 Yu-xiao Liu
Yu-xiao Liu Yang Yu3†
Yang Yu3† Yong-ming Yao
Yong-ming Yao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 27 June 2022
Sec. Neurocritical and Neurohospitalist Care
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.892480
This article is part of the Research Topic Causative Mechanism and Potential Pharmacological and Non-pharmacological Interventions in Sepsis-Associated Encephalopathy View all 7 articles
Sepsis-associated encephalopathy (SAE), the most popular cause of coma in the intensive care unit (ICU), is the diffuse cerebral damage caused by the septic challenge. SAE is closely related to high mortality and extended cognitive impairment in patients in septic shock. At present, many studies have demonstrated that SAE might be mainly associated with blood–brain barrier damage, abnormal neurotransmitter secretion, oxidative stress, and neuroimmune dysfunction. Nevertheless, the precise mechanism which initiates SAE and contributes to the long-term cognitive impairment remains largely unknown. Recently, a growing body of evidence has indicated that there is close crosstalk between SAE and peripheral immunity. The excessive migration of peripheral immune cells to the brain, the activation of glia, and resulting dysfunction of the central immune system are the main causes of septic nerve damage. This study reviews the update on the pathogenesis of septic encephalopathy, focusing on the over-activation of immune cells in the central nervous system (CNS) and the “neurocentral–endocrine–immune” networks in the development of SAE, aiming to further understand the potential mechanism of SAE and provide new targets for diagnosis and management of septic complications.
Sepsis is a common complication in patients with burns or wound injury, infection, shock, and major surgery. It is a dysregulated host response induced by severe infection, which may develop into multiple organ failure and eventually lead to death. In 2020, Rudd et al. (1) analyzed the onset and mortality of sepsis from 1997 to 2017 and found that there were about 48.9 million patients in septic shock in the world. Now, sepsis is regarded as a prominent unsolved problem around the world due to its high incidence, fatality rate, and medical cost.
The multiple organ dysfunction induced by sepsis results in an imbalance of the immune response, which may aggravate the clinical symptoms of sepsis. As the primary organ affected by inflammation in sepsis, the brain is not only critically involved in immune regulatory response but also vulnerable to injury, which may lead to sepsis-associated encephalopathy (SAE). Of note, SAE is a diffuse brain dysfunction secondary to sepsis, with an incidence of more than 70% in patients in septic shock, especially in the elderly people, neonates, and patients with a chronic illness (2). SAE can lead to long-term neurological damage, such as anxiety, memory impairment, and consciousness disorders. It was reported that the mortality rate of patients with septic encephalopathy appeared to be much higher than that of patients without encephalopathy (3). However, the diagnostic criteria of septic encephalopathy have not been well established until now due to the clinical use of sedatives and the existence of a potential neurological disorder. Therefore, it is of great significance to further investigate the pathogenesis of SAE and recognize early the development of SAE.
With marked advancements in neuroscience, the potential role of the neuroendocrine–immune network in the immune response of sepsis has been gradually revealed. It is documented that circulating inflammatory mediators and hormones cause dysfunction of the central nervous system (CNS) in sepsis by inducing the activation of glia and the death of neural cells. In turn, the CNS reacts to the peripheral immune system through the neuroendocrine–immune network, which forms a feedback regulatory loop in the central–peripheral immune system. Then, the CNS triggers a neural reflex and regulates neurotransmitters, neurohormones, and cytokines produced by synapses. Meanwhile, the nervous system extends the nerve fibers to the peripheral organs, which may regulate the secretion function of internal or external secretory glands and maintain the homeostasis of the environment. The central–peripheral immune system regulates the inflammatory response mainly by the hypothalamic–pituitary–adrenal (HPA) axis and the cholinergic anti-inflammatory pathway (CAP), which cooperate to maintain a moderate immune state according to the needs of the body (4). For example, α7 nicotinic acetylcholine receptors (α7 nAChRs), the important component of the CAP, play key roles in central–peripheral immune regulation in the setting of sepsis. The activation of α7 nAChRs inhibits the central and peripheral inflammatory responses and prevents immunosuppression by affecting the differentiation of microglia/macrophages and decreasing the production of inflammatory cytokines. These processes are involved in several signaling pathways, including Janus kinase (JAK)2/signal transducer and activator of transcription (STAT)3 signaling and toll-like receptor 4 (TLR4)/nuclear factor kappa-B (NF-κB) signaling (5–8). However, the disruption of the neuroendocrine–immune network results in an aberrant immune response and aggravates the dysfunction of sepsis. For example, Boomer et al. (9) reported that SAE caused aggressive immunosuppression through the HPA axis, which might induce multiple organ failure and give rise to a vicious circle of immune dysfunction. Souza pointed out that the downregulation of α7 nAChRs in the hypothalamus induced by lipopolysaccharide (LPS) aggravated the neuroinflammatory and metabolic disorders in the brain (10). As an important regulator of the neuroendocrine–immune network, the CNS might be a useful target for the management of septic complications.
Here, we summarize the main physiological alterations of the brain in SAE and discuss the underlying mechanisms that might accelerate the sepsis-induced brain damage, especially focusing on the function of the neuroendocrine–immune network in the pathogenesis of SAE.
SAE is regarded as a cognitive dysfunction induced by sepsis without obvious nervous system infection or structural injury. Due to the lack of a consensus definition of the SAE, the incidence of SAE varied widely among the studies. It has been reported that 8% to more than 70% of patients in septic shock in the ICU may have encephalopathy, only 19% of the patients in the ICU have a normal electroencephalogram (EEG) with alpha rhythm, and 80% of them show epileptic or abnormal EEG (11). The symptoms of septic encephalopathy include memory decline, attention loss, orientation, irritability, and trance. To be specific, SAE may cause long-term neurological deficits, with 10–20% of patients in septic shock showing cognitive deficits, 10–30% of them showing anxiety and stress disorders, and 31–70% of patients having chronic pain. In addition, SAE may result in epileptic seizures, delirium, mild or deep unconsciousness, and even coma (11–13).
The severity of SAE ranges from temporary to permanent brain dysfunction. According to the recent observations of the symptoms, SAE can be divided into acute, sub-acute, or chronic types. The acute SAE can be controlled with patient improvement (14). It is considered a sub-acute or chronic type when the symptoms persist for months or years, and the psychological and cognitive deficits induced by SAE greatly affect the life quality of patients for a long time. Previous studies indicated that mild neurological symptoms in 20–40% of patients with SAE, such as memory changes, depression, anxiety, or cognitive disorders, might persist for 1 year (15, 16). Even worse, the long-term neurological abnormalities not only increase sepsis-induced fatal outcomes but also induce suicidal behaviors within 2 years of recovery (17, 18). Moreover, SAE has an impact on the link between the brain parenchymal and peripheral circulations, which may decrease the blood vessel and capillary densities and cause microcirculation abnormalities (19).
It is believed that the pathophysiological factors of cerebral dysfunction secondary to sepsis mainly include neuroinflammation, blood–brain barrier (BBB) impairment, disorders of brain perfusion, alterations in neurotransmitters, and changes in neuroanatomy and cell death (20, 21). Among them, four types of factors are especially involved with brain dysfunction: the aggressive inflammation of the brain, ischemic injury, the changes in neuroanatomy, and the death of neural cells (Figures 1A,B).
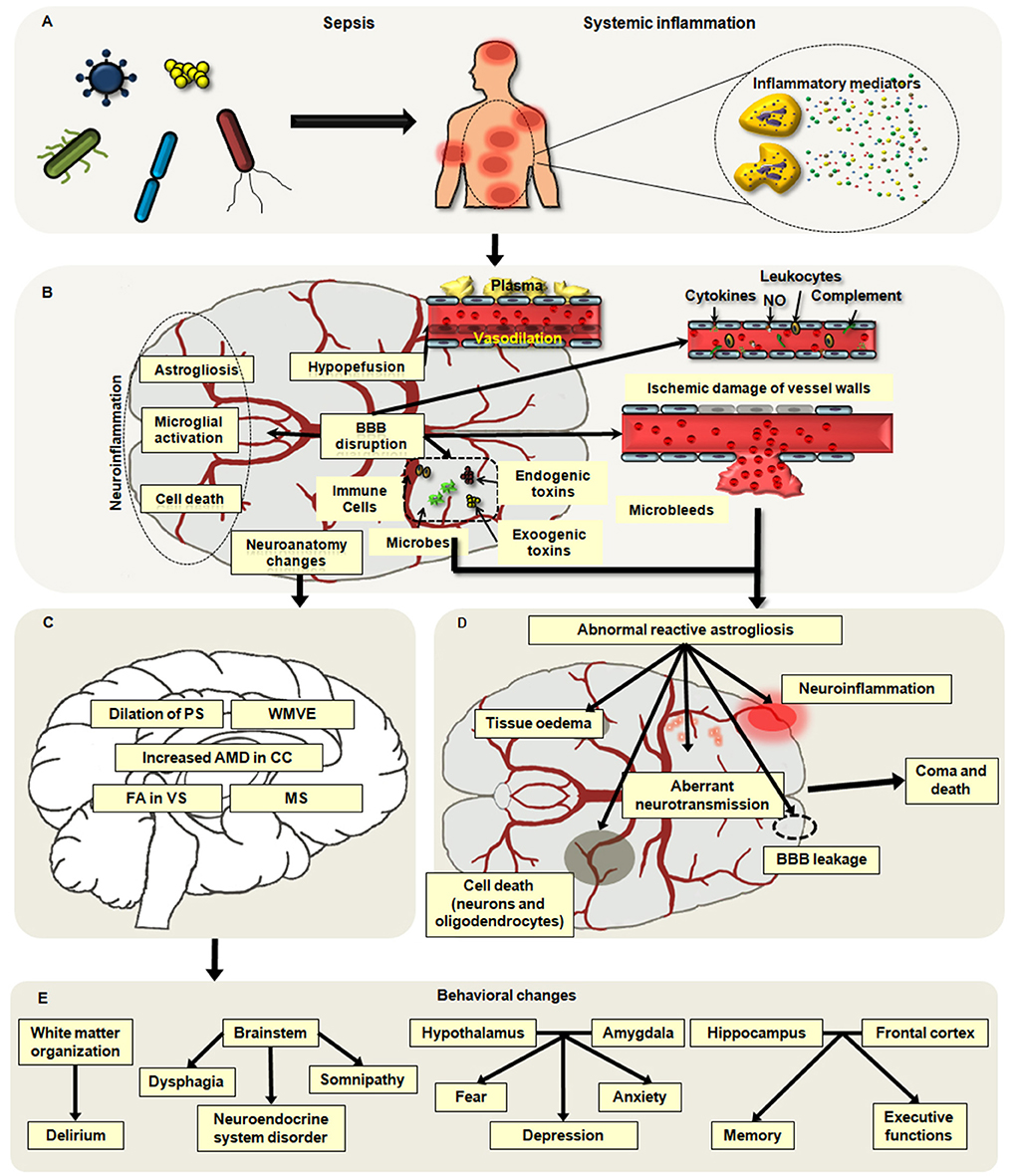
Figure 1. Main pathophysiological alterations in sepsis-associated encephalopathy (SAE). (A,B) Neuroinflammation, hypoperfusion, neuroanatomy changes, and neuronal death are major causes of SAE. Neuroinflammation mainly includes the infiltration of neutrophils and the activation of microglia and astrocytes, which may aggravate brain excitotoxicity and blood–brain barrier (BBB) disruption. Hypoperfusion is frequently observed in patients with SAE, and it can be induced by abnormal vasodilation, impaired autoregulation of the cerebral blood flow (CBF), and inflammatory insults. Apoptosis and pyroptosis are common ways of neuronal cell death associated with brain dysfunction in sepsis. (C) The changes in neuroanatomy including the white matter vasogenic edema (WMVE), myelin separation (MS), dilation of the perivascular spaces (PC), increased axial water diffusion (AWD) in the corpus callosum (CC), and fractional anisotropy (FA) in the ventral striatum (VS) are noticed in patients with SAE. (D) The abnormal astroglial reactivity is regarded as a key contributor to the development of SEA. In the setting of sepsis, the BBB is disrupted by severe systemic inflammation, which may result in ischemic damage of vessel walls, microabscess formation, and infiltration of microbes, immune cells, and toxins into the brain parenchyma. These alterations can cause the abnormal reactivation of astrocytes, which will deteriorate brain injuries including BBB breakdown, neuroinflammation, tissue edema, cell death, and aberrant neuro-transmission. (E) Disorder in various regions of the brain may lead to different behavior alterations secondary to septic challenges.
