
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 10 June 2022
Sec. Neurogenetics
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.899644
This article is part of the Research Topic Genome Analysis for Alzheimer’s Disease View all 6 articles
 Jaya Bagaria1†
Jaya Bagaria1† Yeonsil Moon2†
Yeonsil Moon2† Eva Bagyinszky3
Eva Bagyinszky3 Kyu Hwan Shim1
Kyu Hwan Shim1 Seong Soo A. An1*
Seong Soo A. An1* SangYun Kim4
SangYun Kim4 Seol Heui Han2*
Seol Heui Han2*Apolipoprotein (APOE) is implicated and verified as the main risk factor for early-onset Alzheimer's disease (AD). APOE is a protein that binds to lipids and is involved in cholesterol stability. Our paper reports a case of a sporadic early-onset AD (sEOAD) patient of a 54-year-old Korean man, where a novel APOE Leu159Pro heterozygous mutation was revealed upon Whole Exome Sequence analysis. The proband's CSF showed downregulated levels of Aβ42, with unchanged Tau levels. The mutation is in the Low-Density Lipoprotein Receptor (LDLR) region of the APOE gene, which mediates the clearance of APOE lipoproteins. LDLR works as a high-affinity point for APOE. Studies suggest that APOE-LDLR interplay could have varying effects. The LDLR receptor pathway has been previously suggested as a therapeutic target to treat tauopathy. However, the APOE-LDLR interaction has also shown a significant correlation with memory retention. Leu159Pro could be an interesting mutation that could be responsible for a less damaging pattern of AD by suppressing tau-association neurodegeneration while affecting the patient's memory retention and cognitive performance.
Alzheimer's disease (AD) is prominent dementia instigating factor that affects cognitive abilities resulting in abnormalities like discrete diminution in thinking, memory, attention, or judgment (1). It accounts for 50–75% of dementia worldwide, affecting 44 million people. With the growing population in Asia, aging became an issue, and South Korea is one of the leading countries to witness such an irreversible form of illness in its population (2). While EOAD comprises 1–6% of all AD cases, sporadic EOAD (sEOAD) accounts for only 2% of all AD cases (2). There is a gap in understanding how familial EOAD (fEOAD) differs from sEOAD, however, studies show there is a strong genetic predisposition in fEOAD, which lacks in sEOAD cases despite showing a gradual memory decline that starts at a very early age. fEOAD results from mutations in one of the three genes: APP, PSEN1, and PSEN2. However, sEOAD does not have a deterministic risk factor that explains the development of AD at an early age. Four major genetic loci have been related to AD, APOE (Apolipoprotein E) on chromosome 19, APP (Amyloid precursor protein) on chromosome 21 (3), PSEN1 (Presenilin-1), and PSEN2 (presenilin-2) on chromosome 14 and 1, respectively (4–8). Among these mutations, APP, PSEN1, and PSEN2 mutations represent only a minority of all patient's early-onset AD (EOAD) cases with a familial inheritance, suggesting that APOE (apolipoprotein E) on chromosome 19, may play an important role in disease onset in sporadic cases of EOAD (9, 10). EOAD has a clinical manifestation before 65 years of age. Although there is an overlap of phenotypes between LOAD and EOAD, EOAD shows more symptoms of language and visuospatial impairment, and executive dysfunction (11). APOE has two polymorph sites: Cys112 and Arg158. Their mutations could result in three main alleles of APOE: E2 (Cys112, Cys112), E3 (Cys112, Arg158), and E4 (Arg158, Arg158), and their heterozygous combinations (12, 13). The level of Aß42 in the cerebrospinal fluid determines the onset of preclinical AD and is associated with a strong predictive marker of cognitive decline in normal subjects (14–17), whereas total-tau and phosphorylated-tau are late-onset markers of AD (18). Low levels of Aβ42 indicate the early onset of AD even in cognitively normal people, making it the earliest marker for preclinical prediction of sEOAD (19–21).
The proband patient was referred to the memory clinic of Konkuk University Medical Center at the age of 54, with a 2–3-month history of memory loss, executive dysfunction, and language dysfunction. He experienced difficulties in writing, planning tasks, and discussing a new topic. He started to forget topics he had discussed the previous day. Neuropsychological tests were conducted, and he scored 26 on the Mini-mental status examination (MMSE), 5 on the Clinical dementia rating scale (CDR), 4 on the Clinical Dementia Rating-Sum of Box score (CDR-SB), and 3 on the Global Deterioration Scale (GDS). Memory impairment with frontal/executive dysfunction and mild language dysfunction were revealed. Analysis of his APOE polymorphism revealed APOE 3/3. Brain magnetic resonance imaging (MRI) demonstrated diffuse atrophy of the brain, with more predominant medial temporal lobes, which is consistent with AD (Figure 1A). Because of his young age and clinical presentation with findings, EOAD was the most likely clinical diagnosis. About 5 mg of Donepezil was recommended with a gradual increase to 10 mg. Follow-up neuropsychological test revealed additional decreased attention and mild visuospatial dysfunction, and memantine 10 mg bid was added to his prescription. After 7 months of diagnosis, his activities of daily living started to further depreciate compared to the first visit. He often missed his usual subway stop at the university, which interfered with his daily duty of teaching or discussion. His long-term spatial memory started to impair to the point that he had difficulty remembering the nursing home, where his mother was living, and had to be accompanied by friends or other family members. Changes in behavior and personality started to get more pronounced. He would often scream and use profanity at his sister. He became more stubborn and would not let anybody interfere with his financial management and would often lose a check, or credit cards were suspended due to insufficient bank balance. He showed agitation and poor hygiene. Global cognitive assessments revealed declined daily activities of living; MMSE 25, CDR 1, CDR-SB 5.5, and GDS 4. After 18 months since the first diagnosis, the memory impairment of the patient worsened. The language function was further degraded, and only very short answers with mild dysarthria were possible. The MMSE, CDR, CDRS-SB, and GDS scores were 23, 1, 9, and 5, respectively. Follow-up MRI revealed interval progression of diffuse (mainly frontotemporal) brain atrophy and marked medial temporal lobar atrophy. A marked increase in the size of signal void in the cerebral aqueduct and a decrease in the size of basal ganglia were also compared to the MRI of the first visit, which supported the progression of the disease (Figure 1B).
