 Prem Jareonsettasin
Prem Jareonsettasin Claudia Zeicu1,2†
Claudia Zeicu1,2† Beate Diehl
Beate Diehl Ronald M. Harper
Ronald M. Harper Rónan Astin
Rónan Astin- 1Department of Clinical and Experimental Epilepsy, Queen Square Institute of Neurology, University College London, London, United Kingdom
- 2Division of Medical Specialties, University College London Hospitals NHS Foundation Trust, London, United Kingdom
- 3Department of Clinical Neurophysiology, University College London Hospitals NHS Foundation Trust National Hospital for Neurology and Neurosurgery, London, United Kingdom
- 4Department of Neurobiology and the Brain Research Institute, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, United States
- 5NIHR University College London Hospitals Biomedical Research Centre, London, United Kingdom
Background: The clinical presentation of COVID-19 suggests altered breathing control - tachypnoea, relative lack of dyspnoea, and often a discrepancy between severity of clinical and radiological findings. Few studies characterize and analyse the contribution of breathing drivers and their ventilatory and perceptual responses.
Aim: To establish the prevalence of inappropriate ventilatory and perceptual response in COVID-19, by characterizing the relationships between respiratory rate (RR), dyspnoea and arterial blood gas (ABG) in a cohort of COVID-19 patients at presentation to hospital, and their post-Covid respiratory sequelae at follow-up.
Methods: We conducted a retrospective cohort study including consecutive adult patients admitted to hospital with confirmed COVID-19 between 1st March 2020 and 30th April 2020. In those with concurrent ABG, RR and documented dyspnoea status on presentation, we documented patient characteristics, disease severity, and outcomes at hospital and 6-week post-discharge.
Results: Of 492 admissions, 194 patients met the inclusion criteria. Tachypnoea was present in 75% pronounced (RR>30) in 36%, and persisted during sleep. RR correlated with heart rate (HR) (r = 0.2674), temperature (r = 0.2824), CRP (r = 0.2561), Alveolar-arterial (A-a) gradient (r = 0.4189), and lower PaO2/FiO2 (PF) ratio (r = −0.3636). RR was not correlated with any neurological symptoms. Dyspnoea was correlated with RR (r = 0.2932), A-a gradient (r = 0.1723), and lower PF ratio (r = −0.1914), but not correlated with PaO2 (r = −0.1095), PaCO2 (r = −0.0598) or any recorded neurological symptom except for altered consciousness. Impaired ventilatory homeostatic control of pH/PaCO2 [tachypnoea (RR>20), hypocapnia (PaCO2 <4.6 kPa), and alkalosis (pH>7.45)] was observed in 29%. This group, of which 37% reported no dyspnoea, had more severe respiratory disease (A-a gradient 38.9 vs. 12.4 mmHg; PF ratio 120 vs. 238), and higher prevalence of anosmia (21 vs. 15%), dysgeusia (25 vs. 12%), headache (33 vs. 23%) and nausea (33 vs. 14%) with similar rates of new anxiety/depression (26 vs. 23%), but lower incidence of past neurological or psychiatric diagnoses (5 vs. 21%) compared to appropriate responders. Only 5% had hypoxia sufficiently severe to drive breathing (i.e. PaO2 <6.6 kPa). At 6 weeks post-discharge, 24% (8/34) showed a new breathing pattern disorder with no other neurological findings, nor previous respiratory, neurological, or psychiatric disorder diagnoses.
Conclusions: Impaired homeostatic control of ventilation i.e., tachypnoea, despite hypocapnia to the point of alkalosis appears prevalent in patients admitted to hospital with COVID-19, a finding typically accompanying more severe disease. Tachypnoea prevalence was between 12 and 29%. Data suggest that excessive tachypnoea is driven by both peripheral and central mechanisms, but not hypoxia. Over a third of patients with impaired homeostatic ventilatory control did not experience dyspnoea despite tachypnoea. A subset of followed-up patients developed post-covid breathing pattern disorder.
Introduction
Early descriptive studies of COVID-19 clinical presentations found tachypnoea, a relative lack of dyspnoea, and often a discrepancy between severity of respiratory clinical signs and radiological findings. Some have described the combination of phenomena as “silent” or “happy” hypoxemia (1–4), inferring dysfunctional regulatory breathing mechanisms.
However, data on the nature of breathing control in COVID-19 are lacking. A few previous studies investigated blood gas analysis in hospitalized COVID-19 patients (5–8), but none concurrently assessed arterial blood gases (ABG), respiratory rate and perception of dyspnoea, and therefore could not directly comment on appropriateness of physiological and breathing perception responses, which we attempt to do here.
Various physiological mechanisms control breathing. Hypercapnia/acidosis drives automatic breathing in a negative feedback loop, while hypoxia only drives breathing when severe (i.e., PaO2 <6.6 kPa) (9–11). Additional drives include thermal, emotional, somatosensory, pulmonary afferents, wakefulness-related signals, and conscious volition. Their neural substrates span all levels of the neuraxis, from the periphery to rostral brain areas, overlapping many areas that process smell, taste, emotion, and arousal.
Neurological symptoms, predominantly anosmia, dysgeusia and altered mental status, occur in many COVID-19 patients (12). Accumulating evidence points toward infection of vascular and immune cells, but not CNS neurons, particularly not those of the brainstem and cerebellum (13) – areas where major breathing control sites lie, raising the possibility of altered breathing control during COVID-19 infection due to direct injury to ancillary support areas by the virus.
In this study, our aim was to establish the prevalence of inappropriate ventilatory and perceptual responses in COVID-19, by characterizing breathing responses during acute infection through investigating the relationship between respiratory rate (RR), dyspnoea and ABG in COVID-19 patients at presentation to hospital, their relationship to neurological symptoms and autonomic control dysfunction, and their post-Covid respiratory sequelae at follow-up.
Methods
We retrospectively collected data from electronic medical records (EPIC, Milky Way, Verona, WI, USA) of consecutive patients with a nasopharyngeal PCR-positive COVID-19 diagnosis who presented to the Emergency Department (ED) of University College Hospital, London between 1st March 2020 and 30th April 2020. Excluded patients were those transferred from another hospital, those without ABG results within 4 h of presentation, undocumented dyspnoea status, and patients who were immediately intubated on arrival. All patients had a respiratory presentation of COVID-19 as their main reason for admission. There were no secondary diagnoses, but co-morbidities are listed in Table 2. Clinical data, including RR, HR and ABGs were part of the ED initial evaluation and used for analysis. Dyspnoea status was assigned as positive if any of the following were documented by the clerking clinician: dyspnoea, breathlessness, shortness of breath, air hunger, respiratory discomfort or respiratory distress. Arterial blood samples were processed on ABL90 FLEX gas analysers (Radiometer, Crawley, UK).
Physiological breathing response was considered inappropriate (“Inappropriate Responders”) in those who were simultaneously tachypnoeic (RR>20), hypocapnic (PaCO2 <4.6 kPa) and alkalotic (pH>7.45) (15). Otherwise, the response was considered appropriate (“Appropriate Responders”). No patients had RR <12, which would suggest a deficient respiratory response. Hypoxia was defined as PaO2 <10 kPa. Hypoxia sufficient to stimulate respiratory drive was defined as PaO2 <6.6 kPa. The breathing pattern assessment tool (BPAT) provides a validated score used to grade the severity and make the diagnosis of breathing pattern disorder (BPD), with a BPAT score of 4 or more corresponding to a diagnosis of BPD (16). BPD was assessed using the BPAT for patients who attended face-to-face follow-up appointments.
Correlations between continuous variables were evaluated using Pearson's correlation and described using median and inter-quartile range (IQR); mortality rates between groups were compared using the χ2 test. Data were analyzed using Prism 8 (GraphPad, San Diego, CA, USA). The study was approved by the Westminster Research Ethics Committee (NHS Health Research Authority, IRAS no: 284088).
Results
Of 492 patients admitted with COVID-19 during the study period, 194 had concurrent ABG, RR and documented dyspnoea status, and were therefore included in the study.
Tachypnoea was common and pronounced: 75% (146/194) exhibited RR>20 breaths per minute (bpm), RR>25 bpm in 57% (69/194), RR>30 bpm in 36% (42/194), RR>35 bpm in 22% (42/194), and RR>40 bpm in 8%(16/194) (Figure 1A). RR at 1 and 2 h following arterial blood sampling varied little (average standard deviation: 2.8 bpm). Notably, tachypnoea was maintained with little variation both day and night over the 96 h following admission, which included sleep periods (Figure 1B). No patients in our cohort had RR <12, which would indicate depressed respiratory drive.
