
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 26 September 2022
Sec. Dementia and Neurodegenerative Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.886887
This article is part of the Research TopicHorizon in Frontotemporal Lobar Degeneration Related DisorderView all 14 articles
 Yu Wang1†
Yu Wang1† Xiaohui Duan1†
Xiaohui Duan1† Xiao Zhou1Renbin Wang1Xiangfei Zhang1Zhenhua Cao2Xiaoxia Wang2
Xiao Zhou1Renbin Wang1Xiangfei Zhang1Zhenhua Cao2Xiaoxia Wang2 Zhi Zhou1
Zhi Zhou1 Yu Sun1
Yu Sun1 Dantao Peng1*
Dantao Peng1*Background: The Annexin A11 (ANXA11) gene has been newly identified as a causative gene of amyotrophic lateral sclerosis (ALS) with or without frontotemporal dementia (FTD). The current study aimed to investigate the ANXA11 mutations in a Chinese ALS–FTD or FTD cohort.
Methods: We included ten probands/patients with suspected ALS–FTD or FTD. Mutational analysis of ANXA11 was performed through Next Generation Sequencing (NGS) and Sanger sequencing. We collected and reviewed clinical presentation, neuropsychology test results, brain-imaging findings, and electrophysiological examination findings.
Results: In total, six probands presented with ALS–FTD, and four with behavior variant FTD (bv-FTD). We identified a non-synonymous heterozygous mutation (c.119A>G, p.D40G) of ANXA11 in proband 1, which is associated with ALS. However, this is the first report of the mutation causing ALS–FTD. Proband 1 started with abnormal behavior and progressed to classic upper motor nervous disease. Magnetic resonance imaging (MRI) showed significant bilateral temporal lobe atrophy and bilateral hyperintensities along the corticospinal tracts.18F-AV45-PET imaging showed negative amyloid deposits.
Conclusion: ANXA11-related diseases have high clinical and genetic heterogeneity. Our study confirmed the contribution of ANXA11 mutations to ALS–FTD. The ANXA11 mutations established a complex genotype–phenotype correlation in ALS–FTD. Our research further elucidated the genetic mechanism of ALS–FTD and contributed to setting the foundation of future targeted therapy.
Amyotrophic lateral sclerosis, a lethal progressive neurologic disease, is characterized by selective degeneration of the lower and upper motor neurons. Approximately 5–10% of patients with ALS have a positive family history, suggesting that genetic factors substantially contribute to its pathogenesis. Frontotemporal dementia (FTD) is a spectrum of syndromes characterized by a progressive deterioration in behavior, personality, language, and cognition, associated pathologically with frontotemporal lobar degeneration (FTLD). ALS is closely related to FTD. Up to ~50% of patients with ALS show behavioral dysfunction and/or subtle cognitive impairment, while about 15% meet the psychiatry diagnostic criteria of FTD (termed as ALS–FTD) (1–3). A similar scenario is observed in FTD. Approximately 30% of patients with FTD have motor impairments, and 12.5% meet the diagnostic criteria for ALS (4, 5).
In the past few years, owing to the rapid development of next-generation sequencing, ALS–FTD-associated genes have been progressively identified. For example, mutations of C9orf72, TARDBP, and TBK1 have been identified as major genetic causes of ALS–FTD. The aggregation of TAR DNA-binding protein 43 (TDP-43) in the affected brain regions and motor neurons is a common pathological characteristic of each of these variants (6–10) in up to 97% of ALS and 50% of FTD cases. Beyond that, mutations in CCNF, CHCHD10, FUS, SQSTM1, UBQLN2, and VCP are also associated with ALS–FTD (11). However, the genetic etiology of ALS–FTD in some patients remains unclear. In the current study, mutation in the Annexin A11(AXAN11) gene was proved to be linked to ALS–FTD in a Chinese clinical cohort. We also included a review of previously reported mutations with ALS or ALS–FTD in the AXAN11 gene.
In total, ten probands/patients with suspected ALS–FTD or FTD from the Department of Neurology, China–Japan Friendship Hospital in Beijing, were enrolled in the study from July 2019 to January 2022. The clinical characteristics, brain imaging results, and laboratory profiles were collected. This research was approved by the institutional board of the Ethics Committees of China–Japan Friendship Hospital in Beijing and followed the Declaration of Helsinki.
Genomic DNA was extracted from peripheral blood samples collected from ten suspected patients and healthy volunteers, according to standard procedures. The repeat length of the pathogenic C9orf72 GGGGCC repeat expansion was examined and excluded in these patients using polymerase chain reaction (PCR) amplification combined with microfluidic capillary electrophoresis.