Notably, the disorders of immune responses in the CNS are the major cause of SAE, which play key roles in the dysregulated peripheral immunity and multiple organ dysfunction syndromes (MODS) following a septic challenge. Ischemic injury, the change in the microstructure in the brain, and the neuron death contribute to the brain damage induced by the uncontrolled inflammation and the difficulty of SAE treatment.
Uncontrolled neuroinflammation is a significant indicator of septic encephalopathy. During the process of sepsis, peripheral and local inflammation can induce the migration of neutrophils into the brain and activate the astrocytes and microglia (22–25). Then, the activated immune cells in the CNS may release the excessive proinflammatory mediators, alter oxidative and nitrosative stress, and increase neurotransmitters, which aggravate the central and peripheral inflammatory responses.
The progress of leukocyte recruitment includes trapping, slow rolling, adhesion to endovascular crawling, and migrating. As the hallmark of inflammation, the infiltration of neutrophils is found in the CNS at the early stage of sepsis, while few of them are observed in the normal brain. The neutrophils are induced to damage tissue sites by a variety of inflammatory mediators, including interleukins (ILs), tumor necrosis factor (TNFs), angiotensin, and the complement cascade factors (26–29). In this process, neutrophils migrate and adhere to endothelial cells by selectins and integrins. Then, they activate the cell adhesion molecules on vascular endothelial cells, crawl on the vascular endothelium, and search for interendothelial junctions to migrate through the endothelial barrier. Thereafter, the neutrophils pass through the glia limitans and interact with glial cells at the parenchyma. The accumulation of neutrophils in the CNS may damage the cerebrovascular function and the neural cells by releasing oxygen-free radicals and increasing the volume of erythrocytes (30, 31).
Norman et al. (32) reported that neutrophils were significantly recruited by the brain during the acute inflammatory phase of sepsis, and the transmigration of neutrophils across the endothelial barrier was the main cause of vascular barrier breakdown. Recent studies documented that SB225002 (the antagonist of CXCR2) or kynurenic acid could protect the brain against neutrophil activation and BBB permeability changes in septic animals (33, 34). However, neutrophil recruitment was not involved in cognitive impairment (35). The activated microglia and astroglial cells may be responsible for the dysfunctions of the synaptic and intracerebral communication networks.
Microglia accounts for 6%-18% of human brain cells (36). As the resident immune cells in the brain, the microglia is the first cell to respond to neuroinflammation. They regulate the functions and inflammation of the brain by secreting cytokines and interacting with other neural cells.
In normal conditions, the microglia with branched morphology is resting, which is responsible for micro-environment surveillance. After activation by pathological insults, microglia reveals quick and prominent alterations in morphology, metabolism, and function. A series of studies suggested that microglia was consistently activated in animal models and patients of sepsis (11, 37). For example, the increasing microglia with amoeboid shape was observed in the hippocampus and cortex of the patients who died of septic shock (38, 39).
It has been documented that peripheral inflammation activates microglia with the help of the inflammatory mediators, the interaction between microglia and adjacent cells, and neurotransmitters (40). In the development of sepsis, cytokines produced by peripheral immune cells such as TNF-α and IL-1β transmit signals through the damaged BBB and activate microglia. For example, Ye et al. reported that during sepsis, IL-17A secreted by peripheral immune cells could activate microglia, and then the inflammatory mediators produced by activated microglia enhanced the release of IL-17A from immune cells, which created a vicious cycle to amplify the brain inflammation (41). Adjacent cells including leptomeningeal cells or vascular endothelial cells also augmented the activation of microglia, and the activated endothelial cell stimulated and guided microglia into the inflammatory brain during sepsis by the upregulation of CX3CL1 (42). Moreover, the neurotransmitters such as acetylcholine (Ach), nicotine, and glutamate regulated LPS-induced microglial activation (43).
A series of studies have indicated that the activation of microglia in sepsis results in brain dysfunctions including delirium, memory impairment, acute brain oxidative damage, and long-term cognitive impairment by secreting a large quantity of proinflammatory cytokines, upregulating the expression of multiple enzymes and neurotransmitters, and changing the brain energy metabolism in sepsis (41, 43, 44). For example, a mild level of glutamate is produced by neural cells to maintain the homeostatic state of the brain, while activated microglia may release toxic amounts of glutamate and induce long-term cognitive dysfunction after sepsis (45). Furthermore, microglia can significantly promote glucose uptake and glycolysis by expressing glucose transporters 1 (GLUT), increasing the production of IL-1β, and upregulating oxidative/nitrosative enzymes in inflammation (46).
The growing evidence has proved that the difference in the microenvironment may lead to the heterogeneity of microglia. There are two kinds of activated phenotypes of microglia named M1 and M2 subpopulations, respectively. The microglia with different phenotypes show either adverse or advantageous effects on neurological function (47). It is well known that M2 microglia can protect the neural cells from inflammatory insults, while the activated M1 microglia can aggravate neuroinflammation and oxidative stress by increasing inflammatory mediators and decreasing mitochondrial oxygen consumption and ATP production in sepsis. Thus, the increase in M2 microglia or the inhibition of M1 microglia may alleviate inflammation and brain dysfunction following septic challenge. Yan et al. (47) found that the induction of microglia from the M1 to the M2 phenotype by IL-4/IL-13 stimulation inhibited the release of inflammatory cytokines and alleviated the brain dysfunction in SAE by increasing the mitochondrial content. Thus, it could be a promising therapeutic method to improve the long-term cognitive impairment induced by sepsis by regulating the polarization of microglia and inhibiting the secretion of proinflammatory mediators.
Astrocytes are of great importance to maintain the homeostasis of the brain, which is associated with BBB permeability, neurotransmitter metabolism, synapse connectivity and plasticity, and brain fluid balance (48). Under physiological conditions, the morphology and function of astrocytes are region-dependent and heterogeneous. Astrocytes are essential components of the BBB and the glia-neurovascular barrier in the gray matter protoplasm, and they regulate the neurotransmitter catabolism by specific transporters and provide neurotransmitter precursors for neurons. Importantly, astrocytes are responsible for the formation of the endocranial secretory system, which releases neuromodulators, trophic factors, and hormones.
The pathological changes of astrocytes mainly include astrogliosis and astrodegeneration. It has been demonstrated that reactive astrogliosis is caused by brain injury, and the main alterations of activated astroglial cells are hypertrophic and upregulated expression of the glial fibrillary acidic protein (GFAP) and vimentin (48–50). The degree of astrogliosis relies on the severity of the damage, which is regulated by cellular signaling pathways such as the JAK/STAT and NF-κB pathways (51). The proliferation of astrocytes in the cerebral cortex is one of the common histopathologic changes in the brain secondary to septic insults. Several studies confirmed that reactive astrogliosis was observed in the brain tissues during neuroinflammation induced by LPS exposure, which was associated with synaptic deficits and depressive-like behaviors in mice (25, 52). The activation of astrocytes enhanced the release of inflammatory mediators and aggravated CNS inflammation, and it was regulated by expressions of p21, NF-κB, and inducible nitric oxide synthase (iNOS) in astrocytes from septic animals. Likely, astrocytes might take part in the formation of the brain microabscesses and the infiltration of inflammatory cells in SAE and CNS injury (25, 53). Astrocytes induced the migration of leukocytes into the damaged sites of the brain by upregulating the leukocyte adhesion molecules, destroying the integrity of tight junction (TJ), and promoting the formation of transendothelial cell channels (53, 54). Moreover, astrocytes are responsible for the collapse of the BBB. In the setting of sepsis, cytokines released by astrocytes could inhibit the expression of TJ proteins in endothelial cells and thus aggravate BBB breakdown (55). In LPS-induced animal models, the morphology, transcriptional profile, and phenotype of astrocytes were altered, including early reactivity, astrocytic end-feet remodeling, and even astrocytic loss, thereby resulting in the disruption of BBB (56–58). Being a crucial part of the central immune system, the abnormality of astrocytes accelerates the development of neuroinflammation and aggravates brain dysfunction (Figure 1D). Astrocytes determine the severity and level of brain damage, and astroglial reactivity is regarded as a defining factor in the pathophysiology of SAE (25). Nevertheless, the process of SAE in humans is more complicated than that in animal models, which is associated with age and pathological background. So, deeper exploration should be carried out to clarify the function of the astrocytic network in SAE.
Many studies have demonstrated that ischemic injury often occurs in the brain in the setting of sepsis (59–61). The clinical data have been shown that cerebral blood flow (CBF) is significantly reduced in patients with SAE (59). For instance, Sharshar et al. (60) analyzed the brain tissues of the patients dying from septic shock and found the incidence of cerebral ischemia in septic shock to be 100%. Szilárd et al. (62) reported that the CBF rate was slower in patients with septic encephalopathy than in normal controls by transcranial Doppler ultrasound. Increasing evidence proved that the intravenous injection of bacterial LPS to human volunteers markedly resulted in the reduced global CBF and the damaged cerebral autoregulation of the middle cerebral artery (MCA) blood velocity, in turn contributing to the sepsis-associated delirium and edema (61, 63, 64).
Strikingly, the results from the experimental models of sepsis are controversial. Several studies revealed the decreased blood perfusion distribution in animals after systemic administration of LPS, while others reported the increased CBF in cortical MCA territories after LPS challenge (61, 65). It is speculated that these alterations in CBF during sepsis are due to the impaired autoregulation of CBF, and the discrepancy in different studies might be associated with the model-building methods and the dose of drugs.