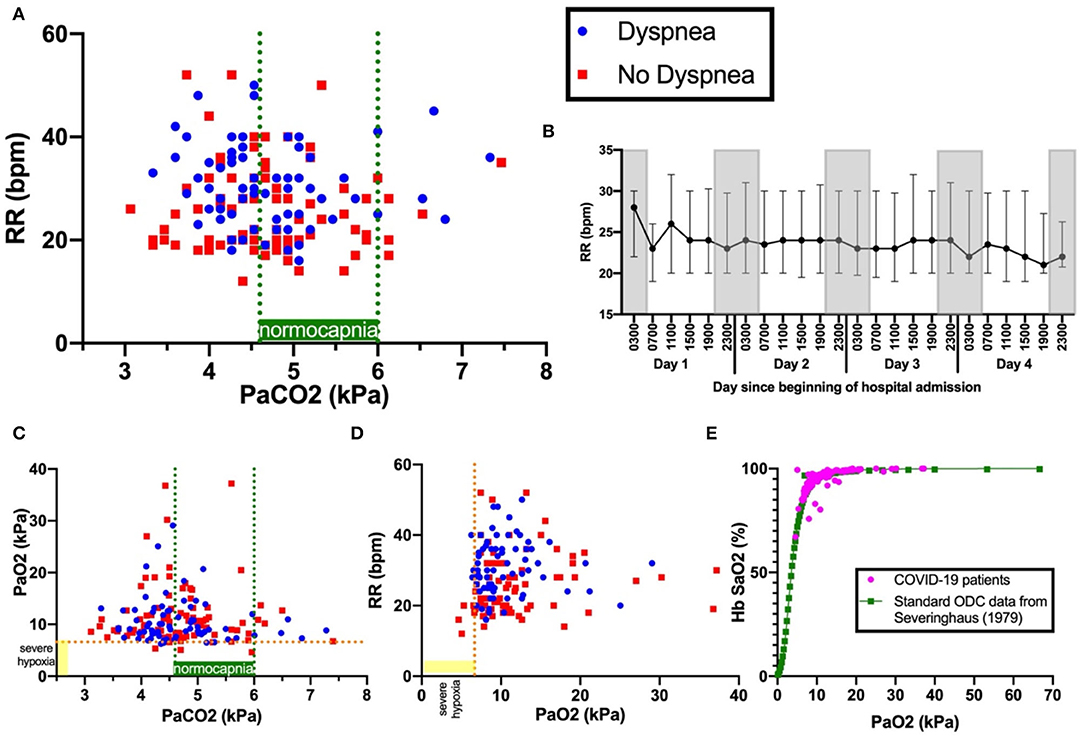
Figure 1. Panel (A) shows PaCO2 versus RR. Blue dots indicate patients with dyspnoea; red squares, patients with no dyspnoea. Dashed green lines indicate boundaries of normocapnia (4.6kPa and 6.0kPa). Dashed orange line indicates severe hypoxia at 6.6kPa, a level sufficient to drive ventilation. Panel (B) shows the 4-hourly RR (median and interquartile range) over the first 96 hours since admission. Gray-shaded boxes indicate night. Panels (C) show PaCO2 versus PaO2 and (D)PaCO2 versus RR. Panel (E) shows the oxygen dissociation curve (ODC) of Covid-19 patients in this cohort plotted against standard human ODC data from (14), showing no shift.
RR correlated with HR (r = 0.267, p < 0.001), temperature (r = 0.282, p < 0.001), CRP (r = 0.256, p < 0.001), A-a gradient (r = 0.419, p < 0.001), and inversely correlated with PF ratio (r = −0.363, p < 0.001). RR did not correlate with mortality (r = 0.0319, p = 0.692), mean arterial pressure (MAP) (r-0.113, p = 0.115), anosmia, dysgeusia, headache, dizziness, altered consciousness, nausea, seizure, new anxiety or depression (Table 1). There was no shift in the oxygen dissociation curve in our cohort (Figure 1E).
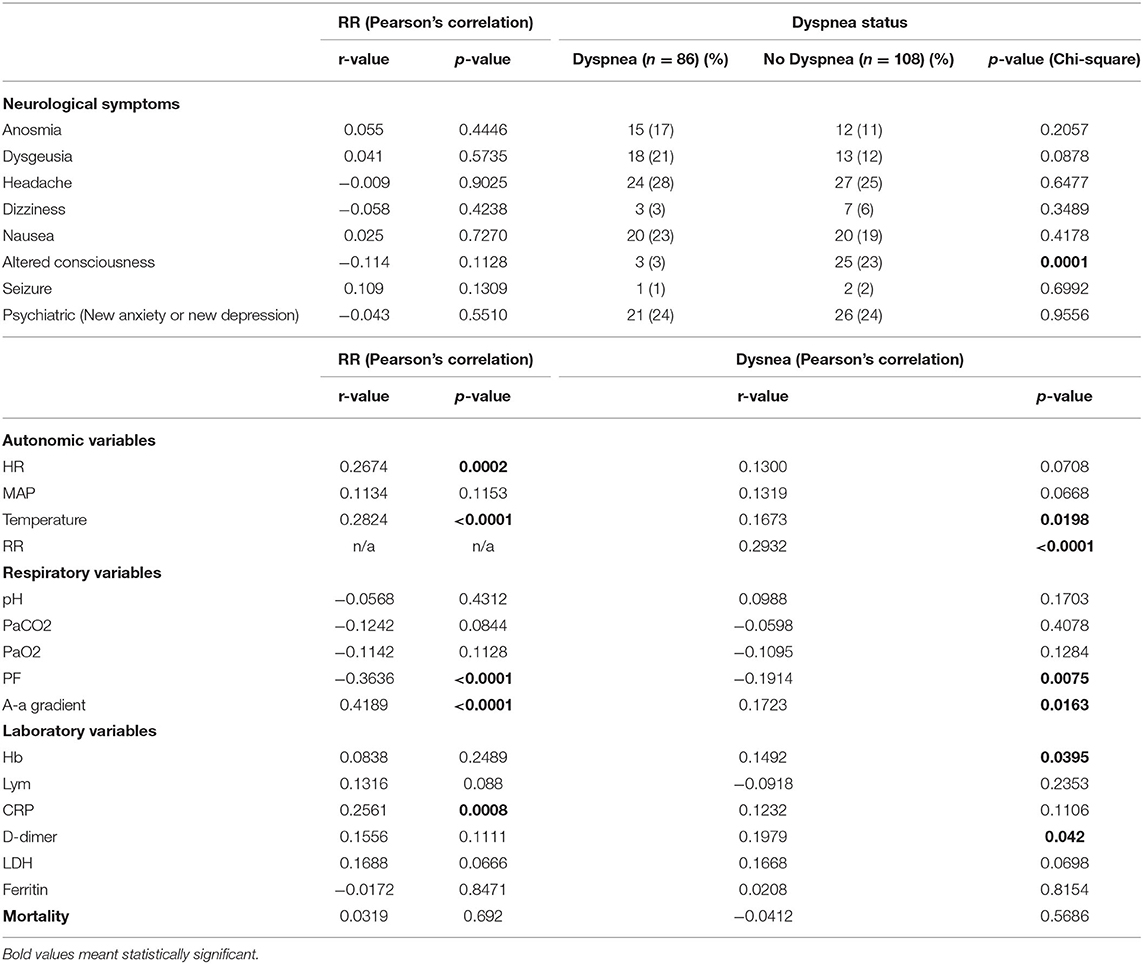
Table 1. Relationship between neurological, autonomic, biochemical variables and RR and dyspnea.
Dyspnoea was present in 44% (86/194) of all patients, 48% (48/101) of those with hypoxia (PaO2 < 10 kPa), but only 33% (4/12) of those with hypoxia sufficient to stimulate respiratory drive (PaO2 < 6.7kPa) (Figures 1C,D). Dyspnoea was weakly correlated with RR (r = 0.293, p < 0.001), temperature (r = 0.167, p = 0.020), A-a gradient (r = 0.172, p = 0.016), Hb (r = 0.149, p = 0.040) and D-dimer (r = 0.198, p = 0.042). Dyspnoea was not correlated with pH (r = 0.099, p-0.170), PaCO2 (r = −0.060, p = 0.408) or PaO2 (r = −0.110, p = 0.128), nor mortality (r = −0.041, p = 0.569). Dyspnoea was weakly inversely correlated to PF ratio (r = −0.191, p = 0.008). Except for altered consciousness (p=0.002), dyspnoea was not correlated with other neurological symptoms.