Whole-exome sequencing was performed following the Illumina specifications. The isolated DNAs were firstly fragmented into 200–250 bp lengths by sonication. Then, DNA libraries were built using the KAPA Library Preparation Kit (Kapa Biosystems, KR0453) and sequenced via the Illumina Noveseq s4 platform (Illumina, San Diego, USA) with 150-bp paired-end reads. The human reference genome (UCSC hg19) was applied to the filter and aligned with the raw data using the Burrows-Wheeler Alignment tool (BWA-0.7.12, http://bio-bwa.sourceforge.net/). GATK software (www.broadinstitute.org/gatk) was used to identify single-nucleotide polymorphisms (SNPs), insertions, and deletions (indels). VEP [Ensemble Variant Effect Predictor, McLaren et al. (12)] was used to annotate all the variants, including the genetic position, type, allele frequency, conservation prediction, etc.
All the variants were filtered first against the 1,000 genomes project database, for a minor allele frequency (MAF) ≥ 1%, and ExAC hom AC ≥3. The obtained variants were further selected according to co-segregation, the genetic model, and an MAF <1% in three databases (1,000 genomes project_EAS, ExAC, and gnomAD_EAS). We then focused on analyzing variants of the ALS-related genes, which were included in the OMIM database. All the candidate pathogenic variants were confirmed by Sanger sequencing and classified according to the American College of Medical Genetics and Genomics (ACMG) standards (13). Finally, the ANXA11 mutations were selected based on their clinical relevance and pathogenicity.
For electrophysiological profiles, examinations were conducted using conventional equipment and according to the standard methods, with skin temperatures maintained between 32 and 34°C. Nerve conduction and needle electromyography (EMG) examinations were conducted on 10 patients.
