 Mohamed R. Khalife
Mohamed R. Khalife Rod C. Scott
Rod C. Scott Amanda E. Hernan
Amanda E. Hernan- 1Division of Neuroscience, Nemours Children's Health, Wilmington, DE, United States
- 2Psychological and Brain Sciences, University of Delaware, Newark, DE, United States
- 3Institute of Child Health, Neurosciences Unit University College London, London, United Kingdom
There has been a major emphasis on defining the role of seizures in the causation of cognitive impairments like memory deficits in epilepsy. Here we focus on an alternative hypothesis behind these deficits, emphasizing the mechanisms of information processing underlying healthy cognition characterized as rate, temporal and population coding. We discuss the role of the underlying etiology of epilepsy in altering neural networks thereby leading to both the propensity for seizures and the associated cognitive impairments. In addition, we address potential treatments that can recover the network function in the context of a diseased brain, thereby improving both seizure and cognitive outcomes simultaneously. This review shows the importance of moving beyond seizures and approaching the deficits from a system-level perspective with the guidance of network neuroscience.
Introduction
Epilepsy is a disorder of the brain characterized by an enduring predisposition to generate seizures and by the neurobiological, cognitive, psychological, and social consequences of this condition (1). Although seizures are an important part of the definition, the associated cognitive and behavioral impairments and learning and memory problems are also important determinants of quality of life (2, 3). The quality of life is highly affected in patients with epilepsy especially if they are in adolescence, affecting the patient's self-esteem and sense of coherence (4). Self-esteem is a main contributor to psychosocial wellbeing, personal reflection, and positive attitude (5), and coherence is the ability to recognize stressors as manageable and solvable (4, 6). In a 5-year follow up study, it was shown that sense of coherence decreased in adolescents with epilepsy. Adolescents who still had seizures showed a greater decrease compared to seizure-free teens, with no effect of seizure frequency on sense of coherence (4). Self-esteem was also decreased in adolescents with epilepsy, however, self-esteem was affected by the seizure frequency where higher seizure frequency was associated with lower self-esteem (4). This indicates that psychosocial wellbeing is affected in adolescents with epilepsy seen by the decrease of both self-esteem and sense of coherence.
Gauffin et al. (7) examined the experience of living with epilepsy and cognitive decline. Their study found out that cognitive decline is persistently present in adults with intractable epilepsy and living with epilepsy and cognitive deficits affected education, employment, self-esteem, social life, and future plans (7). The cognitive deficits are seen in both focal and generalized seizures. Focal seizures occur in a lateralized network in contrast to generalized seizures occurring in a widespread network encompassing both hemispheres (8), however similar cognitive impairments are seen in both epilepsies (9). Patients with focal epilepsy experience various cognitive impairments such as language abnormalities, executive dysfunction, attention deficit and long-term episodic and semantic memory deficits (9–11). Patients with generalized epilepsy experience the same cognitive impairments in addition to acquired knowledge deficits and long-term information processing and retrieval impairment (9, 12, 13).
A common pathophysiological argument for the mechanisms of the relationship between epilepsy and cognition is that the seizure and epileptiform discharges in the EEG directly injure neural networks that are the normal substrate for cognitive function. An alternative hypothesis is that the relationship is indirect; both the seizures and the additional morbidities arise from neural networks that have been disrupted by the etiology of the epilepsy e.g., single gene disorders, malformations of cortical development, or traumatic brain injury. Our starting position is that the action potential is the fundamental unit of information processing in the brain, and that sequences of action potential firing in neuronal populations over time are therefore considered to be mechanisms of cognition, as cognitive function is explicitly about information processing (14, 15). Our goal in the current article is to explore the latter hypothesis in detail, starting at the systems level physiology of brain function and relating this exploration to our understanding of epilepsy. We will describe these system level mechanisms in physiology (Figure 1A) and discuss how these mechanisms are changed or altered in brain diseases associated with epilepsy (Figure 1B), especially in the hippocampus and neocortex, which are important structures involved in memory.
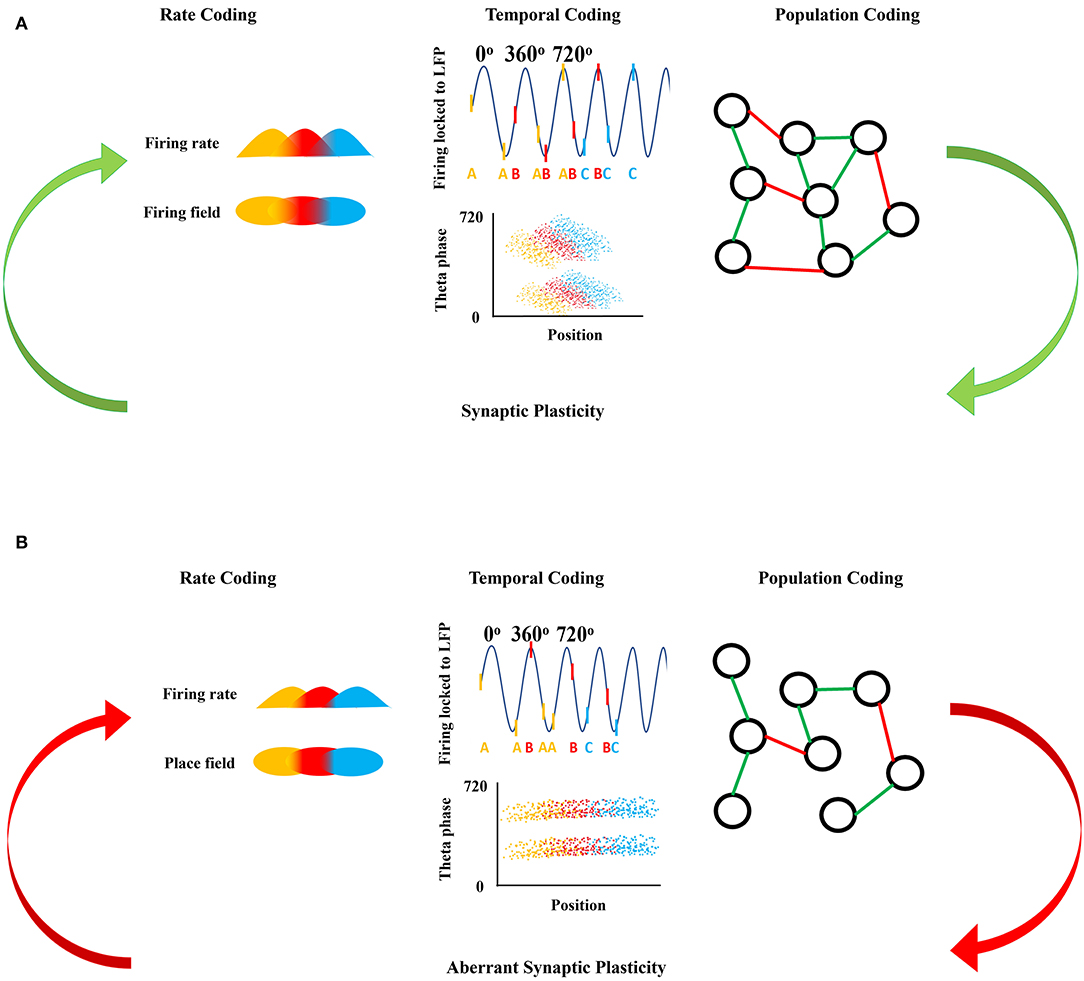
Figure 1. Neuronal firing mechanisms and information processing. (A) Shows the normal information processing and synaptic plasticity feedback loop based on the neural coding mechanisms. Rate coding is shown as the firing rate in time (top) with respect to each firing field in space (bottom) coded by the same color. Temporal coding shows the action potentials as raster ticks riding on the LFP with respect to theta oscillation indicating theta phase precession with each successive peak of theta, with action potentials also represented as a letter at the bottom of the LFP representing firing from the same neuron over time (top). Over many LFP cycles, theta phase precession can be seen in the downward slope of the clouds of dots in the bottom panel, each representing an action potential from three representative neurons show in the top panel. Population coding shows the connections between neurons forming a population network, green representing an excitatory connection between two neurons and red representing an inhibitory connection between two neurons. Synaptic plasticity refines and is refined by these firing mechanisms. (B) Shows the disrupted information processing and synaptic plasticity feedback loop based on the neural coding mechanisms in epileptic brain. In epilepsy, rate coding is disrupted shown here as a decreased firing rate in time with respect to each firing field in space, with decreased overlap in the firing and place fields from each of the three color-coded neurons. Temporal coding is also altered; firing of the colored neurons with respect to theta oscillation is disorganized and there is an absence of theta precession as shown by a flat relationship between the clouds of action potentials from each of the three colored neurons. Population coding shows fewer connections between neurons forming a smaller population network, with potentially different proportions of positive (green) and negative (red) connections in the epileptic brain compared to controls in (A). Aberrant synaptic plasticity occurs as a result of the aberrant firing dynamics.
Rate Coding
The firing rate of neurons in the brain is a mechanism of information transfer (15). For example, in motor neurons, the degree of muscle flexion depends on the number of action potentials per unit time (14). Tactile and texture perception in rodents is also, at least in part, a function of rate coding (16). In hippocampus and entorhinal cortex, rate coding is illustrated by place, time, and grid cells, respectively (17, 18).
Place cells are hippocampal pyramidal cells that fire when an animal visits a specific region of the environment: the cell's “place field” (19, 20). In any given environment, place fields cover the entire space to create a hippocampal cognitive map that is a representation of that space (19, 20). Repeated recordings of place cell populations have shown that the same cells are activated whenever the animal visits the same region in physical space, which suggests that the cells' representation is held in the network once the animal explores that specific field (21). This representation allows recollection of specific spaces and accurate navigation through the environment. This is further supported by the observation that lesioning the hippocampus results in loss of spatial memory (22). Similarly to place cells, time cells in the hippocampus fire at specific times in a task (e.g., the beginning, middle, or end) called time fields (23). These cells can be time locked to an external stimulus, like a tone, or intrinsically by a neural circuit or an oscillation (14, 15).
Grid cells are entorhinal cortical cells that provide activity-based maps of speed and direction in a certain environment (24). Grid cells fire in different locations in an environment forming a triangular grid. Recording different cells at the same location indicates that these cells have the same orientation to the environment (24, 25). It is important to note that the orientation of the grid relative to the environment is dependent on the hippocampal place cell map (20, 25, 26). Interestingly, the map formed by the grid cells is based on external cues, however the formed map persists even in the absence of these cues (24). Together with the place cell map, the grid cell map is believed to be part of the greater hippocampal cognitive map (20). Given what we know from work on place and grid cells, further experiments have shown that firing rate variations in CA3 place cells depended on signals from the lateral entorhinal cortex (24, 27). Lesioning the lateral entorhinal cortex impairs the hippocampal rate remapping upon changing the configuration of the environment (24, 27) suggesting that inputs from the entorhinal cortex are important for hippocampal rate coding in the formation of the spatial memory and cognitive map.
Population Coding
Rate coding as measured in individual neurons is essential for information processing. However, neurons are functionally connected into a network and interactions between the neurons is also critically important (15, 28). This is known as population coding. Population coding increases robustness of network function and minimizes the effects of noise carried by an individual neuron, ensuring that the signal and processing times are not affected (15). For example, damage to one cell will not have a devastating effect on the information being carried since it is carried by many cells (28). Population coding is common in the nervous system and is illustrated in mammalian visual pathways (29), primary motor cortex activity in cats and monkeys (30), owl auditory cortex, cricket nervous system (31), and mice visual cortex (32–34). Neurons in the visual cortex in cats and monkeys and the auditory cortex in owls have shown the ability to synchronize their firing on a few milliseconds time scale through time cells (14, 35). Synchrony seen in the visual cortex of cats and monkeys takes place when the neurons are activated by one stimulus (35), and this synchrony is lost when the neurons are activated by two independent stimuli. Once two neurons synchronize to represent a certain stimulus, these two neurons always synchronize to represent the same stimulus and will desynchronize when representing two independent stimuli (14).
Population coding influences information processing in mice visual systems. Whole cell recordings in layers 2/3 (L2/3) of awake mice have shown that the excitation/inhibition ratio changes based on the visual stimulus (33). Different studies revealed that patterns of excitation and inhibition are generated in response to various visual stimuli (36, 37). For example, as the stimulus contrast or size increases, the excitation/inhibition ratio decreases. This change in ratio is important for tuning and sharpening of the information processing in the visual system (38) and is thought to be controlled by the somatostatin (SOM) neurons and parvalbumin (PV)-expressing cells. SOM neuron suppression was able to enhance the excitation and inhibition for size tuning, indicating that contrast and size in the visual system in mice depends on excitation/inhibition ratio and tuning of total synaptic input (33). PV cell manipulation was shown to modulate L2/3 pyramidal cells spikes in response to visual stimuli without affecting the cells tuning properties suggesting that PV cells create a connection between certain neuron types and specific computations during sensory processing (32, 39). Furthermore, the excitation/inhibition ratio for the same set of stimuli differs between anesthetized and awake mice and even differs between various behavioral states (33). This change in the ratio is influenced by the recruitment of SOM and vasoactive intestinal peptide-expressing (VIP) inhibitory cells in V1 in both wakefulness and alertness for instance (40–42). The Stabilized Supralinear Network (SSN) model proposes that cortical dynamics can change the excitation/inhibition ratio based on single neurons input/output supralinear relationships, strong recurrent excitation, and feedback inhibition (33, 43). This suggests that the dynamic change of excitation/inhibition ratio depends on the dynamic connections between different cortical regions and the feedback loops between the recruited regions. Further support to this suggestion was seen in experiments investigating the rhythmic activity across cortical layers. Population of excitatory neurons in the mouse's primary visual cortex expressed gamma band oscillation following layer-specific optogenetic stimulation (34). Importantly, rhythm-generating circuits in each layer were able to provide layer-specific excitation/inhibition balances hence influencing the information flow between cortical layers (34). These studies provide evidence that neuronal populations can be recruited through precise synchrony contributing to information processing at a system level.
Notably, place cells in the hippocampus are an excellent example of not only rate coding, but also population dynamics. We previously mentioned that the place cells fire whenever the animal is in a specific place field and these cells are distributed within the hippocampus in a way that covers the whole environment the animal occupies. This distribution within the hippocampus will cause a certain degree of overlap between the place fields, thus a population of cells will respond when the animal goes into the field rather than an individual cell since the cells in the hippocampus are receiving multiple sensory inputs to encode a multidimensional map (44).
Another interesting example of population coding is pattern separation. Pattern separation depends on the discrimination between two closely related places, episodes, or spatial configurations based on experience and can influence successful memory encoding. Pattern separation involves different brain regions where experiences are represented by neural populations. Notably, if the same population or neural pattern established during encoding is activated during retrieval, it can lead to a successful memory retrieval (45). For instance, both the posterior occipitotemporal cortex (OTC) and the hippocampus were recorded while participants performed item recognition tasks. Upon retrieval, both regions showed encoding-specific high frequency activity (HFA) where the strength of this activity was associated with enhanced retrieval, however the discrimination between similar items required a hippocampal activity (45) as the pattern separation mechanism is based on orthogonalizing similar input during encoding thus enabling the distinguishing between highly similar memories with minimal interference (46). Animal and human studies have shown that dentate gyrus (DG) and its projection to CA3 underlie the pattern separation process (47–52).
A final relevant example of population coding is working memory in the prefrontal cortex. Working memory is the temporary maintenance of information involving specialized components of cognition that allows retaining immediate past-experience, supporting new knowledge acquisition, solving problems, reasoning, and planning (53). Early models of working memory suggested that persistent firing activity of the neurons in the prefrontal cortex (PFC) throughout the delay phase of the working memory task was required to maintain information in working memory, however, due to the heterogeneity of neurons within the PFC, recent work has shown that the persistent activity of PFC can be weak or absent (54–56). This was seen during the delay phase of an image-sequence matching task in monkeys and humans. In this task, spiking activity in the PFC decreased during the delay phase of the task in monkeys, and BOLD signal on fMRI decreased during the delay phase in humans performing the task, thus challenging the persistent firing working memory model (57, 58). However, the spiking activity and the BOLD signal re-emerge during image presentation and testing period, indicating that, despite a decrease in firing rate throughout the task, the working memory information is maintained in the collective synaptic weights of populations of neurons in the PFC. The heterogeneity of the neurons allows different neurons to signal during different task events as well. For example, parvalbumin-positive neurons will respond to sensory cues and trial outcomes while somatostatin-positive neurons will respond to a motor action like licking (59). These neurons can fire together in working memory maintenance through population coding when a robust mnemonic stimulus is present (60) as seen in monkeys performing oculomotor delayed response and vibrotactile delayed discrimination, which are working memory tasks, while recording single neurons of lateral PFC (60).