Inappropriate responders accounted for 29% (57/194). Respiratory alkalosis was associated with more severe respiratory disease; higher FiO2 (0.60 vs. 0.32, p < 0.001), greater A-a gradient (38.9 vs. 12.4 mmHg, p = 0.002), and lower PF ratio (120 vs. 238, p = 0.002).Markers of inflammation were also higher in this group [LDH (498 vs. 386 IU/L, p < 0.001), Ferritin (1,430 vs. 948 ug/L, p = 0.018)], but no significant difference in mortality was observed (30 vs. 40%, p = 0.195). The prevalence of severe hypoxia was low and similar in both groups (5 vs. 4%). However, inappropriate responders had higher rates of supplemental oxygen use (84 vs. 66%) indicating a higher level of underlying pre-hospital hypoxia that was immediately corrected on admission. Anosmia (21 vs. 11%), dysgeusia (25 vs. 12%), headache (33 vs. 23%), nausea (33 vs. 14%) were more prevalent in inappropriate responders. There were no differences in new anxiety or depression (26 vs. 23%), and past neurological or psychiatric diagnoses were less prevalent (5 vs. 21%) in inappropriate responders. The two groups did not differ significantly in age, sex, BMI, ethnicity, cardiovascular and respiratory co-morbidities. Though inappropriate responders had higher rates of dyspnoea (63 vs. 36%), 37% did not report dyspnoea (Table 2).
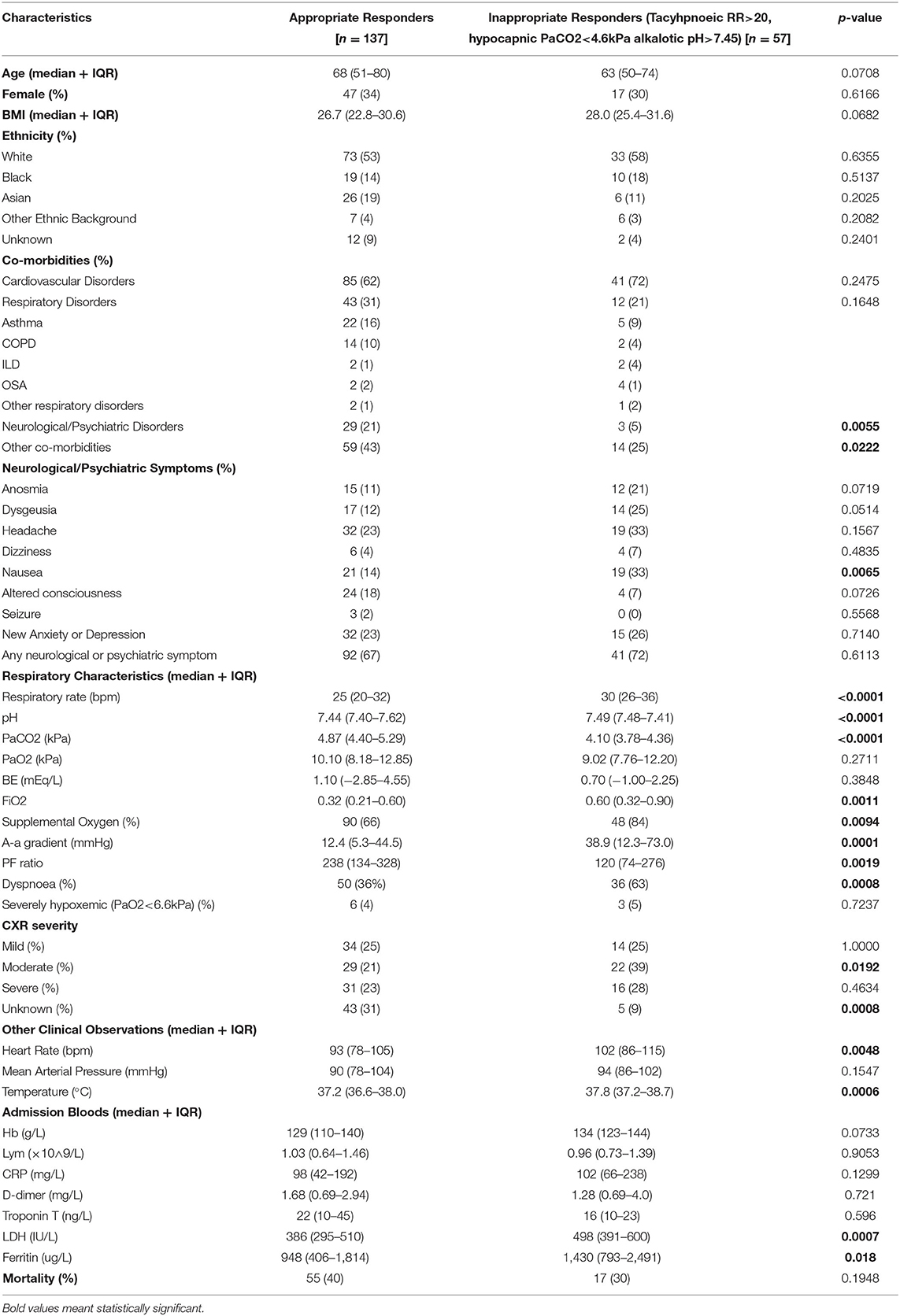
Table 2. Characteristics of appropriate vs inappropriate responders.
Of the total cohort, 38% (74/194), had a 6-week follow-up after hospital discharge, of which 34 (17.5%) were face-to-face consultations which allowed full clinical assessment. A new diagnosis of breathing pattern disorder (BPD) was made in 23.5% (8/34), with a BPAT score of 6 (5–7). None had a past medical history of respiratory, neurological or psychiatric disorder. None had other focal neurological findings on examination. 62.5% (5/8) were in the inappropriate responders group. All eight patients with BPD had 6-month follow-up, of which three still had BPD, with a BPAT score of 2.5 (3–5). Five of the eight patients attended 12-month follow-up, of which one had BPD, with a BPAT score of 2 (2, 3) (Figure 2).
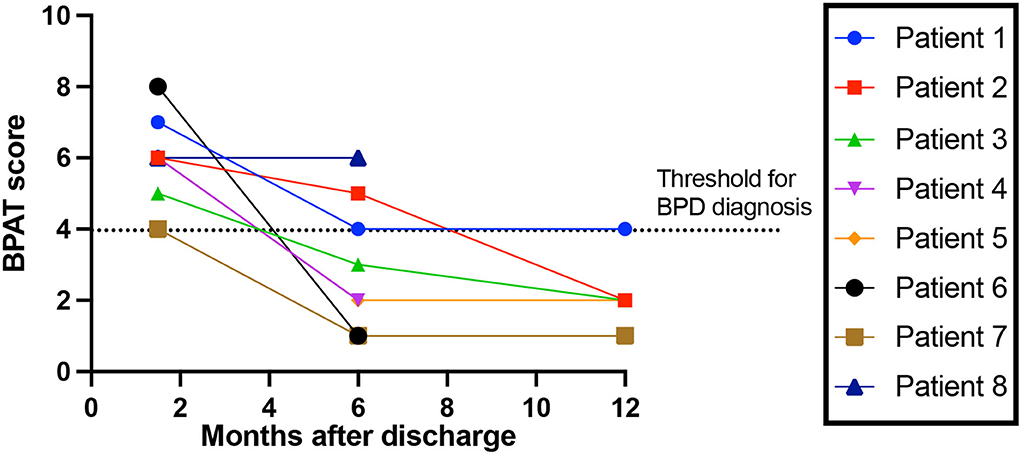
Figure 2. Breathing Pattern Disorder severity over time. Breathing Pattern Assessment Tool score to rate breathing pattern disorder (BPD) severity, over time since discharge from hospital. Threshold for BPD diagnosis is a score of 4 or more (16).
Discussion
Nearly a third of the patients in this study demonstrated impaired homeostatic control of ventilation i.e., tachypnoea, despite hypocapnia to the point of alkalosis, a finding accompanying more severe disease (as evidenced by worse physiological (higher FiO2, greater A-a gradient, and lower PF ratio) and inflammatory markers). Over a third showed a reduced dyspnoeic response to tachypnoea.
The prevalence of inappropriate responders is consistent with estimates of respiratory alkalosis in 28.7–55.4% from other studies that investigated blood gas analysis in hospitalized COVID-19 patients [(5) (55.4%); (6) (28.7%); (7) (30.3%); (8) (40.4%)]. Wu et al. (6) found higher inflammatory markers in respiratory alkalosis group, and other studies corroborate admission hypocapnia as a marker of severe disease (17, 18).