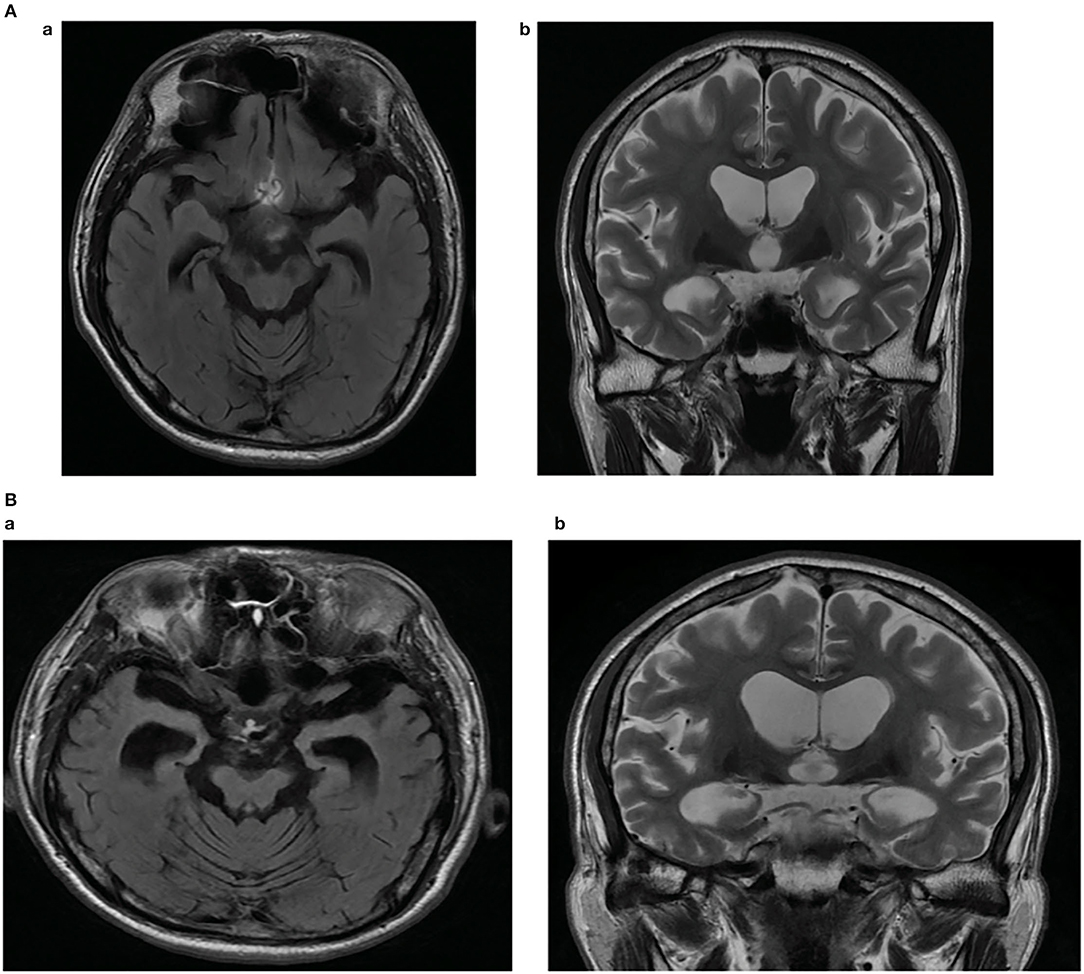
Figure 1. (A) Brain magnetic resonance imaging of the patient showing diffuse atrophy of the brain, more predominant medial temporal lobes. Fluid-attenuated inversion recovery (FLAIR) axial image (a), T2-weighted coronal image (b). (B) Brain magnetic resonance imaging (MRI) of the patient showing interval progression of diffuse (mainly frontotemporal) brain atrophy and marked medial temporal lobar atrophy after 18 months. Marked increased in size of with signal void in cerebral aqueduct, decreased in size of both basal ganglia compared to the MRI of the first visit are revealed. Fluid-attenuated inversion recovery (FLAIR) axial image (a), T2-weighted coronal image (b).
Our patient scored 12(144) on the K-NPI (Korean version of Neuropsychiatric Inventory) test, while it decreased to 9(144) mid-year, and then saw a sharp increase to 44(144) in 2014 when he was diagnosed with EOAD. The detailed test scores are given in the table below (Table 1).
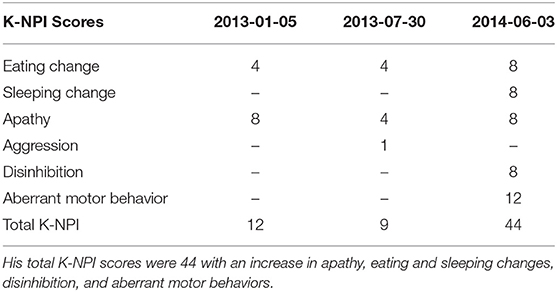
Table 1. Neuropsychiatric scores of the patient were marked as eating and sleeping changes, aberrant motor behavior, aggression, apathy, and disinhibition were identified over the course of his diagnosis.
The proband's mother (I-2) had been diagnosed with subcortical ischemic vascular dementia (SIVD) and the father (I-1) was deceased. The patient survived with an older sister (II-1) and a younger brother (II-3), both are currently unaffected (Figure 2).
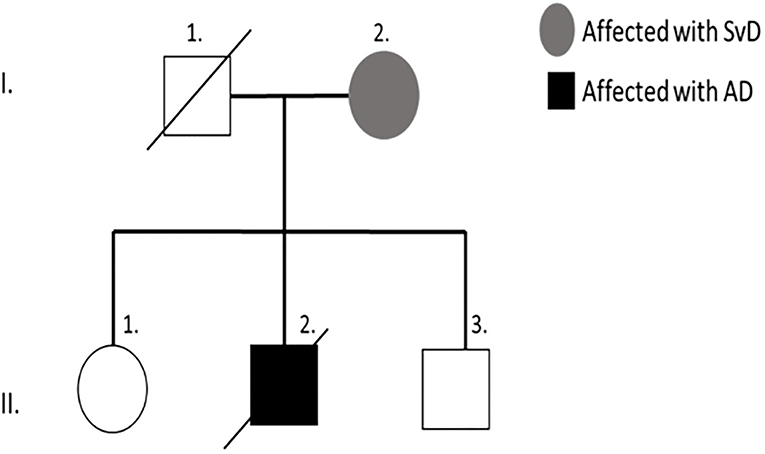
Figure 2. Family tree of proband patient (II-2).
After 19 months of initial diagnosis of dementia, the patient, unfortunately, suffered a heatstroke followed by subsequent pneumonia and died after 10 months following complications of bacterial meningitis and respiratory difficulty.