Temporal Coding and Oscillatory Firing
Oscillatory activity is divided into frequency bands as described in Table 1: infra-slow oscillations (0.5–1 Hz), delta (1.5–4 Hz), theta (4–8, 10 Hz), alpha (8, 10–12 Hz), beta (15–30 Hz), gamma (30–80 Hz), in addition to fast (80–200 Hz), and ultra-fast (200–600 Hz) ripples (61). Each band is thought to be related to specific aspects of cognition (Table 1). Both in vivo and in vitro experiments suggest that synaptic inhibition plays a role in generating neuronal oscillations through two different mechanisms, either through interneuron network activity or reciprocal excitatory-inhibitory loops (62). Theta, as previously discussed, and gamma are two important readouts of the hippocampal function and function of connected regions.

Table 1. Frequency bands and cognitive processes.
Theta rhythm indicates a network that is actively involved in spatial navigation, working memory, and temporal coding (63, 64). It is important to note that theta oscillations and phase-locked neuron discharges with respect to theta oscillations are seen in theta non-generating regions like entorhinal cortex, perirhinal cortex, cingulate cortex, subicular complex, and amygdala (63, 65–67). Lesion or inactivation of the medial septum-diagonal band of Broca neurons recognized as the theta rhythm generators eliminates theta oscillation in all connected cortical regions (68) and leads to spatial and working memory deficits (69–71).
Gamma oscillations ride on top of theta in the hippocampus and dynamically couple hippocampal networks to specific behavioral demands (39, 72, 73). The gamma oscillation is thought to be a readout of information transfer from CA3 to CA1, where CA3 is influencing the activity of CA1 for hippocampal memory retrieval to underlie memory encoding, consolidation, and episodic memory retrieval. Movement sequences, memory encoding and formation, sensory processing and planned trajectories underlying spatial navigation are thought to be temporally organized through timing mechanisms seen through gamma oscillations (74, 75).
The rate of populations of neuronal firing is also modulated in time in respect to theta frequency. For example, in the hippocampus, temporal modulation is manifested as burst firing with bursts occurring at theta frequency (4–12 Hz). The fidelity of burst firing within theta, termed theta modulation, is important for phenomena such as phase precession, phase preference and hippocampal replay, which are believed to allow encoding of space with higher resolution than is possible in the absence of modulation. Phase preference refers to the phenomenon that neuronal firing in the hippocampus is often locked in time with respect to ongoing hippocampal inputs in the theta frequency of the local field potential (LFP). Specific cells fire preferentially at specific phases in the ongoing theta oscillation. For example, directly after the peak of the theta oscillation, PV+ basket cells in CA2/3 area fire at the same phase as pyramidal cells in CA3, but later than basket cells in CA1 (76). CA1 pyramidal cells preferentially fire at the trough of theta (77, 78). Phase precession describes the observation that place cells will discharge whenever the rodent is crossing a place field and this firing occurs at an earlier phase in theta with progressive theta cycle (79, 80). This phase precession is believed to be an important component of information processing. Theta-phase precession could be an indication of item-context associations through spike timing-dependent plasticity (80, 81) given that synaptic inputs need to be precisely synchronized within 5 ms so that EPSPs from different locations will be able to induce postsynaptic firing (15). Umbach et al. showed that neurons in the hippocampus and entorhinal cortex not only fire for space, but also for time. Interestingly, time cells also exhibited theta-phase precession during memory encoding, and the activity of these cells correlates with the use of temporal location during the retrieval phase of the task (82). Neuronal firing coordination with the LFP, like phase-locking and phase precession, offers a key glimpse at the relative timing of inputs in the LFP with outputs of the information processing as the neuronal firing, but neural oscillations are also readouts of synchronized behavior of the network and, as such, are on their own important mesoscale mechanisms of cognition, memory, and behavior.
Coordination between oscillations seen in the LFP or EEG can be an indicator of communication between different brain regions (83, 84). Coherence is a measure of this synchronization with values that range between 0 and 1. The higher the coherence, the more synchronized the regions are (84). The coherence value differs between different brain regions depending on the task performed, for example, theta coherence between hippocampus and striatum during periods of decision is high (>0.8) which indicate learning (84, 85). Not only theta coherence changes during a task, but also gamma as well. Attention tasks in monkeys have shown that gamma coherence increases between the parietal and prefrontal areas (86). Furthermore, CA1 can become coherent with the entorhinal cortex or CA3 through fast or slow gamma characteristics of the entorhinal cortex or CA3, respectively (87). Elevation of hippocampal-entorhinal cortex synchrony was shown to be important for declarative memory formation in epileptic patients performing a memorization task (88). This led to the hypothesis that synchronized brain activity in the gamma range might be an important indicator of controlled flow and routing of information (83, 89), because the rules guiding synaptic plasticity dictate that inputs will be most effective whenever they coincide with peaks of oscillatory network excitability (83). Finally, neural coherence alterations were associated with different disorders like schizophrenia, attention-deficit hyperactivity disorder (ADHD), Alzheimer's disease (AD), and temporal lobe epilepsy (TLE) (90–95) (Table 2).
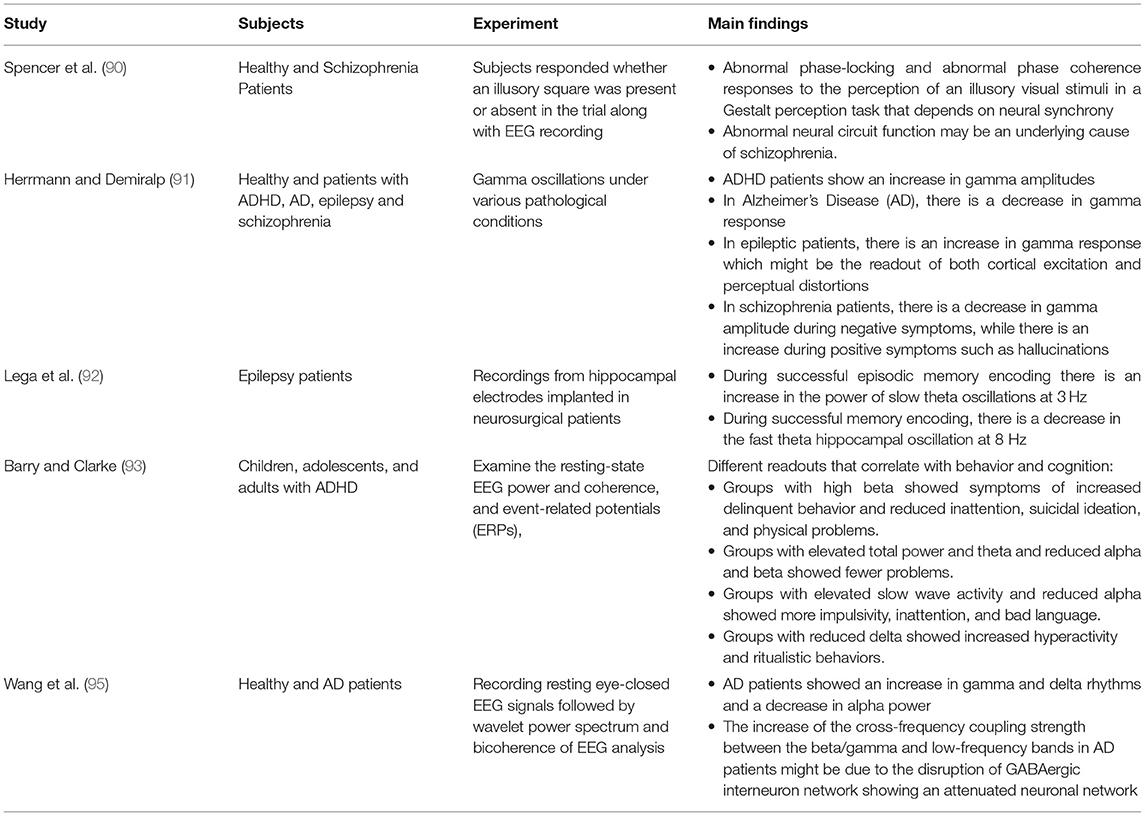
Table 2. Summary of cited clinical literature.
Firing Dynamics Support Plasticity
Temporal, population, and rate coding facilitate plasticity shaped through experiences that enable the brain to adapt to new information. These mechanisms underlie the careful coordination of information between synapses and neurons in the brain that is necessary to promote synaptic plasticity and ensure efficient flow of information between different brain regions required for cognition. In 1949, Donald Hebb postulated that synapse strength can change based on previous activity, which led to what we now know as long-term potentiation (LTP) and long-term depression (LTD), fundamental to network communication. LTP strengthens synaptic transmission through high frequency stimulation of synapses. The first stage of LTP depends on the NMDA and AMPA glutamate receptors (96, 97). The second and third stages of LTP depend on protein synthesis to maintain changes in synaptic strength (96, 97). Maintenance of LTP is essential for place cell stability (98–101). Even though neural plasticity is not a determinant of place cell spatial specificity, rats with neural plasticity deficits had unstable place fields upon revisiting the same environment (102, 103). This shows that neural plasticity is playing a role in the organization of place cells and long-term maintenance of this representation.
The frequency of action potential timing matters; low frequency firing induces LTD, which decreases synaptic efficacy. LTD is also essential for memory formation as it counteracts the LTP to allow new memories to form. Recent evidence has shown that LTD may be involved in formation and maintenance of place fields (104), supporting previous experiments showing that a decrease in the expression of LTD impairs spatial memory retention and consolidation (105, 106). It is worth noting that plastic synapses can form positive feedback loops on the rate, temporal, and population coding mechanisms where this positive loop aids the mechanisms in refining and precisely timing the neuronal firing leading to a more efficient information processing. Another form of plasticity known as short-term plasticity (STP) takes place on a millisecond to minutes timescale and depends on presynaptic calcium accumulation and vesicle depletion (107). This form of plasticity is thought to play a role in information transfer across synaptic connections, activity-dependent synaptic efficacy modulation, promoting synchronization and working memory (107–109). Careful coordination of the firing of populations of neurons in time supports appropriate short and long-term forms of plasticity that are critical for information processing, learning and memory.
Epilepsy and Memory
Cognitive impairments in people with epilepsy are extremely common and have a major negative influence on quality of life. Patients with focal epilepsy have shown to have a significant decrease in their quality of life compared to patients with generalized epilepsy and healthy controls, however, both focal and generalized epilepsy patients have a decreased self-esteem and increased anxiety compared to healthy controls (9). Identification of mechanisms of cognitive impairment is important as this will help to guide development of novel therapeutic strategies to improve outcomes. A major emphasis has been on studying the impact of seizures on cognition and exploring the epileptic encephalopathy hypothesis. Researchers have focused on the time of onset and frequency of seizures, however, there are few clear correlations between seizure characteristics and cognitive outcomes. Taken into consideration that cognitive functions are dependent on complex brain networks and both focal and generalized epilepsy groups share the same cognitive impairments (9), we can say that seizure location is less of a determinant of the cognitive impairments than an altered or dysfunctional network in the brain. This suggests that there must be other factors influencing cognitive outcomes in epilepsy. This assertion is supported by several lines of evidence. Studies assessing the effect of age of onset on cognitive impairment have shown that cognitive impairment already exists at pretreatment baseline in newly diagnosed children (3, 110–112). This led some researchers to question whether cognitive deficits could be present even before seizure onset (113) and this might suggest that the pre-existing impairments could be a result of the same dysregulation that underlies the seizures in the first place. Indeed, the need for special educational services prior to epilepsy onset is more common in children who were later diagnosed than those who weren't (114), suggesting that impairment in cognitive function may be present even before the first seizure. The best predictor of cognitive outcome up to 3 years after the diagnosis of epilepsy in infants is the initial cognitive profile, not any seizure or medication related factor. In adults, these impairments extend to deficits in visual motor tasks, mental flexibility, memory, reaction times, and attention (3, 115).
Others have studied the effects of the disorder duration on cognition, finding a negative correlation between the number of years of the disorder and brain volume (116, 117). This has been interpreted by some to mean that the length time since disease onset is related to the amount or significance of cognitive impairment. However, an alternate interpretation is that early onset of an epilepsy disorder is an indication of a more fundamentally dysfunctional network, leading to early development of recurrent seizures. Similarly, seizure frequency has been noted to be associated with a detrimental effect on cognition, with higher seizure frequency being correlated with lower performance on cognitive tasks and vice versa (9, 118). This was always interpreted to mean that seizures themselves were detrimental to cognition, rather than more frequent seizures being an indicator of a more abnormal neural network that underlies abnormal functioning during the interictal period manifesting as cognitive impairment. Hence, these impairments might not be due to age of onset or frequency of seizures, instead it could be attributed to the fact that children in this situation have a brain disease that presented earlier, and this difference in disease presentation may be an indicator of more severe network dysfunction, and more severe impairments. Further investigation of memory dysfunction in patients with epilepsy showed that people with epilepsy had significant deficits in both semantic and episodic autobiographical memory (9, 119, 120). This deficit was associated with young age at onset, more frequent seizures, and reduced working memory in early-onset epilepsy patients. In contrast, the same deficit was related to depression and lesion (3, 9, 120). The presence of both neurobiological and psychological factors suggests that information processing mechanisms might be altered (121).
Information processing through the mechanisms discussed in this review is shown to be altered in epilepsy and associated disorders. CA1 place cells are unstable in epileptic mice and undergo remapping a few weeks after pilocarpine-induced temporal lobe epilepsy (TLE). The number of place cells decreases, and the spatial tuning curve is less stable over time (122, 123). Prolonged recording over days from populations of neurons in CA1 and dentate gyrus has shown desynchronized interneuron firing between these two areas (124), which suggests that disruption of spatial coding is due to the loss of information processing control by interneurons. The desynchronized interneuron firing can affect the timing of the inputs being sent to the CA1. This was supported by the observation of theta rhythm temporal coordination loss in the dentate gyrus, where these neurons were firing at inconsistent phases of the CA1 theta rhythm (124). Spatial memory alteration was previously shown to be present even during the latent, seizure-free, period after either the pilocarpine-induced status epilepticus (SE) or early life seizures during the 1st weeks of life (39, 125, 126). These deficits were associated with a decrease in the power of theta oscillations (125). It is important to note that spontaneous seizures did not modify or affect any of the spatial deficits that were already present (125). Interestingly, Shuman et al. (124), found that place cell deterioration and place coding alteration occurred several weeks after pilocarpine induction, showing that the development of seizures is not solely responsible for place cell deterioration. Notably, place coding alteration, place cell deterioration, more dispersed place fields, and fewer place field responses were also seen after silencing either CA3, entorhinal cortex or both (124, 127, 128).
In addition, we and others have shown dysregulated population coding in epilepsy models. In-vivo single-unit recording showed that CA1 pyramidal cells are functionally connected to other pyramidal cells and fire in a coordinated fashion during spatial memory tasks; this connectivity is altered in TLE where neuronal reactivation and synchrony predicts the behavioral outcome in a TLE model (129). Population coding functional connectivity is also crucial within the hippocampus and between the hippocampus and PFC to underlie spatial working memory (SWM) (130, 131). During a SWM task, the hippocampal-PFC network shows a distributed dynamic code, seen through temporally regulated firing within and between brain regions, which is needed to combine separate processes together to execute a SWM task (131). The coordinated firing of cells in time is important for several components like attention, decision making and long-term memory, which can predict task performance. The temporal modulation of populations of neurons predicted SWM accuracy in a delayed non-match-to-sample task in control rats and rats with a cortical malformation that, in humans, is an important etiology in epilepsy. Animals with cortical malformations showed deficits in hippocampal firing modulation in addition to decreased functional connectivity between neurons (131).
Furthermore, population coding and neural dynamics are important for pattern separation and this process has been shown to be altered in hippocampal injury and epilepsy. The pattern separation depends on a network spanning different brain regions other than hippocampus, like the dorsal medial prefrontal cortex (dmPFC), however the hippocampus and the parahippocampal cortex serve as a hub for this network (132), thus it is expected that a hippocampal injury will alter the network communication causing pattern separation deficits (47, 133, 134). TLE patients and amnestic mild cognitive impairment (aMCI) patients have pattern separation deficits, and this could be due to hippocampal dysfunction involving DG and CA3 (46, 135). Another reason could be due to the failure of separating similar information during encoding by the hippocampus, hence memories will not be accurately encoded or retrieved. Studies investigating aMCI and TLE patients have shown that aMCI patients have an excess activation of the DG/CA3 area in fMRI compared to control groups and this excess activation is correlated with poor performance on pattern separation tasks. The same poor performance was seen in TLE patients performing the Mnemonic Similarity Task (MST). TLE patients demonstrated poor pattern separation performance compared to controls, however, it is important to note seizure and hippocampal sclerosis did not affect the performance of patients in this task (46, 133). Following studies showing that TLE patients have spatial mnemonic discrimination impairment and that TLE mice have DG-dependent object location memory deficits (50, 136), Madar et al. (52) tested pattern separation in TLE patients and mice with TLE, and then used mouse brain slices to record the spiking patterns of single granule cells (GC) in the dentate gyrus. TLE patients performing object recognition-based MST had a significant deficit in identifying similar but not identical objects suggesting that TLE might be impairing the DG-dependent mnemonic discrimination. Similar deficits were seen in mice with TLE as the mice had a decrease in object-location mnemonic discrimination compared to control mice (52). Slice electrophysiology in the same mice utlized inputs mimicking the same recorded inputs during behavior, and indicated that the output spike-trains of GCs had a higher average correlation compared to input correlation, which signifies a deficit in pattern separation in mice with TLE. Different input ranges demonstrated decreased pattern separation and convergence in DG at multiple timescale levels (52). This shows the importance of population dynamics underlying spatial deficits and signals the importance of assessing functional connectivity.