No previous study concurrently assessed ABG, respiratory rate and perception of dyspnoea, and therefore could not directly comment on appropriateness of physiological and perception response, which is a unique aspect here.
Outside the setting of COVID-19, few studies characterize and analyse the ventilatory response in the context of respiratory infection. Although a few studies report prevalence of hypocapnia in community-acquired pneumonia (19–21), none, to our knowledge, report the prevalence of hypocapnia to the extent of alkalosis (PaCO2 <4.6kPa and pH>7.45); therefore, those findings cannot be directly compared.
Profound hypocapnia is found in the context of critical illness, and when prolonged, may adversely influence outcome (15). However, most critically ill patients with abnormal ventilatory responses present with insufficiency (i.e. with acidemia) rather than excessiveness (22). Inappropriate perception of dyspnea is even less well studied. Impaired perception of dypsnoea is more often found in patients with a history of near fatal asthma (23–25), and is associated with impaired chemosensitivity (23, 26) and downregulation of insular activity (27).
We also explored the potential processes that drive excessive tachypnoea in the inappropriate responders group, as well as the impaired perception of dyspnoea to these breathing patterns, and how these acute findings relate to post-covid syndrome.
What Drives Excessive Tachypnoea?
Breathing is controlled by various physiological mechanisms. At a systems level, hypercapnia (increase in PaCO2) and acidosis are the principal chemical drivers of spontaneous automatic breathing, while hypoxia drives breathing only at severe hypoxemia (i.e. PaO2 < 6.6 kPa). In addition, thermal, peripheral pulmonary afferents, sympathetic, emotional and somatosensory drives provide adaptive value for particular situations. When awake, further signals provide wakefulness-related drive to breath which underlies why hypocapnia during wakefulness, but not sleep or anesthesia, does not cause apnoea (28). At the highest level, we can voluntarily control breathing through top-down influence of lower breathing centers.
At a biological level, these physiological mechanisms span all levels of the neuraxis from the periphery to central areas illustrated in Figure 3. These drivers interact in a complex non-linear fashion across large-scale neural networks that control breathing. The final common downstream pathway includes central rhythm generators (pre-Botzinger, parafacial respiratory group, and post-inspiratory complex) that output to the central pattern generators (rostral and caudal ventral respiratory group) and then onwards to spinal and cranial motor nuclei and their neuromuscular efferent arm (29).
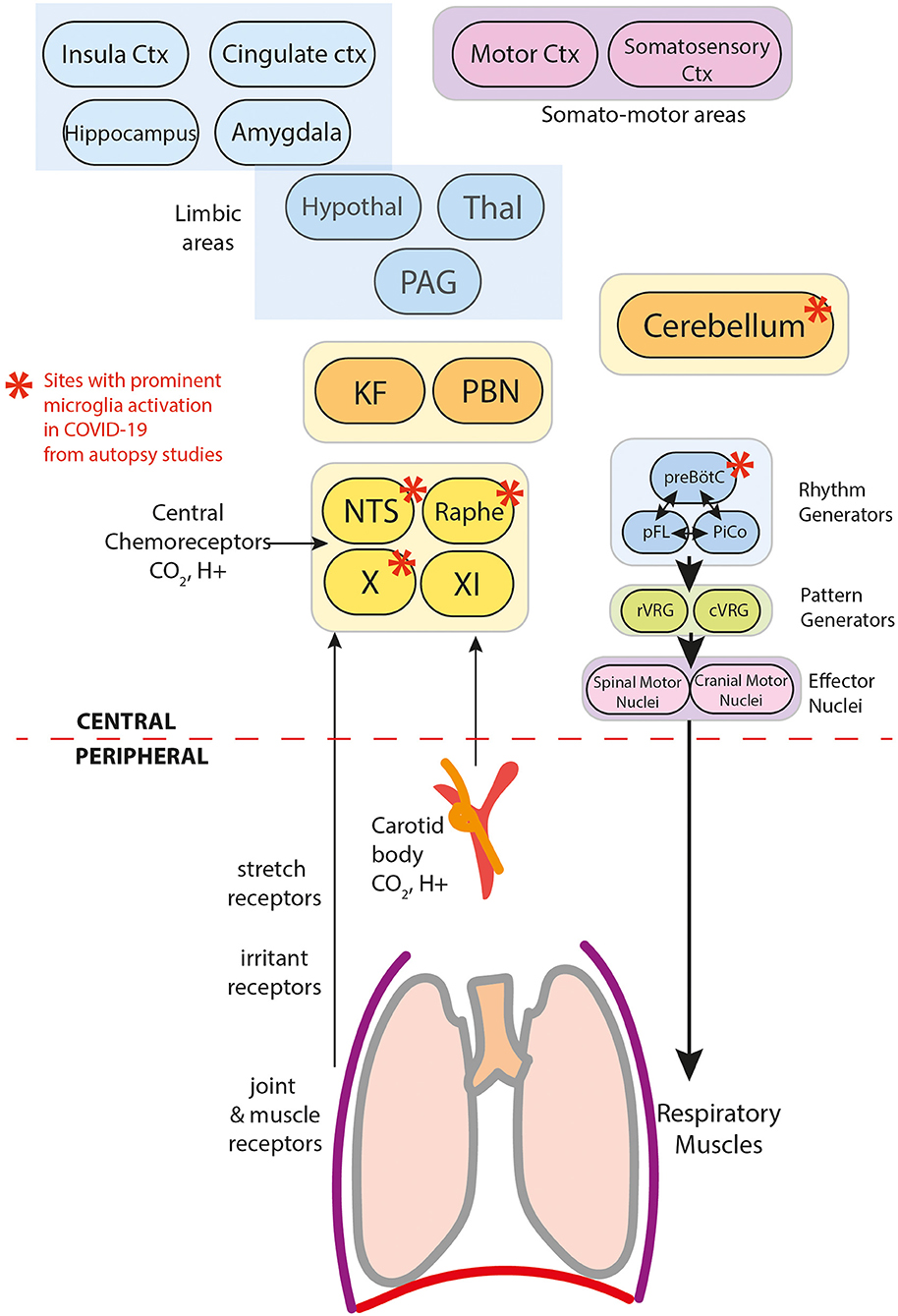
Figure 3. Components of breathing control in the context of COVID-19.
The effects of COVID-19 on breathing control can occur at multiple levels of regulatory control. Here, we explore evidence of COVID-19 effects at various planes, and document disruption inferred from these data.
Hypoxic Drive
Severe hypoxemia (i.e. PaO2 < 6.6kPa) drives breathing through the hypoxic ventilatory reflex (HVR) (9–11). In the periphery, arterial chemoreceptors located in the carotid bodies (CB) (Figure 3) sense PaO2. Aortic bodies play a minimal role, except when carotid bodies are impaired (ref). Oxygen-regulating cells are also present centrally in the caudal hypothalamus, posterior thalamus, periaqueductal gray, nucleus tactus solitarii (NTS) and the rostral ventrolateral medulla (RVLM)(ref). The CB provide the dominant drive for the HVR, since CB denervation significantly attenuates the response (ref). Central oxygen sensors play a role in severe hypoxia-induced tachypnoea in animal models of carotid-deafferented animals (30).
Given that O2-sensing glomus cells express ACE2 (a SARS-CoV-2 receptor), direct infection and impairment of the carotid body could either abolish the HVR or cause abnormal excitability resulting in excessive CB-driven HVR.
However, our results do not support that scenario, since very few patients (5%) had sufficient hypoxemia to drive ventilation. Additionally, hypocapnia observed in our patients would further blunt the HVR.
We cannot exclude pre-hospital periods of sustained severe hypoxia, which could sensitize the CB. Such sensitization results in hyperventilation and increased sympathetic activity that is sustained even after reversal of sustained hypoxic insult, and slowly declines over a few days (31). That more patients in the inappropriate responder group required supplemental oxygen on admission, suggests that this group had higher rates of pre-hospital hypoxia which could sensitize the CB.
CO2 and pH Homeostasis
Central chemoreceptors that sense PaCO2 and pH, are found in the brainstem, cerebellum, hypothalamus and midbrain (32). Those sensors monitor brain interstitial pH, which reflects the integration of PaCO2, cerebral blood flow (CBF), and cerebral metabolic rate. CBF itself responds to changes in PaCO2 (cerebral autoregulation). A set-point exists which keeps PaCO2 and pH in a relatively narrow range. Hypercapnia (PaCO2>4.6kPa) or acidosis (pH <7.35) drives hyperventilation. Conversely, hypocapnia (PaCO2 <4.6kPa) or alkalosis (pH >7.45) drives hypoventilation. The inappropriate responders group showed hypocapnia and alkalosis, which should cause hypoventilation (RR <12), yet they paradoxically are hyperventilating (RR>20). This disturbance in normal homeostasis requires explanation.