This study was performed with the approval of the Institutional Review Board of Konkuk University School of Medicine and Konkuk University Medical Center (IRB no. KUH2021-03-016). Written informed consent was collected from the patient under study to allow clinical and genetic data to be utilized for medical research purposes. A probable diagnosis of early-onset Alzheimer's disease (EOAD) was actualized by the clinicians. The white blood cell and CSF samples of the patient were received in our laboratory. The concentrations of CSF Aβ42, T-tau, and P-tau181 were quantified using commercial ELISA kits (INNOTEST β-AMYLOID (1–42), INNOTEST hTAU- Ag, and INNOTEST PHOSPHO-TAU (181P), Fujirebio Europe, Gent, Belgium) according to the manufacturer's instructions. The patient's blood sample was purified from the Qiagen blood kit. Whole exome sequencing (WES) was performed on the patient by Novogene Inc (https://en.novogene.com; Beijing, China). DNA of the patient was extracted from the whole blood sample using standard laboratory protocols. DNA purification was checked using the NanoPhotometer spectrophotometer. Whole exome sequencing (WES) was performed on the Novaseq6000 platform with a pair-end 150 bp read length. The average coverage was 100X. The intricate genetic assessment was performed by NGS. The targeted gene panel test was reviewed for several causal and risk genes involved in different neurodegenerative diseases like Alzheimer's Disease, Parkinson's Disease, frontotemporal dementia, and prion disease (Supplementary Table 1) Standard Sanger sequencing was performed to validate the NGS records by Bioneer Inc., (http://eng.bioneer.com/home.aspx, Bioneer Inc. Daejeon, Korea). Data alignment was done using National Center for Biotechnology Information (NCBI) Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi) The variant was checked in the NCBI database genome browser for humans (https://www.ncbi.nlm.nih.gov/genome/gdv/) for its uniqueness. It was also monitored in 1,000 Genomes (https://www.internationalgenome.org/), GnomAD browser (https://gnomad.broadinstitute.org/), and in the Korean reference genome database (KRGDB) amongst 1,722 healthy Korean individuals. Mutations were screened by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), Sorting Intolerant from Tolerant (SIFT; http://sift.jcvi.org/), and PROVEAN (http://provean.jcvi.org/index.php) software, which performs predictions on the potential pathogenic nature of variants. Analysis by the ExPASy server (https://www.expasy.org/) was also carried out on three parameters: bulkiness, polarity, and hydrophobicity (Kyte and Doolittle scale). Crystal structure of APOE was downloaded from the Protein Data Bank (PDB; http://www.rcsb.org/pdb/) and mutation was generated by Discovery Studio 3.5 Visualizer software, designed by Accelrys. Normal (E3) and mutant (Leu159Pro) APOE were compared based on structural changes and intramolecular interactions.
Cerebrospinal fluid (CSF) analysis on the patient revealed very low concentrations of amyloid-beta 42, suggesting substantial progression of amyloid pathology (Table 2). However, his total tau and phosphorylated tau were not increased (Table 2). This could suggest possible protection against tauopathy.
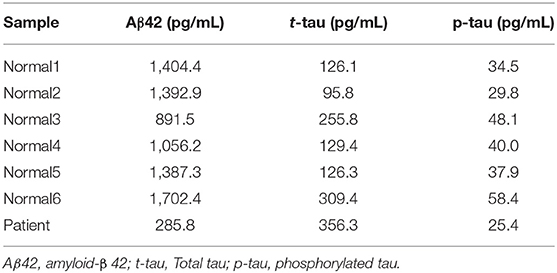
Table 2. Cerebrospinal fluid (CSF) analysis on the patient compared to 6 cognitive normal samples revealed downregulated Aβ42; Total-tau and Phosphorylated-tau showed decreased values, suggesting tauopathy did not start.
Whole Exome Sequencing (WES) revealed no pathogenic mutations in APP, PSEN1, or PSEN2 genes. No rare variants were found in AD risk genes either, such as SORL1, ABCA7, or CLU (Supplementary Table 2). In addition, no pathogenic variants appeared in other disease-associated genes including PD, FTD, and Prion disease. A novel heterozygous T>C exchange at chromosome 19 g.19:45412083, c.530T>C; Leu159Pro was recognized and validated in the APOE-coding region with WES, as well as Sanger Sequencing (Figure 3).
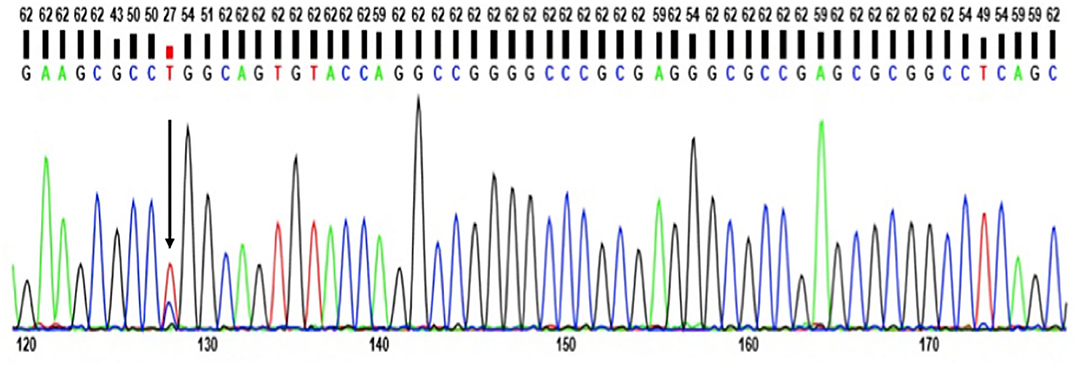
Figure 3. Standard sequencing of APOE Leu159Pro mutation showing it is a heterozygous mutation.
This is a Leucine to Proline substitution in APOE exon 4 at position 159, which was not found in either of the databases like KRGDB, 1,000 Genome, or ExAC. APOE Leu159Pro was suggested as deleterious or damaging by MutationTaster, PolyPhen2, and SIFT tools. SIFT and PolyPhen2 revealed this mutation as pathogenic with HumDiv and HumVar scores, whereas MutationTaster predicted this as a disease-causing mutation with a probability value of prediction security of 0.999. Multiple sequence alignment by PolyPhen2 suggested that Leucine159 is conservative among several primate species, including Hylobates leucogenys, Macaca fascicularis, Papio Anubis, or Callithrix jacchus. The majority of the mammalian species also had leucine in the same position: California sea lion (Zalophus californianus), Naked mole-rat (Heterocephalus glaber), and mouse (Mus Musculus). Proline was not found at the homologous position of APOE-like sequences of any species; however, Methionine and Isoleucine were observed in Rabbit (Oryctolagus cuniculus) and Duckbill platypus (Ornithorhynchus anatinus), respectively.
The ExPasy tool showed that scores of bulkiness significantly dropped from 15.903 to 15.462 at Leu159Pro, and nearby amino acids were also affected as result (Figure 4A). Kyte and Doolittle Hydrophobicity scores revealed that the score of hydropathicity on Pro159 also dropped from −1.011 to −0.411 (Pro159), suggesting that this mutation is less hydrophobic (Figure 4B). Structure prediction of APOE3 Leu159Pro revealed that proline could disturb the structure of Helix-4 since it could result in a kink inside the alpha-helix (Figures 5A,B).