Imaging and histological experiments showed that structural and functional connectivity were altered in TLE patients as well (3, 137). Histological changes have been observed in the amygdala, entorhinal and parahippocampal cortices in TLE patients (3, 138–142). MRI images investigating hippocampal sclerosis associated with TLE, found that in addition to hippocampus, atrophy is present in the adjacent mesiotemporal, temporopolar structures, and thalamus (3, 143–146), and this atrophy increases over time (147–149). Experiments investigating tissue microstructure and structural covariance indicate that structural connectivity was impacted in TLE. Diffusion tensor MRI showed a disorganization in fiber arrangement in temporolimbic and adjacent regions (3, 150–153). Structural covariance such as cortical thickness or gray matter volume was altered between the mesiotemporal and neocortical regions and within the corticocortical networks (154–156). Resting state functional connectivity revealed a deficit in network connectivity in TLE patients compared to healthy controls. TLE patients had a decrease in ipsilateral mesiotemporal networks connectivity and ipsilateral and contralateral hippocampi connectivity (3, 153, 157–159). This decrease in connectivity extends beyond the temporal lobes into the posterior cingulate, inferior parietal, and medial prefrontal cortices disrupting the default mode network (DMN) (160–164). These changes and deficits suggest that structural connectivity is impacted in TLE patients and that TLE is also associated with functional connectivity deficits and reorganization.
Early stage TLE patients experience functional connectivity deficits mainly in the ipsilateral hemisphere (162, 165) in addition to disturbed interhemispheric connections (3, 158, 166). However, in patients with generalized epilepsy, there is an increase in the interhemispheric connectivity in addition to reduced functional connectivity (167–171). fMRI studies investigating the network connections in epileptic brains showed an increase in functional connectivity within the temporal lobe, alongside a decrease between temporal and other regions. Also, there is a decrease in the connection probability between neighboring brain regions, known as the clustering coefficient, within the DMN (161, 172). This decrease in clustering coefficient as well as increased path length, i.e., distance between one node and another, was revealed to be associated with cognitive decline in patients with cryptogenic epilepsy and only seen in patients with cognitive decline (172–174). The decreased cluster coefficient within the DMN could underlie the language impairment in patients with generalized epilepsy without focal brain damage. Gauffin et al. (175) conducted an experiment where patients with generalized epilepsy without focal damage performed a sentence-reading task while going through fMRI. Patients with generalized epilepsy took longer time to read both congruent (simple) and incongruent (complex) sentences compared to healthy controls with no reading time difference between congruent and incongruent sentences in the patients group which suggests that patients perceived both types as complex (175). BOLD fMRI indicated the activation of a left-lateralized frontotemporal network, anterior cingulate cortex and occipital cortex in both patients and controls upon reading both types of sentences (175). However, patients with generalized epilepsy had reduced DMN suppression compared to healthy controls (175). Further lack of suppression was seen in the left anterior temporal lobe and the posterior cingulate cortex, in addition to irregular activation of the right hippocampus proper and right parahippocampal gyrus (175). The reduced DMN activity suppression can be due to reduced functional segregation of the DMN in generalized epilepsy patients (170) where this can alter the balance between activated and deactivated neural networks hence disturbing the cognitive function (176, 177). Further evidence of network alteration in TLE patients was seen by Bernhardt et al. (178) upon analyzing hub nodes between controls and TLE patients. Hubs are also known as nodes that have multiple connections within a network with one central position and the connections formed by the hub nodes are essential for communication and network synchronization (179). Hub nodes in TLE patients were mainly located in the limbic and temporal association cortices instead of being evenly distributed between different lobes and this was thought to be due to connectivity disturbances between the temporolimbic and extratemporal neocortical structures (178) providing evidence that epileptic brains express decreased integration and enhanced segregation (172). It is also important to note that memory impairments are present in patients who don't show a lesion with MRI (180) which further supports the notion that cognitive impairments depend on the affected network rather than a structural lesion (9). These studies emphasize the necessity to move beyond the classical lesion model into a network approach which can provide several advantages by helping track or predict cognitive decline in epilepsy patients, improving diagnosis, and developing more accurate resection surgeries by targeting the areas where the hub nodes are mostly concentrated.
It is critical to note that experimental designs that induce an underlying disorder associated with epilepsy, but in which there are no overt seizures, and no other subclinical epileptiform activity was noted, show changes in information processing and behavioral deficits. Loss of function of sodium channels Nav1.1 associated with human epilepsy in CA1 can cause disruptions to place cells and spatial cognition without producing seizures (181). Nav1.1 knockdown in the medial septum causes alterations in temporal and rate coding in those neurons, and deficits in working memory that are correlated with the degree of LFP alteration in the hippocampus rather than seizure frequency (39, 182). Similar effects are seen in animals with a malformation of cortical development where no overt or subclinical seizures were noted. These animals have reduced fidelity of place cells, reduction in the magnitude of theta modulation, and disrupted population coding in addition to spatial and working memory deficits. The addition of induced seizures in this model did not make the behavioral deficits worse, indicating that the main contributor to the cognitive impairment was the underlying brain substrate and not seizures (183).
Notably, subclinical epileptiform activity or inter-ictal spikes (IIS) can disrupt cognitive function; however the number of spikes is not a reliable indicator of the associated cognitive impairment. Several studies have previously shown that patients with benign epilepsy with centro-temporal spikes experience IQ and school performance deficits (184, 185). These deficits were correlated with the frequency of IIS but not seizure frequency (184, 185). However, this may be related to timing of the IIS relative to ongoing cognitive processing, as the presence of the IIS may be an indicator that the brain is not in a state where it can be performing cognitive computations. Kleen et al. (186) investigated the effect of focal IIS on hippocampus in TLE. They showed that rats with unilateral intrahippocampal pilocarpine infusion developed hippocampal spikes that caused a response latency deficit in hippocampal-dependent operant behavior task, delayed-match-to-sample (186). However, the hippocampal spikes only altered the cognitive performance when they occur at the same time during memory retrieval; spikes occurring during memory encoding or maintenance did not affect the cognitive performance and overall IIS frequency during a trial was not predictive of accuracy during that trial (186). Similar results were seen in patients with refractory seizures performing Sternberg task, a delayed information task that depends on short-term memory processes, along with EEG recordings (187). Contralateral or bilateral to seizure focus hippocampal interictal epileptiform discharges (IED) during memory retrieval disrupted memory retrieval, and bilateral IED during memory maintenance was able to disrupt that process, however no effect was seen on memory encoding (187). These studies show that focal IIS and hippocampal IED are associated with disruptions in memory maintenance and retrieval only when they occur during the same time window as the memory processes. Taken together, this suggests that IIS/IED are indicators of disrupted network processing underlying cognition.
In addition to deficits in rate, temporal and population coding, plasticity deficits are also present in epilepsy, in accordance with the view that these neural coding mechanisms support plasticity. Kainic-acid induced status epilepticus (SE) model in rats shows a significant decrease in hippocampal LTP in addition to cell loss, and signs of hippocampal sclerosis (188). These rats also have deficits in the hippocampal-dependent novel object recognition spatial memory task that positively correlated with LTP magnitudes (188). These findings were also seen in the pilocarpine model where the mice showed a significant decrease in the hippocampal synaptopodin acting-binding protein in CA1 region which alters the ability of the neurons to express synaptic plasticity leading to a decrease in LTP induction in Schaffer collateral-CA1 synapses (189). STP and working memory are also altered in kainic acid-induced SE. Following kainic acid-induced SE, there was a decrease in STP, reduced LTP capacity, impaired spatial learning, and increased inhibition in the dentate gyrus (190). STP was altered in a model with recurrent hyperexcitability leading to seizures during development (191), as well as a model with aberrant GABA signaling during development leading to frequent interictal discharges. Animals with frequent IDs in the developing PFC showed a decrease in attention, and sociability alongside these changes in STP (192). The growing evidence on neural networks and epilepsy shows that these disrupted neural networks are likely responsible for the cognitive impairments seen with the disease and that the underlying etiology is the cause of both the disease and coding impairments seen in epilepsy animals and patients as well. The corollary is that recovering neural networks toward normal has potential for recovering cognitive impairments (Tables 3, 4).
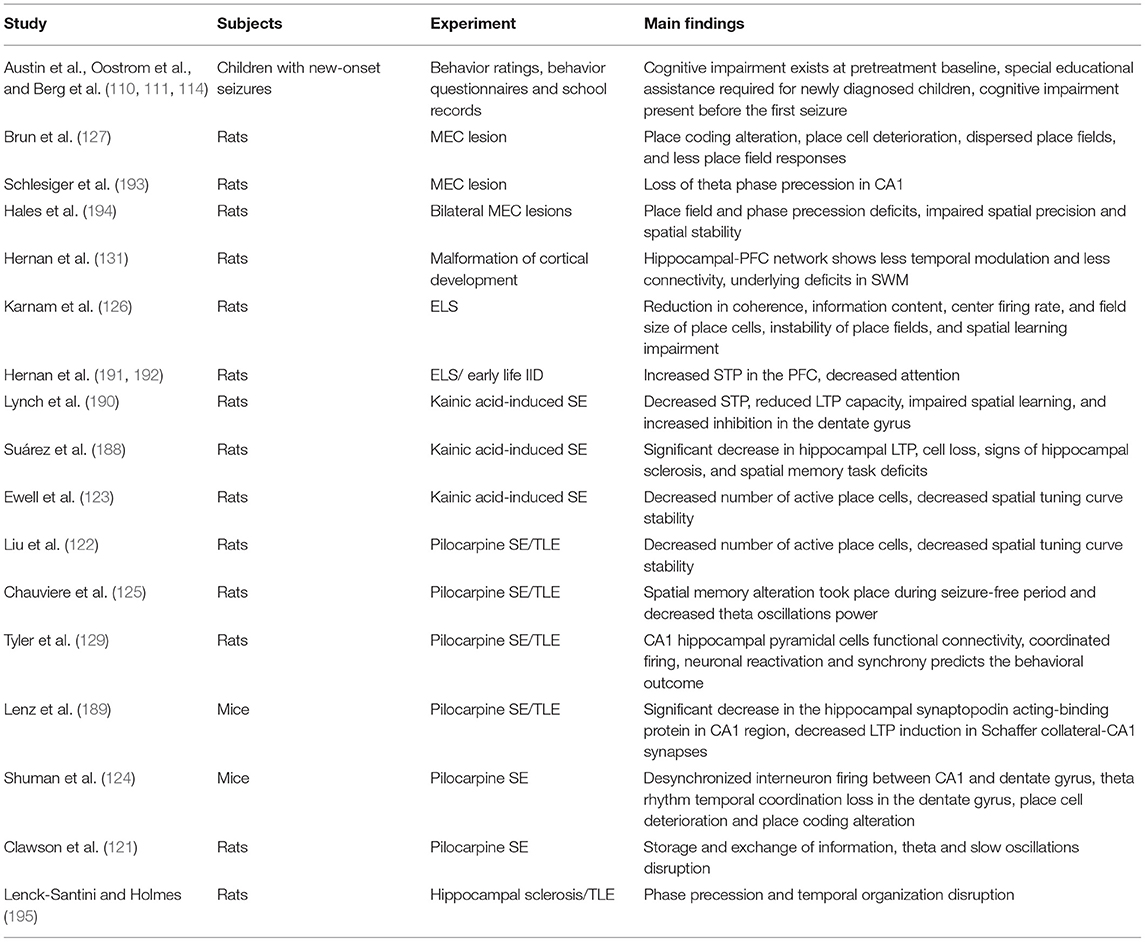
Table 3. Overview of cited preclinical research.
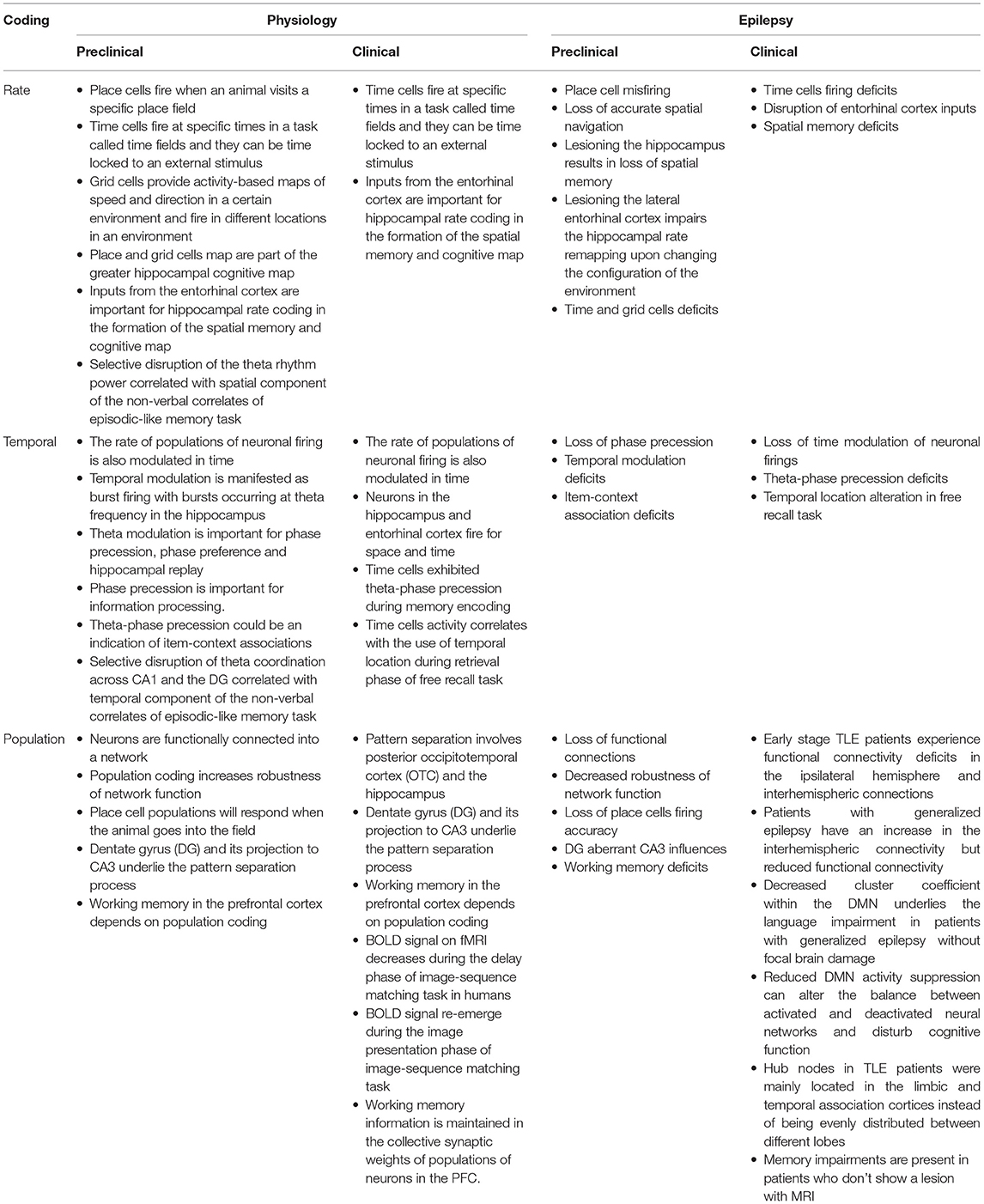
Table 4. Main takeaways for the pre-clinical sections and associated clinical relevance.
Therapeutic Strategies
The main issue with finding the appropriate treatment is whether neural network function can be recovered even in the context of a diseased brain. Here we will discuss potential therapeutic approaches that might influence the neural network function.
Gene Therapy
Rett Syndrome (RTT) is a progressive neurodevelopmental disorder mainly affecting females in early childhood (196, 197). Development starts deteriorating at 6–18 months of age leading to neurological and neurobehavioral alterations and epilepsy (198). Loss of function mutations in the X-linked gene encoding the methyl-CpG-binding protein 2 (MeCP2) involved in transcriptional silencing and activation and RNA splicing modulation is thought to contribute to the pathophysiology of RTT (199, 200).
RTT is associated with significant behavioral abnormalities: motor discoordination and social interaction deficits as well as deficits in cognitive abilities like learning and memory (201–203). MeCP2 knockout mice show reduced neuronal activity in cortical and hippocampal areas (204) as well as deficits in LTP expression in the hippocampus (197). Epilepsy has been reported in 60–80% of RTT patients (205–207). Although children with RTT often have seizures, it is widely accepted that the main driver of the cognitive impairments is a function of the genetic cause. Deficits in LTP, reduction in neuronal activity and seizures indicate that behavioral and cognitive deficits extend to a network problem that involves several mechanisms underlying neuronal activity and plasticity.