All the patients underwent 3.0T MRI with a device using eight-channel head coils (Discovery MR750 scanner; GE Medical Systems, United States) in the China–Japan Friendship Hospital. The sequences performed included T1- and T2-weighted fluid-attenuated inversion recovery (FLAIR) and standard coronal T2-weighted sequences.
In total, five patients were selected for 18F-AV45 PET scans using the Discovery Elite scanner (GE Healthcare) at the Tiantan Hospital. 18F-AV45 PET was performed at 20 min and 50 min postinjection of 248 ± 58 MBq. 18F-AV45 PET profiles were analyzed using an ordered subset expectation maximization algorithm with weighted attenuation. Images were smoothed using a 5 mm Gaussian kernel with scatter correction and evaluated prior to the analysis of patient motion and adequacy of statistical counts. Finally, the standardized uptake value ratios (SUVRs) were computed and normalized according to the cerebellar gray matter reference region and the mean activity, from 50 to 70 min.
We searched and reviewed published reports of ANXA11 mutations using PubMed. Clinical, biochemical, neuroimaging, and genetic data from individual references were sourced and compared with the corresponding results of our research.
The current cohort included 10 patients with behavioral variant FTD (bv-FTD). In total, six had probable bv-FTD with ALS according to the Rascovsky criteria. The clinical characteristics of the current Chinese clinical cohort are displayed in Table 1.
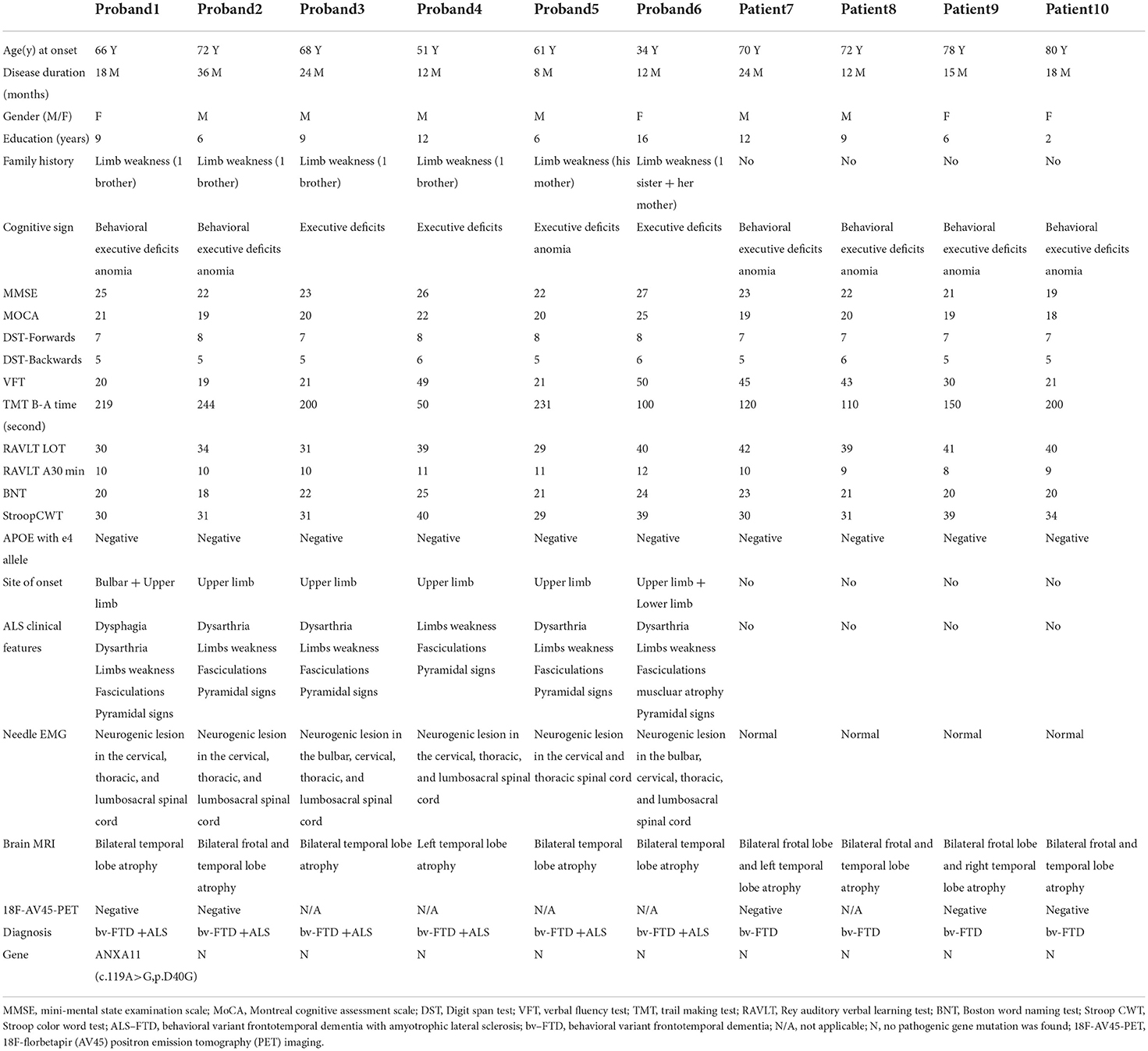
Table 1. Clinical features of ten probands/patients.