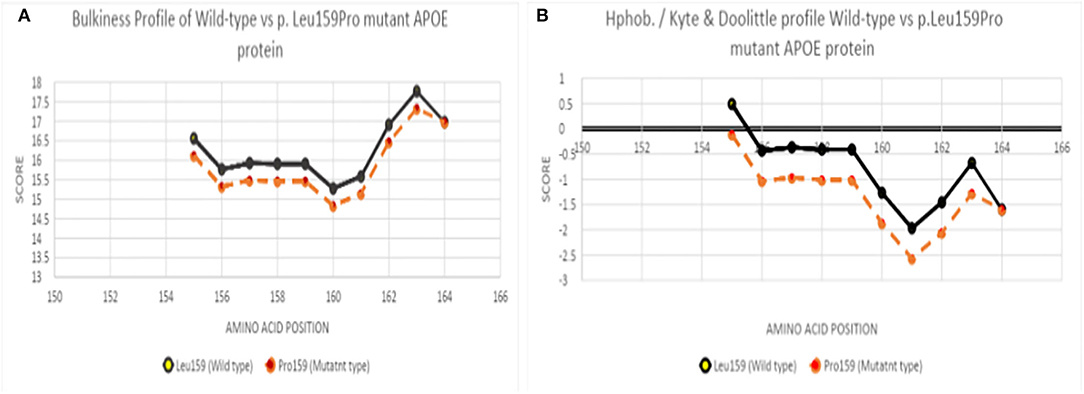
Figure 4. (A) Bulkiness profile of wild-type and Leu159Pro mutant APOE protein, (B) Hydrophobicity profile of wild-type and Leu159Pro APOE mutant protein.
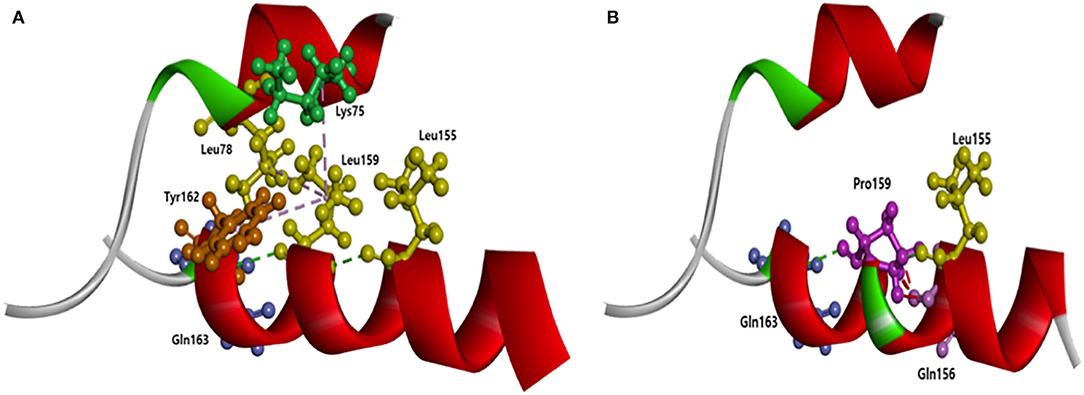
Figure 5. Intramolecular interactions of APOE with Leu159, compared to the APOE with Pro159. Pro159 may significantly disturb the intramolecular structure of APOE by the loss of contact between Helix-1 and Helix-4. (A) Normal APOE with Leu159; (B) Mutant APOE with Pro159.
This kink may change the shape of the helix, resulting in an abnormal helical motion. Intramolecular interactions could be significantly changed due to APOE3 Leu159Pro. In normal APOE3, Leu 159 could form hydrogen bonds with two nearby residues: Leu155 and Gln163. Additionally, Leu159 could form hydrophobic interactions with Tyr162 and two residues in helix-1 (Leu78 and Lys75). Pro159 could destroy the contact between Helix-4 and Helix-1 since hydrophobic interactions with Leu18 and Lys75 would be lost. In addition, Pro159 could form strong hydrogen bonds with Gln156. The hydrogen bonds between Pro159, Leu155, and Gln163 remain, but the distance between Pro159 and these residues may alter.
The ClueGo pathway analysis was performed to predict putative interaction among neurodegenerative genes and their possible common pathways. Among the gene interactions, APOE was found to play a central role in three pathways (Supplementary Figure 1). First, along with ABCA7, SORL1, and PON1, APOE may play a role in the positive regulation of cholesterol efflux. Second, APOE could interact with several genes (including EPHA1, DBN1, GIGYF2, NOTCH3, FIG1, OPTN, PTK2B, SETX, and MAPT), and play a role in the negative regulation of fatty acid oxidation. Finally, along with ABCA7, LRRK2, HIP1R, SINJ1, and APLP2, APOE may play a role in the positive regulation of endocytosis.
APOE is a 299 amino acids long single-chain polypeptide glycoprotein, that binds to the receptors of the cell surface in the low-density lipoprotein (LDL) receptor family. It helps in transporting lipid and lipid metabolism (22). Human APOE has three main polymorphic variants APOE2, APOE3, and APOE4 depending on the Cys-Arg substitute residues at positions 112 and 158 (23). APOE, a two-domain structure protein, activates the LDL receptor (24). The N-terminal domain consists of residues 1–191 and the C-terminal domain consists of 192–299. These two domains relate to a hinge. The N-terminal binds to the LDL receptor while the C-terminal binds to hyper-triglyceridemic very low-density lipoprotein (HVLDL) (25). Site 1–191 contains the receptor-bindingding domain in the Helix4 at residues 130–161 (26). Patients with APOE E4 allele were found to have amyloid depositions an in early age (40s−50s), while also resulting in glucose hypermetabolism in preclinical age of 30s (27, 28). The Association of APOE and EOAD remained unclear, but its role in EOAD may not be ruled out, as mutant APOE along with other putative genetic variants may result in an early onset of the disease formation (29). For example, APOE E4 along with PSEN1 Glu280Ala resulted in earlier disease onset (30), however, APOE E2 with PSEN1 Glu280Ala delayed the age of onset of the disease (31).