Hippocampal place cells are impaired in RTT mice (203). Normally, place fields become refined as the animal-environment experience increases and are stabilized during memory consolidation in sleep. This process involves synchronous re-activation within high-frequency short time-scale windows, known as sharp-wave ripples (208), which is associated with synaptic plasticity transforming short-term memories into long-term ones (209). This process is disrupted in RTT mice as these mice show deficits in experience-dependent refinement of spatial information in addition to increased place cell baseline firing synchrony during sleep (203). Neural oscillations are also impaired in RTT. Organoids developed from stem cells of RTT patients, demonstrated individual neuron firing at a rapid and persistent rate, diminished or reduced gamma oscillation in addition to epileptiform-appearing spikes and high-frequency oscillations (210, 211). Rett mice show desynchronized and reduced theta oscillations during exploratory behavior (210, 212), underscoring impaired temporal coding underlying cognitive and behavioral deficits in RTT mice.
MeCP2 gene therapy has been shown to improve the survival and improve some behavioral deficits seen in RTT (196). Treated mice showed normalized gene expression in addition to better mobility and more exploratory behavior in the open field (213, 214), which could involve normalized place cell activity. This improvement was accompanied by a normalization of neuronal nuclear volume in MeCP2 transduced cells in the dentate gyrus (215). MeCP2 is a master transcriptional regulator of activity-dependent gene expression; recovering it may restore the brain's ability to respond plastically, thereby allowing the network to be in a state where it is ready to receive new information.
Rett syndrome is a very specific disorder whose pathophysiology seems to be directly related to MeCP2. Other causes of epilepsy are less straightforward and may require other gene therapy strategies. One such strategy is targeting the hyperexcitable granule cells in the dentate gyrus in TLE (216). Reducing granule cell hyperactivity via inhibitory chemogenetic receptors, DREADDs (CamKIIα-hM4Di), was able to normalize performance in the spatial object recognition task, reduce seizures and restore the dentate gyrus information coding process (216). Over-expression of the voltage-gated potassium channel Kv1.1 via lentiviral vector or AAV significantly reduced the seizure frequency in rats with focal neocortical epilepsy (FNE) or TLE, respectively (217). Evidence from behavioral and cognitive studies in epilepsy emphasize the need for a new gene therapy strategies. Cognitive and behavioral deficits vary among epilepsy patients, even patients with the same type of epilepsy as this could be due to different genomic factors (3, 218). Different genetic variants are associated with various comorbidities. For example, executive dysfunction was associated with catechol-O-methyltransferase (COMT), methylenetetrahydrofolate reductase (MTHFR) and BDNF in TLE and pediatric epilepsy (219, 220), memory impairment was associated with apolipoprotein E (APOE) and BDNF in TLE (221, 222), impaired working memory was associated with COMT and MTHFR in pediatric epilepsy (220), decreased information processing was associated with RE1- silencing transcription factor (REST) (219), and anxiety and depression were associated with BDNF and COMT (223). Further investigation of epigenomic, transcriptomic, and proteomic changes in epilepsy along with understanding the functional recovery mechanisms seen with gene therapy in RTT, TLE, and FNE at the level of rate, population and temporal coding will allow us to explore the possibility of treating diseased brains.
Environmental Enrichment
The efficacy of simple environmental enrichment (EE) strategies on improving cognition was first noted by Donald Hebb in 1947. He found that the rats he took home with him performed better on behavioral tasks than rats housed in the lab (224). This spurred Hebb's hypothesis that frequent pairing of neuronal firing leads to more efficient excitation in the future; that exposure to a more enriched environment during development critical periods might be influencing the behavior in adulthood. Hebb's observations were the first to connect environmental influences to plasticity. Today, EE paradigms involve exposure to different housing conditions that enable sensory, motor, and cognitive stimulation (225, 226).
EE has been studied in different neurological diseases like Parkinson's and Alzheimer's diseases. EE has been shown to slow cognitive decline in Alzheimer's disease (227). To investigate the effects of EE on epilepsy, it was shown that EE can reduce cognitive deficits, increase neural plasticity, improve motor coordination, and reduce the frequency of seizures (228). We will focus on the effects of EE on information processing mechanisms.
There is a strong link between EE, plasticity, and the mechanisms underlying plasticity. Housing young rats in an enriched environment for 30 days was shown to increase synaptophysin and post-synaptic density (PSD) in the cortex, hippocampus, thalamus, and hypothalamus (226). This suggests that the enriched environment was able to stimulate the formation of new functional synapses in these brain regions. Hippocampal gamma power increases during theta states in rats housed in enriched environments (229). This occurs alongside an increase in interhemispheric coherence of gamma oscillations after EE (229). Mice housed in EE conditions also had an increase in CA1 gamma oscillations (230).
EE also affects rate and population coding. Prolonged exposure to an enriched environment was able to increase the selectivity of CA1 place cells to a particular area in the arena in a way where fewer place cells are activated after brief exposure to a novel environment, along with an increase in global remapping efficiency and this was further supported by the increased expression of the activation protein Arc in CA1 and dentate gyrus (231). This might suggest that the exposure to an enriched environment might be changing how place cells process information by recruiting more or new populations of neurons leading to a more efficient population coding mechanism.
Due to the various effects of EE on these important mechanisms, interest has been growing in investigating the effects of EE on epilepsy. Exposing rats with absence epilepsy to an enriched environment resulted in fewer seizures in adulthood, reduced seizure frequency, and reduced anxiety levels in adulthood (232). The beneficial effects of EE were also seen in TLE rats in the lithium/pilocarpine model. EE was able to alleviate depression and hyperactivity in addition to restoring theta LFP power in the CA1 region (233). The positive effects of EE were further seen in rats with malformation of cortical development (MCD). Rats with MCD had a disruption in their fine spike timing and place-modulated rate coding in CA1 region, which was improved upon with EE exposure (234).
These studies show that EE have a positive impact on rate and population coding. This is important as these processes are disrupted in epilepsy where these are essential for information processing and plasticity. This opens the door for future investigations on how EE can possibly modulate the brain network in ways that make it less susceptible to insults and improves outcome in patients with epilepsy.
Brain Stimulation
Brain stimulation is another therapeutic option for improving cognitive deficits associated with a variety of neurological diseases. Brain stimulation can either activate or inhibit the brain activity in a specific region which gives the ability to modulate cognitive functions. Various types of brain stimulation exist, deep brain stimulation (DBS) is an invasive technique that involves direct implantation of electrodes in the brain while transcranial magnetic stimulation (TMS) is a non-invasive technique that uses magnetic fields applied to the head (235). Brain stimulation techniques have been mainly studied in Alzheimer's disease (AD) and Parkinson Disease (PD).
DBS was tested in AD for the first time in 1984, and while this study did not show any memory or cognitive improvements, it was able to partially stop the left frontal lobe deterioration (236). In 2010, DBS went into phase I trial to investigate its effect on AD patients, and it was shown that after DBS of the fornix/hypothalamus, the patients had improved memory, reduced cognitive decline, enhanced mental state and social performance in addition to increased hippocampal volume (237–239). Further experiments exploring DBS and AD took place after this trial, and the experiments showed the positive effects of DBS on stabilizing cognitive performance (240), influencing cognitive function and disease progression depending on the disease stage and brain region being stimulated (241). For example, nucleus basalis of Meynert (NBM) DBS had a positive effect on sensory gating of auditory information into memory (242). Repetitive TMS (rTMS) was also applied for AD patients. rTMS delivers trains of pulses at the same intensity over a period of time. It mainly uses high frequency (≥5 Hz) for cortical excitability, low-frequency ( ≤ 1 Hz) for cortical inhibition or theta-burst stimulation (TBS) (243). Several trials have shown that rTMS enhanced cognitive function in AD patients when applied to the bilateral dorsolateral prefrontal cortices (DLPFCs) (244–247). Animal studies also investigated the effect of DBS on AD. Acute fornix DBS was able to improve learning and long-term memory in the triple transgenic AD mouse (3 × Tg) model (248). Also, bilateral intermittent NBM DBS enhanced and maintained spatial memory tasks in AD rats (249). Similar results were seen with single rostral intralaminar thalamic (ILN) DBS, in addition to preservation of dendritic spine density in the mPFC and hippocampus and enhanced expression of PSD-95 (250).
In PD, bilateral subthalamic nucleus (STN) and internal globus pallidus (GPi) DBS was able to significantly reduce dyskinesia and improve motor symptoms with long-term benefit (251–253). Additional studies have shown overall improvement in quality of life and continued efficacy in patients that lasted more than 10 years (254, 255). Although most studies agree on the positive effects of DBS on motor function and quality of life, there is contradictory evidence on the positive effects of DBS on cognition and attention in PD patients. Some studies have found that PD patients continued to experience PD-associated declines in executive function, visuospatial reasoning and memory, and verbal memory after DBS (256–258). However, other studies have shown that DBS groups performed better than control groups in memory functions and visuospatial tasks (259, 260). The contradictory results seen with DBS on cognition in PD patients could be due to the stimulated brain regions and using on paradigm for all patients. STN and GPi are the most studied regions in PD due to their importance in dyskinesia and motor coordination, however these regions are not directly involved in memory per se.
DBS is used for epilepsy patients to control and manage refractory seizures; however, DBS may also be beneficial for the cognitive deficits seen in the patients. Ezzyat et al. developed a subject-based approach to investigate the effect of DBS on memory facilitation if performed in a timely manner. Taking into account the disrupted memory network in epilepsy patients, interfering at the right time can reverse the dysfunctional activity of memory encoding. The team was able to differentiate low from high encoding states which indicate neural activity and either stimulating a single medial temporal lobe (MTL) structure like hippocampus or structure involved in memory encoding like prefrontal cortex in the learning session (261). Studies stimulating a single MTL region had contradictory conclusions, indicating both memory facilitation (262, 263) and memory disruption (264, 265). Interestingly, the stimulation was able to increase the encoding-state and memory recall when performed during low-encoding states (261) and this suggests that the accurate stimulation of a single MTL structure or a region involved in memory encoding can reverse the deficits if done at a specific time of memory process. Pilocarpine rats showed a decrease in hippocampal theta power and percentage of time oscillating in theta (266), however, continuous stimulation through Barnes maze task or pre-task stimulation of the medial septum at 7.7 Hz was able to prevent theta oscillations reductions, improve spatial navigation and search strategy during the task. This cognitive improvement was accompanied by significant increase in seizure threshold in these rats. This shows that theta stimulation of the septum has potential to rescue cognitive impairments and increase seizure threshold, further supporting a mechanistic link upstream of both of these symptoms of epilepsy (266, 267). The same stimulation paradigm was used with rats after a traumatic brain injury (TBI) and it was shown that these rats had improved spatial learning and object exploration in addition to increased hippocampal theta oscillations (268). Taken together, these data show that neuronal stimulation approaches may be effective in restoring normal network function and improving cognition broadly.
Interneuron Implantation
Interneuron implantation is another possible treatment that can potentially recover the network function given the importance of interneurons in balancing the inhibition-excitation, controlling gamma and theta oscillations, and sharp wave ripples in the hippocampus. Interneuron precursor implantation into the prefrontal cortex of Pten mutant mice, an autism mouse model, was able to reverse the social behavior deficits seen in these mice; however, the implantation did not normalize baseline and social interaction-evoked EEG signals, but did modify inhibitory signaling in the PFC, underscoring a complex relationship between etiology and circuit restoration underlying behavioral improvement in disease (269). Interneuron implantation has been shown to be beneficial in epilepsy as well. Implantation in TLE, absence epilepsy, and generalized epilepsy models in rodents was able to increase seizure threshold, reduce seizure frequency and duration, reduce network excitability, and improve behavioral deficits (270–273). Implanting interneurons derived from human induced pluripotent stem cell (hiPSC) into the hippocampus of TLE rat model was able to reduce spontaneous seizures frequency after status epilepticus (274–276) which shows translational significance from rodents to humans. In addition to reducing seizures frequency, there was a decrease in the aberrant mossy fiber sprouting, and improved cognition and mood. The implanted rats showed an improvement in hippocampal dependent tasks like object recognition and improvement in pattern separation and novel object recognition (276), which suggests that the implantation might be recovering the communication between different regions or reactivating the DG/CA3 connections required for pattern separation. Integration of interneurons into the CA3 network may be how the new interneurons are affecting the network. region of the hippocampus of epileptic mice was able to improve the working memory in Y-maze test and spatial memory in water maze, however both tasks depend on the PFC (270, 274, 277). This raises the question of how locally implanted interneurons can enhance tasks that are dependent on different brain regions as well. Given the crucial role for interneurons in the timing of the action potential firing, these local connections are likely refining the signal from hippocampus to the PFC. Interestingly, MGE implantation was able to increase memory precision in mice with traumatic brain injury (TBI) as well. Implanted mice performed better in object location task and contextual fear memory where both tasks depend on hippocampus and hippocampal interneurons, respectively (278). Based on these data, GABAergic interneurons transplants may be a promising therapeutic approach for different diseases, however, further investigations are needed to determine the right time and location of implantation for the different investigated diseases.
Conclusion
In this review, we addressed the role of neuronal dynamics in supporting proper cognition, learning and memory, and discussed how these dynamics are altered in epilepsy. The data suggest that cognitive impairments seen in patients with epilepsy and preclinical models of epilepsy are likely due to plasticity changes, alterations to neuronal coding regimes, desynchronization, and functional connectivity disruptions from the effect of underlying etiology, rather than seizures themselves. Although we accept that the seizures could also have some negative impact on network behaviors, we strongly argue that the seizure effect is very small when compared to the etiology effect. We therefore suggest that these deficits should be approached from a systems neuroscience perspective, while being informed by mechanisms needed for normal cognitive function and development in a dynamic experience-dependent and plastic network. Importantly, this calls us to move beyond seizures into network science that is guiding possible treatments and defining new pathophysiology. This might help advance the epilepsy research forward and open the door potentially to answer unsolved questions in the field.
Author Contributions
MK, AH, and RS: draft manuscript writing and editing. All authors reviewed the final draft manuscript. All authors contributed to the article and approved the submitted version.
Funding
AH and MK were funded by an NIH NINDS K22NS104230. RS was funded by an NIH NINDS R21NS117112.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. (2014) 55:475–82. doi: 10.1111/epi.12550
2. Cano-López I, Hampel KG, Garcés M, Villanueva V, González-Bono E. Quality of life in drug-resistant epilepsy: relationships with negative affectivity, memory, somatic symptoms and social support. J Psychosom Res. (2018) 114:31–7. doi: 10.1016/j.jpsychores.2018.09.001
3. Hermann BP, Struck AF, Busch RM, Reyes A, Kaestner E, McDonald CR. Neurobehavioural comorbidities of epilepsy: towards a network-based precision taxonomy. Nat Rev Neurol. (2021) 17:731–46. doi: 10.1038/s41582-021-00555-z
4. Gauffin H, Landtblom A-M, Räty L. Self-esteem and sense of coherence in young people with uncomplicated epilepsy: a 5-year follow-up. Epilepsy Behav. (2010) 17:520–4. doi: 10.1016/j.yebeh.2010.01.167
5. Winter DG. Personality: Analysis and Interpretation of Lives. New York, NY: McGraw-Hill (1996). p. 678.
6. Antonovsky A. Unraveling the Mystery of Health: How People Manage Stress and Stay Well. San Francisco, CA: Jossey-Bass (1987). p. 218.
7. Gauffin H, Flensner G, Landtblom A-M. Living with epilepsy accompanied by cognitive difficulties: young adults' experiences. Epilepsy Behav. (2011) 22:750–8. doi: 10.1016/j.yebeh.2011.09.007
8. Fisher RS. The new classification of seizures by the international league against epilepsy 2017. Curr Neurol Neurosci Rep. (2017) 17:48. doi: 10.1007/s11910-017-0758-6
9. Gauffin H, Landtblom A.-M., Vigren P, Frick A, Engström M, McAllister A, et al. Similar profile and magnitude of cognitive impairments in focal and generalized epilepsy: a pilot study. Front Neurol. (2022) 12:746381. doi: 10.3389/fneur.2021.746381
10. Bell B, Lin JJ, Seidenberg M, Hermann B. The neurobiology of cognitive disorders in temporal lobe epilepsy. Nat Rev Neurol. (2011) 7:154–64. doi: 10.1038/nrneurol.2011.3
11. Bartha-Doering L, Trinka E. The interictal language profile in adult epilepsy. Epilepsia. (2014) 55:1512–25. doi: 10.1111/epi.12743
12. Loughman A, Bowden SC, D'Souza WJ. A comprehensive assessment of cognitive function in the common genetic generalized epilepsy syndromes. Eur J. Neurol. (2017) 24:453–60. doi: 10.1111/ene.13232
13. Simani L, Roozbeh M, Rostami M, Pakdaman H, Ramezani M, Asadollahi M. Attention and inhibitory control deficits in patients with genetic generalized epilepsy and psychogenic nonepileptic seizure. Epilepsy Behav. (2020) 102:106672. doi: 10.1016/j.yebeh.2019.106672
14. Gerstner W, Kreiter AK, Markram H, Herz AVM. Neural codes: firing rates and beyond. Proc Nat Acad Sci USA. (1997) 94:12740–1. doi: 10.1073/pnas.94.24.12740
15. Lenck-Santini P-P, Scott RC. Mechanisms responsible for cognitive impairment in epilepsy. Cold Spring Harb Perspect Med. (2015) 5:a022772. doi: 10.1101/cshperspect.a022772
16. Zuo Y, Safaai H, Notaro G, Mazzoni A, Panzeri S, Diamond ME. Complementary contributions of spike timing and spike rate to perceptual decisions in rat S1 and S2 cortex. Curr Biol. (2015) 25:357–63. doi: 10.1016/j.cub.2014.11.065
17. Dragoi G, Harris KD, Buzsáki G. Place representation within hippocampal networks is modified by long-term potentiation. Neuron. (2003) 39:843–53. doi: 10.1016/S0896-6273(03)00465-3
18. Lu L, Leutgeb JK, Tsao A, Henriksen EJ, Leutgeb S, Barnes CA, et al. Impaired hippocampal rate coding after lesions of the lateral entorhinal cortex. Nat Neurosci. (2013) 16:1085–93. doi: 10.1038/nn.3462
19. Tolman EC. Cognitive maps in rats and men. Psychol Rev. (1948) 55:189–208. doi: 10.1037/h0061626
20. O'Keefe J, Nadel L. The Hippocampus as a Cognitive Map. Oxford; New York, NY: Clarendon Press; Oxford University Press (1978).