Due to the reversibility of SAE, most studies on histopathology of SAE did not observe the obvious changes in deeper structures and the spinal cord of the brain (66). Nevertheless, emerging evidence has documented that the disorder of cerebral autoregulation may contribute to edema and white matter change in sepsis (67, 68). Alterations in neuroanatomy have been noted in the patients with SAE including the white matter vasogenic edema, myelin separation, dilation of the perivascular spaces, incremental axial water diffusion in the corpus callosum, and fractional anisotropy in the ventral striatum (39, 68, 69). The dysfunction in various regions of the brain may cause different behavior disorders. The impairment of white matter organization causes delirium (70). The malfunction of the brainstem can lead to dysphagia, neuroendocrine system dysregulation, and somnipathy, which appear to be associated with multiple organ dysfunction and higher mortality in patients (71–73). The disorder of the hypothalamus or the amygdala is related to behavioral changes including depression, fear, and anxiety (74, 75). As is known, the hippocampus and the frontal cortex play vital roles in memory formation; thus, the disruption of the hippocampus and the frontal cortex causes the deterioration of memory and executive functions (76–78) (Figures 1C,E). Until now, the sepsis-induced alterations in the brain microstructure are largely unknown, but it is predictable that these changes may contribute to the development of brain dysfunction and long-term cognitive impairment. Therefore, further understanding of the neuroanatomical approach is of significance to explore the clinical features and psychocognitive disorders of SAE.
Increasing studies have indicated that cell death is an important contributor to brain dysfunction during SAE. The apoptosis and pyroptosis of brain cells induced by inflammation are critically involved in the onset and progression of SAE.
Apoptosis is a sort of regulated cell death accompanied by contraction of the cells and foam cell formation in the cell membrane. It is well accepted that mitochondria are major apoptosis-associated organelles. In the development of sepsis, mitochondrial dysfunction is found in various regions of the brain, especially in the hippocampus, which may cause the imbalance of oxygen/nitrogen reactive species, hippocampus cell apoptosis, and severe neurocognitive impairment. Elevated neuronal apoptosis is characterized by enhanced pro-apoptotic proteins, which are released from mitochondrial cleaved caspase levels. Omi/HtrA2, a serine protease in mitochondria, regulates apoptosis of hippocampus cells by translocation from mitochondria into the cytoplasm in cecal ligation and puncture (CLP)-induced sepsis (79). More recently, the mitochondrial isomerase cyclophilin D (CypD) was found to be associated with mitochondrial dysfunction and cell apoptosis in SAE. The knockout of CypD could protect the brain cells from apoptosis by decreasing the opening mitochondrial permeability transition pore and inhibiting the free radicals from mitochondria, which were able to alleviate the brain damage and improve the survival of CLP-induced SAE (80). Similarly, the inhibition of ceramide significantly attenuated neuronal apoptosis and cognitive impairment of septic animals (81).
There is an obvious link between the apoptosis of neuronal cells and the abnormality of proinflammatory signaling pathways, including NF-κB, mitogen-activated protein kinase (MAPK), and brain-derived neurotrophic factor (BDNF)/tyrosine kinase (Trk)B during SAE. The activation of NF-κB is regarded as a central event in sepsis, which may trigger the inflammatory mediator networks. Some studies reported that the activation of NF-κB was related to increases in the mortality rate and poor clinical outcomes in sepsis. Accordingly, the inhibitors of NF-κB could attenuate LPS-induced long-term potentiation in the dentate gyrus and ischemia-induced neuronal apoptosis (82). Furthermore, p65, a subunit of the NF-κB heterodimer, can induce the transcription of microglia Nod-like receptor protein 3 (NLRP3) by directly binding to the promoter region of NLRP3 in SAE. As an important component of the NLRP3 inflammasome, the activation of NLRP3 induces cell apoptosis by regulating the expression of BCL and Bax. There is accumulating evidence demonstrating that downregulated activation of the NF-κB/p65-induced NLRP3 inflammasome inhibits the neuron apoptosis induced by LPS in vivo and in vitro (83–85).
MAPK plays a key role in inflammation-induced apoptosis. p38 MAPK is activated in the brain accompanied by the upregulated phosphorylation of MAPKAPK2 upon sepsis. Cell apoptosis in the cortex and the hippocampus of CLP animals could be alleviated by p38 MAPK inhibitors (86). Zhou et al. (87) found that the neuronal apoptosis and autophagy were highly related to the p38 MAPK signaling pathway in the CLP-induced sepsis model, while immunity-related GTPase M1 (IRGM1) partially ameliorated neuronal apoptosis by the p38 MAPK signaling pathway. BDNF is regarded as an important component to retain activity-dependent synaptic plasticity. It has been documented that the inactivation of BDNF/TrkB signaling following a brain injury is involved in apoptosis and autophagy of neuronal cells. For instance, the activation of BDNF/TrkB alleviated brain damage induced by SAE, while the abnormality of BDNF/TrkB signaling was associated with brain dysfunction in aging mice (88, 89). Likely, treatment with emodin, a chemical compound from rhubarb, obviously decreased the apoptosis of hippocampal neurons in CLP mice through the activation of the BDNF/TrkB pathway (90).
Pyroptosis is defined as regulated necrosis with rapid damage to the plasma membrane and the release of intracellular contents (85), and it is a common pattern of cell death in sepsis, which can aggravate inflammation. Many studies have confirmed the pyroptosis of peripheral immune cells in patients in septic shock (91, 92). Recently, we observed pyroptosis of splenic dendritic cells (DCs) in CLP-induced septic mice (93). Similarly, Xu et al. noticed the pyroptosis of neural cells in septic mice and found that the inhibition of pyroptosis could alleviate brain damage and cognitive impairment in SAE. They speculated that pyroptosis of neural cells was induced by inflammatory mediators, which were secreted by the peripheral blood into the CNS during SAE (94). The pyroptosis can be mediated through the canonical or non-canonical signaling pathway. In the non-canonical signaling pathway, LPS can directly activate caspase-11,−4, and−5 to cleave the critical pyroptosis executioner, gasdermin D (GSDMD), and form N-terminal GSDMD (GSDMD-NT). Then, GSDMD-NT becomes a ring-like structure and forms holes in the cell membrane, which causes the production of IL-1β and IL-18, and leads to cell lysis. The canonical signaling pathway plays a potential role in the pathogenesis of SAE, and the inhibition of pyroptosis may be a useful therapeutic target for SAE (95, 96).
The canonical signaling pathway relies on the inflammasome pre-activation. Then, the inflammasome can activate caspase-1 by the adaptor apoptosis speck-like protein, which cleaves GSDMD to form GSDMD-NT (97, 98). As the pivotal canonical inflammasome, the NLRP3 inflammasome is highly associated with brain damage during SAE. With this regard, expressions of NLRP3, GSDMD-NT, and cleaved caspase-1 were augmented in the brain of septic mice, and the upregulation of NLRP3, caspase-1, and gasdermin-D in the hippocampus resulted in the cognitive deficits in the mice model of SAE by inducing pyroptosis (99). Recently, we found that sestrin 2 improved the outcome of sepsis by decreasing the NLRP3/caspase-1-dependent pyroptosis, and it was closely related to the protein kinase RNA (PER)-like ER kinase (PERK)-activating transcription factor (ATF)4-C/EBP homologous protein (CHOP) pathway (95). Moreover, Lei et al. (100) explored the nexus between autophagy and pyroptosis in mice with SAE, and the results showed that both autophagy and pyroptosis were involved in the development of SAE in mice, and pannexin-1 decreased the occurrence of pyroptosis by autophagy. Collectively, it was suggested that both the canonical and non-canonical pathways of cell pyroptosis are associated with cell apoptosis induced by LPS (95). However, the exact functions of the canonical and non-canonical pyroptosis together with the networks among pyroptosis, apoptosis, and autophagy in SAE are required in further studies.
Reciprocal interactions between the central and peripheral immune systems are regarded as crucial parts of the host's response to septic insults. When systemic inflammatory responses are induced by severe trauma or infection, the peripheral inflammatory signal is sent to the CNS. The CNS regulates immune system compensatory changes and maintains homeostasis after perception and integration of peripheral signals through sympathetic, parasympathetic, and HPA gland axial. The aberrant responses of neuroendocrine–immune networks are responsible for the immune disruption and the worse outcome during sepsis. It is well known that the neural central–endocrine–immune networks are mainly composed of HPA and CAP.
Basically, the HPA axis is a classical anti-inflammatory pathway of the nervous system. Upon septic exposure, the CNS regulates the peripheral inflammatory immune response mainly through the HPA axis. Endotoxin or inflammatory mediators activate the HPA axis through the vagus nerve or humoral pathway, and the hypothalamus releases corticotropin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH), which will promote the production of glucocorticoid (GC) released by adrenal cortical cells and avoid damage and dysfunction of organs induced by excessive inflammation (101–103). The regulation of the endocrine system based on the HPA axis is relatively slower and longer (Figure 2).