All 10 patients (6 men and 4 women) diagnosed with bv-FTD were from the Chinese mainland. The onset of symptoms occurred at the age of 34–80 years, median (IQR) is 69 (58.5–73.5). All 10 patients showed behavioral and executive deficits, and anomia. There were six patients with positive family histories. In total, one proband initially presented with dysarthria, and five probands presented with limb weakness as the initial symptom. Initially, Proband 1 presented with euphoria, loss of manners, impulsiveness, rash behavior, and difficulty cooking at the age of 66 years. A few months later, her speech became slurred, and she had difficulties in expressing and naming. The patient also had gradual weakness in both upper limbs. Fasciculations, hyperreflexia, and positive Babinski sign of the limbs were observed. EMG demonstrated a neurogenic lesion in the cervical, thoracic, and lumbosacral spinal cord. Brain MRI showed bilateral temporal lobe atrophy and bilateral signal hyperintensities along the corticospinal tracts (Figure 1). 18F-AV45-PET imaging showed negative amyloid deposits. The patient was diagnosed as having ALS with bv-FTD. She had an older brother who developed limb atrophy and weakness at 55 years of age and died at 67 years without providing a peripheral blood sample.
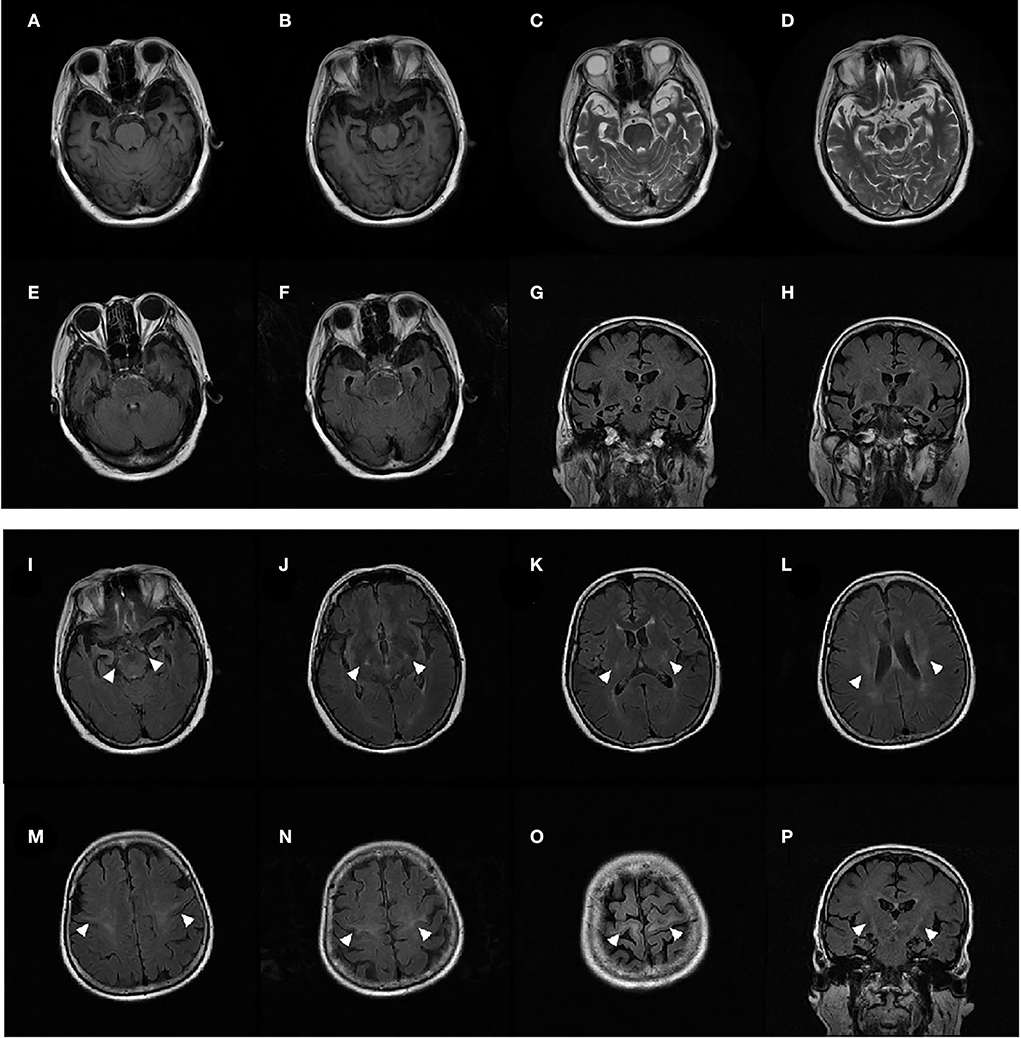
Figure 1. MRI of patient 1 showed significant bilateral temporal lobe atrophy (A–H). T2-weighted fluid-attenuated inversion recovery (FLAIR) coronal and axial MRI displayed bilateral signal hyperintensities along the corticospinal tracts in the primary motor cortex, centrum semiovale, posterior limb of the internal capsule, and the cerebral peduncle (I–P) (arrows).
We identified one non-synonymous heterozygous mutation (c.119A>G, p.D40G) in ANXA11, which was previously reported to be associated with ALS, but to our knowledge, this is the first time that has been found in ALS–FTD. By reviewing previous literature in the Human Gene Mutation Database (HGMD), we found out thirty-two different ANXA11 variants have been identified in ALS and/or ALS–FTD, including patients from the United Kingdom, Southern Africans, Brazil, France, German, Korea, Spain, Japan, and China (Table 2) (14–26). To further investigate the correlation between phenotype and genotype, we reviewed and summarized all the studies on ANXA11 mutations (Figure 2).