21. Davidson TJ, Kloosterman F, Wilson MA. Hippocampal replay of extended experience. Neuron. (2009) 63:497–507. doi: 10.1016/j.neuron.2009.07.027
22. Clark RE, Broadbent NJ, Squire LR. Impaired remote spatial memory after hippocampal lesions despite extensive training beginning early in life. Hippocampus. (2005) 15:340–6. doi: 10.1002/hipo.20076
23. Pastalkova E, Itskov V, Amarasingham A, Buzsáki G. Internally generated cell assembly sequences in the rat hippocampus. Science. (2008) 321:1322–7. doi: 10.1126/science.1159775
24. Hafting T, Fyhn M, Molden S, Moser M-B, Moser EI. Microstructure of a spatial map in the entorhinal cortex. Nature. (2005) 436:801–6. doi: 10.1038/nature03721
25. O'Keefe J, Burgess N. Dual phase and rate coding in hippocampal place cells: theoretical significance and relationship to entorhinal grid cells. Hippocampus. (2005) 15:853–66. doi: 10.1002/hipo.20115
26. Muller R, Kubie J. The effects of changes in the environment on the spatial firing of hippocampal complex-spike cells. J Neurosci. (1987) 7:1951–68. doi: 10.1523/JNEUROSCI.07-07-01951.1987
27. Ruth RE, Collier TJ, Routtenberg A. Topographical relationship between the entorhinal cortex and the septotemporal axis of the dentate gyrus in rats: II. Cells projecting from lateral entorhinal subdivision. J Comparat. Neurol. (1988) 270:506–16. doi: 10.1002/cne.902700404
28. Pouget A, Dayan P, Zemel R. Information processing with population codes. Nat Rev Neurosci. (2000) 1:125–32. doi: 10.1038/35039062
29. Usrey WM, Reid RC. Synchronous activity in the visual system. Annu Rev Physiol. (1999) 61:435–56. doi: 10.1146/annurev.physiol.61.1.435
30. Tolhurst DJ, Movshon JA, Dean AF. The statistical reliability of signals in single neurons in cat and monkey visual cortex. Vision Res. (1983) 23:775–85. doi: 10.1016/0042-6989(83)90200-6
31. Salinas E, Abbott LF. Vector reconstruction from firing rates. J Comput Neurosci. (1994) 1:89–107. doi: 10.1007/BF00962720
32. Atallah BV, Bruns W, Carandini M, Scanziani M. Parvalbumin-expressing interneurons linearly transform cortical responses to visual stimuli. Neuron. (2012) 73:159–70. doi: 10.1016/j.neuron.2011.12.013
33. Adesnik H. Synaptic mechanisms of feature coding in the visual cortex of awake mice. Neuron. (2017) 95:1147–59.e4. doi: 10.1016/j.neuron.2017.08.014
34. Adesnik H. Layer-specific excitation/inhibition balances during neuronal synchronization in the visual cortex. J Physiol. (2018) 596:1639–57. doi: 10.1113/JP274986
35. Aertsen A, Braitenberg V. Brain Theory: Biological Basis Computational Principles. Amsterdam; New York, NY: Elsevier. (1996). Available online at: http://site.ebrary.com/id/10254720 (accessed February 14, 2022).
36. Anderson JS, Carandini M, Ferster D. Orientation tuning of input conductance, excitation, and inhibition in cat primary visual cortex. J Neurophysiol. (2000) 84:909–26. doi: 10.1152/jn.2000.84.2.909
37. Li Y, Liu B, Chou X, Zhang LI, Tao HW. Synaptic basis for differential orientation selectivity between complex and simple cells in mouse visual cortex. J Neurosci. (2015) 35:11081–93. doi: 10.1523/JNEUROSCI.5246-14.2015
38. Li Y, Ma W, Pan C, Zhang LI, Tao HW. Broadening of cortical inhibition mediates developmental sharpening of orientation selectivity. J Neurosci. (2012) 32:3981–91. doi: 10.1523/JNEUROSCI.5514-11.2012
39. Trevelyan AJ, Bruns W, Mann EO, Crepel V, Scanziani M. The information content of physiological and epileptic brain activity. J Physiol. (2013) 591:799–805. doi: 10.1113/jphysiol.2012.240358
40. Fu Y, Tucciarone JM, Espinosa JS, Sheng N, Darcy DP, Nicoll RA, et al. A cortical circuit for gain control by behavioral state. Cell. (2014) 156:1139–52. doi: 10.1016/j.cell.2014.01.050
41. Pakan JM, Lowe SC, Dylda E, Keemink SW, Currie SP, Coutts CA, et al. Behavioral-state modulation of inhibition is context-dependent and cell type specific in mouse visual cortex. Elife. (2016) 5:e14985. doi: 10.7554/eLife.14985
42. Millman DJ, Ocker GK, Caldejon S, Kato I, Larkin JD, Lee EK, et al. VIP interneurons in mouse primary visual cortex selectively enhance responses to weak but specific stimuli. eLife. (2020) 9:e55130. doi: 10.7554/eLife.55130
43. Rubin DB, Van Hooser SD, Miller KD. The stabilized supralinear network: a unifying circuit motif underlying multi-input integration in sensory cortex. Neuron. (2015) 85:402–17. doi: 10.1016/j.neuron.2014.12.026
44. França TFA, Monserrat JM. Hippocampal place cells are topographically organized, but physical space has nothing to do with it. Brain Struct Funct. (2019) 224:3019–29. doi: 10.1007/s00429-019-01968-9
45. Lohnas LJ, Duncan K, Doyle WK, Thesen T, Devinsky O, Davachi L. Time-resolved neural reinstatement and pattern separation during memory decisions in human hippocampus. Proc Nat Acad Sci USA. (2018) 115:E7418–27. doi: 10.1073/pnas.1717088115
46. Lalani SJ, Reyes A, Kaestner E, Stark SM, Stark CEL, Lee D, et al. Impaired behavioral pattern separation in refractory temporal lobe epilepsy and mild cognitive impairment. J Int Neuropsychol Soc. (2021) 2021:1–13. doi: 10.1017/S1355617721000734
47. Leutgeb JK, Leutgeb S, Moser M-B, Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science. (2007) 315:961–6. doi: 10.1126/science.1135801
48. Bakker A, Kirwan CB, Miller M, Stark CEL. Pattern separation in the human hippocampal CA3 and dentate gyrus. Science. (2008) 319:1640–2. doi: 10.1126/science.1152882
49. Wesnes KA, Annas P, Basun H, Edgar C, Blennow K. Performance on a pattern separation task by Alzheimer's patients shows possible links between disrupted dentate gyrus activity and apolipoprotein E 4 status and cerebrospinal fluid amyloid-β42 levels. Alzheimer's Res Ther. (2014) 6:20. doi: 10.1186/alzrt250
50. Reyes A, Holden HM, Chang Y-HA, Uttarwar VS, Sheppard DP, DeFord NE, et al. Impaired spatial pattern separation performance in temporal lobe epilepsy is associated with visuospatial memory deficits and hippocampal volume loss. Neuropsychologia. (2018) 111:209–15. doi: 10.1016/j.neuropsychologia.2018.02.009
51. Madar AD, Ewell LA, Jones MV. Pattern separation of spiketrains in hippocampal neurons. Sci Rep. (2019) 9:5282. doi: 10.1038/s41598-019-41503-8
52. Madar AD, Pfammatter JA, Bordenave J, Plumley EI, Ravi S, Cowie M, et al. Deficits in behavioral and neuronal pattern separation in temporal lobe epilepsy. bioRxiv:2020.02.13.948364. (2020). doi: 10.1101/2020.02.13.948364
53. Baddeley AD, Logie RH. Working memory: the multiple-component model. In: A Miyake, P Shah, editors, Models of Working Memory. 1st ed. Cambridge: Cambridge University Press (1999). p. 28–61. doi: 10.1017/CBO9781139174909.005
54. Sreenivasan KK, Curtis CE, D'Esposito M. Revisiting the role of persistent neural activity during working memory. Trends Cogn Sci. (2014) 18:82–9. doi: 10.1016/j.tics.2013.12.001
55. Sarma A, Masse NY, Wang X-J, Freedman DJ. Task-specific versus generalized mnemonic representations in parietal and prefrontal cortices. Nat Neurosci. (2016) 19:143–9. doi: 10.1038/nn.4168
56. Masse NY, Hodnefield JM, Freedman DJ. Mnemonic encoding and cortical organization in parietal and prefrontal cortices. J Neurosci. (2017) 37:6098–112. doi: 10.1523/JNEUROSCI.3903-16.2017
57. Emrich SM, Riggall AC, LaRocque JJ, Postle BR. Distributed patterns of activity in sensory cortex reflect the precision of multiple items maintained in visual short-term memory. J Neurosci. (2013) 33:6516–23. doi: 10.1523/JNEUROSCI.5732-12.2013
58. Wasmuht DF, Spaak E, Buschman TJ, Miller EK, Stokes MG. Intrinsic neuronal dynamics predict distinct functional roles during working memory. Nat Commun. (2018) 9:3499. doi: 10.1038/s41467-018-05961-4
59. Pinto L, Dan Y. Cell-type-specific activity in prefrontal cortex during goal-directed behavior. Neuron. (2015) 87:437–50. doi: 10.1016/j.neuron.2015.06.021
60. Murray JD, Bernacchia A, Roy NA, Constantinidis C, Romo R, Wang X-J. Stable population coding for working memory coexists with heterogeneous neural dynamics in prefrontal cortex. Proc Nat Acad Sci USA. (2017) 114:394–9. doi: 10.1073/pnas.1619449114
61. Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science. (2004) 304:1926–9. doi: 10.1126/science.1099745
62. Wang X-J. Neurophysiological and computational principles of cortical rhythms in cognition. Physiol Rev. (2010) 90:1195–268. doi: 10.1152/physrev.00035.2008
63. Buzsáki G. Theta oscillations in the hippocampus. Neuron. (2002) 33:325–40. doi: 10.1016/S0896-6273(02)00586-X
64. Buzsáki G. Theta rhythm of navigation: link between path integration and landmark navigation, episodic and semantic memory. Hippocampus. (2005) 15:827–40. doi: 10.1002/hipo.20113
65. Mitchell SJ, Ranck JB. Generation of theta rhythm in medial entorhinal cortex of freely moving rats. Brain Res. (1980) 189:49–66. doi: 10.1016/0006-8993(80)90006-2
66. Leung L-WS, Borst JGG. Electrical activity of the cingulate cortex. I. Generating mechanisms and relations to behavior. Brain Res. (1987) 407:68–80. doi: 10.1016/0006-8993(87)91220-0
67. Paré D, Collins DR. Neuronal correlates of fear in the lateral amygdala: multiple extracellular recordings in conscious cats. J Neurosci. (2000) 20:2701–10. doi: 10.1523/JNEUROSCI.20-07-02701.2000
68. Petsche H, Stumpf C, Gogolak G. The significance of the rabbit's septum as a relay station between the midbrain and the hippocampus I. The control of hippocampus arousal activity by the septum cells. Electroencephal Clin Neurophysiol. (1962) 14:202–11. doi: 10.1016/0013-4694(62)90030-5
69. Smith HR, Pang KCH. Orexin-saporin lesions of the medial septum impair spatial memory. Neuroscience. (2005) 132:261–71. doi: 10.1016/j.neuroscience.2004.12.037
70. Dwyer TA, Servatius RJ, Pang KCH. Noncholinergic lesions of the medial septum impair sequential learning of different spatial locations. J Neurosci. (2007) 27:299–303. doi: 10.1523/JNEUROSCI.4189-06.2007
71. Cañas A, Juncadella M, Lau R, Gabarrós A, Hernández M. Working memory deficits after lesions involving the supplementary motor area. Front Psychol. (2018) 9:765. doi: 10.3389/fpsyg.2018.00765
72. Montgomery SM, Buzsaki G. Gamma oscillations dynamically couple hippocampal CA3 and CA1 regions during memory task performance. Proc Nat Acad Sci USA. (2007) 104:14495–500. doi: 10.1073/pnas.0701826104
73. Buzsáki G, Wang X-J. Mechanisms of gamma oscillations. Annu Rev Neurosci. (2012) 35:203–25. doi: 10.1146/annurev-neuro-062111-150444
74. Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. (2007) 8:45–56. doi: 10.1038/nrn2044
75. Nuñez A, Buño W. The theta rhythm of the hippocampus: from neuronal and circuit mechanisms to behavior. Front Cell Neurosci. (2021) 15:649262. doi: 10.3389/fncel.2021.649262
76. Tukker JJ, Lasztoczi B, Katona L, Roberts JDB, Pissadaki EK, Dalezios Y, et al. Distinct dendritic arborization and in vivo firing patterns of parvalbumin-expressing basket cells in the hippocampal area CA3. J Neurosci. (2013) 33:6809–25. doi: 10.1523/JNEUROSCI.5052-12.2013
77. Harris KD, Henze DA, Hirase H, Leinekugel X, Dragoi G, Czurkó A, et al. Spike train dynamics predicts theta-related phase precession in hippocampal pyramidal cells. Nature. (2002) 417:738–41. doi: 10.1038/nature00808
78. Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. (2008) 321:53–7. doi: 10.1126/science.1149381
79. O'Keefe J, Recce ML. Phase relationship between hippocampal place units and the EEG theta rhythm. Hippocampus. (1993) 3:317–30. doi: 10.1002/hipo.450030307
80. Skaggs WE, McNaughton BL, Wilson MA, Barnes CA. Theta phase precession in hippocampal neuronal populations and the compression of temporal sequences. Hippocampus. (1996) 6:149–72. doi: 10.1002/(SICI)1098-1063(1996)6:2<149::AID-HIPO6>3.0.CO;2-K
81. Magee JC, Johnston D. A synaptically controlled, associative signal for hebbian plasticity in hippocampal neurons. Science. (1997) 275:209–13. doi: 10.1126/science.275.5297.209
82. Umbach G, Kantak P, Jacobs J, Kahana M, Pfeiffer BE, Sperling M, et al. Time cells in the human hippocampus and entorhinal cortex support episodic memory. Proc Nat Acad Sci USA. (2020) 117:28463–74. doi: 10.1073/pnas.2013250117
83. Fries P. A mechanism for cognitive dynamics: neuronal communication through neuronal coherence. Trends Cogn Sci. (2005) 9:474–80. doi: 10.1016/j.tics.2005.08.011
84. Buzsáki G. Neural syntax: cell assemblies, synapsembles, and readers. Neuron. (2010) 68:362–85. doi: 10.1016/j.neuron.2010.09.023
85. DeCoteau WE, Thorn C, Gibson DJ, Courtemanche R, Mitra P, Kubota Y, et al. Learning-related coordination of striatal and hippocampal theta rhythms during acquisition of a procedural maze task. Proc Nat Acad Sci USA. (2007) 104:5644–9. doi: 10.1073/pnas.0700818104
86. Gregoriou GG, Gotts SJ, Zhou H, Desimone R. High-frequency, long-range coupling between prefrontal and visual cortex during attention. Science. (2009) 324:1207–10. doi: 10.1126/science.1171402
87. Colgin LL, Denninger T, Fyhn M, Hafting T, Bonnevie T, Jensen O, et al. Frequency of gamma oscillations routes flow of information in the hippocampus. Nature. (2009) 462:353–7. doi: 10.1038/nature08573
88. Fell J, Klaver P, Lehnertz K, Grunwald T, Schaller C, Elger CE, et al. Human memory formation is accompanied by rhinal–hippocampal coupling and decoupling. Nat Neurosci. (2001) 4:1259–64. doi: 10.1038/nn759
89. Siegel M, Donner TH, Engel AK. Spectral fingerprints of large-scale neuronal interactions. Nat Rev Neurosci. (2012) 13:121–34. doi: 10.1038/nrn3137
90. Spencer KM, Nestor PG, Niznikiewicz MA, Salisbury DF, Shenton ME, McCarley RW. Abnormal neural synchrony in schizophrenia. J Neurosci. (2003) 23:7407–11. doi: 10.1523/JNEUROSCI.23-19-07407.2003
91. Herrmann C, Demiralp T. Human EEG gamma oscillations in neuropsychiatric disorders. Clin Neurophysiol. (2005) 116:2719–33. doi: 10.1016/j.clinph.2005.07.007
92. Lega BC, Jacobs J, Kahana M. Human hippocampal theta oscillations and the formation of episodic memories. Hippocampus. (2012) 22:748–61. doi: 10.1002/hipo.20937
93. Barry RJ, Clarke AR. Resting state brain oscillations and symptom profiles in attention deficit/hyperactivity disorder. Clin Neurophysiol. (2013) 17:275–87. doi: 10.1016/B978-0-7020-5307-8.00017-X
94. Inostroza M, Brotons-Mas JR, Laurent F, Cid E, de la Prida LM. Specific impairment of “what-where-when” episodic-like memory in experimental models of temporal lobe epilepsy. J Neurosci. (2013) 33:17749–62. doi: 10.1523/JNEUROSCI.0957-13.2013
95. Wang J, Fang Y, Wang X, Yang H, Yu X, Wang H. Enhanced gamma activity and cross-frequency interaction of resting-state electroencephalographic oscillations in patients with Alzheimer's disease. Front Aging Neurosci. (2017) 9:243. doi: 10.3389/fnagi.2017.00243
97. Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. (2008) 33:18–41. doi: 10.1038/sj.npp.1301559
98. Rotenberg A, Mayford M, Hawkins RD, Kandel ER, Muller RU. Mice expressing activated CaMKII lack low frequency LTP and do not form stable place cells in the CA1 region of the hippocampus. Cell. (1996) 87:1351–61. doi: 10.1016/S0092-8674(00)81829-2
