Andravizou A, Dardiotis E, Artemiadis A, Sokratous M, Siokas V, Tsouris Z, et al. Brain atrophy in multiple sclerosis: mechanisms, clinical relevance and treatment options. Autoimmun Highlights. (2019) 10:7. doi: 10.1186/s13317-019-0117-5
108. De Stefano N, Arnold DL. Towards a better understanding of pseudoatrophy in the brain of multiple sclerosis patients. Mult Scler. (2015) 21:675–6. doi: 10.1177/1352458514564494
109. Lagana M, Pelizzari L, Baglio F. Relationship between MRI perfusion and clinical severity in multiple sclerosis. Neural Regen Res. (2020) 15:646–52. doi: 10.4103/1673-5374.266906
110. Lapointe E, Li DKB, Traboulsee AL, Rauscher A. What have we learned from perfusion MRI in multiple sclerosis? Am J Neuroradiol. (2018) 39:994–1000. doi: 10.3174/ajnr.A5504
111. Mutsaerts HJMM, van Osch MJP, Zelaya FO, Wang DJJ, Nordhøy W, Wang Y, et al. Multi-Vendor reliability of arterial spin labeling perfusion MRI using a near-identical sequence: Implications for multi-center studies. Neuroimage. (2015) 113:143–52. doi: 10.1016/j.neuroimage.2015.03.043
112. Alsop DC, Detre JA, Golay X, Günther M, Hendrikse J, Hernandez-Garcia L, et al. Recommended implementation of arterial spin labeled perfusion MRI for clinical applications: a consensus of the ISMRM perfusion study group and the European consortium for ASL in Dementia. Magn Reson Med. (2015) 73:102–16. doi: 10.1002/mrm.25197
113. Chiaravalloti ND, Hillary FG, Ricker JH, Christodoulou C, Kalnin AJ, Liu WC, et al. Cerebral activation patterns during working memory performance in multiple sclerosis using fMRI. J Clin Exp Neuropsychol. (2005) 27:33–54. doi: 10.1080/138033990513609
114. Staffen W, Mair A, Zauner H, Unterrainer J, Niederhofer H, Kutzelnigg A, et al. Cognitive function and fMRI in patients with multiple sclerosis: Evidence for compensatory cortical activation during an attention task. Brain. (2002) 125:1275–82. doi: 10.1093/brain/awf125
115. Mainero C, Caramia F, Pozzilli C, Pisani A, Pestalozza I, Borriello G, et al. fMRI evidence of brain reorganization during attention and memory tasks in multiple sclerosis. Neuroimage. (2004) 21:858–67. doi: 10.1016/j.neuroimage.2003.10.004
116. Ouyang Q, Wang Y, Zhang YW Yu M, Wang X. Change in functional brain activation patterns induced by olfactory stimulation in multiple sclerosis. Neuropsychiatr Dis Treat. (2020) 16:1451–8. doi: 10.2147/NDT.S252933
117. White AT, Lee JN, Light AR, Light KC. Brain activation in multiple sclerosis: A BOLD fMRI study of the effects of fatiguing hand exercise. Mult Scler. (2009) 15:580–6. doi: 10.1177/1352458508100034
118. Reddy H, Narayanan S, Arnoutelis R, Jenkinson M, Antel J, Matthews PM, et al. Evidence for adaptive functional changes in the cerebral cortex with axonal injury from multiple sclerosis. Brain. (2000) 123:2314–20. doi: 10.1093/brain/123.11.2314
119. Pantano P, Mainero C, Lenzi D, Caramia F, Iannetti GD, Piattella MC, et al. longitudinal fMRI study on motor activity in patients with multiple sclerosis. Brain. (2005) 128:2146–53. doi: 10.1093/brain/awh549
Keywords: cerebral blood flow (CBF), cerebrovascular disease, cerebrovascular hemodynamics, cerebrovascular reactivity (CVR), multiple sclerosis, neurovascular coupling (NVC)
Citation: Vestergaard MB, Frederiksen JL, Larsson HBW and Cramer SP (2022) Cerebrovascular Reactivity and Neurovascular Coupling in Multiple Sclerosis—A Systematic Review. Front. Neurol. 13:912828. doi: 10.3389/fneur.2022.912828
Received: 04 April 2022; Accepted: 04 May 2022;
Published: 01 June 2022.
Edited by:
Bruno Stankoff, Sorbonne Universités, FranceReviewed by:
Ivan G. Milanov, Multiprofile Hospital for Active Treatment in Neurology and Psychiatry St. Naum, BulgariaErmelinda De Meo, San Raffaele Hospital (IRCCS), Italy
Copyright © 2022 Vestergaard, Frederiksen, Larsson and Cramer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark B. Vestergaard, bWFyay5iaXRzY2gudmVzdGVyZ2FhcmRAcmVnaW9uaC5kaw==
†These authors have contributed equally to this work and share last authorship