PCR-positive SARS-CoV-2 is present at autopsy in the brainstem and cerebellum, specifically in vascular and glial cells, but not neurons, along with activated microglia and evidence of secondary neuronal damage in chemosensitive areas. Specific affected areas include CN X, NTS, dorsal raphe nuclei and cerebellum (13, 33–36) (Figure 3). As such, dysfunction in this redundant network of chemoreceptors appears plausible. Serotonergic neurons of the dorsal raphe, with their extensive projections to motor and respiratory regulatory areas, especially to the cerebellum, are of particular concern.
Two possible mechanisms are conceivable:
First is rheostasis – i.e., shifting the setpoint lower such that hyperventilation is driven by lower PaCO2 than the normal 4.6 kPa. Figure 4 shows that lowering the threshold at which PaCO2 drives breathing (i.e., lower than the normal set-point/threshold of 4.6 kPa), lowers the proportion of inappropriate responders i.e., those who still have simultaneous tachypnoea (RR>20) and alkalosis (pH>7.45) at the new PaCO2 threshold. Rheostasis is normally an adaptive process in homeostatic systems to a sustained change in the environment, such as increased core temperature setpoint during infections, or vestibular-ocular reflex set-points after prolonged stimulation or imbalance in vestibular input (37). Supportive evidence for this hypothesis predicts a rebound hypoventilation when the setpoint returns to normal after the acute insult is removed; that possibility has not been tested in COVID-19 patients.
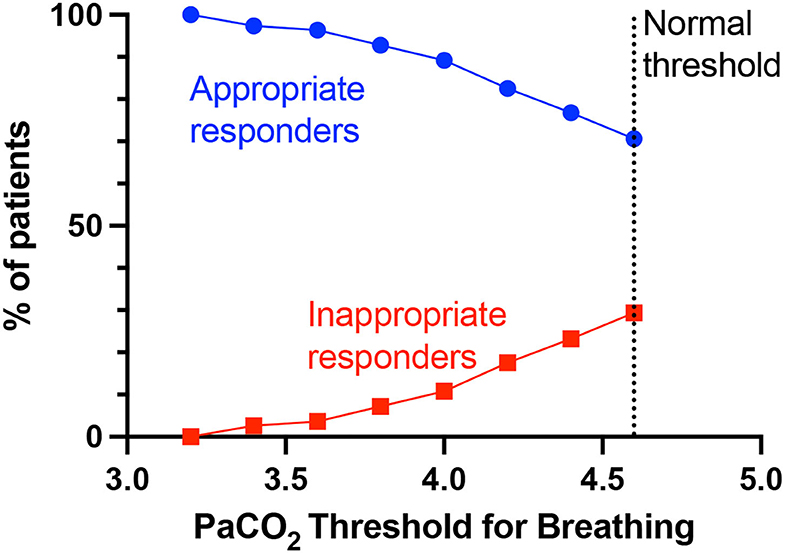
Figure 4. PaCO2 thresholds for breathing and % appropriate and inappropriate responders. The normal PaCO2 threshold for breathing is >4.6 kPa, below which PaCO2 as a breathing drive would be suppressed. Lowering the PaCO2 threshold (set-point) for breathing, will decrease the proportion of patients who are considered inappropriate responders for that particular PaCO2 threshold i.e., still simultaneously have tachypnoea (RR>20) and alkalosis (pH>7.45).
A second potential mechanism is added bias/additional ventilatory drives.
Aberrant Peripheral Sensing
Peripheral drivers via pulmonary vagal C-fibers and slow-adapting mechanoreceptors (SARS) provide sensory feedback to central respiratory centers on local chemical and mechanical conditions. Pulmonary vagal C-fibers fibers are sensitive to inflammatory mediators (including histamine, bradykinin, and prostaglandins), and are consistently activated in lung oedema (38) and experimental acute lung injury (39). These C-fibers can modulate ventilation (increase RR and decrease tidal volume) (40), possibly through vagally-mediated cytokine release in the brainstem (41).
Slow-adapting mechanoreceptors are normally activated by lung inflation, and inhibit central chemoreception (42). Peripheral drives from these sensors may explain hyperventilation in pulmonary oedema, pulmonary fibrosis and pulmonary embolism which persists in the absence of hypercapnia or severe hypoxemia (43).
Our data support a role for these peripheral receptors. This inference is based on the observation of a higher prevalence of more-severe lung disease in the hypocapnic group. Acute respiratory distress syndrome increases RR before impairing gas exchange in rodent models, suggesting an initial role for peripheral afferent stimulation. The acute lung inflammation found in COVID-19 would be expected to stimulate SARs in a similar manner.
Other Breathing Drives
Thermal Drive
Animals can regulate their body temperature with an increase of core temperature by 1°C, triggering hyperventilation to induce heat loss (44). Our data, showing a correlation between temperature and RR, support this relationship. The significantly higher temperature in the inappropriate responder group suggests a contribution of thermal drive to tachypnoea.
Diminished “Higher” Drivers of Breathing
Normally, higher brain centers influence breathing to allow flexible control of breathing with emotion, experience and context, and provide signals involved in the “wakefulness drive to breathe” (28). Our study assessed measures of breathing throughout the day and night. A remarkable finding was that little change in tachypnoea was found in sleeping periods. Although classification of sleep states was unavailable, the data indicate that the normal slowing of respiratory rates with quiet sleep did not occur. That finding is significant, since it points to an abolition of the descending brain influences that mediate control of breathing during sleep states. Although descending limbic and thalamic drives, such as airflow, olfactory or temperature influences may be exerting timing effects, the timing influences that normally slow breathing during sleep appear to be ineffective.
Systemic Inflammation
Systemic inflammation itself increases respiratory drive. Our data support greater hyperinflammation in the inappropriate responder group. Hypocapnia is seen in critically ill patients, systemic inflammatory response syndrome, and liver failure (45). In COVID-19, hyperinflammatory responses contribute to disease severity and mortality (46).
Central Neurogenic Hyperventilation
In the inappropriate responders group, we found a higher prevalence of anosmia (21 vs. 15%), dysgeusia (25 vs. 12%), headache (33 vs. 23%) and nausea (33 vs. 14%) with similar rates of new anxiety/depression (26 vs. 23%), but a lower incidence of past neurological or psychiatric diagnoses (5 vs. 21%) compared to appropriate responders. These findings warrant exploration of possible central neurogenic contributions to hyperventilation.
The demonstration of COVID-19 influences on the olfactory apparatus (36) and the role of those structures on sensing CO2 and other aspects of air passage, as well as the known injury to the amygdala and other limbic structures mediating taste and drive to respiratory phase switching areas of the parabrachial pons (47) and thus, respiratory rate, provide a number of potential central mechanisms to mediate the findings here. Central neurogenic hyperventilation has been reported in other conditions involving immune dysfunction, including multiple sclerosis (48), anti-NMDA receptor encephalitis (49), neuro-Behcet's (50) and Bickerstaff encephalitis (51). Interestingly, marked tachypnoea was the predominant respiratory phenotype of the 1918–1925 epidemic of encephalitis lethargica (52), a disease with recent evidence suggestive of an immune-mediated pathogenesis (53).
Anosmia and dysgeusia are the most prevalent neurological symptoms in COVID-19, suggesting key roles for forebrain limbic structures (Figure 3), particularly olfactory and amygdala structures. However, the weight of evidence supports an interpretation that anosmia results predominantly from SARS-CoV-2 infection of non-neuronal cells in the olfactory epithelium and olfactory bulb (54), and dysgeusia more likely results from peripheral damage to ACE-2-expressing cells of taste buds and peripheral chemoreceptors, or cranial nerves responsible for gustation (CN VII, IX, or X) (55).
Centrally, olfaction is processed by multiple cortical and subcortical regions (56), in particular temporal lobe areas, including the piriform and entorhinal cortex, hippocampus, parahippocampus, amygdala and extra-temporal areas such as the orbitofrontal cortex. Amongst these structures, the hippocampus and amygdala are critical subcortical structures controlling breathing (57, 58).
Central gustatory areas include the NTS, parabrachial nucleus, gustatory thalamus (ventropostero-medial nucleus), amygdala basolateral nucleus and central nucleus, insula cortex, orbitofrontal cortex and anterior cingulate cortex. Among these structures, the NTS and parabrachial nuclei are chemosensitive brainstem structures or receive afferent signals mediating control of breathing. The higher prevalence of nausea in the inappropriate responder group supports involvement of the NTS and parabrachial nuclei. Of interest, the nausea finding is corroborated by the significantly higher prevalence of vomiting in the respiratory alkalosis group (21.2%) compared to the non-respiratory alkalosis group (7.3%) in a previous study (6).