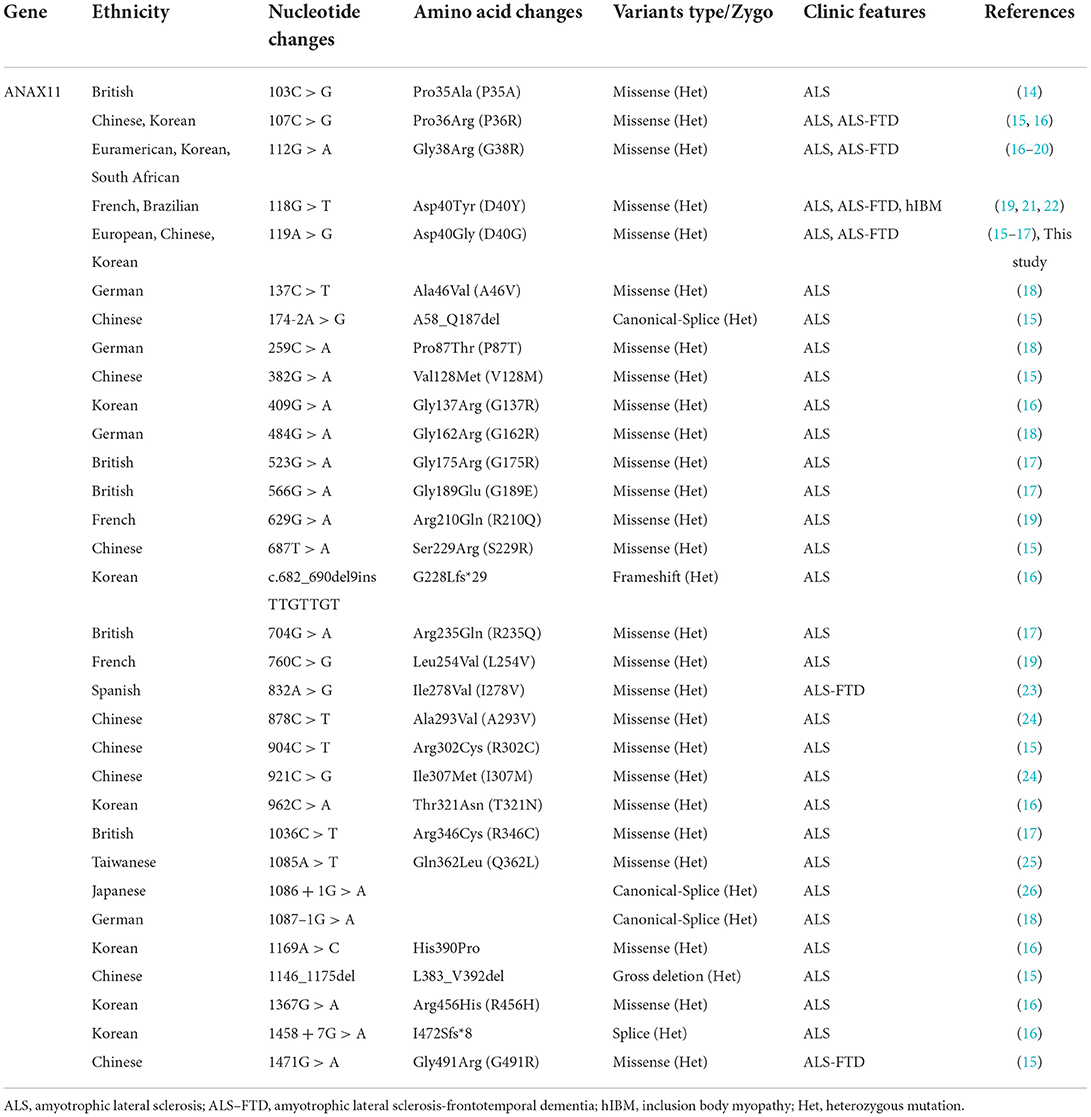
Table 2. Clinical and genetic characteristics of ANXA11-related diseases.
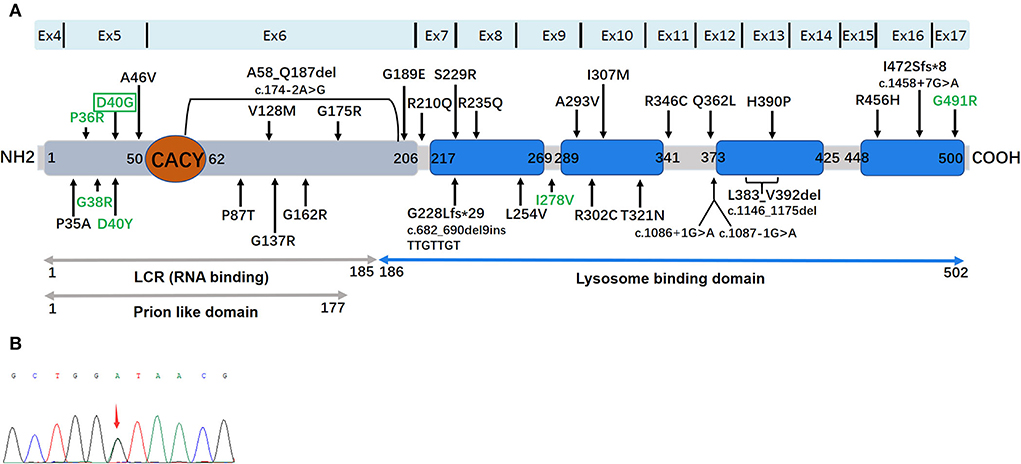
Figure 2. (A) The location and distribution of 32 mutations in a 2D schematic representation of the ANXA11 protein. ANXA11 protein and functional domains: a Prion-like domain (gray) was determined by the software PLAAC (Prion Like Amino Acid Composition). RNA (gray) and lysosome (blue) binding domains are represented. The binding site with Calcyclin/S100A6 (CACY) is located at N terminal (orange). The four highly conserved annexin domains are represented by blue square. Reported ALS-related mutations are displayed in black, and ALS/ALS-FTD-related mutations pointed by green. D4OG was detected in the present study are displayed in green by green box. (B) Sequence chromatograms of polymerase chain reaction (PCR) products show the heterozygous c.119A>G (p.D40G) mutation in this study.
Located on the human chromosome 10q22.3, the ANXA11 gene encodes the 505 amino acid annexin A11 protein, which is a member of a calcium-dependent phospholipid-binding annexin protein family. The primary function of the annexin protein family is to bind Ca2+, RNA, other proteins, and lipid membranes. Unlike other family members, ANXA11 shows a uniquely long N-terminal domain that contains the calcyclin binding site (residues 50–62). Calcyclin can mediate ubiquitination and proteasome degradation of many target proteins (27). In total, four conserved annexin domains, including annexin1-4, constitute the conserved C terminus (28).