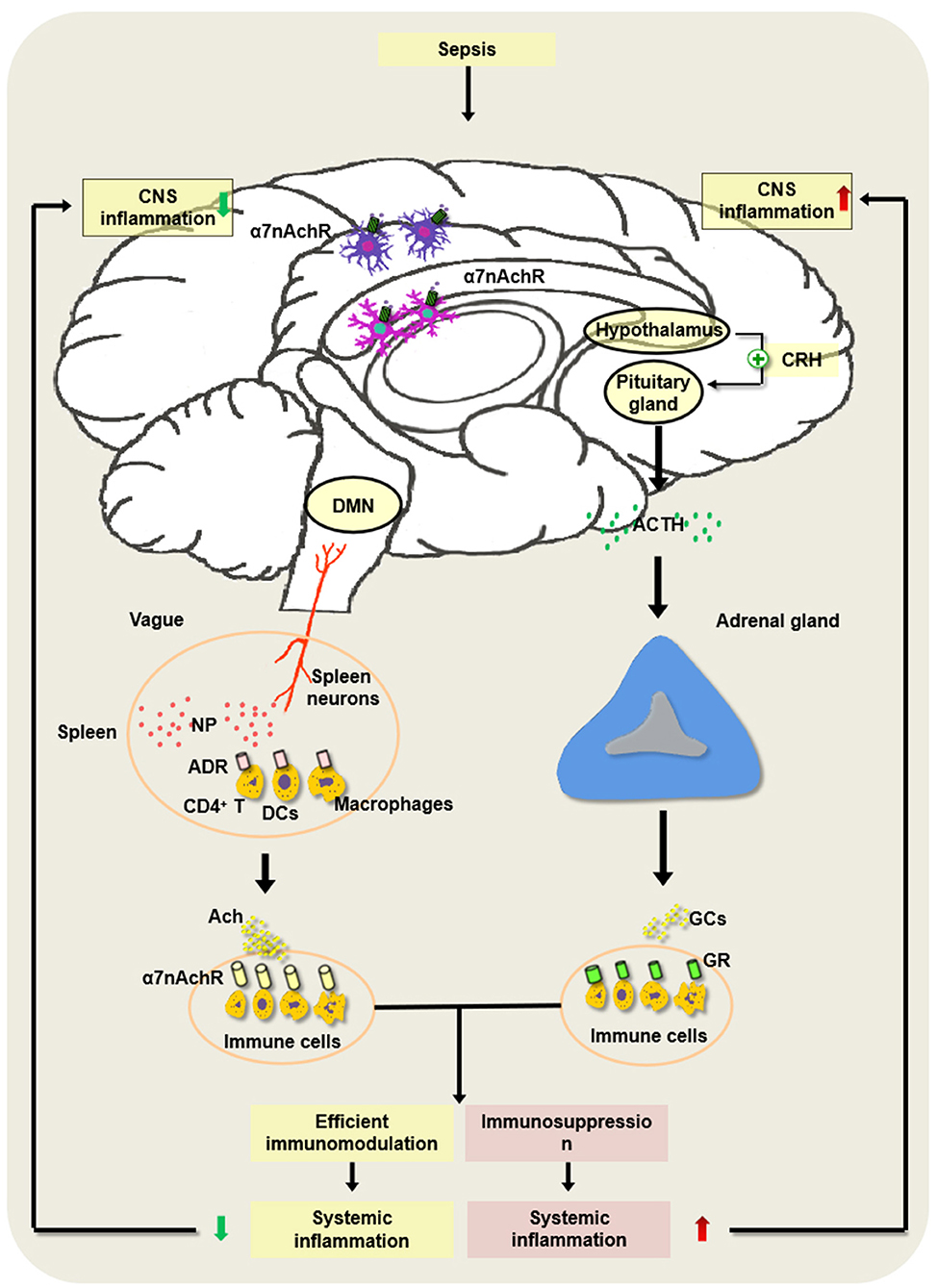
Figure 2. Regulatory mechanism underlying hypothalamic–pituitary–adrenal (HPA) axis and cholinergic anti-inflammatory pathway (CAP) in the development of sepsis. In the pathogenesis of sepsis, the HPA axis is activated via the vagus nerve or the humoral pathway, and the hypothalamus and pituitary gland release corticotropin-releasing hormone (CRH) as well as adrenocorticotropic hormone (ACTH). Then, it can augment the production of glucocorticoids (GCs) released by adrenal cortical cells and inhibit excessive inflammation, which may alleviate the sepsis-induced CNS inflammation. However, severe sepsis can result in damage to the HPA axis and the abnormal induction of GC, thereby contributing to immunosuppression and intractable inflammation of the CNS. Of note, the constant brain damage aggravates the abnormality of the HPA axis and forms a vicious cycle under septic exposure. In addition, the activated splenic nerve can release norepinephrine (NP), which binds to the adrenaline receptor (ADR) on immune cells such as CD4+ T cells, dendritic cells (DCs), and macrophages to produce ACh. ACh binds to α7 nAChR on the inflammatory cells and suppresses the inflammatory response in the spleen. Nevertheless, the persistent activation of the vagus nerve may lead to the development of host immune suppression following septic insults.
The damage to the HPA axis and the abnormal production of adrenocortical GC appear to be associated with the occurrence and development of sepsis. First, the change in the adrenal microenvironment is an important factor affecting the prognosis of sepsis. An integrated adrenal stress response is critical for the survival of patients with septic shock. Under physiological states, the cortical cells, medulla cells, and epithelial cells in the adrenal gland interact with inflammatory immune cells to form a complex adrenal microenvironment, which plays a key regulatory role in the synthesis and secretion of corticosteroids in sepsis and septic shock (104, 105). A variety of immune cells, including lymphocytes, neutrophils, and macrophages, are associated with adrenal insufficiency and the dysfunction of the HPA axis in the setting of sepsis (106). Under the stimulation of LPS and pathogenic microorganisms, a large number of neutrophils are recruited into the infected sites in adrenal glands by adhesion molecules and chemokines, which may alter the subset of adrenal tissues, leading to dysfunction of the HPA axis and abnormal secretion of adrenal hormones. A study by Carla et al. (107) observed a significant increase in leukocyte infiltration in adrenal glands during sepsis, which was associated with the high mortality of septic animals.
The surface receptors on the adrenal cortical cells and immune cells, such as scavenger receptors, TLR2, and TLR4, are related to the dysfunction of the adrenal glands secondary to sepsis. Kanczkowski et al. found systemic but not adrenocortical-specific knockout of myeloid differentiation protein 88 (MyD88), a TLR adaptor protein, reduced adrenal inflammatory response and HPA axis activation in LPS-induced sepsis (108). A recent study revealed that type B type I scavenger receptors were highly expressed on adrenal cortical cells, which promoted cortisol synthesis by capturing esterified cholesterol; loss of the type I scavenger receptor gene led to adrenal cortical dysfunction, thereby increasing sepsis-induced mortality in mice (109).
GCs produced by the HPA axis are of great value to maintain homeostasis and protect the host from the life threat caused by sepsis (110). The dissonance of the adrenal gland might increase the mortality of patients in septic shock and septic animals because of the disorder in GC hormone metabolism (110, 111). The activated HPA axis enhances the release of GCs from the adrenal gland, which binds to the cytosolic GC receptor (GR) on the cell membrane and participates in many cellular and physiological contexts including interferon (IFN)-γ production, inflammation, and LPS-induced mortality by regulating a series of gene networks. Severe sepsis per se can induce the disorder of the corticosteroid system, which results in intractable immunosuppression together with excessive inflammation.
GR is distributed in various kinds of immune cells, e.g., DCs, myeloid cells, macrophages, T cells, and natural killer cells (NKs). It protected the mice from shock induced by LPS and CLP through the upregulation of anti-inflammatory genes, such as glucocorticoid-induced leucine zipper (GILZ), MAPK phosphatase-1 (MKP-1), and programmed death 1 (PD-1), or the inhibition of the proinflammatory genes, including STAT1, IL-1β, and IL-12, in a dimer-dependent way (111–118). Disorder of the HPA axis by modulation of GR might alter the sensitivity of mice to septic challenge. The abnormality of the GR signaling induced by sepsis deteriorated the pathophysiology by decreasing the lactate clearance and increasing the sensitivity of the mice to lactate-mediated toxicity, while the upregulation of GR inhibited the inflammatory response and improved the survival following endotoxic shock (119, 120). Additionally, GR deletion in T cells was associated with the immunosuppressive state in mice subjected to CLP (121).
Recently, several studies have indicated that GC/GR signaling pathways contribute to promoting the capacity of monocytes/macrophages to kill the various particles and bacteria (122, 123). Injection of low-dose GCs or the upregulation of the GILZ markedly protected animals against CLP-induced sepsis by augmenting bacterial clearance (124–126). Of note, GCs help to clean out apoptotic cells and cell debris, which is crucial to tissue repair. It has been shown that the CNS (cerebral cortex, basal forebrain, midbrain, and brainstem) is critically associated with the regulation of immunity and inflammation; moreover, the hypothalamus and the limbic system play central roles in the regulation of neuroendocrine and autonomic nervous systems (127). The CNS can directly innervate the immune organs through the HPA axis and regulate the peripheral immune system. Likely, the information with regard to the peripheral immune–inflammatory response can be transmitted into the CNS through the nervous and humoral pathways to affect neuronal activity (128, 129). The persistent excitation of the HPA axis has negative impacts on the body, and the insufficient HPA axis function also causes adverse effects. Moderate excitation and timely termination of the neuroendocrine system have an important influence on the course of sepsis. However, the precise role and underlying mechanism of the HPA axis in the central–peripheral immune nets are not well understood and need to be further explored.
In 2003, Tracey et al. (130) first found that the excitation of the vagus nerve protected the brain from endotoxemia and ischemia–reperfusion injury by inhibiting the release of both early and late cytokines such as TNF-α and high-mobility group box-1 protein (HMGB1). This neural pathway is named the CAP. The cholinergic neural pathways exist between the central and peripheral immune systems, which consist of the vagus, the ACh receptor, the spleen, and the splenic nerves (102, 130).
The vagal parasympathetic fibers start from the vagus dorsal motor nucleus (DMN) and stop at the parasympathetic ganglia of the vagal plexus. The emitted postganglionic fibers are distributed in the thoracic and abdominal organs that control the activity of smooth muscles, cardiac muscles, and glands. Strikingly, the vagus nerve can regulate the generation of proinflammatory cytokines. When the vagal nerves sense the stimulation of inflammatory insults, they transmit inflammatory signals to the center neural system. At the same time, the dorsal nucleus of the vagal nerve delivers anti-inflammatory signals to the endothelial reticulate system by ganglion fibers to enhance the synthesis and release of ACh and inhibit the synthesis of proinflammatory cytokines (131–135).
CAP exerts anti-inflammatory effects dependent on the spleen and the splenic nerves. The spleen is an important secondary lymphoid organ. The splenic nerves originate from supraceliac mesenteric neurons and are distributed to the spleen. It was reported that the activation of the vagus nerve or the splenic nerve significantly inhibited the production of TNF-α released by the red pulp and the marginal zone (132, 133, 136). However, the mechanism by which stimulating the vagus or the splenic nerve produces ACh in the spleen is controversial. A previous study speculated that ACh was released from vagal neurons, and it interacted with α7 nAChR on immune cells and ameliorated the inflammatory lesions (137). However, a series of studies implicate that, as an adrenergic nerve, the splenic nerve does not release ACh directly. The stimulated splenic nerve produces norepinephrine (NP), which combines with the adrenaline receptor (ADR) on the surface of CD4+ChAT+ T cells to initiate the synthesis of ACh, and ACh acts on the inflammatory cells expressing α7 nAChR in the spleen and suppresses the inflammatory response in the spleen (101). The stimulation of the vagus nerve did not reduce proinflammatory mediators in the plasma of sepsis in nude mice lacking T cells. In addition, β-adrenergic receptors on T cells are the important components of splenic nerve signaling (138, 139). Recent studies showed that DCs and macrophages were positively expressed choline acetyltransferase (ChAT), and their activation promoted the ACh synthesis (137) (Figure 2).
The cholinergic pathway regulates systemic inflammation more rapidly and significantly than the HPA axis. The most prominent feature of the CAP is mediated by α7 nAChR. In the CNS, α7 nAChR is located on the surface of various nerve cells including astrocytes, microglia cells, interneurons, and immature granule cells, which are distributed in the basal ganglia, the hippocampus, and the brain gray matter (140–142). It is believed that α7 nAChR can be involved in many psychiatric and neurological disorders including neuroinflammation, Parkinson's disease, and Alzheimer's disease by regulating neurotransmitter release, synaptic plasticity, and signal transduction (143–145). The functions of α7nAChR in the nervous system are determined by location, time, and context. α7 nAChR in hippocampal interneurons is associated with hippocampus-dependent memory, and its activation on excitatory synapses appears to be involved in synaptic activities. However, α7 nAChR on microglia and astrocytes is responsible for the development of neuroinflammation. After being activated by ACh and choline, α7 nAChR can inhibit the expression of proinflammatory mediators by downregulating the phosphorylation of JAK2/STAT3 and reducing nuclear translocation of NF-κB. Li et al. (146) found that ACh protected neurons from inflammatory and apoptotic effects of activated microglia by α7 nAChR. Similarly, it was reported that α7 nAChR activation on astrocytes had a neuroprotective impact via inhibiting IL-6 as well as TNF-α secretion and decreasing apoptosis and toxicity of neurons upon LPS stimulation (147–149).