99. Kentros C, Hargreaves E, Hawkins RD, Kandel ER, Shapiro M, Muller RV. Abolition of long-term stability of new hippocampal place cell maps by NMDA receptor blockade. Science. (1998) 280:2121–6. doi: 10.1126/science.280.5372.2121
100. Barry JM, Rivard B, Fox SE, Fenton AA, Sacktor TC, Muller RU. Inhibition of protein kinase M disrupts the stable spatial discharge of hippocampal place cells in a familiar environment. J Neurosci. (2012) 32:13753–62. doi: 10.1523/JNEUROSCI.0319-12.2012
101. Schoenenberger P, O'Neill J, Csicsvari J. Activity-dependent plasticity of hippocampal place maps. Nat Commun. (2016) 7:11824. doi: 10.1038/ncomms11824
102. Knierim JJ. Synaptic plasticity and place cell formation. Encycl Neurosci. (2009) 5:735–40. doi: 10.1016/B978-008045046-9.00830-5
103. Derdikman D, Moser EI. A manifold of spatial maps in the brain. Space Time Number Brain. (2011) 9:41–57. doi: 10.1016/B978-0-12-385948-8.00004-9
104. Ashby DM, Floresco SB, Phillips AG, McGirr A, Seamans JK, Wang YT. LTD is involved in the formation and maintenance of rat hippocampal CA1 place-cell fields. Nat Commun. (2021) 12:100. doi: 10.1038/s41467-020-20317-7
105. Sun L, Liu SY, Zhou XW, Wang XC, Liu R, Wang Q, et al. Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience. (2003) 118:1175–82. doi: 10.1016/S0306-4522(02)00697-8
106. Ge Y, Dong Z, Bagot RC, Howland JG, Phillips AG, Wong TP, et al. Hippocampal long-term depression is required for the consolidation of spatial memory. Proc Nat Acad Sci USA. (2010) 107:16697–702. doi: 10.1073/pnas.1008200107
107. Blackman AV, Abrahamsson T, Costa RP, Lalanne T, Sjöström PJ. Target-cell-specific short-term plasticity in local circuits. Front Synaptic Neurosci. (2013) 5:11. doi: 10.3389/fnsyn.2013.00011
108. Tsodyks M, Uziel A, Markram H. Synchrony generation in recurrent networks with frequency-dependent synapses. J Neurosci. (2000) 20:RC50. doi: 10.1523/JNEUROSCI.20-01-j0003.2000
109. Deng P-Y, Klyachko VA. The diverse functions of short-term plasticity components in synaptic computations. Commun Integr Biol. (2011) 4:543–8. doi: 10.4161/cib.15870
110. Austin JK, Dunn DW, Caffrey HM, Perkins SM, Harezlak J, Rose DF. Recurrent seizures and behavior problems in children with first recognized seizures: a prospective study. Epilepsia. (2002) 43:1564–73. doi: 10.1046/j.1528-1157.2002.26002.x
111. Oostrom KJ, Smeets-Schouten A, Kruitwagen CLJJ, Peters ACB, Jennekens-Schinkel A, for for the Dutch Study Group of Epilepsy in Childhood. Not only a matter of epilepsy: early problems of cognition and behavior in children with “epilepsy only”—a prospective, longitudinal, controlled study starting at diagnosis. Pediatrics. (2003) 112:1338–44. doi: 10.1542/peds.112.6.1338
112. Keezer MR, Sisodiya SM, Sander JW. Comorbidities of epilepsy: current concepts and future perspectives. Lancet Neurol. (2016) 15:106–15. doi: 10.1016/S1474-4422(15)00225-2
113. Elger CE, Helmstaedter C, Kurthen M. Chronic epilepsy and cognition. Lancet Neurol. (2004) 3:663–72. doi: 10.1016/S1474-4422(04)00906-8
114. Berg AT, Smith SN, Frobish D, Levy SR, Testa FM, Beckerman B, et al. Special education needs of children with newly diagnosed epilepsy. Dev Med Child Neurol. (2005) 47:749. doi: 10.1017/S001216220500157X
115. Pulliainen V, Kuikka P, Jokelainen M. Motor and cognitive functions in newly diagnosed adult seizure patients before antiepileptic medication: cognitive functions in newly diagnosed seizure patients. Acta Neurol Scand. (2000) 101:73–8. doi: 10.1034/j.1600-0404.2000.101002073.x
116. Lawson JA, Vogrin S, Bleasel AF, Cook MJ, Burns L, McAnally L, et al. Predictors of hippocampal, cerebral, and cerebellar volume reduction in childhood epilepsy. Epilepsia. (2000) 41:1540–5. doi: 10.1111/j.1528-1157.2000.tb00122.x
117. Keller SS, Roberts N. Voxel-based morphometry of temporal lobe epilepsy: an introduction and review of the literature. Epilepsia. (2008) 49:741–57. doi: 10.1111/j.1528-1167.2007.01485.x
118. Seidenberg M, O'leary DS, Giordani B, Berent S, Boll TJ. Test-retest IQ changes of epilepsy patients: assessing the influence of practice effects. J Clin Neuropsychol. (1981) 3:237–55. doi: 10.1080/01688638108403128
119. Oyegbile TO, Dow C, Jones J, Bell B, Rutecki P, Sheth R, et al. The nature and course of neuropsychological morbidity in chronic temporal lobe epilepsy. Neurology. (2004) 62:1736–42. doi: 10.1212/01.WNL.0000125186.04867.34
120. Rayner G, Jackson GD, Wilson SJ. Mechanisms of memory impairment in epilepsy depend on age at disease onset. Neurology. (2016) 87:1642–9. doi: 10.1212/WNL.0000000000003231
121. Clawson W, Madec T, Ghestem A, Quilichini PP, Battaglia D, Bernard C. Disordered information processing dynamics in experimental epilepsy. preprint. Neuroscience. (2021) 2021:430768. doi: 10.1101/2021.02.11.430768
122. Liu X, Muller RU, Huang L-T, Kubie JL, Rotenberg A, Rivard B, et al. Seizure-induced changes in place cell physiology: relationship to spatial memory. J Neurosci. (2003) 23:11505–15. doi: 10.1523/JNEUROSCI.23-37-11505.2003
123. Ewell LA, Fischer KB, Leibold C, Leutgeb S, Leutgeb JK. The impact of pathological high-frequency oscillations on hippocampal network activity in rats with chronic epilepsy. Elife. (2019) 8:e42148. doi: 10.7554/eLife.42148
124. Shuman T, Aharoni D, Cai DJ, Lee CR, Chavlis S, Page-Harley L, et al. Breakdown of spatial coding and interneuron synchronization in epileptic mice. Nat Neurosci. (2020) 23:229–38. doi: 10.1038/s41593-019-0559-0
125. Chauviere L, Rafrafi N, Thinus-Blanc C, Bartolomei F, Esclapez M, Bernard C. Early deficits in spatial memory and theta rhythm in experimental temporal lobe epilepsy. J Neurosci. (2009) 29:5402–10. doi: 10.1523/JNEUROSCI.4699-08.2009
126. Karnam HB, Zhou J-L, Huang L-T, Zhao Q, Shatskikh T, Holmes GL. Early life seizures cause long-standing impairment of the hippocampal map. Exp Neurol. (2009) 217:378–87. doi: 10.1016/j.expneurol.2009.03.028
127. Brun VH, Leutgeb S, Wu H-Q, Schwarcz R, Witter MP, Moser EI, et al. Impaired spatial representation in CA1 after lesion of direct input from entorhinal cortex. Neuron. (2008) 57:290–302. doi: 10.1016/j.neuron.2007.11.034
128. Davoudi H, Foster DJ. Acute silencing of hippocampal CA3 reveals a dominant role in place field responses. Nat Neurosci. (2019) 22:337–42. doi: 10.1038/s41593-018-0321-z
129. Tyler AL, Mahoney JM, Richard GR, Holmes GL, Lenck-Santini P-P, Scott RC. Functional network changes in hippocampal CA1 after status epilepticus predict spatial memory deficits in rats. J Neurosci. (2012) 32:11365–76. doi: 10.1523/JNEUROSCI.1516-12.2012
130. Yu JY, Frank LM. Hippocampal–cortical interaction in decision making. Neurobiol Learn Mem. (2015) 117:34–41. doi: 10.1016/j.nlm.2014.02.002
131. Hernan A, Mahoney J, Curry W, Mawe S, Scott R. Distributed dynamic coding for spatial working memory in hippocampal-prefrontal networks. Neuroscience. (2019) 2019:630673. doi: 10.1101/630673
132. Nash MI, Hodges CB, Muncy NM, Kirwan CB. Pattern separation beyond the hippocampus: a high-resolution whole-brain investigation of mnemonic discrimination in healthy adults. Hippocampus. (2021) 31:408–21. doi: 10.1002/hipo.23299
133. Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, et al. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. (2012) 74:467–74. doi: 10.1016/j.neuron.2012.03.023
134. Brock Kirwan C, Hartshorn A, Stark SM, Goodrich-Hunsaker NJ, Hopkins RO, Stark CEL. Pattern separation deficits following damage to the hippocampus. Neuropsychologia. (2012) 50:2408–14. doi: 10.1016/j.neuropsychologia.2012.06.011
135. Yassa MA, Stark SM, Bakker A, Albert MS, Gallagher M, Stark CEL. High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic Mild Cognitive Impairment. Neuroimage. (2010) 51:1242–52. doi: 10.1016/j.neuroimage.2010.03.040
136. Bui AD, Nguyen TM, Limouse C, Kim HK, Szabo GG, Felong S, et al. Dentate gyrus mossy cells control spontaneous convulsive seizures and spatial memory. Science. (2018) 359:787–90. doi: 10.1126/science.aan4074
137. Bernhardt BC, Bonilha L, Gross DW. Network analysis for a network disorder: the emerging role of graph theory in the study of epilepsy. Epilepsy Behav. (2015) 50:162–70. doi: 10.1016/j.yebeh.2015.06.005
138. Yilmazer-Hanke DM, Wolf HK, Schramm J, Elger CE, Wiestler OD, Blümcke I. Subregional pathology of the amygdala complex and entorhinal region in surgical specimens from patients with pharmacoresistant temporal lobe epilepsy. J Neuropathol Exp Neurol. (2000) 59:907–20. doi: 10.1093/jnen/59.10.907
139. Bothwell S, Meredith GE, Phillips J, Staunton H, Doherty C, Grigorenko E, et al. Neuronal hypertrophy in the neocortex of patients with temporal lobe epilepsy. J Neurosci. (2001) 21:4789–800. doi: 10.1523/JNEUROSCI.21-13-04789.2001
140. Blanc F, Martinian L, Liagkouras I, Catarino C, Sisodiya SM, Thom M. Investigation of widespread neocortical pathology associated with hippocampal sclerosis in epilepsy: a postmortem study. Epilepsia. (2011) 52:10–21. doi: 10.1111/j.1528-1167.2010.02773.x
141. Sinjab B, Martinian L, Sisodiya SM, Thom M. Regional thalamic neuropathology in patients with hippocampal sclerosis and epilepsy: a postmortem study. Epilepsia. (2013) 54:2125–33. doi: 10.1111/epi.12403
142. Tai XY, Bernhardt B, Thom M, Thompson P, Baxendale S, Koepp M, et al. Review: neurodegenerative processes in temporal lobe epilepsy with hippocampal sclerosis: clinical, pathological and neuroimaging evidence. Neuropathol Appl Neurobiol. (2018) 44:70–90. doi: 10.1111/nan.12458
143. Cendes F, Andermann F, Gloor P, Evans A, Jones-Gotman M, Watson C, et al. MRI volumetric measurement of amygdala and hippocampus in temporal lobe epilepsy. Neurology. (1993) 43:719–719. doi: 10.1212/WNL.43.4.719
144. Bernasconi N, Bernasconi A, Caramanos Z, Dubeau F, Richardson J, Andermann F, et al. Entorhinal cortex atrophy in epilepsy patients exhibiting normal hippocampal volumes. Neurology. (2001) 56:1335–9. doi: 10.1212/WNL.56.10.1335
145. Bernasconi N, Bernasconi A, Caramanos Z, Antel SB, Andermann F, Arnold DL. Mesial temporal damage in temporal lobe epilepsy: a volumetric MRI study of the hippocampus, amygdala and parahippocampal region. Brain. (2003) 126:462–9. doi: 10.1093/brain/awg034
146. Bernhardt BC, Hong S-J, Bernasconi A, Bernasconi N. Magnetic resonance imaging pattern learning in temporal lobe epilepsy: classification and prognostics. Ann Neurol. (2015) 77:436–46. doi: 10.1002/ana.24341
147. Bernhardt BC, Worsley KJ, Kim H, Evans AC, Bernasconi A, Bernasconi N. Longitudinal and cross-sectional analysis of atrophy in pharmacoresistant temporal lobe epilepsy. Neurology. (2009) 72:1747–54. doi: 10.1212/01.wnl.0000345969.57574.f5
148. Coan AC, Appenzeller S, Bonilha L, Li LM, Cendes F. Seizure frequency and lateralization affect progression of atrophy in temporal lobe epilepsy. Neurology. (2009) 73:834–42. doi: 10.1212/WNL.0b013e3181b783dd
149. Bernhardt BC, Kim H, Bernasconi N. Patterns of subregional mesiotemporal disease progression in temporal lobe epilepsy. Neurology. (2013) 81:1840–7. doi: 10.1212/01.wnl.0000436069.20513.92
150. Concha L, Beaulieu C, Gross DW. Bilateral limbic diffusion abnormalities in unilateral temporal lobe epilepsy. Ann Neurol. (2005) 57:188–96. doi: 10.1002/ana.20334
151. Yogarajah M, Duncan JS. Diffusion-based magnetic resonance imaging and tractography in epilepsy. Epilepsia. (2008) 49:189–200. doi: 10.1111/j.1528-1167.2007.01378.x
152. Bonilha L, Edwards JC, Kinsman SL, Morgan PS, Fridriksson J, Rorden C, et al. Extrahippocampal gray matter loss and hippocampal deafferentation in patients with temporal lobe epilepsy. Epilepsia. (2010) 51:519–28. doi: 10.1111/j.1528-1167.2009.02506.x
153. Hermann B, Conant LL, Cook CJ, Hwang G, Garcia-Ramos C, Dabbs K, et al. Network, clinical and sociodemographic features of cognitive phenotypes in temporal lobe epilepsy. NeuroImage. (2020) 27:102341. doi: 10.1016/j.nicl.2020.102341
154. Bernhardt BC, Worsley KJ, Besson P, Concha L, Lerch JP, Evans AC, et al. Mapping limbic network organization in temporal lobe epilepsy using morphometric correlations: insights on the relation between mesiotemporal connectivity and cortical atrophy. Neuroimage. (2008) 42:515–24. doi: 10.1016/j.neuroimage.2008.04.261
155. Mueller SG, Laxer KD, Barakos J, Cheong I, Garcia P, Weiner MW. Widespread neocortical abnormalities in temporal lobe epilepsy with and without mesial sclerosis. Neuroimage. (2009) 46:353–9. doi: 10.1016/j.neuroimage.2009.02.020
156. Mueller SG, Laxer KD, Barakos J, Cheong I, Finlay D, Garcia P, et al. Involvement of the thalamocortical network in TLE with and without mesiotemporal sclerosis: thalamocortical Network in TLE. Epilepsia. (2009) 51:1436–45. doi: 10.1111/j.1528-1167.2009.02413.x
157. Bettus G, Bartolomei F, Confort-Gouny S, Guedj E, Chauvel P, Cozzone PJ, et al. Role of resting state functional connectivity MRI in presurgical investigation of mesial temporal lobe epilepsy. J Neurol Neurosurg Psychiatry. (2010) 81:1147–54. doi: 10.1136/jnnp.2009.191460
158. Pereira FRS, Alessio A, Sercheli MS, Pedro T, Bilevicius E, Rondina JM, et al. Asymmetrical hippocampal connectivity in mesial temporal lobe epilepsy: evidence from resting state fMRI. BMC Neurosci. (2010) 11:66. doi: 10.1186/1471-2202-11-66
159. Zhang C, Yang H, Qin W, Liu C, Qi Z, Chen N, et al. Characteristics of resting-state functional connectivity in intractable unilateral temporal lobe epilepsy patients with impaired executive control function. Front Hum Neurosci. (2017) 11:609. doi: 10.3389/fnhum.2017.00609
160. Zhang Z, Lu G, Zhong Y, Tan Q, Liao W, Wang Z, et al. Altered spontaneous neuronal activity of the default-mode network in mesial temporal lobe epilepsy. Brain Res. (2010) 1323:152–60. doi: 10.1016/j.brainres.2010.01.042
161. Liao W, Zhang Z, Pan Z, Mantini D, Ding J, Duan X, et al. Default mode network abnormalities in mesial temporal lobe epilepsy: a study combining fMRI and DTI. Hum Brain Mapp. (2011) 32:883–95. doi: 10.1002/hbm.21076
162. Pittau F, Grova C, Moeller F, Dubeau F, Gotman J. Patterns of altered functional connectivity in mesial temporal lobe epilepsy: functional connectivity in MTLE. Epilepsia. (2012) 53:1013–23. doi: 10.1111/j.1528-1167.2012.03464.x
163. Haneef Z, Lenartowicz A, Yeh HJ, Levin HS, Engel J, Stern JM. Functional connectivity of hippocampal networks in temporal lobe epilepsy. Epilepsia. (2014) 55:137–45. doi: 10.1111/epi.12476
164. Zhang C, Zhang H, Xu K, Yang H, Liu C, Yu T, et al. Impaired prefrontal cortex-thalamus pathway in intractable temporal lobe epilepsy with aberrant executive control function: MRI evidence. Clin Neurophysiol. (2019) 130:484–90. doi: 10.1016/j.clinph.2018.12.007
165. Bettus G, Guedj E, Joyeux F, Confort-Gouny S, Soulier E, Laguitton V, et al. Decreased basal fMRI functional connectivity in epileptogenic networks and contralateral compensatory mechanisms. Hum Brain Mapp. (2009) 30:1580–91. doi: 10.1002/hbm.20625