ANXA11-related ALS was initially identified in 2017 by whole-exome sequencing in 180 sporadic-ALS (SALS) cases and 751 European familial-ALS (FALS) (17). Smith et al. identified six ANXA11 mutations (G38R, D40G, G175R, G189E, R235Q, and R346C) in 9 patients from 6 families, and 3 SALS cases without FTD. In the aforementioned study, the D40G mutation was found to be the most common mutation. Patients carrying the D40G mutation presented a delayed-onset of classical ALS symptoms, with 5/6 cases having the bulbar-onset disease. Subsequently, a study in a non-Caucasian population supported the pathogenicity of D40G in the ANXA11 mutation associated with ALS. Of note, a sporadic ALS case was found once in a Chinese mainland cohort of 383 patients with ALS or ALS–FTD (15). There was also another reported study of 500 Korean patients with SALS (16). Liu et al. failed to discover D40G; instead, they found two rare heterozygous missense variants, namely, c.878C>T (p.A293V) and c.921C>G (p.I307M), in another Chinese cohort with 434 patients with SALS and 50 patients who had the index FALS (24). If the results of the two Chinese cohorts are combined, the D40G mutation rate is rarely low (0.12%, 1/867) in the Chinese patients with ALS or ALS–FTD. The aforementioned results suggest that p.D40G mutation is not the primary cause of ALS in the Chinese population (24).
According to the functional analysis, p.D40G being located near the calcyclin-binding region could cause abnormal binding of calcyclin. Analyses from a postmortem p.D40G ALS case showed profuse annexin A11-positive aggregates in neurons and neuropil of the neocortex and hippocampus, and motor neurons of the spinal cord (17).
In the current study, patients with the same D40G mutation have different clinical symptoms: (1) five of six European patients and one Korean patient who carried the mutation initially showed difficulty in swallowing and speaking (bulbar-onset ALS) (17); (2) a Chinese patient initially displayed left arm weakness at the age of 59 years (15); (3) in the present study, proband 1 with the ANXA11 p.D40G mutation initially presented abnormal behaviors, executive deficits, and anomia, and later progressed to classic upper motor nervous system damage in the bulbar and limbs. MRI showed significant bilateral temporal lobe atrophy and bilateral signal hyperintensities along the corticospinal tracts. The patient was diagnosed with ALS with bv-FTD. To our knowledge, this study is the first to associate the D40G mutation with ALS–FTD. Our results provided more genetic support for ALS and FTD.
Reviewing the literature, the spectrum of genotypes and phenotypes associated with ANXA11-related diseases has expanded as follows: (14–26) (i) late-onset or early-onset ALS (black mutations in Figure 2); (ii) ALS with FTD (P36R, G38R, D40Y, D40G, I278V, and G491R); (iii) inclusion body myopathy (hIBM), isolated or in combination with ALS/FTD (D40Y). In addition, the ordinary single nucleotide polymorphism (rs1049550, C>T, p.R230C, and MAF 0.44) in ANXA11 may enhance the risk of sarcoidosis (29). Furthermore, the rs1049550T in the ANXA11 allele plays a protective role for sarcoidosis in the Chinese Han nationality (30). Like other multisystem proteinopathies (MSP), ANXA11-related disorders possess a high clinical heterogeneity (Table 2), suggesting that diverse phenotypes driven by the ANXA11 mutations require long-term patient follow-ups. Of the six mutations, four mutations that were related to the ALS–FTD phenotype were clustered in ANXA11 within the long N terminus. The P36R, G38R, D40Y, and D40G mutations are near the calcyclin-binding domain in annexin 11, indicating the functional importance of this region. We know that calcyclin forms a regulatory complex with the calcyclin-binding protein (CACYBP) and RING-type E3 ubiquitin ligase SIAH-1, thereby regulating the ubiquitination and degradation of many proteins, including β-catenin (27). Therefore, calcyclin plays a critical role in proteostasis. However, the pathogenetic mechanism of ANXA11 mutations leading to ALS–FTD is unclear. Teyssou et al. performed the neuropathological analysis for the G38R case and revealed that FTLD–TDP type A allocations were elicited by the deposition of a mass of TDP-43 lesions in the cortex (31). In patients with ALS, TDP-43 lesion allocations are common because it is associated with a pure FTD phenotype or behavior, related to non-fluent aphasia, or linked to the GRN or C9orf72 mutation (32). Currently, in vivo and in vitro experiments are warranted to further this area of research.
In conclusion, this study confirmed the essential role of ANXA11 mutations in ALS and ALS–FTD. Our results enhanced the understanding of the clinical spectrum and the underlying mechanisms of ANXA11-related diseases, including typical ALS, hIBM, FTD, and their combinations.