Headache as a symptom has no specific localization, but hints at involvement of CN V (trigeminal nuclei). Another study of hospitalized COVID-19 patients identified the presence of new-onset headache in those presenting without dyspnoea, who also presented earlier (4). This finding raises the possibility of early activation of the trigeminal-vascular system, a concept supported by neuropathological studies showing neuroinvasive potential of SARS-CoV-2 to the brainstem (36). CN V plays a major role in respiratory timing through airflow receptors in the nasal and oral cavities and motor activation of the upper airway musculature (59). These timing roles are especially important for preventing obstructive apnea and maintaining appropriate coordination of cerebellar and pontine respiratory timing circuitry through airflow and thermal afferent activity to the parabrachial pons, a major site of respiratory phase switching (and thus, respiratory timing). The thermal role can be readily demonstrated through cold water facial immersion, which results in immediate apnea, while warming results in tachypnoea and panting.
The amygdala, insula and anterior cingulate cortex, all injured in COVID-19, also serve critical respiratory roles, integrating afferent input from a wide range of receptors and sending projections to other amygdala structures and the hypothalamus; the central nucleus of the amygdala has prominent projections to the parabrachial pons and can influence respiratory rate, even to the point of pacing inspiratory efforts (60). The hypothalamus provides substantial thermal drive to breathing, perhaps influencing the significant role we found for breathing rate and temperature.
Autopsy reports in COVID-19 indicate local immune-mediated activity in the brainstem and cerebellum (13). The cerebellum plays a critical role in respiratory timing, coordinating afferent stimuli from multiple somatic and vascular sites and essential timing circuitry with the parabrachial pons. The cerebellar fastigial nuclei are particularly important in these ventilatory roles, specifically during chemical stress and not during eupnoea. Injury to the fastigial nuclei, such as in Central Congenital Hypoventilation Syndrome or heart failure patients, distorts both amplitude and timing to ventilatory and blood pressure challenges (61, 62).
Sensitization of the Efferent Arm
A possible source of hyperventilation lies in the efferent arm. There is no evidence to suggest dysfunction in the motor nuclei, motor neurons or muscles. Nevertheless, pre-admission sustained hypoxia could centrally sensitize motor neurons driving the phrenic nerves, enhancing phrenic output. However, the principal findings suggest a timing dysfunction, i.e., a rate, not motor effort, issue.
Overall, the data presented here suggest that tachypnoea was driven by both peripheral and central mechanisms, but not hypoxia.
What Drives Impaired Perception of Dyspnoea to Tachypnoea?
Increased afferent feedback from chest wall mechanoreceptors and muscle stretch receptors with increased RR is usually perceived as breathlessness (63). It is abnormal that over a third of patients in the inappropriate responder group had reduced dyspnoeic response to tachypnoea.
First, neuromechanical coupling may be maintained in this COVID-19 cohort due to relatively preserved lung compliance (64). This coupling is unusual for most disorders that lead to acute lung injury. To a large degree, this interpretation explains the lack of dyspnoea, because the mechanoreceptor activity should continue to be proportional to the predicted activity from a given motor signal that drives ventilation. Therefore, there should not be an error signal, which should indicate no dyspnoea. This possibility has been supported by others (64, 65).
Secondly, interruptions in central processes that compare expected consequences of (breathing) motor commands and the actual consequences (feedback from periphery) may occur. Normally the “error” signal generated from a mismatch between these expected and actual consequences would generate the dyspnoeic perception of “increased work of breathing” (66). Both the cerebellum and insula play major roles in the perception of dyspnoea (67), as well as their aforementioned roles in control of ventilation. Damage to the cerebellum could impair gain and timing of these signals. Alternatively, a shift in setpoint to a higher threshold for dyspnoea perception would require a higher RR to perceive dyspnoea. Here, one possibility is a downregulation of insula activity. In patients with asthma, downregulation of affect-related insula cortex activity correlates with blunted perception of dyspnoea (68). Lesions in the right insular cortex are associated with blunted dyspnoea (69).
How Do These Acute Findings Relate to Post-COVID Syndrome in a Subset of Patients?
The prevalence of breathing pattern disorder (BPD) at 6 weeks post-discharge was (24%) and in the absence of other neurological findings, or previous respiratory, neurological, or psychiatric disorder diagnoses. Notably, most patients recovered over time. The pathophysiology of breathing pattern disorder is poorly understood, but involves abnormal breathing rate, pattern and inappropriate dyspnoea. The neural mechanisms underlying the recovery are not understood.
Whether mechanisms of post-Covid breathing pattern disorder can be inferred from our data is unclear. Only 62.5% of patients who had BPD were inappropriate responders in the acute phase – for this group, rheostasis may be explanatory– a shift in set-point during the acute phase to a higher state. Such a shift is likely followed by a resetting after the acute illness that disturbs breathing perception and results in the high prevalence of breathing disorder found in our cohort. Our data suggest that by 1 year after the acute insult, the set-point has reset to its pre-Covid state.
Future work should focus on prospective cohort studies of hospitalized COVID-19 patients, with an emphasis on gathering more objective respiratory rate using wearable devices, more quantitative measures of perception of dyspnea over multiple intervals from admission to discharge, and prolonged follow-up. Correlation of respiratory patterning with cardiovascular changes would also be useful. Determination of respiratory patterning during the normally short-lasting periods of rapid eye movement sleep would help differentiate whether COVID-19 impacts breathing differently during that state, thus helping to determine abberant influences. Additional functional neuroimaging of subsets of patients with impaired ventilatory and perceptual response would further mechanistic understanding. More broadly, it remains unanswered whether the phenomena we observe here are unique to COVID-19 or are found in other respiratory conditions – further studies are needed.
Clinical Implications
We show (1) that dyspnoea alone poorly correlates with disease severity or degree of hypoxia, despite its inclusion in many severity triage scoring systems; (2) tachypnoea appears to be a more useful clinical marker, as it is common, and correlated with more severe pulmonary disease; (3) our study supports the use of early blood gas analysis - with hypocapnia and respiratory alkalosis being of particular concern, because this group has more severe disease; (4) we suggest that acute impairment in breathing control may lead to dysfunctional breathing that is prolonged, but will likely resolve by 1 year. The unresponsiveness of control mechanisms to extreme values in pH and oxygenation mandate further studies into processes mediating disruption of sensory, integrative central processing, and motor output on respiration, and the activities underlying recovery of longer-term effects of COVID-19 on breathing control.