In the periphery immune system, the α7 nicotinic receptor is almost expressed in all kinds of immune cells. The activation of α7 nAChR is evident to suppress the induction of proinflammatory cytokines secondary to the activation of TLRs including TLR2, TLR3, and TLR9 (150). Similarly, the activation of α7 nAChR on macrophages significantly alleviates the LPS-mediated inflammatory processes by downregulating NF-κB-mediated transcription. Moreover, α7 nAChRs on T cells and antigen-presenting cells (APCs) regulate the differentiation of CD4+ T cells. Mashimo et al. (151) reported that activated α7 nAChRs on T cells promoted T-cell differentiation, while the activation of α7nAChRs on APCs suppressed T-cell differentiation by decreasing antigen processing.
With the development of the genome-wide association study, a series of studies confirmed that the CHRNA7 genes encoded for the α7 nAChRs in the human brain and the variations within CHRNA7 were associated with the abnormal response of CAP (140, 152). In addition, chaperone proteins such as resistance to inhibitors of cholinesterase-3 (RIC3) Ly6h and NACHO could greatly affect the expression and function of nAChRs (143, 153, 154).
Thus, evidence shows that the CAP can abate both systemic and cerebral inflammations resulting from septic challenges. However, immune dysfunction in sepsis appears to be bipolar, accompanied by the excessive inflammatory response or immune suppression at the early or later stage. The constitutive activation of the vagus nerve may lead to immune impairment and infection in septic survivors (155). In the previous studies, we found that HMGB1 in the CNS could induce immune depression of DCs by triggering the hyperactivation of the CAP in CLP-induced septic mice, and the inhibition of cerebral HMGB1 significantly protected the brain against sepsis-induced injury and improved the immune function of splenic T cells (156–158). These data suggest that the nervous system plays an important role in immune surveillance and inflammation control in the bidirectional communication between the brain and the immune system. The efferent signal of the vagal nerve provides a direct pathway to the neural–immune response, and the CNS can use the CNS–peripheral immune axis through the cholinergic neural pathway to modulate the immune response and control the course of sepsis. Nevertheless, the current mechanistic study of the cholinergic neural pathway in the central–peripheral immune regulation of sepsis is still in its infancy.
As the center of neuroendocrine immune networks, the CNS plays a pivotal role in maintaining the balance between inflammatory response and immunosuppression. In the development of sepsis, uncontrolled neuroinflammation may induce an over-activated inflammatory response or inactivation of peripheral immune cells in sepsis via HPA or CAP. At the same time, the peripheral immune system can be fed back to the center and aggravate the progress of SAE, which may form a vicious cycle and cause the disorder of the host immune system. Therefore, it may be a potential target for treating sepsis by alleviating the disharmonious interaction between the central and peripheral immune systems. Many studies from our group and others have revealed that the inhibition of cerebral HMGB1 may attenuate the sepsis-induced brain injury and improve the T-cell-mediated immunity by CPA. However, it remains a new field to explore the management of sepsis by central–peripheral immune mechanisms, and further studies are needed to elucidate the close crosstalk between the CNS and peripheral immune systems following sepsis and septic shock.
Y-xL and YY drafted the manuscript. J-pL, W-jL, and YC drew the illustrations. R-mY revised the manuscript. Y-mY conceived and designed the review. All authors contributed to the article and approved the final manuscript.
The present work was supported by grants from the National Natural Science Foundation of China (82130062 and 82172124) and the Key Medical Innovation Program of the Chinese People's Liberation Army (18CXZ026).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet. (2020) 395:200–11. doi: 10.1016/S0140-6736(19)32989-7
2. Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol. (2012) 8:557–66. doi: 10.1038/nrneurol.2012.183
3. Sonneville R, de Montmollin E, Poujade J, Garrouste-Orgeas M, Souweine B, Darmon M, et al. Potentially modifiable factors contributing to sepsis-associated encephalopathy. Intensive Care Med. (2017) 43:1075–84. doi: 10.1007/s00134-017-4807-z
4. Hu S, Wang Y, Li H. The Regulation effect of alpha7nAChRs and M1AChRs on inflammation and immunity in sepsis. Mediators Inflamm. (2021) 2021:9059601. doi: 10.1155/2021/9059601
5. Siniavin AE, Streltsova MA, Kudryavtsev DS, Shelukhina IV, Utkin YN, Tsetlin VI. Activation of alpha7 nicotinic acetylcholine receptor upregulates HLA-DR and macrophage receptors: potential role in adaptive immunity and in preventing immunosuppression. Biomolecules. (2020) 10:507. doi: 10.3390/biom10040507
6. Yang YH Li DL, Bi XY, Sun L, Yu XJ, Fang HL, et al. Acetylcholine inhibits LPS-induced MMP-9 production and cell migration via the alpha7 nAChR-JAK2/STAT3 pathway in RAW2647 cells. Cell Physiol Biochem. (2015) 36:2025–38. doi: 10.1159/000430170
7. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. (2003) 421:384–8. doi: 10.1038/nature01339
8. Han YG, Qin X, Zhang T, Lei M, Sun FY, Sun JJ, et al. Electroacupuncture prevents cognitive impairment induced by lipopolysaccharide via inhibition of oxidative stress and neuroinflammation. Neurosci Lett. (2018) 683:190–5. doi: 10.1016/j.neulet.2018.06.003
9. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. (2011) 306:2594–605. doi: 10.1001/jama.2011.1829
10. Souza ACP, Souza CM, Amaral CL, Lemes SF, Santucci LF, Milanski M, et al. Short-term high-fat diet consumption reduces hypothalamic expression of the nicotinic acetylcholine receptor alpha7 subunit (alpha7nAChR) and affects the anti-inflammatory response in a mouse model of sepsis. Front Immunol. (2019) 10:565. doi: 10.3389/fimmu.2019.00565
11. Mazeraud A, Righy C, Bouchereau E, Benghanem S, Bozza FA, Sharshar T. Septic-associated encephalopathy: a comprehensive review. Neurotherapeutics. (2020) 17:392–403. doi: 10.1007/s13311-020-00862-1
12. Carpenter KC, Hakenjos JM, Fry CD, Nemzek JA. The influence of pain and analgesia in rodent models of sepsis. Comp Med. (2019) 69:546–54. doi: 10.30802/AALAS-CM-19-000004
13. Helbing DL, Bohm L, Witte OW. Sepsis-associated encephalopathy. CMAJ. (2018) 190:E1083. doi: 10.1503/cmaj.180454
14. Calsavara AJC, Nobre V, Barichello T, Teixeira AL. Post-sepsis cognitive impairment and associated risk factors: a systematic review. Aust Crit Care. (2018) 31:242–53. doi: 10.1016/j.aucc.2017.06.001
15. Barichello T, Sayana P, Giridharan VV, Arumanayagam AS, Narendran B, Della Giustina A, et al. Long-term cognitive outcomes after sepsis: a translational systematic review. Mol Neurobiol. (2019) 56:186–251. doi: 10.1007/s12035-018-1048-2
16. Catarina AV, Branchini G, Bettoni L, De Oliveira JR, Nunes FB. Sepsis-associated encephalopathy: from pathophysiology to progress in experimental studies. Mol Neurobiol. (2021) 58:2770–9. doi: 10.1007/s12035-021-02303-2
17. Bedirli N, Bagriacik EU, Yilmaz G, Ozkose Z, Kavutcu M, Cavunt Bayraktar A, et al. Sevoflurane exerts brain-protective effects against sepsis-associated encephalopathy and memory impairment through caspase 3/9 and Bax/Bcl signaling pathway in a rat model of sepsis. J Int Med Res. (2018) 46:2828–42. doi: 10.1177/0300060518773265
18. Lund-Sorensen H, Benros ME, Madsen T, Sorensen HJ, Eaton WW, Postolache TT, et al. A nationwide Cohort study of the association between hospitalization with infection and risk of death by suicide. JAMA Psychiatry. (2016) 73:912–9. doi: 10.1001/jamapsychiatry.2016.1594
19. Nwafor DC, Brichacek AL, Mohammad AS, Griffith J, Lucke-Wold BP, Benkovic SA, et al. Targeting the blood-brain barrier to prevent sepsis-associated cognitive impairment. J Cent Nerv Syst Dis. (2019) 11:1179573519840652. doi: 10.1177/1179573519840652
20. Ehler J, Barrett LK, Taylor V, Groves M, Scaravilli F, Wittstock M, et al. Translational evidence for two distinct patterns of neuroaxonal injury in sepsis: a longitudinal, prospective translational study. Crit Care. (2017) 21:262. doi: 10.1186/s13054-017-1850-7
21. Westhoff D, Engelen-Lee JY, Hoogland ICM, Aronica EMA, van Westerloo DJ, van de Beek D, et al. Systemic infection and microglia activation: a prospective postmortem study in sepsis patients. Immun Ageing. (2019) 16:18. doi: 10.1186/s12979-019-0158-7
22. Michels M, Vieira AS, Vuolo F, Zapelini HG, Mendonca B, Mina F, et al. The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav Immun. (2015) 43:54–9. doi: 10.1016/j.bbi.2014.07.002
23. Comim CM, Vilela MC, Constantino LS, Petronilho F, Vuolo F, Lacerda-Queiroz N, et al. Traffic of leukocytes and cytokine up-regulation in the central nervous system in sepsis. Intensive Care Med. (2011) 37:711–8. doi: 10.1007/s00134-011-2151-2
24. Greenhalgh AD, David S, Bennett FC. Immune cell regulation of glia during CNS injury and disease. Nat Rev Neurosci. (2020) 21:139–52. doi: 10.1038/s41583-020-0263-9
25. Shulyatnikova T, Verkhratsky A. Astroglia in sepsis associated encephalopathy. Neurochem Res. (2020) 45:83–99. doi: 10.1007/s11064-019-02743-2
26. Rosales C. Neutrophil: A cell with many roles in inflammation or several cell types? Front Physiol. (2018) 9:113. doi: 10.3389/fphys.2018.00113