166. Morgan VL, Rogers BP, Sonmezturk HH, Gore JC, Abou-Khalil B. Cross hippocampal influence in mesial temporal lobe epilepsy measured with high temporal resolution functional magnetic resonance imaging. Epilepsia. (2011) 52:1741–9. doi: 10.1111/j.1528-1167.2011.03196.x
167. Bai X, Guo J, Killory B, Vestal M, Berman R, Negishi M, et al. Resting functional connectivity between the hemispheres in childhood absence epilepsy. Neurology. (2011) 76:1960–7. doi: 10.1212/WNL.0b013e31821e54de
168. Luo C, Li Q, Xia Y, Lei X, Xue K, Yao Z, et al. Resting state basal ganglia network in idiopathic generalized epilepsy. Hum Brain Mapp. (2012) 33:1279–94. doi: 10.1002/hbm.21286
169. Masterton RA, Carney PW, Jackson GD. Cortical and thalamic resting-state functional connectivity is altered in childhood absence epilepsy. Epilepsy Res. (2012) 99:327–34. doi: 10.1016/j.eplepsyres.2011.12.014
170. McGill ML, Devinsky O, Kelly C, Milham M, Castellanos FX, Quinn BT, et al. Default mode network abnormalities in idiopathic generalized epilepsy. Epilepsy Behav. (2012) 23:353–9. doi: 10.1016/j.yebeh.2012.01.013
171. Yang T, Luo C, Li Q, Guo Z, Liu L, Gong Q, et al. Altered resting-state connectivity during interictal generalized spike-wave discharges in drug-naïve childhood absence epilepsy. Hum Brain Mapp. (2013) 34:1761–7. doi: 10.1002/hbm.22025
172. Diessen E, van Zweiphenning WJEM, Jansen FE, Stam CJ, Braun KPJ, Otte WM. Brain network organization in focal epilepsy: a systematic review and meta-analysis. PLoS ONE. (2014) 9:e114606. doi: 10.1371/journal.pone.0114606
173. Vlooswijk MCG, Vaessen MJ, Jansen JFA, de Krom MCFTM, Majoie HJM, Hofman P, et al. Loss of network efficiency associated with cognitive decline in chronic epilepsy. Neurology. (2011) 77:938–44. doi: 10.1212/WNL.0b013e31822cfc2f
174. Vaessen MJ, Jansen JFA, Vlooswijk MCG, Hofman PAM, Majoie HJM, Aldenkamp AP, et al. White matter network abnormalities are associated with cognitive decline in chronic epilepsy. Cerebr Cortex. (2012) 22:2139–47. doi: 10.1093/cercor/bhr298
175. Gauffin H, van Ettinger-Veenstra H, Landtblom A-M, Ulrici D, McAllister A, Karlsson T, et al. Impaired language function in generalized epilepsy: inadequate suppression of the default mode network. Epilepsy Behav. (2013) 28:26–35. doi: 10.1016/j.yebeh.2013.04.001
176. Hampson M, Driesen NR, Skudlarski P, Gore JC, Constable RT. Brain connectivity related to working memory performance. J Neurosci. (2006) 26:13338–43. doi: 10.1523/JNEUROSCI.3408-06.2006
177. Kelly AMC, Uddin LQ, Biswal BB, Castellanos FX, Milham MP. Competition between functional brain networks mediates behavioral variability. Neuroimage. (2008) 39:527–37. doi: 10.1016/j.neuroimage.2007.08.008
178. Bernhardt BC, Chen Z, He Y, Evans AC, Bernasconi N. Graph-theoretical analysis reveals disrupted small-world organization of cortical thickness correlation networks in temporal lobe epilepsy. Cerebr Cortex. (2011) 21:2147–57. doi: 10.1093/cercor/bhq291
179. Diessen E, van Diederen SJH, Braun KPJ, Jansen FE, Stam CJ. Functional and structural brain networks in epilepsy: what have we learned? Epilepsia. (2013) 54:1855–65. doi: 10.1111/epi.12350
180. Rayner G, Tailby C, Jackson G, Wilson S. Looking beyond lesions for causes of neuropsychological impairment in epilepsy. Neurology. (2019) 92:e680–9. doi: 10.1212/WNL.0000000000006905
181. Sakkaki S, Barrière S, Bender AC, Scott RC, Lenck-Santini PP. Focal dorsal hippocampal Nav1.1 knock down alters place cell temporal coordination and spatial behavior. Cerebr Cortex. (2020) 30:5049–66. doi: 10.1093/cercor/bhaa101
182. Bender AC, Luikart BW, Lenck-Santini PP. Cognitive deficits associated with Nav1.1 alterations: involvement of neuronal firing dynamics and oscillations. PLoS ONE. (2016) 11:e0151538. doi: 10.1371/journal.pone.0151538
183. Lucas MM, Lenck-Santini P-P, Holmes GL, Scott RC. Impaired cognition in rats with cortical dysplasia: additional impact of early-life seizures. Brain. (2011) 134:1684–93. doi: 10.1093/brain/awr087
184. Weglage J, Demsky A, Pietsch M, Kurlemann G. Neuropsychological, intellectual, and behavioral findings in patients with centrotemporal spikes with and without seizures. Dev Med Child Neurol. (1997) 39:646–51. doi: 10.1111/j.1469-8749.1997.tb07357.x
185. Fonseca LC, Tedrus GMAS, Tonelotto JM, Antunes T, Chiodi MG. School performance in children with benign childhood epilepsy with centrotemporal spikes. Arquivos De Neuro-Psiquiatria. (2004) 62:459–62. doi: 10.1590/S0004-282X2004000300015
186. Kleen JK, Scott RC, Holmes GL, Lenck-Santini PP. Hippocampal interictal spikes disrupt cognition in rats. Ann Neurol. (2010) 67:250–7. doi: 10.1002/ana.21896
187. Kleen JK, Scott RC, Holmes GL, Roberts DW, Rundle MM, Testorf M, et al. Hippocampal interictal epileptiform activity disrupts cognition in humans. Neurology. (2013) 81:18–24. doi: 10.1212/WNL.0b013e318297ee50
188. Suárez LM, Cid E, Gal B, Inostroza M, Brotons-Mas JR, Gómez-Domínguez D, et al. Systemic injection of kainic acid differently affects LTP magnitude depending on its epileptogenic efficiency. PLoS ONE. (2012) 7:e48128. doi: 10.1371/journal.pone.0048128
189. Lenz M, Ben Shimon M, Deller T, Vlachos A, Maggio N. Pilocarpine-induced status epilepticus is associated with changes in the actin-modulating protein synaptopodin and alterations in long-term potentiation in the mouse hippocampus. Neural Plast. (2017) 2017:1–7. doi: 10.1155/2017/2652560
190. Lynch M, Sayin Ü, Bownds J, Janumpalli S, Sutula T. Long-term consequences of early postnatal seizures on hippocampal learning and plasticity: developmental plasticity in the hippocampus. Eur J Neurosci. (2000) 12:2252–64. doi: 10.1046/j.1460-9568.2000.00117.x
191. Hernan AE, Holmes GL, Isaev D, Scott RC, Isaeva E. Altered short-term plasticity in the prefrontal cortex after early life seizures. Neurobiol Dis. (2013) 50:120–6. doi: 10.1016/j.nbd.2012.10.007
192. Hernan AE, Alexander A, Jenks KR, Barry J, Lenck-Santini P-P, Isaeva E, et al. Focal epileptiform activity in the prefrontal cortex is associated with long-term attention and sociability deficits. Neurobiol Dis. (2014) 63:25–34. doi: 10.1016/j.nbd.2013.11.012
193. Schlesiger MI, Cannova CC, Boublil BL, Hales JB, Mankin EA, Brandon MP, et al. The medial entorhinal cortex is necessary for temporal organization of hippocampal neuronal activity. Nat Neurosci. (2015) 18:1123–32. doi: 10.1038/nn.4056
194. Hales JB, Schlesiger MI, Leutgeb JK, Squire LR, Leutgeb S, Clark RE. Medial entorhinal cortex lesions only partially disrupt hippocampal place cells and hippocampus-dependent place memory. Cell Rep. (2014) 9:893–901. doi: 10.1016/j.celrep.2014.10.009
195. Lenck-Santini P-P, Holmes GL. Altered phase precession and compression of temporal sequences by place cells in epileptic rats. J Neurosci. (2008) 28:5053–62. doi: 10.1523/JNEUROSCI.5024-07.2008
196. Powers S, Miranda C, Dennys-Rivers C, Huffenberger A, Braun L, Rinaldi F, et al. Rett syndrome gene therapy improves survival and ameliorates behavioral phenotypes in MeCP2 null (S51.002). Neurology. (2019) 92(15Suppl.):S51.002.
197. Moretti P M., Levenson J, Battaglia F, Atkinson R, Teague R, et al. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. (2006) 26:319–27. doi: 10.1523/JNEUROSCI.2623-05.2006
198. Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol. (1983) 14:471–9. doi: 10.1002/ana.410140412
199. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. (1999) 23:185–8. doi: 10.1038/13810
200. Chahrour M, Jung SY, Shaw C, Zhou X, Wong STC, Qin J, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. (2008) 320:1224–9. doi: 10.1126/science.1153252
201. Moretti P, Bouwknecht JA, Teague R, Paylor R, Zoghbi HY. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum Mol Genet. (2005) 14:205–20. doi: 10.1093/hmg/ddi016
202. Stearns NA, Schaevitz LR, Bowling H, Nag N, Berger UV, Berger-Sweeney J. Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neuroscience. (2007) 146:907–21. doi: 10.1016/j.neuroscience.2007.02.009
203. Kee SE, Mou X, Zoghbi HY, Ji D. Impaired spatial memory codes in a mouse model of Rett syndrome. Elife. (2018) 7:e31451. doi: 10.7554/eLife.31451
204. Asaka Y, Jugloff DGM, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. (2006) 21:217–27. doi: 10.1016/j.nbd.2005.07.005
205. Cardoza B, Clarke A, Wilcox J, Gibbon F, Smith PEM, Archer H, et al. Epilepsy in Rett syndrome: association between phenotype and genotype, and implications for practice. Seizure. (2011) 20:646–9. doi: 10.1016/j.seizure.2011.06.010
206. Ito-Ishida A, Ure K, Chen H, Swann JW, Zoghbi HY. Loss of MeCP2 in parvalbumin-and somatostatin-expressing neurons in mice leads to distinct Rett syndrome-like phenotypes. Neuron. (2015) 88:651–8. doi: 10.1016/j.neuron.2015.10.029
207. Operto FF, Mazza R, Pastorino GMG, Verrotti A, Coppola G. Epilepsy and genetic in Rett syndrome: a review. Brain Behav. (2019) 9:e01250. doi: 10.1002/brb3.1250
208. Buzsaki G, Horvath Z, Urioste R, Hetke J, Wise K. High-frequency network oscillation in the hippocampus. Science. (1992) 256:1025–7. doi: 10.1126/science.1589772
209. Buzsáki G. Two-stage model of memory trace formation: a role for “noisy” brain states. Neuroscience. (1989) 31:551–70. doi: 10.1016/0306-4522(89)90423-5
210. Lang M, Wither RG, Brotchie JM, Wu C, Zhang L, Eubanks JH. Selective preservation of MeCP2 in catecholaminergic cells is sufficient to improve the behavioral phenotype of male and female Mecp2-deficient mice. Hum Mol Genet. (2013) 22:358–71. doi: 10.1093/hmg/dds433
211. Samarasinghe RA, Miranda OA, Buth JE, Mitchell S, Ferando I, Watanabe M, et al. Identification of neural oscillations and epileptiform changes in human brain organoids. Nat Neurosci. (2021) 24:1488–500. doi: 10.1038/s41593-021-00906-5
212. D'Cruz JA, Wu C, Zahid T, El-Hayek Y, Zhang L, Eubanks JH. Alterations of cortical and hippocampal EEG activity in MeCP2-deficient mice. Neurobiol Dis. (2010) 38:8–16. doi: 10.1016/j.nbd.2009.12.018
213. Kosai K, Kusaga A, Isagai T, Hirata K, Nagano S, Murofushi Y, et al. Rett syndrome is reversible and treatable by MeCP2 gene therapy into the striatum in mice. Mol Ther. (2005) 11:S24. doi: 10.1016/j.ymthe.2005.06.086
214. Luoni M, Giannelli S, Indrigo MT, Niro A, Massimino L, Iannielli A, et al. Whole brain delivery of an instability-prone Mecp2 transgene improves behavioral and molecular pathological defects in mouse models of Rett syndrome. Elife. (2020) 9:e52629. doi: 10.7554/eLife.52629
215. Gadalla KK, Bailey ME, Spike RC, Ross PD, Woodard KT, Kalburgi SN, et al. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther. (2013) 21:18–30. doi: 10.1038/mt.2012.200
216. Kahn JB, Port RG, Yue C, Takano H, Coulter DA. Circuit-based interventions in the dentate gyrus rescue epilepsy-associated cognitive dysfunction. Brain. (2019) 142:2705–21. doi: 10.1093/brain/awz209
217. Snowball A, Chabrol E, Wykes RC, Shekh-Ahmad T, Cornford JH, Lieb A, et al. Epilepsy gene therapy using an engineered potassium channel. J Neurosci. (2019) 39:3159–69. doi: 10.1523/JNEUROSCI.1143-18.2019
218. Hermann B, Seidenberg M, Lee E-J, Chan F, Rutecki P. Cognitive phenotypes in temporal lobe epilepsy. J Int Neuropsychol Soc. (2007) 13:12–20. doi: 10.1017/S135561770707004X
219. Warburton A, Miyajima F, Shazadi K, Crossley J, Johnson MR, Marson AG, et al. NRSF and BDNF polymorphisms as biomarkers of cognitive dysfunction in adults with newly diagnosed epilepsy. Epilepsy Behav. (2016) 54:117–27. doi: 10.1016/j.yebeh.2015.11.013
220. Colliva C, Ferrari M, Benatti C, Guerra A, Tascedda F, Blom JMC. Executive functioning in children with epilepsy: genes matter. Epilepsy Behav. (2019) 95:137–47. doi: 10.1016/j.yebeh.2019.02.019
221. Busch RM, Lineweaver TT, Naugle RI, Kim KH, Gong Y, Tilelli CQ, et al. ApoE-ε4 is associated with reduced memory in long-standing intractable temporal lobe epilepsy. Neurology. (2007) 68:409–14. doi: 10.1212/01.wnl.0000253021.60887.db
222. Sidhu MK, Thompson PJ, Wandschneider B, Foulkes A, de Tisi J, Stretton J, et al. The impact of brain-derived neurotrophic factor Val66Met polymorphism on cognition and functional brain networks in patients with intractable partial epilepsy. CNS Neurosci Ther. (2019) 25:223–32. doi: 10.1111/cns.13003
223. Doherty C, Hogue O, Floden DP, Altemus JB, Najm IM, Eng C, et al. BDNF and COMT, but not APOE, alleles are associated with psychiatric symptoms in refractory epilepsy. Epilepsy Behav. (2019) 94:131–6. doi: 10.1016/j.yebeh.2019.02.032
225. Rosenzweig MR, Bennett EL. Cerebral changes in rats exposed individually to an enriched environment. J Comparat Physiol Psychol. (1972) 80:304–13. doi: 10.1037/h0032978
226. Nithianantharajah J. Environmental enrichment results in cortical and subcortical changes in levels of synaptophysin and PSD-95 proteins. Neurobiol Learn Mem. (2004) 81:200–10. doi: 10.1016/j.nlm.2004.02.002
227. Xu J, Sun J, Xue Z, Li X. An enriched environment reduces the stress level and locomotor activity induced by acute morphine treatment and by saline after chronic morphine treatment in mice. Neuroreport. (2014) 25:701–9. doi: 10.1097/WNR.0000000000000164
228. Akyuz E, Eroglu E. Envisioning the crosstalk between environmental enrichment and epilepsy: a novel perspective. Epilepsy Behav. (2021) 115:107660. doi: 10.1016/j.yebeh.2020.107660
229. Shinohara Y, Hosoya A, Hirase H. Experience enhances gamma oscillations and interhemispheric asymmetry in the hippocampus. Nat Commun. (2013) 4:1652. doi: 10.1038/ncomms2658
230. Tanaka M, Wang X, Mikoshiba K, Hirase H, Shinohara Y. Rearing-environment-dependent hippocampal local field potential differences in wild-type and inositol trisphosphate receptor type 2 knockout mice: experience-dependent hippocampal LFP patterns in WT and IP3 R2-KO mice. J Physiol. (2017) 595:6557–68. doi: 10.1113/JP274573
231. Bilkey DK, Cheyne KR, Eckert MJ, Lu X, Chowdhury S, Worley PF, et al. Exposure to complex environments results in more sparse representations of space in the hippocampus. Hippocampus. (2017) 27:1178–91. doi: 10.1002/hipo.22762
232. Dezsi G, Ozturk E, Salzberg MR, Morris M, O'Brien TJ, Jones NC. Environmental enrichment imparts disease-modifying and transgenerational effects on genetically-determined epilepsy and anxiety. Neurobiol Dis. (2016) 93:129–36. doi: 10.1016/j.nbd.2016.05.005
233. Vrinda M, Sasidharan A, Aparna S, Srikumar BN, Kutty BM, Shankaranarayana Rao BS. Enriched environment attenuates behavioral seizures and depression in chronic temporal lobe epilepsy. Epilepsia. (2017) 58:1148–58. doi: 10.1111/epi.13767
234. Hernan AE, Mahoney JM, Curry W, Richard G, Lucas MM, Massey A, et al. Environmental enrichment normalizes hippocampal timing coding in a malformed hippocampus. PLoS ONE. (2018) 13:e0191488. doi: 10.1371/journal.pone.0191488
235. The National Institute of Mental Health. Brain Stimulation Therapies. (NIMH). (2016). Available online at: https://www.nimh.nih.gov/health/topics/brain-stimulation-therapies/brain-stimulation-therapies (accessed February 14, 2022).