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found at: National Center for Biotechnology Information (NCBI) BioProject, https://www.ncbi.nlm.nih.gov/bioproject/, PRJNA832024.
The studies involving human participants were reviewed and approved by the Ethics Committee of China-Japan Friendship Hospital (2021-1-Y0). The patients/participants provided their written informed consent to participate in this study.
YW, XD, and DP designed the study. YW, XD, XZho, RW, and DP contributed patient material and clinical data. XW, ZC, XZho, ZZ, XZha, and YS carried out the experiments. YW, XD, DP, and RW analyzed and interpreted the data. YW and XD wrote the manuscript. All authors have made significant contributions and have approved the final version of this manuscript.
This study received funding from Deutsche Herzstiftung e.V. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. All authors declare no other competing interests. Constanze Pfitzer is participant in the BIH Charité Clinician Scientist Program funded by the Charité—Universitätsmedizin Berlin and the Berlin Institute of Health. This work was supported by the Competence Network for Congenital Heart Defects (Federal Ministry of Education and Research/grant number 01GI0601) and the National Register for Congenital Heart Defects (Federal Ministry of Education and Research/grant number 01KX2140). We acknowledge financial support by Land Schleswig- Holstein within the funding program Open Access Publikationsfonds.
Authors ZC and XW are employed by Running Gene Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ringholz G, Appel S, Bradshaw M, Cooke N, Mosnik D, Schulz P. Prevalence and patterns of cognitive impairment in sporadic ALS. Digest World Core Med J. (2006) 65:586–90. doi: 10.1212/01.wnl.0000172911.39167.b6
2. Wheaton M, Salamone A, Mosnik D, Mcdonald R, Appel S, Schmolck H, et al. Cognitive impairment in familial ALS. Neurology. (2007) 69:1411–7. doi: 10.1212/01.wnl.0000277422.11236.2c
3. Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. (2013) 14:248–64. doi: 10.1038/nrn3430
4. Burrell J, Kiernan M, Vucic S, Hodges J. Motor neuron dysfunction in frontotemporal dementia. Brain. (2011) 134:2582–94. doi: 10.1093/brain/awr195
5. Van Langenhove T, Piguet O, Burrell J, Leyton C, Foxe D, Abela M, et al. Predicting development of amyotrophic lateral sclerosis in frontotemporal dementia. J Alzheimers Dis. (2017) 58:163–70. doi: 10.3233/JAD-161272
6. Van der Zee J, Gijselinck I, Van Mossevelde S, Perrone F, Dillen L, Heeman B, et al. TBK1 mutation spectrum in an extended European patient cohort with frontotemporal dementia and amyotrophic lateral sclerosis. Hum Mutat. (2017) 38:297–309. doi: 10.1002/humu.23161
7. DeJesus-Hernandez M, Mackenzie I, Boeve B, Boxer A, Baker M, Rutherfoed N, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
8. Renton A, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs J, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
9. Kabashi E, Valdmanis P, Dion P, Spiegelman D, McConkey B, Velde C, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. (2008) 40:572–4. doi: 10.1038/ng.132
10. Sreedharan J, Blair I, Tripathi V, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. (2008) 319:1668–72. doi: 10.1126/science.1154584
11. Crook A, Williams K, Adams L, Blair I, Rowe D. Predictive genetic testing for amyotrophic lateral sclerosis and frontotemporal dementia: genetic counselling considerations. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:475–85. doi: 10.1080/21678421.2017.1332079
12. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. (2016) 17:122. doi: 10.1186/s13059-016-0974-4
13. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
14. Shepheard S, Parker M, Cooper-Knock J, Verber N, Tuddenham L, Heath P, et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (2021) 92:510–8. doi: 10.1136/jnnp-2020-325014
15. Zhang K, Liu Q, Liu K, Shen D, Tai H, Shu S, et al. ANXA11 mutations prevail in Chinese ALS patients with and without cognitive dementia. Neurol Genet. (2018) 4:e237. doi: 10.1212/NXG.0000000000000237
16. Nahm M, Lim S, Kim Y, Park J, Noh M, Lee S, et al. ANXA11 mutations in ALS cause dysregulation of calcium homeostasis and stress granule dynamics. Sci Transl Med. (2020) 12:3993. doi: 10.1126/scitranslmed.aax3993