27. Rosales C. Neutrophils at the crossroads of innate and adaptive immunity. J Leukoc Biol. (2020) 108:377–96. doi: 10.1002/JLB.4MIR0220-574RR
28. Rungelrath V, Kobayashi SD, DeLeo FR. Neutrophils in innate immunity and systems biology-level approaches. Wiley Interdiscip Rev Syst Biol Med. (2020) 12:e1458. doi: 10.1002/wsbm.1458
29. Wu F, Liu L, Zhou H. Endothelial cell activation in central nervous system inflammation. J Leukoc Biol. (2017) 101:1119–32. doi: 10.1189/jlb.3RU0816-352RR
30. Norman MU, James WG, Hickey MJ. Differential roles of ICAM-1 and VCAM-1 in leukocyte-endothelial cell interactions in skin and brain of MRL/faslpr mice. J Leukoc Biol. (2008) 84:68–76. doi: 10.1189/jlb.1107796
31. Boos L, Szalai AJ, Barnum SR. C3a expressed in the central nervous system protects against LPS-induced shock. Neurosci Lett. (2005) 387:68–71. doi: 10.1016/j.neulet.2005.07.015
32. Petri B, Kaur J, Long EM Li H, Parsons SA, Butz S, et al. Endothelial LSP1 is involved in endothelial dome formation, minimizing vascular permeability changes during neutrophil transmigration in vivo. Blood. (2011) 117:942–52. doi: 10.1182/blood-2010-02-270561
33. Wu F, Chen X, Zhai L, Wang H, Sun M, Song C, et al. CXCR2 antagonist attenuates neutrophil transmigration into brain in a murine model of LPS induced neuroinflammation. Biochem Biophys Res Commun. (2020) 529:839–45. doi: 10.1016/j.bbrc.2020.05.124
34. Poles MZ, Naszai A, Gulacsi L, Czako BL, Gal KG, Glenz RJ, et al. Kynurenic acid and its synthetic derivatives protect against sepsis-associated neutrophil activation and brain mitochondrial dysfunction in rats. Front Immunol. (2021) 12:717157. doi: 10.3389/fimmu.2021.717157
35. Andonegui G, Zelinski EL, Schubert CL, Knight D, Craig LA, Winston BW, et al. Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI Insight. (2018) 3:e99364. doi: 10.1172/jci.insight.99364
36. Pelvig DP, Pakkenberg H, Stark AK, Pakkenberg B. Neocortical glial cell numbers in human brains. Neurobiol Aging. (2008) 29:1754–62. doi: 10.1016/j.neurobiolaging.2007.04.013
37. Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D. Systemic inflammation and microglia activation: systematic review of animal experiments. J Neuroinflammation. (2015) 12:114. doi: 10.1186/s12974-015-0332-6
38. Polito A, Brouland JP, Porcher R, Sonneville R, Siami S, Stevens RD, et al. Hyperglycaemia and apoptosis of microglia cells in human septic shock. Crit Care. (2011) 15:R131. doi: 10.1186/cc10244
39. Lemstra AW, Groen in't Woud JC, Hoozemans JJ, van Haastert ES, Rozemuller AJ, Eikelenboom P, van Gool WA. Microglia activation in sepsis: a case-control study. J Neuroinflammation. (2007) 4:4. doi: 10.1186/1742-2094-4-4
40. Li Y, Yin L, Fan Z, Su B, Chen Y, Ma Y, et al. Microglia: a potential therapeutic target for sepsis-associated encephalopathy and sepsis-associated chronic pain. Front Pharmacol. (2020) 11:600421. doi: 10.3389/fphar.2020.600421
41. Ye B, Tao T, Zhao A, Wen L, He X, Liu Y, et al. Blockade of IL-17A/IL-17R pathway protected mice from sepsis-associated encephalopathy by inhibition of microglia activation. Mediators Inflamm. (2019) 2019:8461725. doi: 10.1155/2019/8461725
42. Wang H, Hong LJ, Huang JY, Jiang Q, Tao RR, Tan C, et al. P2RX7 sensitizes Mac-1/ICAM-1-dependent leukocyte-endothelial adhesion and promotes neurovascular injury during septic encephalopathy. Cell Res. (2015) 25:674–90. doi: 10.1038/cr.2015.61
43. Tauber SC, Djukic M, Gossner J, Eiffert H, Bruck W, Nau R. Sepsis-associated encephalopathy and septic encephalitis: an update. Expert Rev Anti Infect Ther. (2021) 19:215–31. doi: 10.1080/14787210.2020.1812384
44. Kawakami M, Hattori M, Ohashi W, Fujimori T, Hattori K, Takebe M, et al. Role of G protein-coupled receptor kinase 2 in oxidative and nitrosative stress-related neurohistopathological changes in a mouse model of sepsis-associated encephalopathy. J Neurochem. (2018) 145:474–88. doi: 10.1111/jnc.14329
45. Michels M, Steckert AV, Quevedo J, Barichello T, Dal-Pizzol F. Mechanisms of long-term cognitive dysfunction of sepsis: from blood-borne leukocytes to glial cells. Intensive Care Med Exp. (2015) 3:30. doi: 10.1186/s40635-015-0066-x
46. Lauro C, Limatola C. Metabolic reprograming of microglia in the regulation of the innate inflammatory response. Front Immunol. (2020) 11:493. doi: 10.3389/fimmu.2020.00493
47. Yan C, Ma Z, Ma H, Li Q, Zhai Q, Jiang T, et al. Mitochondrial transplantation attenuates brain dysfunction in sepsis by driving microglia M2 polarization. Mol Neurobiol. (2020) 57:3875–90. doi: 10.1007/s12035-020-01994-3
48. Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev. (2018) 98:239–389. doi: 10.1152/physrev.00042.2016
49. Pekny M, Pekna M, Messing A, Steinhauser C, Lee JM, Parpura V, et al. Astrocytes: a central element in neurological diseases. Acta Neuropathol. (2016) 131:323–45. doi: 10.1007/s00401-015-1513-1
50. Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. (2017) 46:957–67. doi: 10.1016/j.immuni.2017.06.006
51. Sofroniew MV. Astrogliosis. Cold Spring Harb Perspect Biol. (2014) 7:a020420. doi: 10.1101/cshperspect.a020420
52. Moraes CA, Santos G, de Sampaio e Spohr TC, D'Avila JC, Lima FR, Benjamim CF, et al. Activated microglia-induced deficits in excitatory synapses through IL-1beta: implications for cognitive impairment in sepsis. Mol Neurobiol. (2015) 52:653–63. doi: 10.1007/s12035-014-8868-5
53. Gimenez MA, Sim JE, Russell JH. TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J Neuroimmunol. (2004) 151:116–25. doi: 10.1016/j.jneuroim.2004.02.012
54. Lossinsky AS, Shivers RR. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Review. Histol Histopathol. (2004)19:535–64. doi: 10.14670/HH-19.535
55. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
56. Cardoso FL, Herz J, Fernandes A, Rocha J, Sepodes B, Brito MA, et al. Systemic inflammation in early neonatal mice induces transient and lasting neurodegenerative effects. J Neuroinflammation. (2015) 12:82. doi: 10.1186/s12974-015-0299-3
57. Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci. (2012) 32:6391–410. doi: 10.1523/JNEUROSCI.6221-11.2012
58. Biesmans S, Meert TF, Bouwknecht JA, Acton PD, Davoodi N, De Haes P, et al. Systemic immune activation leads to neuroinflammation and sickness behavior in mice. Mediators Inflamm. (2013) 2013:271359. doi: 10.1155/2013/271359
59. Pierrakos C, Attou R, Decorte L, Kolyviras A, Malinverni S, Gottignies P, et al. Transcranial Doppler to assess sepsis-associated encephalopathy in critically ill patients. BMC Anesthesiol. (2014) 14:45. doi: 10.1186/1471-2253-14-45
60. Sharshar T, Annane D, de la Grandmaison GL, Brouland JP, Hopkinson NS, Francoise G. The neuropathology of septic shock. Brain Pathol. (2004) 14:21–33. doi: 10.1111/j.1750-3639.2004.tb00494.x
61. Villega F, Delpech JC, Griton M, Andre C, Franconi JM, Miraux S, et al. Circulating bacterial lipopolysaccharide-induced inflammation reduces flow in brain-irrigating arteries independently from cerebrovascular prostaglandin production. Neuroscience. (2017) 346:160–72. doi: 10.1016/j.neuroscience.2017.01.018
62. Szatmari S, Vegh T, Csomos A, Hallay J, Takacs I, Molnar C, et al. Impaired cerebrovascular reactivity in sepsis-associated encephalopathy studied by acetazolamide test. Crit Care. (2010) 14:R50. doi: 10.1186/cc8939
63. Pfister D, Siegemund M, Dell-Kuster S, Smielewski P, Ruegg S, Strebel SP, et al. Cerebral perfusion in sepsis-associated delirium. Crit Care. (2008) 12:R63. doi: 10.1186/cc6891
64. Schramm P, Klein KU, Falkenberg L, Berres M, Closhen D, Werhahn KJ, et al. Impaired cerebrovascular autoregulation in patients with severe sepsis and sepsis-associated delirium. Crit Care. (2012) 16:R181. doi: 10.1186/cc11665
65. Rosengarten B, Hecht M, Wolff S, Kaps M. Autoregulative function in the brain in an endotoxic rat shock model. Inflamm Res. (2008) 57:542–6. doi: 10.1007/s00011-008-7199-2
66. Molnar L, Fulesdi B, Nemeth N, Molnar C. Sepsis-associated encephalopathy: a review of literature. Neurol India. (2018) 66:352–61. doi: 10.4103/0028-3886.227299
67. Griton M, Dhaya I, Nicolas R, Raffard G, Periot O, Hiba B, et al. Experimental sepsis-associated encephalopathy is accompanied by altered cerebral blood perfusion and water diffusion and related to changes in cyclooxygenase-2 expression and glial cell morphology but not to blood-brain barrier breakdown. Brain Behav Immun. (2020) 83:200–13. doi: 10.1016/j.bbi.2019.10.012
68. Piazza O, Cotena S, De Robertis E, Caranci F, Tufano R. Sepsis associated encephalopathy studied by MRI and cerebral spinal fluid S100B measurement. Neurochem Res. (2009) 34:1289–92. doi: 10.1007/s11064-008-9907-2
69. Zrzavy T, Hoftberger R, Berger T, Rauschka H, Butovsky O, Weiner H, et al. Pro-inflammatory activation of microglia in the brain of patients with sepsis. Neuropathol Appl Neurobiol. (2019) 45:278–90. doi: 10.1111/nan.12502
70. Sharshar T, Carlier R, Bernard F, Guidoux C, Brouland JP, Nardi O, et al. Brain lesions in septic shock: a magnetic resonance imaging study. Intensive Care Med. (2007) 33:798–806. doi: 10.1007/s00134-007-0598-y
71. Azabou E, Rohaut B, Heming N, Magalhaes E, Morizot-Koutlidis R, Kandelman S, et al. Early impairment of intracranial conduction time predicts mortality in deeply sedated critically ill patients: a prospective observational pilot study. Ann Intensive Care. (2017) 7:63. doi: 10.1186/s13613-017-0290-5