Limitations of the Study
The limitations include a relatively small number of subjects, and that the data are derived from a single center. However, the study is from a geographical location with a highly heterogenous population, providing a wide representation of physiological presentation. The study is also a retrospective design, and therefore, no formal protocolised assessment of dyspnoea was available, nor were comments on hyperpnoea. Not unique to the study is the difficulty of counting RR in clinical situations. However, the persistence of tachypnoea over multiple recordings argues for the validity of the data. Inherently, the perception of dyspnoea is subjective and multi-dimensional – but our inclusion criteria for recording dyspnoea covers these multi-dimensional descriptors. We also had limited detailed data on other autonomic aspects including cardiac patterning. We had no neuropsychometric assessment, for practical reasons during that phase of the COVID-19 pandemic which may have revealed subtler psychological localisable deficits. Although we note that nearly a third of patients in our study had impaired homeostatic control of ventilation, the study only included 194 (who met the inclusion criteria) out of the 492 patients admitted. Therefore, the lowest bound of the prevalence estimate would be 11.6% (57/492). Finally, the number of patients who attended follow-up appointment was low, which limited inferences of post-acute Covid effects.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and the study was approved by the Westminster Research Ethics Committee (NHS Health Research Authority, IRAS no: 284088). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
PJ, CZ, RH, and RA: acquisition and analysis or interpretation of data. PJ, RH, and RA: drafting of the manuscript, administrative, and technical or material support. PJ: statistical analysis. RH and RA: obtained funding and supervision. All authors: concept and design and critical revision of the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
Funding
This research was supported in part by the Fidelity Charitable Nancy Adams and Scott Schoen Fund and the Kraig and Linda Kupiec Family Trust.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tobin MJ, Laghi F, Jubran A. Why COVID-19 silent hypoxemia is baffling to physicians. Am J Respir Crit Care Med. (2020) 202:356–60. doi: 10.1164/rccm.202006-2157CP
2. Brouqui P, Amrane S, Million M, Cortaredona S, Parola P, Lagier JC, et al. Asymptomatic hypoxia in COVID-19 is associated with poor outcome. Int J Infect Dis. (2021) 102:233–8. doi: 10.1016/j.ijid.2020.10.067
3. Busana M, Gasperetti A, Giosa L, Forleo GB, Schiavone M, Mitacchione G, et al. Prevalence and outcome of silent hypoxemia in COVID-19. Minerva Anestesiol. (2021) 87:325–33. doi: 10.23736/S0375-9393.21.15245-9
4. García-Grimshaw M, Flores-Silva FD, Chiquete E, Cantú-Brito C, Michel-Chávez A, Vigueras-Hernández AP, et al. Characteristics and predictors for silent hypoxemia in a cohort of hospitalized COVID-19 patients. Auton Neurosci. (2021) 235:102855. doi: 10.1016/j.autneu.2021.102855
5. Mondal S, Das TK, Bhattacharya S, Banerjee S, Hazra D. Blood gas analysis among COVID-19 patients: a single centre retrospective observational study. J Clin Diagn Res. (2021) 3:1–4. doi: 10.7860/JCDR/2021/49835.15185
6. Wu C, Wang G, Zhang Q, Yu B, Lv J, Zhang S, et al. Association Between Respiratory Alkalosis and the Prognosis of COVID-19 Patients. Front Med. (2021) 8:6–11. doi: 10.3389/fmed.2021.564635
7. Alfano G, Fontana F, Mori G, Giaroni F, Ferrari A, Giovanella S, et al. Acid base disorders in patients with COVID-19. Int Urol Nephrol. (2022) 54:405–10. doi: 10.1007/s11255-021-02855-1
8. Chiumello D, Pozzi T, Fratti I, Modafferi L, Montante M, Papa GF. et al. Acid-base disorders in COVID-19 patients with acute respiratory distress syndrome. J Clin Med. (2022) 11:2093. doi: 10.3390/jcm11082093
9. Weil JV, Byrne-Quinn E, Sodal IE, Friesen WO, Underhill B, Filley GF., et al. Hypoxic ventilatory drive in normal man. J Clin Investig. (1970) 49:1061–72. doi: 10.1172/JCI106322
10. Mohan R, Duffin J. The effect of hypoxia on the ventilatory response to carbon dioxide in man. Respir Physiol. (1997) 108:101–15. doi: 10.1016/S0034-5687(97)00024-8
11. Moosavi SH, Golestanian E, Binks AP, Lansing RW, Brown R, Banzett RB. Hypoxic and hypercapnic drives to breathe generate equivalent levels of air hunger in humans. J Appl Physiol. (2003) 94:141–54. doi: 10.1152/japplphysiol.00594.2002
12. Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, et al. Neurological associations of COVID-19. Lancet Neurol. (2020) 19:767–83. doi: 10.1016/S1474-4422(20)30221-0
13. Solomon T. Neurological infection with SARS-CoV-2 - the story so far. Nat Rev Neurol. (2021) 17:65–6. doi: 10.1038/s41582-020-00453-w
14. Severinghaus JW. Simple, accurate equations for human blood O2 dissociation computations. J Appl Physiol. (1979) 46:599–602.
16. Todd S, Walsted ES, Grillo L, Livingston R, Menzies-Gow A, Hull JH. Novel assessment tool to detect breathing pattern disorder in patients with refractory asthma. Respirology. (2018) 23:284–90. doi: 10.1111/resp.13173
17. Turcato G, Panebianco L, Zaboli A, Scheurer C, Ausserhofer D, Wieser A, et al. Correlation between arterial blood gas and CT volumetry in patients with SARS-CoV-2 in the emergency department. Int J Infect Dis. (2020) 97:233–5. doi: 10.1016/j.ijid.2020.06.033
18. Jain P, Sinha N, Prasad M, Padole V. Clinical and laboratory profile of COVID-19 patients admitted at a tertiary care center in New Delhi and assessment of factors predicting disease severity. Indian J Med Spec. (2021) 12:59–63. doi: 10.4103/injms.injms_158_20
19. Sin DD, Man SFP, Marrie TJ. Arterial carbon dioxide tension on admission as a marker of in-hospital mortality in community-acquired pneumonia. Am J Med. (2005) 118:145–50. doi: 10.1016/j.amjmed.2004.10.014
20. Laserna E, Sibila O, Aguilar PR, Mortensen EM, Anzueto A, Blanquer JM, et al. Hypocapnia and Hypercapnia Are Predictors for ICU Admission and Mortality in Hospitalized Patients With Community-Acquired Pneumonia. Chest. (2012) 142:1193–9. doi: 10.1378/chest.12-0576
21. Yassin Z, Saadat M, Abtahi H, Rahimi Foroushani A, Peiman S. Prognostic value of on admission arterial PCO(2) in hospitalized patients with community-acquired pneumonia. J Thorac Dis. (2016) 8:2765–71. doi: 10.21037/jtd.2016.10.21
22. Jung B, Rimmele T, Le Goff C, Chanques G, Corne P, Jonquet O, et al. Severe metabolic or mixed acidemia on intensive care unit admission: incidence, prognosis and administration of buffer therapy. a prospective, multiple-center study. Crit Care. (2011) 15:R238. doi: 10.1186/cc10487
23. Kikuchi Y, Okabe S, Tamura G, Hida W, Homma M, Shirato K, et al. Chemosensitivity and Perception of Dyspnea in Patients with a History of Near-Fatal Asthma. N Engl J Med. (1994) 330:1329–34. doi: 10.1056/NEJM199405123301901
24. Martínez-Moragón E, Perpi, ñá M, Fullana J, Macián V, Lloris A, Belloch A.. [Perception of dyspnea and treatment adherence in asthmatic patients]. Arch Bronconeumol. (2008) 44:459–463. doi: 10.1016/S1579-2129(08)60083-X
25. Barnes PJ, Szefler SJ, Reddel HK, Chipps BE. Symptoms and perception of airway obstruction in asthmatic patients: clinical implications for use of reliever medications. J Allergy Clin Immunol. (2019) 144:1180–6. doi: 10.1016/j.jaci.2019.06.040
26. Chang KC, Morrill CG, Chai H. Impaired response to hypoxia after bilateral carotid body resection for treatment of bronchial asthma. Chest. (1978) 73:667–9. doi: 10.1378/chest.73.5.667
27. von Leupoldt A, Sommer T, Kegat S, Eippert F, Baumann H, Klose H, et al. Down-regulation of insular cortex responses to dyspnea and pain in asthma. Am J Respir Crit Care Med. (2009) 180:232–8. doi: 10.1164/rccm.200902-0300OC
28. Dubois M, Chenivesse C, Raux M, Morales-Robles A, Nierat M-C, Garcia G, et al. Neurophysiological evidence for a cortical contribution to the wakefulness-related drive to breathe explaining hypocapnia-resistant ventilation in humans. J Neurosci. (2016) 36:10673–82. doi: 10.1523/JNEUROSCI.2376-16.2016
29. Del Negro CA, Funk GD, Feldman JL. Breathing matters. Nat Rev Neurosci. (2018) 19:351–67. doi: 10.1038/s41583-018-0003-6