Oikawa et. al discovered APOE Sendai, a G>C point mutation at residue codon 145, with an Arginine to Proline substitution (32). Sam et al. reported APOE Chicago, a G>C point mutation at residue codon 147, with an Arginine to Proline substitution (33). Kinomura et al. reported APOE Okayama, a G>C substitution at codon 150, with an Arginine to Glycine replacement (22). Cautero et al. described a novel APOE Modena, a C>T homozygous mutation at residue codon 150, with an Arginine to Cysteine change (34). Luo et al. identified APOE Guangzhou, a G>C nucleotide substitution at residue codon 150, with an Arginine to Proline exchange (35). Bomback et al. presented a novel APOE Las Vegas mutation, a C>A point nucleotide change at codon 152, with an Alanine to Asparagine exchange (36). Tokura et al. reported a case of APOE2 Kurashiki novel mutation, a G>C substitution at codon 158 with Arginine to Proline change, while Mitani et al. reported a case of APOE3 Osaka novel mutation, also a G>C point mutation at codon 158 with Arginine to Proline substitution (37, 38). APOE plays a crucial role in mediating lipoprotein clearance by binding to cell surface receptors of LDLR (39). Until now, most of the mutations reported on the LDLR site of 135–160 (Figure 6) are associated with a rare disease called Lipoprotein Glomerulopathy (LPG) (40) which results from an increased APOE serum level (41). There are several reported cases with APOE but asymptomatic for LPG or renal failure (42–44). Leu159Pro lies next to APOE Osaka mutation in the lipid-binding region and alters the conformation of APOE. It might result in increased aggregation of Aβ as well as specific effects like cholesterol efflux on lipid metabolism.
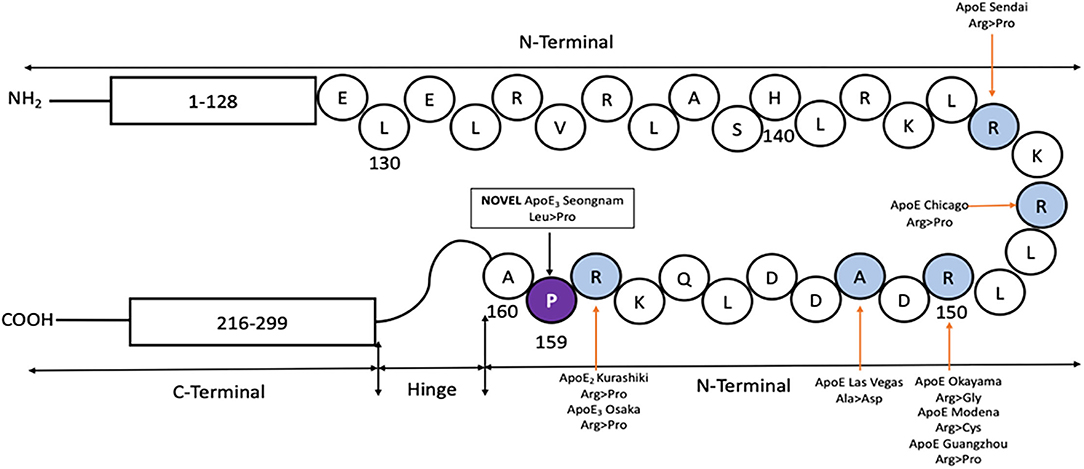
Figure 6. Protein map of APOE, showing the N and C terminal connected with a hinge. Previously reported mutations in the LDLR site (shaded in blue) are shown, along-with the novel APOE3 Leu159Pro (shaded in violet with the mutated codon) found in a Korean male patient with EOAD.
Several common variants were found among the analyzed genes, included in our gene panel (Supplementary Tables 1, 2). Even though the majority of these variants were common, their impact on disease onset may not be ruled out. It may be possible that these variants in risk genes may interact and possibly act as risk modifiers (45, 46). ClueGo mapping was performed on mutation-carrying genes to predict their possible interactions. APOE may interact with several genes and was predicted to play a role in three pathways: (1) positive regulation of cholesterol reflux (2) negative regulation of fatty acid oxidation, and (3) positive regulation of endocytosis.
APOE may highly impact in positive regulation of cholesterol efflux along with ABCA7, SORL1, PON1, and APOC4. In ABCA7, three common missense mutations were found: Glu188Gly, Gly1527Ala, and Ala2045Ser. Among these variants, Gly1527Ala was found to reduce the ABCA7 expression and could play a role as a risk modifier (47, 48). Mutant ABCA7 and APOE were suggested to play a role in cognitive dysfunctions, amyloid generation, and senile plaque formation (49, 50). Both ABCA7 and APOE were confirmed to play a role in lipid homeostasis and cholesterol metabolism. Lower ABCA7 expression near blood-brain barrier (BBB) may reduce the APOE expression. Lower ABCA7 and APOE expression may result in decreased cholesterol exchange across blood-brain barrier (51, 52). Two common variants were found in SORL, Gln1074Glu, and Val1967Ile, both were suggested to be common benign variants (53). SORL1 could play a role as an APOE receptor and induce the cholesterol intake into neurons (54, 55). PON1 was also found to interact with APOE through the cholesterol pathway. APOE was suggested to enhance PON1 activity and stability. Their interaction may result in anti-atherogenic effects, such as inhibiting LDL cholesterol oxidation or enhancing the macrophage cholesterol efflux (56). Further investigations are needed on how APOE and APOC4 could interact to impact cholesterol metabolism (57). However, abnormal expression of APOC genes may be involved in human hyperlipidemias (58).
The APOE also interacts with several proteins to play a role in fatty acid oxidation. APOE dysfunctions may impair fatty acid transport, and reduce the fatty acid oxidation, resulting in fatty acid aggregation in astrocytes and brain (59). The third function APOE could be involved in the regulation of endocytosis (60). The role of common variants in the case of APOE Leu159Pro mutation may not be ruled out as these variants could interact as risk modifiers and impact the disease-related pathways.
It was previously reported that the APOE4 allele increases susceptibility to AD, as well as to an earlier onset of the disease (61). However, the consideration that APOE3 is neutral compared to APOE4, is not based on their contributions to pathogenesis. Holtzman et al. demonstrate how any allele of APOE can interpose pronounced tauopathy (62). AD is a slow developing disease starts with the accumulation of amyloid plaques in the brain which reduces Abeta42 in cerebrospinal fluid (CSF) (63). Tauopathy occurs over several years. Mutations in the LDLR domain of the APOE gene may disrupt the protein's ability to traffic lipids.
In this report, we described a Korean patient with sporadic EOAD who had long-term spatial memory impairment along with executive dysfunction, and language impairments. Mutations in other AD-related genes were absent, but there's a novel mutation Leu159Pro in the LDLR domain of APOE. His CSF analysis revealed a pronounced downregulation of Aβ42 levels, however his t-tau, and p-tau levels were not upregulated. This would infer that the patient had significant amyloid pathogenesis which contributed to his dementia, and long-term spatial memory impairment, however, tauopathy did not start. This could be possible due to the Leu159Pro mutation in the LDLR domain of APOE. The LDLR receptor interacts with all the different isoforms of APOE in the brain with different binding attractions (64, 65). The interplay of APOE and LDLR affects spatial memory, independent of APOE isoforms, associated with AD (66). While APOE has been studied profoundly about its disease-modifying mechanism in AD, the effect of APOE mutations in the LDLR domain in the brain has been understudied.