72. Azabou E, Navarro V, Kubis N, Gavaret M, Heming N, Cariou A, et al. Value and mechanisms of EEG reactivity in the prognosis of patients with impaired consciousness: a systematic review. Crit Care. (2018) 22:184. doi: 10.1186/s13054-018-2104-z
73. Rohaut B, Porcher R, Hissem T, Heming N, Chillet P, Djedaini K, et al. Brainstem response patterns in deeply-sedated critically-ill patients predict 28-day mortality. PLoS ONE. (2017) 12:e0176012. doi: 10.1371/journal.pone.0176012
74. Cai H, Haubensak W, Anthony TE, Anderson DJ. Central amygdala PKC-delta(+) neurons mediate the influence of multiple anorexigenic signals. Nat Neurosci. (2014) 17:1240–8. doi: 10.1038/nn.3767
75. Tovote P, Fadok JP, Luthi A. Neuronal circuits for fear and anxiety. Nat Rev Neurosci. (2015) 16:317–31. doi: 10.1038/nrn3945
76. Semmler A, Widmann CN, Okulla T, Urbach H, Kaiser M, Widman G, et al. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry. (2013) 84:62–9. doi: 10.1136/jnnp-2012-302883
77. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. (2010) 304:1787–94. doi: 10.1001/jama.2010.1553
78. Zhu F, Zheng Y, Ding YQ, Liu Y, Zhang X, Wu R, et al. Minocycline and risperidone prevent microglia activation and rescue behavioral deficits induced by neonatal intrahippocampal injection of lipopolysaccharide in rats. PLoS ONE. (2014) 9:e93966. doi: 10.1371/journal.pone.0093966
79. Wang P, Hu Y, Yao D, Li Y. Omi/HtrA2 regulates a mitochondria-dependent apoptotic pathway in a murine model of septic encephalopathy. Cell Physiol Biochem. (2018) 49:2163–73. doi: 10.1159/000493819
80. Kobayashi T, Uchino H, Elmer E, Ogihara Y, Fujita H, Sekine S, et al. Disease outcome and brain metabolomics of cyclophilin-D knockout mice in sepsis. Int J Mol Sci. (2022) 23:961. doi: 10.3390/ijms23020961
81. Zhang L, Jiang Y, Deng S, Mo Y, Huang Y, Li W, et al. S100B/RAGE/Ceramide signaling pathway is involved in sepsis-associated encephalopathy. Life Sci. (2021) 277:119490. doi: 10.1016/j.lfs.2021.119490
82. Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. (2006) 290:L622–45. doi: 10.1152/ajplung.00477.2005
83. Chen S, Tang C, Ding H, Wang Z, Liu X, Chai Y, et al. Maf1 Ameliorates sepsis-associated encephalopathy by suppressing the NF-kB/NLRP3 inflammasome signaling pathway. Front Immunol. (2020) 11:594071. doi: 10.3389/fimmu.2020.594071
84. Lin CC, Hsieh HL, Shih RH, Chi PL, Cheng SE, Yang CM. Up-regulation of COX-2/PGE2 by endothelin-1 via MAPK-dependent NF-kappaB pathway in mouse brain microvascular endothelial cells. Cell Commun Signal. (2013) 11:8. doi: 10.1186/1478-811X-11-8
85. Tsuchiya K. Inflammasome-associated cell death: Pyroptosis, apoptosis, and physiological implications. Microbiol Immunol. (2020) 64:252–69. doi: 10.1111/1348-0421.12771
86. Gu M, Mei XL, Zhao YN. Sepsis and cerebral dysfunction: BBB damage, neuroinflammation, oxidative stress, apoptosis and autophagy as key mediators and the potential therapeutic approaches. Neurotox Res. (2021) 39:489–503. doi: 10.1007/s12640-020-00270-5
87. Zhou RX Li YY, Qu Y, Huang Q, Sun XM, Mu DZ, Li XH. Regulation of hippocampal neuronal apoptosis and autophagy in mice with sepsis-associated encephalopathy by immunity-related GTPase M1. CNS Neurosci Ther. (2020) 26:177–88. doi: 10.1111/cns.13229
88. Wu J, Dong L, Zhang M, Jia M, Zhang G, Qiu L, et al. Class I histone deacetylase inhibitor valproic acid reverses cognitive deficits in a mouse model of septic encephalopathy. Neurochem Res. (2013) 38:2440–9. doi: 10.1007/s11064-013-1159-0
89. Qiu LL, Pan W, Luo D, Zhang GF, Zhou ZQ, Sun XY, et al. Dysregulation of BDNF/TrkB signaling mediated by NMDAR/Ca2+/calpain might contribute to postoperative cognitive dysfunction in aging mice. J Neuroinflammation. (2020) 17:23. doi: 10.1186/s12974-019-1695-x
90. Gao LL, Wang ZH, Mu YH, Liu ZL, Pang L. Emodin promotes autophagy and prevents apoptosis in sepsis-associated encephalopathy through activating BDNF/TrkB signaling. Pathobiology. (2022) 89:135–45. doi: 10.1159/000520281
91. Wang YC, Liu QX, Liu T, Xu XE, Gao W, Bai XJ Li ZF. Caspase-1-dependent pyroptosis of peripheral blood mononuclear cells predicts the development of sepsis in severe trauma patients: a prospective observational study. Medicine. (2018) 97:e9859. doi: 10.1097/MD.0000000000009859
92. Chen N, Ou Z, Zhang W, Zhu X, Li P, Gong J. Cathepsin B regulates non-canonical NLRP3 inflammasome pathway by modulating activation of caspase-11 in Kupffer cells. Cell Prolif. (2018) 51:e12487. doi: 10.1111/cpr.12487
93. Wang LX, Ren C, Yao RQ, Luo YN, Yin Y, Wu Y, et al. Sestrin2 protects against lethal sepsis by suppressing the pyroptosis of dendritic cells. Cell Mol Life Sci. (2021) 78:8209–27. doi: 10.1007/s00018-021-03970-z
94. Xu XE, Liu L, Wang YC, Wang CT, Zheng Q, Liu QX, et al. Caspase-1 inhibitor exerts brain-protective effects against sepsis-associated encephalopathy and cognitive impairments in a mouse model of sepsis. Brain Behav Immun. (2019) 80:859–70. doi: 10.1016/j.bbi.2019.05.038
95. Wang Y, Liu X, Wang Q, Yang X. Roles of the pyroptosis signaling pathway in a sepsis-associated encephalopathy cell model. J Int Med Res. (2020) 48:300060520949767. doi: 10.1177/0300060520949767
96. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. (2011) 479:117–1121. doi: 10.1038/nature10558
97. Evavold CL, Kagan JC. Inflammasomes: threat-assessment organelles of the innate immune system. Immunity. (2019) 51:609–24. doi: 10.1016/j.immuni.2019.08.005
98. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
99. Fu Q, Wu J, Zhou XY Ji MH, Mao QH Li Q, et al. NLRP3/Caspase-1 pathway-induced pyroptosis mediated cognitive deficits in a mouse model of sepsis-associated encephalopathy. Inflammation. (2019) 42:306–18. doi: 10.1007/s10753-018-0894-4
100. Lei Y, Zhou R, Sun X, Tang F, Gao H, Chen L, et al. The pannexin-1 channel regulates pyroptosis through autophagy in a mouse model of sepsis-associated encephalopathy. Ann Transl Med. (2021) 9:1802. doi: 10.21037/atm-21-6579
101. Quan N, Banks WA. Brain-immune communication pathways. Brain Behav Immun. (2007) 21:727–35. doi: 10.1016/j.bbi.2007.05.005
102. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. (2000) 405:458–62. doi: 10.1038/35013070
103. Andersson U, Tracey KJ. Neural reflexes in inflammation and immunity. J Exp Med. (2012) 209:1057–68. doi: 10.1084/jem.20120571
104. Kanczkowski W, Sue M, Zacharowski K, Reincke M, Bornstein SR. The role of adrenal gland microenvironment in the HPA axis function and dysfunction during sepsis. Mol Cell Endocrinol. (2015) 408:241–8. doi: 10.1016/j.mce.2014.12.019
105. Kanczkowski W, Sue M, Bornstein SR. Adrenal gland microenvironment and its involvement in the regulation of stress-induced hormone secretion during sepsis. Front Endocrinol. (2016) 7:156. doi: 10.3389/fendo.2016.00156
106. Kanczkowski W, Chatzigeorgiou A, Samus M, Tran N, Zacharowski K, Chavakis T, et al. Characterization of the LPS-induced inflammation of the adrenal gland in mice. Mol Cell Endocrinol. (2013) 371:228–35. doi: 10.1016/j.mce.2012.12.020
107. Jennewein C, Tran N, Kanczkowski W, Heerdegen L, Kantharajah A, Drose S, et al. Mortality of septic mice strongly correlates with adrenal gland inflammation. Crit Care Med. (2016) 44:e190–199. doi: 10.1097/CCM.0000000000001373
108. Kanczkowski W, Alexaki VI, Tran N, Grossklaus S, Zacharowski K, Martinez A, et al. Hypothalamo-pituitary and immune-dependent adrenal regulation during systemic inflammation. Proc Natl Acad Sci USA. (2013) 110:14801–6. doi: 10.1073/pnas.1313945110
109. Gilibert S, Galle-Treger L, Moreau M, Saint-Charles F, Costa S, Ballaire R, et al. Adrenocortical scavenger receptor class B type I deficiency exacerbates endotoxic shock and precipitates sepsis-induced mortality in mice. J Immunol. (2014) 193:817–26. doi: 10.4049/jimmunol.1303164
110. Vandewalle J, Libert C. Glucocorticoids in sepsis: to be or not to be. Front Immunol. (2020) 11:1318. doi: 10.3389/fimmu.2020.01318
111. Chen LS, Singh SP, Muller G, Bornstein SR, Kanczkowski W. Transcriptional analysis of sepsis-induced activation and damage of the adrenal endothelial microvascular cells. Front Endocrinol. (2019) 10:944. doi: 10.3389/fendo.2019.00944
112. Van Looveren K, Timmermans S, Vanderhaeghen T, Wallaeys C, Ballegeer M, Souffriau J, et al. Glucocorticoids limit lipopolysaccharide-induced lethal inflammation by a double control system. EMBO Rep. (2020) 21:e49762. doi: 10.15252/embr.201949762
113. Quatrini L, Wieduwild E, Escaliere B, Filtjens J, Chasson L, Laprie C, et al. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat Immunol. (2018) 19:954–62. doi: 10.1038/s41590-018-0185-0
114. Ng HP, Jennings S, Nelson S, Wang G. Short-chain alcohols upregulate GILZ gene expression and attenuate LPS-induced septic immune response. Front Immunol. (2020) 11:53. doi: 10.3389/fimmu.2020.00053
115. Li CC, Munitic I, Mittelstadt PR, Castro E, Ashwell JD. Suppression of dendritic cell-derived IL-12 by endogenous glucocorticoids is protective in LPS-induced sepsis. PLoS Biol. (2015) 13:e1002269. doi: 10.1371/journal.pbio.1002269
116. Kleiman A, Hubner S, Rodriguez Parkitna JM, Neumann A, Hofer S, Weigand MA, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J. (2012) 26:722–9. doi: 10.1096/fj.11-192112
117. Guo L, Zheng Z, Ai J, Howatt DA, Mittelstadt PR, Thacker S, et al. Scavenger receptor BI and high-density lipoprotein regulate thymocyte apoptosis in sepsis. Arterioscler Thromb Vasc Biol. (2014) 34:966–75. doi: 10.1161/ATVBAHA.113.302484
118. Fudulu DP, Horn G, Hazell G, Lefrancois-Martinez AM, Martinez A, Angelini GD, et al. Co-culture of monocytes and zona fasciculata adrenal cells: An in vitro model to study the immune-adrenal cross-talk. Mol Cell Endocrinol. (2021) 526:111195. doi: 10.1016/j.mce.2021.111195