17. Smith B, Topp S, Fallini C, Shibata H, Chen H, Troakes C, et al. Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci Transl Med. (2017) 9:eaad9157. doi: 10.1126/scitranslmed.aad9157
18. Müller K, Brenner D, Weydt P, Meyer T, Grehl T, Petri S, et al. Comprehensive analysis of the mutation spectrum in 301 German ALS families. J Neurol Neurosurg Psychiatry. (2018) 89:817–27. doi: 10.1136/jnnp-2017-317611
19. Teyssou E, Muratet F, Amador M, Ferrien M, Lautrette G, Machat S, et al. Genetic screening of ANXA11 revealed novel mutations linked to amyotrophic lateral sclerosis. Neurobiol Aging. (2021) 99:102.e11–20. doi: 10.1016/j.neurobiolaging.2020.10.015
20. Nel M, Mahungu A, Monnakgotla N, Botha G, Mulder N, Wu G, et al. Revealing the mutational spectrum in southern Africans with amyotrophic lateral sclerosis. Neurol Genet. (2022) 8:e654. doi: 10.1212/NXG.0000000000000654
21. Nunes Gonçalves J, Leoni T, Martins M, Peluzzo T, Dourado M, Saute J, et al. Genetic epidemiology of familial ALS in Brazil. Neurobiol Aging. (2021) 102:227.e1–4. doi: 10.1016/j.neurobiolaging.2021.01.007
22. Leoni T, González-Salazar C, Rezende T, Hernández A, Mattos A, Neto A, et al. A novel multisystem proteinopathy caused by a missense ANXA11 variant. Ann Neurol. (2021) 90:239–252. doi: 10.1002/ana.26136
23. Dols-Icardo O, García-Redondo A, Rojas-García R, Borrego-Hernández D, Illán-Gala I, Muñoz-Blanco J, et al. Analysis of known amyotrophic lateral sclerosis and frontotemporal dementia genes reveals a substantial genetic burden in patients manifesting both diseases not carrying the C9orf72 expansion mutation. J Neurol Neurosurg Psychiatry. (2018) 89:162–8. doi: 10.1136/jnnp-2017-316820
24. Liu X, Wu C, He J, Zhang N, Fan D. Two rare variants of the ANXA11 gene identified in Chinese patients with amyotrophic lateral sclerosis. Neurobiol Aging. (2019) 74:235.e9–12. doi: 10.1016/j.neurobiolaging.2018.09.020
25. Tsai P, Liao Y, Jih K, Soong B, Lin K, Lee Y. Genetic analysis of ANXA11 variants in a Han Chinese cohort with amyotrophic lateral sclerosis in Taiwan. Neurobiol Aging. (2018) 72:188.e1–2. doi: 10.1016/j.neurobiolaging.2018.07.002
26. Sainouchi M, Hatano Y, Tada M, Ishihara T, Ando S, Kato T, et al. A novel splicing variant of ANXA11 in a patient with amyotrophic lateral sclerosis: histologic and biochemical features. Acta Neuropathol Commun. (2021) 9:106. doi: 10.1186/s40478-021-01202-w
27. Fukushima T, Zapata J, Singha N, Thomas M, Kress C, Krajewska M, et al. Critical function for SIP, a ubiquitin E3 ligase component of the beta-catenin degradation pathway, for thymocyte development and G1 checkpoint. Immunity. (2006) 2:29–39. doi: 10.1016/j.immuni.2005.12.002
28. Wang J, Guo C, Liu S, Qi H, Yin Y, Liang R, et al. Annexin A11 in disease. Clin Chim Acta. (2014) 431:164–8. doi: 10.1016/j.cca.2014.01.031
29. Hofmann S, Franke A, Fischer A, Jacobs G, Nothnagel M, Gaede K, et al. Genome-wide association study identifies ANXA11 as a new susceptibility locus for sarcoidosis. Nat Genet. (2008) 40:1103–6. doi: 10.1038/ng.198
30. Feng X, Zang S, Yang Y, Zhao S, Li Y, Gao X, et al. Annexin A11 (ANXA11) gene polymorphisms are associated with sarcoidosis in a Han Chinese population: a case-control study. BMJ Open. (2014) 4:e004466. doi: 10.1136/bmjopen-2013-004466
31. Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. (2011) 122:111–3. doi: 10.1007/s00401-011-0845-8
Keywords: annexin A11, ANXA11, amyotrophic lateral sclerosis, frontotemporal dementia, genotype, phenotype [mesh]
Citation: Wang Y, Duan X, Zhou X, Wang R, Zhang X, Cao Z, Wang X, Zhou Z, Sun Y and Peng D (2022) ANXA11 mutations are associated with amyotrophic lateral sclerosis–frontotemporal dementia. Front. Neurol. 13:886887. doi: 10.3389/fneur.2022.886887
Received: 01 March 2022; Accepted: 05 August 2022;
Published: 26 September 2022.
Edited by:
Liyong Wu, Capital Medical University, ChinaReviewed by:
Maria Claudia Torrieri, University of Turin, ItalyCopyright © 2022 Wang, Duan, Zhou, Wang, Zhang, Cao, Wang, Zhou, Sun and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dantao Peng, cGVuZ2RhbnRhbzIwMDBAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.