We hypothesize that Leu159Pro promotes lipoprotein metabolism and APOE turnover, by increasing LDLR expression. This results in an intracellular blockage resulting in aberrant accrual of Aβ plaques in the brain. This overexpression of Aβ plaques should lead to tau-related neurodegeneration. However, LDLR has a protective effect on tauopathy (62). Patients with dementia often display metabolic dysfunction with altered liver proteins. However, little is known about their cumulative effect on cognition (10). Our patient showed fewer signs of memory abilities despite exceedingly high levels of amyloid plaques, and low levels of tau build-up, similar to Christchurch homozygote mutation (67), however, it is interesting to note that similar to Christchurch heterozygous mutation, our patient too exhibited signs of spatial disorientation and progressive cognitive decline with an early onset of the disease (68). Leu159Pro may downgrade the ability of APOE to bind to specific factors essential to yielding tau tangles. Clinical information such as postmortem examination, tau-pet imaging, and detailed lipid profile since the onset of the disease condition are not currently available, which limits the understanding of the contribution of this new variant in lipid homeostasis as well as the formation of tau fibrils.
In conclusion, mutations in the LDLR domain of APOE could be a protective target to tau-associated neurodegeneration, while increasing the risk of amyloid load in sEOAD. Additional studies are required to better understand this hypothesis to produce a more definite answer to whether mutations in the LDLR domain of APOE could indeed be an interesting area to study about sporadic EOAD.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: FigShare: https://doi.org/10.6084/m9.figshare.16627975.v2 (Bagaria, Jaya (2021): APOE Leu159Pro Sanger Sequencing.figshare.
The studies involving human participants were reviewed and approved by Institutional Review Board of Konkuk University School of Medicine and Konkuk University Medical Center (IRB no. KUH2021-03-016). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
JB, YM, and SA: conceptualization. YM: data curation. JB: formal analysis, methodology, and software. SA: funding acquisition and resources. JB, YM, and EB: investigation. KS: CSF analysis. YM, SA, and SH: project administration. EB, SA, SK, and SH: supervision and writing—review and editing. YM and EB: validation. EB: visualization. JB and YM: writing—original draft. All authors contributed to the article and approved the submitted version.
This research was funded by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2021R1A6A1A03038996), Brain Research Program of the National Research Foundation (NRF) funded by the Ministry of Science & ICT (NRF-2018M3C7A1056571), and Gachon University research fund of 2021 (GCU-202008480012).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.899644/full#supplementary-material
1. Bature F, Guinn BA, Pang D, Pappas Y. Signs and symptoms preceding the diagnosis of Alzheimer's disease: a systematic scoping review of literature from 1937 to 2016. BMJ Open. (2017) 7:e015746. doi: 10.1136/bmjopen-2016-015746
2. Nardini E, Hogan R, Flamier A, Bernier G. Alzheimer's disease: a tale of two diseases? Neural Regen Res. (2021) 16:1958–64. doi: 10.4103/1673-5374.308070
3. Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. (1991) 349:704–6. doi: 10.1038/349704a0
4. Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. (1995) 375:754–60. doi: 10.1038/375754a0
5. Alzheimer's Disease Collaborative G. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat Genet. (1995) 11:219–22. doi: 10.1038/ng1095-219
6. Cruts M, Backhovens H, Wang SY, Van Gassen G, Theuns J, De Jonghe CD, et al. Molecular genetic analysis of familial early-onset Alzheimer's disease linked to chromosome 14q243. Hum Mol Genet. (1995) 4:2363–71. doi: 10.1093/hmg/4.12.2363
7. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. (1993) 261:921–3. doi: 10.1126/science.8346443
8. Mahley RW, Rall SC Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. (2000) 1:507–37. doi: 10.1146/annurev.genom.1.1.507
9. Bagyinszky E, Youn YC, An SS, Kim S. The genetics of Alzheimer's disease. Clin Interv Aging. (2014) 9:535–51. doi: 10.2147/CIA.S51571
10. Bagaria J, Nho K, An SSA. Importance of GWAS in finding un-targeted genetic association of sporadic Alzheimer's disease. Mol Cell Toxicol. (2021) 17:233–44. doi: 10.1007/s13273-021-00130-z
11. van der Flier WM, Pijnenburg YA, Fox NCvScheltens P. Early-onset versus late-onset Alzheimer's disease: the case of the missing APOE ε4 allele. Lancet Neurol. (2011) 10:280–8. doi: 10.1016/S1474-4422(10)70306-9
12. Giau VV, Bagyinszky E, An SS, Kim SY. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr Dis Treat. (2015) 11:1723–37. doi: 10.2147/NDT.S84266
13. Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. (2009) 63:287–303. doi: 10.1016/j.neuron.2009.06.026
14. Schoonenboom NS, Reesink FE, Verwey NA, Kester MI, Teunissen CE, van de Ven PM, et al. Cerebrospinal fluid markers for differential dementia diagnosis in a large memory clinic cohort. Neurology. (2012) 78:47–54. doi: 10.1212/WNL.0b013e31823ed0f0
15. Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J, et al. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. (2007) 6:734–46. doi: 10.1016/S1474-4422(07)70178-3
16. Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. (2009) 302:385–93. doi: 10.1001/jama.2009.1064
17. Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. (2006) 5:228–34. doi: 10.1016/S1474-4422(06)70355-6
18. Buchhave P, Minthon L, Zetterberg H, Wallin AK, Blennow K, Hansson O. Cerebrospinal fluid levels of β-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry. (2012) 69:98–106. doi: 10.1001/archgenpsychiatry.2011.155
19. Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, et al. Decreased cerebrospinal fluid Abeta (42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. (2009) 65:176–83. doi: 10.1002/ana.21559
20. Izaks GJ, Gansevoort RT, van der Knaap AM, Navis G, Dullaart RP, Slaets JP. The association of APOE genotype with cognitive function in persons aged 35 years or older. PLoS ONE. (2011) 6:e27415. doi: 10.1371/journal.pone.0027415
21. Skoog I, Davidsson P, Aevarsson O, Vanderstichele H, Vanmechelen E, Blennow K. Cerebrospinal fluid beta-amyloid 42 is reduced before the onset of sporadic dementia: a population-based study in 85-year-olds. Dement Geriatr Cogn Disord. (2003) 15:169–76. doi: 10.1159/000068478
22. Kinomura M, Sugiyama H, Saito T, Matsunaga A, Sada KE, Kanzaki M, et al. Novel variant apolipoprotein E Okayama in a patient with lipoprotein glomerulopathy. Nephrol Dial Transplant. (2008) 23:751–6. doi: 10.1093/ndt/gfm675
23. Bennet AM, Di Angelantonio E, Ye Z, Wensley F, Dahlin A, Ahlbom A, et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA. (2007) 298:1300–11. doi: 10.1001/jama.298.11.1300
24. Datta G, Chaddha M, Garber DW, Chung BH, Tytler EM, Dashti N, et al. The receptor binding domain of apolipoprotein E, linked to a model class A amphipathic helix, enhances internalization and degradation of LDL by fibroblasts. Biochemistry. (2000) 39:213–20. doi: 10.1021/bi991209w
25. Bradley WA, Gianturco SH. ApoE is necessary and sufficient for the binding of large triglyceride-rich lipoproteins to the LDL receptor; apoB is unnecessary. J Lipid Res. (1986) 27:40–8. doi: 10.1016/S0022-2275(20)38865-9
26. Wilson C, Wardell M, Weisgraber K, Mahley R, Agard D. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein. Science. (1991) 252:1817–22. doi: 10.1126/science.2063194
27. Fan J, Tao W, Li X, Li H, Zhang J, Wei D, et al. The contribution of genetic factors to cognitive impairment and dementia: apolipoprotein E gene, gene interactions, and polygenic risk. Int J Mol Sci. (2019) 20:8–9. doi: 10.3390/ijms20051177
28. Sando SB, Melquist S, Cannon A, Hutton ML, Sletvold O, Saltvedt I, et al. Epsilon 4 lowers age at onset and is a high risk factor for Alzheimer's disease; a case control study from central Norway. BMC Neurol. (2008) 8:9. doi: 10.1186/1471-2377-8-9
29. Jia L, Xu H, Chen S, Wang X, Yang J, Gong M, et al. The APOE epsilon4 exerts differential effects on familial and other subtypes of Alzheimer's disease. Alzheimers Dement. (2020) 16:1613–23. doi: 10.1002/alz.12153
30. Pastor P, Roe CM, Villegas A, Bedoya G, Chakraverty S, Garcia G, et al. Apolipoprotein Eepsilon4 modifies Alzheimer's disease onset in an E280A PS1 kindred. Ann Neurol. (2003) 54:163–9. doi: 10.1002/ana.10636
31. Velez JI, Lopera F, Sepulveda-Falla D, Patel HR, Johar AS, Chuah A, et al. APOE*E2 allele delays age of onset in PSEN1 E280A Alzheimer's disease. Mol Psychiatry. (2016) 21:916–24. doi: 10.1038/mp.2015.177
32. Oikawa S, Matsunaga A, Saito T, Sato H, Seki T, Hoshi K, et al. Apolipoprotein E Sendai (arginine 145–>proline): a new variant associated with lipoprotein glomerulopathy. J Am Soc Nephrol. (1997) 8:820–3. doi: 10.1681/ASN.V85820
33. Sam R, Wu H, Yue L, Mazzone T, Schwartz MM, Arruda JA, et al. Lipoprotein glomerulopathy: a new apolipoprotein E mutation with enhanced glomerular binding. Am J Kidney Dis. (2006) 47:539–48. doi: 10.1053/j.ajkd.2005.12.031
34. Cautero N, Di Benedetto F, De Ruvo N, Montalti R, Guerrini GP, Ballarin R, et al. Novel genetic mutation in apolipoprotein E2 homozygosis and its implication in organ donation: a case report. Transplant Proc. (2010) 42:1349–51. doi: 10.1016/j.transproceed.2010.03.104
35. Luo B, Huang F, Liu Q, Li X, Chen W, Zhou SF Yu X. Identification of apolipoprotein E Guangzhou (arginine 150 proline), a new variant associated with lipoprotein glomerulopathy. Am J Nephrol. (2008) 28:347–53. doi: 10.1159/000111828
36. Bomback AS, Song H, D'Agati VD, Cohen SD, Neal A, Appel GB, et al. New apolipoprotein E mutation, apoE Las Vegas, in a European-American with lipoprotein glomerulopathy. Nephrol Dial Transplant. (2010) 25:3442–6. doi: 10.1093/ndt/gfq389
37. Mitani A, Ishigami M, Watase K, Minakata T, Yamamura T. A novel apolipoprotein E mutation, ApoE Osaka (Arg158 Pro), in a dyslipidemic patient with lipoprotein glomerulopathy. J Atheroscler Thromb. (2011) 18:531–5. doi: 10.5551/jat.7377
38. Tokura T, Itano S, Kobayashi S, Kuwabara A, Fujimoto S, Horike H, et al. A novel mutation ApoE2 Kurashiki (R158P) in a patient with lipoprotein glomerulopathy. J Atheroscler Thromb. (2011) 18:536–41. doi: 10.5551/jat.8102
39. Ishibashi S, Herz J, Maeda N, Goldstein JL, Brown MS. The two-receptor model of lipoprotein clearance: tests of the hypothesis in “knockout” mice lacking the low density lipoprotein receptor, apolipoprotein E, or both proteins. Proc Natl Acad Sci USA. (1994) 91:4431–5. doi: 10.1073/pnas.91.10.4431
40. Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer's diseases. Neurobiol Dis. (2014) 72 (Pt A):3–12. doi: 10.1016/j.nbd.2014.08.025
41. Magistroni R, Bertolotti M, Furci L, Fano RA, Leonelli M, Pisciotta L, et al. Lipoprotein glomerulopathy associated with a mutation in apolipoprotein e. Clin Med Insights Case Rep. (2013) 6:189–96. doi: 10.4137/CCRep.S12209
42. Saito T, Oikawa S, Sato H, Sasaki J. Lipoprotein glomerulopathy: renal lipidosis induced by novel apolipoprotein E variants. Nephron. (1999) 83:193–201. doi: 10.1159/000045511
43. Toyota K, Hashimoto T, Ogino D, Matsunaga A, Ito M, Masakane I, et al. Founder haplotype of APOE-Sendai mutation associated with lipoprotein glomerulopathy. J Hum Genet. (2013) 58:254–8. doi: 10.1038/jhg.2013.8