119. Vandewalle J, Timmermans S, Paakinaho V, Vancraeynest L, Dewyse L, Vanderhaeghen T, et al. Combined glucocorticoid resistance and hyperlactatemia contributes to lethal shock in sepsis. Cell Metab. (2021) 33:1763–76e5. doi: 10.1016/j.cmet.2021.07.002
120. Dejager L, Pinheiro I, Puimege L, Fan YD, Gremeaux L, Vankelecom H, et al. Increased glucocorticoid receptor expression and activity mediate the LPS resistance of SPRET/EI mice. J Biol Chem. (2010) 285:31073–86. doi: 10.1074/jbc.M110.154484
121. Mittelstadt PR, Monteiro JP, Ashwell JD. Thymocyte responsiveness to endogenous glucocorticoids is required for immunological fitness. J Clin Invest. (2012) 122:2384–94. doi: 10.1172/JCI63067
122. van der Goes A, Hoekstra K, van den Berg TK, Dijkstra CD. Dexamethasone promotes phagocytosis and bacterial killing by human monocytes/macrophages in vitro. J Leukoc Biol. (2000) 67:801–7. doi: 10.1002/jlb.67.6.801
123. Desgeorges T, Caratti G, Mounier R, Tuckermann J, Chazaud B. Glucocorticoids shape Macrophage phenotype for tissue repair. Front Immunol. (2019) 10:1591. doi: 10.3389/fimmu.2019.01591
124. Ellouze M, Vigouroux L, Tcherakian C, Woerther PL, Guguin A, Robert O, et al. Overexpression of GILZ in macrophages limits systemic inflammation while increasing bacterial clearance in sepsis in mice. Eur J Immunol. (2020) 50:589–602. doi: 10.1002/eji.201948278
125. Doulias T, Quickert S, Weis S, Claus RA, Kontopoulou K, Giamarellos-Bourboulis EJ, et al. Low-dose hydrocortisone prolongs survival in a lethal sepsis model in adrenalectomized rats. J Surg Res. (2018) 227:72–80. doi: 10.1016/j.jss.2018.02.011
126. Ballegeer M, Vandewalle J, Eggermont M, Van Isterdael G, Dejager L, De Bus L, et al. Overexpression of Gilz protects mice against lethal septic peritonitis. Shock. (2019) 52:208–14. doi: 10.1097/SHK.0000000000001252
127. Jankord R, Herman JP. Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann N Y Acad Sci. (2008) 1148:64–73. doi: 10.1196/annals.1410.012
128. Spencer-Segal JL, Singer BH, Laborc K, Somayaji K, Watson SJ, Standiford TJ, et al. Sepsis survivor mice exhibit a behavioral endocrine syndrome with ventral hippocampal dysfunction. Psychoneuroendocrinology. (2020) 117:104679. doi: 10.1016/j.psyneuen.2020.104679
129. Singer BH, Newstead MW, Zeng X, Cooke CL, Thompson RC, Singer K, et al. Cecal ligation and puncture results in long-term central nervous system myeloid inflammation. PLoS ONE. (2016) 11:e0149136. doi: 10.1371/journal.pone.0149136
130. Czura CJ, Friedman SG, Tracey KJ. Neural inhibition of inflammation: the cholinergic anti-inflammatory pathway. J Endotoxin Res. (2003) 9:409–13. doi: 10.1179/096805103225002755
131. Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, et al. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci USA. (2008) 105:11008–13. doi: 10.1073/pnas.0803237105
132. Kees MG, Pongratz G, Kees F, Scholmerich J, Straub RH. Via beta-adrenoceptors, stimulation of extrasplenic sympathetic nerve fibers inhibits lipopolysaccharide-induced TNF secretion in perfused rat spleen. J Neuroimmunol. (2003) 145:77–85. doi: 10.1016/j.jneuroim.2003.09.011
133. Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med. (2006) 203:1623–8. doi: 10.1084/jem.20052362
134. Berthoud HR, Powley TL. Characterization of vagal innervation to the rat celiac, suprarenal and mesenteric ganglia. J Auton Nerv Syst. (1993) 42:153–69. doi: 10.1016/0165-1838(93)90046-w
135. Bellinger DL, Felten SY, Lorton D, Felten DL. Origin of noradrenergic innervation of the spleen in rats. Brain Behav Immun. (1989) 3:291–311. doi: 10.1016/0889-1591(89)90029-9
136. Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. (2011) 334:98–101. doi: 10.1126/science.1209985
137. Mashimo M, Takeshima S, Okuyama H, Matsurida A, Murase M, Ono S, et al. alpha7 nAChRs expressed on antigen presenting cells are insensitive to the conventional antagonists alpha-bungarotoxin and methyllycaconitine. Int Immunopharmacol. (2020) 81:106276. doi: 10.1016/j.intimp.2020.106276
138. Riether C, Kavelaars A, Wirth T, Pacheco-Lopez G, Doenlen R, Willemen H, et al. Stimulation of beta(2)-adrenergic receptors inhibits calcineurin activity in CD4+ T cells via PKA-AKAP interaction. Brain Behav Immun. (2011) 25:59–66. doi: 10.1016/j.bbi.2010.07.248
139. Kohm AP, Sanders VM. Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol Rev. (2001) 53:487–525.
140. Sinkus ML, Graw S, Freedman R, Ross RG, Lester HA, Leonard S. The human CHRNA7 and CHRFAM7A genes: a review of the genetics, regulation, and function. Neuropharmacology. (2015) 96(Pt B):274–88. doi: 10.1016/j.neuropharm.2015.02.006
141. Parada E, Egea J, Buendia I, Negredo P, Cunha AC, Cardoso S, et al. The microglia alpha7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid Redox Signal. (2013) 19:1135–48. doi: 10.1089/ars.2012.4671
142. John D, Shelukhina I, Yanagawa Y, Deuchars J, Henderson Z. Functional alpha7 nicotinic receptors are expressed on immature granule cells of the postnatal dentate gyrus. Brain Res. (2015) 1601:15–30. doi: 10.1016/j.brainres.2014.12.041
143. Wu M, Liu CZ, Barrall EA, Rissman RA, Joiner WJ. Unbalanced regulation of alpha7 nAChRs by Ly6h and NACHO contributes to neurotoxicity in Alzheimer's disease. J Neurosci. (2021) 41:8461–74. doi: 10.1523/JNEUROSCI.0494-21.2021
144. Nebrisi EE. Neuroprotective activities of curcumin in Parkinson's disease: a review of the literature. Int J Mol Sci. (2021) 22:11248. doi: 10.3390/ijms222011248
145. Letsinger AC, Gu Z, Yakel JL. Alpha7 nicotinic acetylcholine receptors in the hippocampal circuit: taming complexity. Trends Neurosci. (2022) 45:145–57. doi: 10.1016/j.tins.2021.11.006
146. Li L, Liu Z, Jiang YY, Shen WX, Peng YP, Qiu YH. Acetylcholine suppresses microglia inflammatory response via alpha7nAChR to protect hippocampal neurons. J Integr Neurosci. (2019) 18:51–6. doi: 10.31083/j.jin.2019.01.114
147. Patel H, McIntire J, Ryan S, Dunah A, Loring R. Anti-inflammatory effects of astroglial alpha7 nicotinic acetylcholine receptors are mediated by inhibition of the NF-kappaB pathway and activation of the Nrf2 pathway. J Neuroinflammation. (2017) 14:192. doi: 10.1186/s12974-017-0967-6
148. Di Cesare Mannelli L, Tenci B, Zanardelli M, Failli P, Ghelardini C. Alpha7 nicotinic receptor promotes the neuroprotective functions of astrocytes against oxaliplatin neurotoxicity. Neural Plast. (2015) 2015:396908. doi: 10.1155/2015/396908
149. Cao M, MacDonald JW, Liu HL, Weaver M, Cortes M, Durosier LD, et al. Alpha7 nicotinic acetylcholine receptor signaling modulates ovine fetal brain astrocytes transcriptome in response to endotoxin. Front Immunol. (2019) 10:1063. doi: 10.3389/fimmu.2019.01063
150. Rosas-Ballina M, Goldstein RS, Gallowitsch-Puerta M, Yang L, Valdes-Ferrer SI, Patel NB, et al. The selective alpha7 agonist GTS-21 attenuates cytokine production in human whole blood and human monocytes activated by ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol Med. (2009) 15:195–202. doi: 10.2119/molmed.2009.00039
151. Mashimo M, Komori M, Matsui YY, Murase MX, Fujii T, Takeshima S, et al. Distinct roles of alpha7 nAChRs in antigen-presenting cells and CD4+ T cells in the regulation of T cell differentiation. Front Immunol. (2019) 10:1102. doi: 10.3389/fimmu.2019.01102
152. Gillentine MA, Schaaf CP. The human clinical phenotypes of altered CHRNA7 copy number. Biochem Pharmacol. (2015) 97:352–62. doi: 10.1016/j.bcp.2015.06.012
153. Kweon HJ, Gu S, Witham E, Dhara M, Yu H, Mandon ED, et al. NACHO engages N-glycosylation ER chaperone pathways for alpha7 nicotinic receptor assembly. Cell Rep. (2020) 32:108025. doi: 10.1016/j.celrep.2020.108025
154. Halevi S, McKay J, Palfreyman M, Yassin L, Eshel M, Jorgensen E, Treinin M. The C. elegans ric-3 gene is required for maturation of nicotinic acetylcholine receptors. EMBO J. (2002) 21:1012–20. doi: 10.1093/emboj/21.5.1012
155. Rana M, Fei-Bloom Y, Son M, La Bella A, Ochani M, Levine YA, et al. Constitutive vagus nerve activation modulates immune suppression in sepsis survivors. Front Immunol. (2018) 9:2032. doi: 10.3389/fimmu.2018.02032
156. Ren C, Yao RQ, Wang LX Li JC, Chen KW, Wu Y, et al. Antagonism of cerebral high mobility group box 1 ameliorates dendritic cell dysfunction in sepsis. Front Pharmacol. (2021) 12:665579. doi: 10.3389/fphar.2021.665579
157. Ren C, Tong YL Li JC, Dong N, Hao JW, Zhang QH, Yao YM. Early antagonism of cerebral high mobility group box-1 protein is benefit for sepsis induced brain injury. Oncotarget. (2017) 8:92578–88. doi: 10.18632/oncotarget.21502
Keywords: sepsis-associated encephalopathy, neuroendocrine–immune network, hypothalamic–pituitary–adrenal axis, cholinergic anti-inflammatory pathway, neuroinflammation
Citation: Liu Y-x, Yu Y, Liu J-p, Liu W-j, Cao Y, Yan R-m and Yao Y-m (2022) Neuroimmune Regulation in Sepsis-Associated Encephalopathy: The Interaction Between the Brain and Peripheral Immunity. Front. Neurol. 13:892480. doi: 10.3389/fneur.2022.892480
Received: 09 March 2022; Accepted: 27 May 2022;
Published: 27 June 2022.
Edited by:
Hao Wang, Shandong University, ChinaReviewed by:
Jianfeng Wu, The First Affiliated Hospital of Sun Yat-sen University, ChinaCopyright © 2022 Liu, Yu, Liu, Liu, Cao, Yan and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-ming Yao, Y19mZkBzaW5hLmNvbQ==; Run-min Yan, eWRyb21jOEBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.