236. Turnbull IM, McGeer PL, Beattie L, Calne D, Pate B. Stimulation of the basal nucleus of meynert in senile dementia of Alzheimer's type. Stereotact Funct Neurosurg. (1985) 48:216–21. doi: 10.1159/000101130
237. Laxton AW, Tang-Wai DF, McAndrews MP, Zumsteg D, Wennberg R, Keren R, et al. A phase I trial of deep brain stimulation of memory circuits in Alzheimer's disease. Ann Neurol. (2010) 68:521–34. doi: 10.1002/ana.22089
238. Sankar T, Chakravarty MM, Bescos A, Lara M, Obuchi T, Laxton AW, et al. Deep brain stimulation influences brain structure in Alzheimer's disease. Brain Stimul. (2015) 8:645–54. doi: 10.1016/j.brs.2014.11.020
239. Mao Z-Q, Wang X, Xu X, Cui Z-Q, Pan L-S, Ning X-J, et al. Partial improvement in performance of patients with severe Alzheimer's disease at an early stage of fornix deep brain stimulation. Neural Regen Res. (2018) 13:2164. doi: 10.4103/1673-5374.241468
240. Fontaine D, Deudon A, Lemaire JJ, Razzouk M, Viau P, Darcourt J, et al. Symptomatic treatment of memory decline in Alzheimer's disease by deep brain stimulation: a feasibility study. J Alzheimer's Dis. (2013) 34:315–23. doi: 10.3233/JAD-121579
241. Kuhn J, Hardenacke K, Shubina E, Lenartz D, Visser-Vandewalle V, Zilles K, et al. Deep brain stimulation of the nucleus basalis of meynert in early stage of Alzheimer's dementia. Brain Stimul. (2015) 8:838–9. doi: 10.1016/j.brs.2015.04.002
242. Dürschmid S, Reichert C, Kuhn J, Freund H, Hinrichs H, Heinze H. Deep brain stimulation of the nucleus basalis of Meynert attenuates early EEG components associated with defective sensory gating in patients with Alzheimer disease – a two-case study. Eur J Neurosci. (2020) 51:1201–9. doi: 10.1111/ejn.13749
243. Rossi S, Hallett M, Rossini PM, Pascual-Leone A. Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clin Neurophysiol. (2009) 120:2008–39. doi: 10.1016/j.clinph.2009.08.016
244. Cotelli M, Manenti R, Cappa SF, Geroldi C, Zanetti O, Rossini PM, et al. Effect of transcranial magnetic stimulation on action naming in patients with Alzheimer disease. Arch Neurol. (2006) 63:1602. doi: 10.1001/archneur.63.11.1602
245. Cotelli M, Manenti R, Cappa SF, Zanetti O, Miniussi C. Transcranial magnetic stimulation improves naming in Alzheimer disease patients at different stages of cognitive decline. Eur J Neurol. (2008) 15:1286–92. doi: 10.1111/j.1468-1331.2008.02202.x
246. Bentwich J, Dobronevsky E, Aichenbaum S, Shorer R, Peretz R, Khaigrekht M, et al. Beneficial effect of repetitive transcranial magnetic stimulation combined with cognitive training for the treatment of Alzheimer's disease: a proof of concept study. J Neural Transm. (2011) 118:463–71. doi: 10.1007/s00702-010-0578-1
247. Ahmed MA, Darwish ES, Khedr EM, El Serogy YM, Ali AM. Effects of low versus high frequencies of repetitive transcranial magnetic stimulation on cognitive function and cortical excitability in Alzheimer's dementia. J Neurol. (2012) 259:83–92. doi: 10.1007/s00415-011-6128-4
248. Gallino D, Devenyi GA, Germann J, Guma E, Anastassiadis C, Chakravarty MM. Longitudinal assessment of the neuroanatomical consequences of deep brain stimulation: application of fornical DBS in an Alzheimer's mouse model. Brain Res. (2019) 1715:213–23. doi: 10.1016/j.brainres.2019.03.030
249. Koulousakis P, van den Hove D, Visser-Vandewalle V, Sesia T. Cognitive improvements after intermittent deep brain stimulation of the nucleus basalis of meynert in a transgenic rat model for Alzheimer's disease: a preliminary approach. J Alzheimer's Dis. (2020) 73:461–6. doi: 10.3233/JAD-190919
250. Tsai S-T, Chen S-Y, Lin S-Z, Tseng G-F. Rostral intralaminar thalamic deep brain stimulation ameliorates memory deficits and dendritic regression in β-amyloid-infused rats. Brain Struct Funct. (2020) 225:751–61. doi: 10.1007/s00429-020-02033-6
251. Limousin P, Krack P, Pollak P, Benazzouz A, Ardouin C, Hoffmann D, et al. Electrical stimulation of the subthalamic nucleus in advanced Parkinson's disease. N Engl J Med. (1998) 339:1105–11. doi: 10.1056/NEJM199810153391603
252. Volkmann J, Sturm V, Weiss P, Kappler J, Voges J, Koulousakis A, et al. Bilateral high-frequency stimulation of the internal globus pallidus in advanced Parkinson's disease. Ann Neurol. (1998) 44:953–61. doi: 10.1002/ana.410440615
253. Krack P, Batir A, Van Blercom N, Chabardes S, Fraix V, Ardouin C, et al. Five-year follow-up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson's disease. N Engl J Med. (2003) 349:1925–34. doi: 10.1056/NEJMoa035275
254. Williams A, Gill S, Varma T, Jenkinson C, Quinn N, Mitchell R, et al. Deep brain stimulation plus best medical therapy versus best medical therapy alone for advanced Parkinson's disease (PD SURG trial): a randomised, open-label trial. Lancet Neurol. (2010) 9:581–91. doi: 10.1016/S1474-4422(10)70093-4
255. Castrioto A. Ten-year outcome of subthalamic stimulation in Parkinson's disease: a blinded evaluation. Arch Neurol. (2011) 68:1550. doi: 10.1001/archneurol.2011.182
256. Smeding HMM, Speelman JD, Koning-Haanstra M, Schuurman PR, Nijssen P, van Laar T, et al. Neuropsychological effects of bilateral STN stimulation in Parkinson disease: a controlled study. Neurology. (2006) 66:1830–6. doi: 10.1212/01.wnl.0000234881.77830.66
257. Smeding HMM, Speelman JD, Huizenga HM, Schuurman PR, Schmand B. Predictors of cognitive and psychosocial outcome after STN DBS in Parkinson's Disease. J Neurol Neurosurg Psychiatry. (2011) 82:754–60. doi: 10.1136/jnnp.2007.140012
258. York MK, Dulay M, Macias A, Levin HS, Grossman R, Simpson R, et al. Cognitive declines following bilateral subthalamic nucleus deep brain stimulation for the treatment of Parkinson's disease. J Neurol Neurosurg Psychiatry. (2008) 79:789–95. doi: 10.1136/jnnp.2007.118786
259. Williams AE, Arzola GM, Strutt AM, Simpson R, Jankovic J, York MK. Cognitive outcome and reliable change indices two years following bilateral subthalamic nucleus deep brain stimulation. Parkinsonism Relat Disord. (2011) 17:321–7. doi: 10.1016/j.parkreldis.2011.01.011
260. Sáez-Zea C, Escamilla-Sevilla F, Katati MJ, Mínguez-Castellanos A. Cognitive effects of subthalamic nucleus stimulation in Parkinson's disease: a controlled study. Eur Neurol. (2012) 68:361–6. doi: 10.1159/000341380
261. Ezzyat Y, Kragel JE, Burke JF, Levy DF, Lyalenko A, Wanda P, et al. Direct brain stimulation modulates encoding states and memory performance in humans. Curr Biol. (2017) 27:1251–8. doi: 10.1016/j.cub.2017.03.028
262. Fell J, Staresina BP, Do Lam ATA, Widman G, Helmstaedter C, Elger CE, et al. Memory modulation by weak synchronous deep brain stimulation: a pilot study. Brain Stimul. (2013) 6:270–3. doi: 10.1016/j.brs.2012.08.001
263. Miller JP, Sweet JA, Bailey CM, Munyon CN, Luders HO, Fastenau PS. Visual-spatial memory may be enhanced with theta burst deep brain stimulation of the fornix: a preliminary investigation with four cases. Brain. (2015) 138:1833–42. doi: 10.1093/brain/awv095
264. Perrine K, Devinsky O, Uysal S, Luciano DJ, Dogali M. Left temporal neocortex mediation of verbal memory: evidence from functional mapping with cortical stimulation. Neurology. (1994) 44:1845. doi: 10.1212/WNL.44.10.1845
265. Coleshill SG. Material-specific recognition memory deficits elicited by unilateral hippocampal electrical stimulation. J Neurosci. (2004) 24:1612–6. doi: 10.1523/JNEUROSCI.4352-03.2004
266. Lee DJ, Izadi A, Melnik M, Seidl S, Echeverri A, Shahlaie K, et al. Stimulation of the medial septum improves performance in spatial learning following pilocarpine-induced status epilepticus. Epilepsy Res. (2017) 130:53–63. doi: 10.1016/j.eplepsyres.2017.01.005
267. Izadi A, Pevzner A, Lee DJ, Ekstrom AD, Shahlaie K, Gurkoff GG. Medial septal stimulation increases seizure threshold and improves cognition in epileptic rats. Brain Stimul. (2019) 12:735–42. doi: 10.1016/j.brs.2019.01.005
268. Lee DJ, Gurkoff GG, Izadi A, Seidl SE, Echeverri A, Melnik M, et al. Septohippocampal neuromodulation improves cognition after traumatic brain injury. J Neurotrauma. (2015) 32:1822–32. doi: 10.1089/neu.2014.3744
269. Southwell DG, Seifikar H, Malik R, Lavi K, Vogt D, Rubenstein JL, et al. Interneuron transplantation rescues social behavior deficits without restoring wild-type physiology in a mouse model of autism with excessive synaptic inhibition. J Neurosci. (2020) 40:2215–27. doi: 10.1523/JNEUROSCI.1063-19.2019
270. Hunt RF, Girskis KM, Rubenstein JL, Alvarez-Buylla A, Baraban SC. GABA progenitors grafted into the adult epileptic brain control seizures and abnormal behavior. Nat Neurosci. (2013) 16:692–7. doi: 10.1038/nn.3392
271. Handreck A, Backofen-Wehrhahn B, Bröer S, Löscher W, Gernert M. Anticonvulsant effects by bilateral and unilateral transplantation of GABA-producing cells into the subthalamic nucleus in an acute seizure model. Cell Transplant. (2014) 23:111–32. doi: 10.3727/096368912X658944
272. Henderson KW, Gupta J, Tagliatela S, Litvina E, Zheng X, Van Zandt MA, et al. Long-term seizure suppression and optogenetic analyses of synaptic connectivity in epileptic mice with hippocampal grafts of GABAergic interneurons. J Neurosci. (2014) 34:13492–504. doi: 10.1523/JNEUROSCI.0005-14.2014
273. Hammad M, Schmidt SL, Zhang X, Bray R, Frohlich F, Ghashghaei HT. Transplantation of GABAergic interneurons into the neonatal primary visual cortex reduces absence seizures in stargazer mice. Cerebr Cortex. (2015) 25:2970–9. doi: 10.1093/cercor/bhu094
274. Cunningham M, Cho J-H, Leung A, Savvidis G, Ahn S, Moon M, et al. hPSC-derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell Stem Cell. (2014) 15:559–73. doi: 10.1016/j.stem.2014.10.006
275. Hunt RF, Baraban SC. Interneuron transplantation as a treatment for epilepsy. Cold Spring Harb Perspect Med. (2015) 5:a022376. doi: 10.1101/cshperspect.a022376
276. Upadhya D, Hattiangady B, Castro OW, Shuai B, Kodali M, Attaluri S, et al. Human induced pluripotent stem cell-derived MGE cell grafting after status epilepticus attenuates chronic epilepsy and comorbidities via synaptic integration. Proc Nat Acad Sci USA. (2019) 116:287–96. doi: 10.1073/pnas.1814185115
277. Anderson NC, Van Zandt MA, Shrestha S, Lawrence DB, Gupta J, Chen CY, et al. Pluripotent stem cell-derived interneuron progenitors mature and restore memory deficits but do not suppress seizures in the epileptic mouse brain. Stem Cell Res. (2018) 33:83–94. doi: 10.1016/j.scr.2018.10.007
Keywords: epilepsy, cognition, neural coding, information processing, place cells, population coding, phase precession
Citation: Khalife MR, Scott RC and Hernan AE (2022) Mechanisms for Cognitive Impairment in Epilepsy: Moving Beyond Seizures. Front. Neurol. 13:878991. doi: 10.3389/fneur.2022.878991
Received: 18 February 2022; Accepted: 19 April 2022;
Published: 12 May 2022.
Edited by:
Yvonne Höller, University of Akureyri, IcelandReviewed by:
Anny Reyes, University of California, San Diego, United StatesGia Michele Ratto, National Research Council (CNR), Italy
Anne-Marie Landtblom, Uppsala University, Sweden
Copyright © 2022 Khalife, Scott and Hernan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohamed R. Khalife, bWtoYWxpZmVAdWRlbC5lZHU=