30. Teppema LJ, Dahan A. The ventilatory response to hypoxia in mammals: mechanisms, measurement, and analysis. Physiol Rev. (2010) 90:675–754. doi: 10.1152/physrev.00012.2009
31. Hansen J, Sander M. Sympathetic neural overactivity in healthy humans after prolonged exposure to hypobaric hypoxia. J Physiol. (2003) 546(Pt 3):921–929. doi: 10.1113/jphysiol.2002.031765
32. Nattie E, Li A. Central chemoreceptors: locations and functions. Compr Physiol. (2012) 2:221–54. doi: 10.1002/cphy.c100083
33. Matschke J, Lütgehetmann M, Hagel C, Sperhake JP, Schröder AS, Edler C, et al. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol. (2020) 19:919–29. doi: 10.1016/S1474-4422(20)30308-2
34. von Weyhern C. H., Kaufmann I, Neff F, Kremer M. Early evidence of pronounced brain involvement in fatal COVID-19 outcomes. The Lancet. (2020) 395. doi: 10.1016/S0140-6736(20)31282-4
35. Lee JC, Nallani R, Cass L, Bhalla V, Chiu AG, Villwock JA, et al. Systematic review of the neuropathologic findings of post-viral olfactory dysfunction: implications and novel insight for the COVID-19 pandemic. Am J Rhinol Allergy. (2021) 35:323–33. doi: 10.1177/1945892420957853
36. Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci. (2021) 24:168–75. doi: 10.1038/s41593-020-00758-5
37. Jareonsettasin P, Otero-Millan J, Ward BKBK, Roberts DCDC, Schubert MC, Zee DSDS. Multiple time courses of vestibular set-point adaptation revealed by sustained magnetic field stimulation of the labyrinth. Curr Biol. (2016) 26:1359–66. doi: 10.1016/j.cub.2016.03.066
38. Paintal AS. Vagal sensory receptors and their reflex effects. Physiol Rev. (1973) 53:159–227. doi: 10.1152/physrev.1973.53.1.159
39. Lin RL, Gu Q, Lee LY. Hypersensitivity of vagal pulmonary afferents induced by tumor necrosis factor alpha in mice. Front Physiol. (2017) 8:411. doi: 10.3389/fphys.2017.00411
40. Lee LY, Pisarri TE. Afferent properties and reflex functions of bronchopulmonary C-fibers. Respir Physiol. (2001) 125:47–65. doi: 10.1016/S0034-5687(00)00204-8
41. Jacono FJ, Mayer CA, Hsieh YH, Wilson CG, Dick TE. Lung and brainstem cytokine levels are associated with breathing pattern changes in a rodent model of acute lung injury. Respir Physiol Neurobiol. (2011) 178:429–38. doi: 10.1016/j.resp.2011.04.022
42. Kubin L, Alheid GF, Zuperku EJ, McCrimmon DR. Central pathways of pulmonary and lower airway vagal afferents. J Appl Physiol. (2006) 101:618–627. doi: 10.1152/japplphysiol.00252.2006
43. Jonkman AH, de Vries HJ, Heunks LMA. Physiology of the respiratory drive in ICU patients: implications for diagnosis and treatment. Crit Care. (2020) 24:104. doi: 10.1186/s13054-020-2776-z
44. White MD. Components and mechanisms of thermal hyperpnea. J Appl Physiol. (2006) 101:655–63. doi: 10.1152/japplphysiol.00210.2006
45. Vaporidi K, Akoumianaki E, Telias I, Goligher EC, Brochard L, Georgopoulos D. Respiratory drive in critically Ill patients. Pathophysiology and clinical implications. Am J Respir Crit Care Med. (2019) 201:20–32. doi: 10.1164/rccm.201903-0596SO
46. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. (2020) 395:1033–4. doi: 10.1016/S0140-6736(20)30628-0
47. Hopkins DA, Holstege G. Amygdaloid projections to the mesencephalon, pons and medulla oblongata in the cat. Exp Brain Res. (1978) 32:529–47. doi: 10.1007/BF00239551
48. Takahashi M, Tsunemi T, Miyayosi T, Mizusawa H. Reversible central neurogenic hyperventilation in an awake patient with multiple sclerosis. J Neurol. (2007) 254:1763–4. doi: 10.1007/s00415-007-0662-0
49. Vural A, Arsava EM, Dericioglu N, Topcuoglu MA. Central neurogenic hyperventilation in anti-NMDA receptor encephalitis. Intern Med. (2012) 51:2789–92. doi: 10.2169/internalmedicine.51.8215
50. Alkhachroum AM, Saeed S, Kaur J, Shams T, DeGeorgia MA. A case of neuro-behcet's disease presenting with central neurogenic hyperventilation. Am J Case Rep. (2016) 17:154–9. doi: 10.12659/AJCR.895382
51. Nystad D, Salvesen R, Nielsen EW. Brain stem encephalitis with central neurogenic hyperventilation. J Neurol Neurosurg Psychiatry. (2007) 78:107–8. doi: 10.1136/jnnp.2006.094375
52. Turner WA, Critchley M. Respiratory disorders in epidemic encephalitis. Brain. (1925) 48:72–104. doi: 10.1093/brain/48.1.72
53. Dale RC, Church AJ, Surtees RAH, Lees AJ, Adcock JE, Harding B, et al. Encephalitis lethargica syndrome: 20 new cases and evidence of basal ganglia autoimmunity. Brain. (2004) 127(Pt 1):21–33. doi: 10.1093/brain/awh008
54. Brann DH, Tsukahara T, Weinreb C, Lipovsek M, Van den Berge K, Gong B, et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci Adv. (2020) 6:eabc5801. doi: 10.1126/sciadv.abc5801
55. Lozada-Nur F, Chainani-Wu N, Fortuna G, Sroussi H. Dysgeusia in COVID-19: Possible Mechanisms and Implications. Oral Surg Oral Med Oral Pathol Oral Radiol. (2020) 130:344–6. doi: 10.1016/j.oooo.2020.06.016
56. Fjaeldstad AW, Stiller-Stut F, Gleesborg C, Kringelbach ML, Hummel T, Fernandes HM. Validation of olfactory network based on brain structural connectivity and its association with olfactory test scores. Front Syst Neurosci. (2021) 15:638053. doi: 10.3389/fnsys.2021.638053
57. Harper RM, Poe GR, Rector DM, Kristensen MP. Relationships between hippocampal activity and breathing patterns. Neurosci Biobehav Rev. (1998) 22:233–6. doi: 10.1016/S0149-7634(97)00010-9
58. Nobis WP, González Otárula KA, Templer JW, Gerard EE, VanHaerents S, Lane G, et al. The effect of seizure spread to the amygdala on respiration and onset of ictal central apnea. J Neurosurg. (2019) 132:1313–23. doi: 10.3171/2019.1.JNS183157
59. Koizumi H, Nomura K, Yokota Y, Enomoto A, Yamanishi T, Iida S, et al. Regulation of trigeminal respiratory motor activity in the brainstem. J Dent Res. (2009) 88:1048–53. doi: 10.1177/0022034509345998
60. Harper RM, Frysinger RC, Trelease RB, Marks JD. State-dependent alteration of respiratory cycle timing by stimulation of the central nucleus of the amygdala. Brain Res. (1984) 306:1–8. doi: 10.1016/0006-8993(84)90350-0
61. Ogren JA, Macey PM, Kumar R, Woo MA, Harper RM. Central autonomic regulation in congenital central hypoventilation syndrome. Neuroscience. (2010) 167:1249–56. doi: 10.1016/j.neuroscience.2010.02.078
62. Ogren JA, Macey PM, Kumar R, Fonarow GC, Hamilton MA, Harper RM. et al. Impaired cerebellar and limbic responses to the valsalva maneuver in heart failure. Cerebellum. (2012) 11:931–8. doi: 10.1007/s12311-012-0361-y
64. Gattinoni L, Coppola S, Cressoni M, Busana M, Rossi S, Chiumello D. Covid-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome'. Am J Respir Crit Care Med. (2020) 201:1299–300. doi: 10.1164/rccm.202003-0817LE
65. Marini JJ, Gattinoni L. Management of COVID-19 respiratory distress. JAMA. (2020) 323:2329–30. doi: 10.1001/jama.2020.6825
66. Laviolette L, Laveneziana P. Dyspnoea: a multidimensional and multidisciplinary approach. Eur Respir J. (2014) 43:1750–62. doi: 10.1183/09031936.00092613
67. Banzett RB, O'Donnell CR, Guilfoyle TE, Parshall MB, Schwartzstein RM, Meek PM, et al. Multidimensional Dyspnea Profile: an instrument for clinical and laboratory research. Eur Respir J. (2015) 45:1681–91. doi: 10.1183/09031936.00038914
68. von Leupoldt A, Sommer T, Kegat S, Baumann HJ, Klose H, Dahme B, et al. The Unpleasantness of perceived dyspnea is processed in the anterior insula and amygdala. Am J Respir Crit Care Med. (2008) 177:1026–32. doi: 10.1164/rccm.200712-1821OC
Keywords: ventilation, impaired homeostasis, COVID-19, breathing pattern disorder, dyspnea, post-covid breathing pattern dysfunction
Citation: Jareonsettasin P, Zeicu C, Diehl B, Harper RM and Astin R (2022) Inappropriate Ventilatory Homeostatic Responses in Hospitalized COVID-19 Patients. Front. Neurol. 13:909915. doi: 10.3389/fneur.2022.909915
Received: 31 March 2022; Accepted: 19 May 2022;
Published: 15 June 2022.
Edited by:
Beatrice Paradiso, Dolo Hospital, Venice, ItalyReviewed by:
Miguel García-Grimshaw, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), MexicoFrancesco Turco, University of Pisa, Italy
Copyright © 2022 Jareonsettasin, Zeicu, Diehl, Harper and Astin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prem Jareonsettasin, cC5qYXJlb25zZXR0YXNpbkB1Y2wuYWMudWs=
†These authors have contributed equally to this work and share first authorship
‡These authors share senior authorship