44. Hu Z, Huang S, Wu Y, Liu Y, Liu X, Su D, et al. Hereditary features, treatment, and prognosis of the lipoprotein glomerulopathy in patients with the APOE Kyoto mutation. Kidney Int. (2014) 85:416–24. doi: 10.1038/ki.2013.335
45. Bagyinszky E, Kang MJ, Pyun J, Giau VV, An SSA, Kim S. Early-onset Alzheimer's disease patient with prion (PRNP) pVal180Ile mutation. Neuropsychiatr Dis Treat. (2019) 15:2003–13. doi: 10.2147/NDT.S215277
46. Rosenthal SL, Wang X, Demirci FY, Barmada MM, Ganguli M, Lopez OL, et al. Beta-amyloid toxicity modifier genes and the risk of Alzheimer's disease. Am J Neurodegener Dis. (2012) 1:191–8. Available online at: www.AJND.us/ISSN:2165-591X/AJND1207005
47. Feher A, Juhasz A, Pakaski M, Janka Z, Kalman J. Association study of the ABCA7 rs3752246 polymorphism in Alzheimer's disease. Psychiatry Res. (2019) 279:376–7. doi: 10.1016/j.psychres.2019.01.081
48. Bamji-Mirza M, Li Y, Najem D, Liu QY, Walker D, Lue LF, et al. Genetic variations in ABCA7 can increase secreted levels of amyloid-beta40 and amyloid-beta42 peptides and ABCA7 transcription in cell culture models. J Alzheimers Dis. (2016) 53:875–92. doi: 10.3233/JAD-150965
49. Berg CN, Sinha N, Gluck MA. The effects of APOE and ABCA7 on cognitive function and Alzheimer's disease risk in African Americans: a focused mini review. Front Hum Neurosci. (2019) 13:387. doi: 10.3389/fnhum.2019.00387
50. Chang YT, Hsu SW, Huang SH, Huang CW, Chang WN, Lien CY, et al. ABCA7 polymorphisms correlate with memory impairment and default mode network in patients with APOEepsilon4-associated Alzheimer's disease. Alzheimers Res Ther. (2019) 11:103. doi: 10.1186/s13195-019-0563-3
51. Lamartiniere Y, Boucau MC, Dehouck L, Krohn M, Pahnke J, Candela P, et al. ABCA7 downregulation modifies cellular cholesterol homeostasis and decreases amyloid-beta peptide efflux in an in vitro model of the blood-brain barrier. J Alzheimers Dis. (2018) 64:1195–211. doi: 10.3233/JAD-170883
52. Dib S, Pahnke J, Gosselet F. Role of ABCA7 in human health and in Alzheimer's Disease. Int J Mol Sci. (2021) 22:4–9. doi: 10.3390/ijms22094603
53. Vardarajan BN, Zhang Y, Lee JH, Cheng R, Bohm C, Ghani M, et al. Coding mutations in SORL1 and Alzheimer disease. Ann Neurol. (2015) 77:215–27. doi: 10.1002/ana.24305
54. Barthelson K, Newman M, Lardelli M. Sorting out the role of the sortilin-related receptor 1 in Alzheimer's Disease. J Alzheimers Dis Rep. (2020) 4:123–40. doi: 10.3233/ADR-200177
55. Reitz C. Dyslipidemia and the risk of Alzheimer's disease. Curr Atheroscler Rep. (2013) 15:307. doi: 10.1007/s11883-012-0307-3
56. Gaidukov L, Viji RI, Yacobson S, Rosenblat M, Aviram M, Tawfik DS. ApoE induces serum paraoxonase PON1 activity and stability similar to ApoA-I. Biochemistry. (2010) 49:532–8. doi: 10.1021/bi9013227
57. Allan CM I, Walker D, Segrest JP, Taylor JM. Identification and characterization of a new human gene (APOC4) in the apolipoprotein E, C-I, and C-II gene locus. Genomics. (1995) 28:291–300. doi: 10.1006/geno.1995.1144
58. Jong MC, Hofker MH, Havekes LM. Role of ApoCs in lipoprotein metabolism: functional differences between ApoC1, ApoC2, and ApoC3. Arterioscler Thromb Vasc Biol. (1999) 19:472–84. doi: 10.1161/01.ATV.19.3.472
59. Qi G, Mi Y, Shi X, Gu H, Brinton RD, Yin F. ApoE4 impairs neuron-astrocyte coupling of fatty acid metabolism. Cell Rep. (2021) 34:108572. doi: 10.1016/j.celrep.2020.108572
60. Kockx M, Jessup W, Kritharides L. Regulation of endogenous apolipoprotein E secretion by macrophages. Arterioscler Thromb Vasc Biol. (2008) 28:1060–7. doi: 10.1161/ATVBAHA.108.164350
61. Rocchi A, Pellegrini S, Siciliano G, Murri L. Causative and susceptibility genes for Alzheimer's disease: a review. Brain Res Bull. (2003) 61:1–24. doi: 10.1016/S0361-9230(03)00067-4
62. Shi Y, Andhey PS, Ising C, Wang K, Snipes LL, Boyer K, et al. Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron. (2021) 109:2413–26.e7. doi: 10.1016/j.neuron.2021.05.034
63. Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. (2006) 59:512–9. doi: 10.1002/ana.20730
64. Yamamoto T, Choi HW, Ryan RO. Apolipoprotein E isoform-specific binding to the low-density lipoprotein receptor. Anal Biochem. (2008) 372:222–6. doi: 10.1016/j.ab.2007.09.005
65. Innerarity TL, Friedlander EJ, Rall SC Jr, Weisgraber KH, Mahley RW. The receptor-binding domain of human apolipoprotein Binding of apolipoprotein E fragments. J Biol Chem. (1983) 258:12341–7. doi: 10.1016/S0021-9258(17)44180-9
66. Johnson LA, Olsen RHJ, Merkens LS, DeBarber A, Steiner RD, Sullivan PM, et al. Apolipoprotein E-low density lipoprotein receptor interaction affects spatial memory retention and brain ApoE levels in an isoform-dependent manner. Neurobiol Dis. (2014) 64:150–62. doi: 10.1016/j.nbd.2013.12.016
67. Arboleda-xVelasquez JF, Lopera F, O'Hare M, Delgado-Tirado S, Marino C, Chmielewska N, et al. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. (2019) 25:1680–3. doi: 10.1038/s41591-019-0611-3
Keywords: Low-Density Lipoprotein Receptor (LDLR), APOE, Leu159Pro, early-onset Alzheimer's disease, novel mutation, Whole Exome Sequence (WES) analysis
Citation: Bagaria J, Moon Y, Bagyinszky E, Shim KH, An SSA, Kim S and Han SH (2022) Whole Exome Sequencing Reveals a Novel APOE Mutation in a Patient With Sporadic Early-Onset Alzheimer's Disease. Front. Neurol. 13:899644. doi: 10.3389/fneur.2022.899644
Received: 18 March 2022; Accepted: 04 May 2022;
Published: 10 June 2022.
Edited by:
Na Zhao, Mayo Clinic Florida, United StatesReviewed by:
Tulsi Patel, Icahn School of Medicine at Mount Sinai, United StatesCopyright © 2022 Bagaria, Moon, Bagyinszky, Shim, An, Kim and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seong Soo A. An, c2VvbmcuYW5AZ21haWwuY29t; Seol Heui Han, YWx6ZG9jQGt1aC5hYy5rcg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.