 Jie Rao2
Jie Rao2 Jing Sun
Jing Sun Yan Li
Yan Li Fangwang Fu
Fangwang Fu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 25 April 2022
Sec. Multiple Sclerosis and Neuroimmunology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.872684
This article is part of the Research TopicMultiple Sclerosis and Neuroimmunology – Case Report Collection, Volume IIView all 35 articles
Background and Objectives: To describe a new case of neuromyelitis optica spectrum disorder (NMOSD) induced by the administration of interferon-alpha (IFNα) and to raise awareness of this rare drug-induced disease of IFNα treatment.
Methods: A single case study and comprehensive literature review of eight cases.
Results: A 24-year-old man was diagnosed with cerebral venous thrombosis and essential thrombocythemia. He had been undergoing IFNα treatment (IFNα-2b, 3 million IU per day) without any side effects for 18 months, at which point the patient developed persistent hiccups, nausea, urinary retention, and numbness. Spinal magnetic resonance imaging revealed a longitudinal abnormality extending from the medulla to the entire spinal cord. The patient was positive for anti-aquaporin-4 antibody (AQP4-IgG) in both the serum and cerebrospinal fluid (CSF), which confirmed the diagnosis of NMOSD. Thus, recombinant IFNα-2b was suspended immediately. Because his condition did not improve after 6-day treatment of methylprednisolone pulse therapy (1,000 mg for 3 days, then 500 mg for 3 days), intravenous immunoglobulin (0.4 g/kg/day for 5 days) was administered. The patient gradually improved. Low-dose prednisolone and mycophenolate mofetil were subsequently administered as a long-term treatment. The patient was discharged with subtle limb numbness and their expanded disability status score (EDSS) was 1. At the 1-year follow-up, the patient had not relapsed and tested negative for AQP4-IgG. We further identified the eight patients with IFNα-induced NMOSD. The median onset age was 59 years, and the median time of IFNα exposure was 18 months. Optic neuritis was the most common initial symptom (five, 55.6%), followed by myelitis in three patients and area postrema syndrome in one patient. More than half (five, 55.6%) of the patients were monophasic. After IFNα discontinuation and immunotherapy, most (seven, 77.8%) patients remained relapse-free. However, only one patient was free of sequelae.
Conclusion: This study highlights the potential pathogenic risk of NMOSD of IFNα treatment. Given the high disability rates of this rare drug-induced disease, it is crucial to monitor the early manifestations of NMOSD during IFNα treatment.
Neuromyelitis Optica spectrum disorder (NMOSD) refers to a spectrum of the central nervous system (CNS) neuroinflammatory demyelinating disease that predominantly attacks the spinal cord, optic nerves, and brain (1, 2). The NMOSD is usually associated with a significant reduction in patients' quality of life and heavy economic burden. In contrast to T-lymphocyte-predominant myelinopathy in multiple sclerosis (MS) (3), NMOSD is considered an autoimmune astrocytopathy mediated by anti-aquaporin-4 antibodies (AQP4-IgG) (2). Type I interferons (IFNIs), particularly interferon-alpha (IFNα), are widely used to treat various infectious, immunological, and oncological diseases (4). They are also moderately effective for restricting CNS autoimmunity and are currently recommended as first-line immunomodulatory therapy for MS (5, 6). However, their use in patients with NMOSD is associated with neurologic deterioration and increased AQP4-IgG production (7, 8), which suggests a possible link between the development of NMOSD and IFNI. Although various CNS side effects of IFNI treatment have been reported to date, NMOSD remains largely unrecognized as a drug-induced disease (DID) of IFNI therapy (9). Recently, NMOSD cases associated with IFNα treatment started arising sporadically in patients with hepatitis C (9–14), systemic mastocytosis (9), and malignant melanoma (15). However, the underlying diseases may contribute to the occurrence of NMOSD in such cases. The precise relationship between IFNα and NMOSD remains elusive.
Herein, we present a case where the patient developed NMOSD following the administration of recombinant IFNα-2b for essential thrombocythemia (ET). We also determined the clinical profile and potential mechanism of IFNα-induced NMOSD by reviewing previously published cases.
This study was a single case report with a comprehensive literature review. The ethics committees waived institutional review and ethical approval. The patient provided written informed consent for publication. De-identified data are available upon appropriate request to the corresponding author.
We performed a systematic review of reported cases published between January 1, 2000, and December 31, 2021, by searching PubMed and China National Knowledge Infrastructure databases using the following search strategy: “NMOSD,” “NMO,” “AQP-4 antibody,” “type I interferon,” “interferon-alpha,” and “interferon-beta.” Reference lists of the identified publications were manually searched to find relevant publications not captured by the initial search strategy. Seven reports of eight cases were identified and reviewed.
A 24-year-old Chinese man presented with progressive headaches and right tinnitus for 3 months. He had no medical or family history relevant to his symptoms. He first visited the local hospital 2 weeks after onset, but no neurologic deficit was found. Brain MRI was performed 1 month later, which showed no abnormalities. He was admitted to our hospital after developing nausea and blurred vision. Neurological examination showed neck stiffness and bilateral optic disc swelling (ODS) (Figure 1A). Bilateral enlargement of the physiological blind spot was found in the visual field examinations (Figure 1C). Brain MRI with contrast showed no enhancement of the optic nerve (Figure 1H); however, the patient exhibited several imaging signs of intracranial hypertension (IH), which included the empty-sella sign (Figure 1E), perioptic subarachnoid space distension (Figures 1F,G), optic nerve tortuosity (Figure 1G), and posterior globe flattening (Figures 1F,G). A lumbar puncture was subsequently performed with an opening pressure of over 400 mm H2O. The CSF analysis results were all within normal ranges. Therefore, the ODS was considered a result of IH rather than optic neuritis. Contrast MRI also showed filling defects in the right transverse-sigmoid sinus (Figure 1I), which were confirmed by contrast-enhanced magnetic resonance venography (CE-MRV) (Figure 1J). The diagnosis of cerebral venous sinus thrombosis (CVST) was then established. Thrombosis risk factors were screened, and laboratory analyses for acquired and genetic hypercoagulability states were performed. However, the patient showed no abnormalities except for thrombocytosis (402 × 109 /L). The patient underwent whole-exome sequencing, which revealed a mutation in JAK2-V617F (Figure 2A). Furthermore, a bone marrow biopsy showed increased numbers of megakaryocytes. Thus, ET with high thrombosis risk was established. The patient received low-molecular-weight heparin for 1 month, then, switched to indefinite 3-month warfarin treatment with an INR of 2–3. For patients with ET in the high-risk category, timely initiation of first-line cytoreduction therapies with recombinant IFNα or hydroxyurea are recommended. Therefore, IFNα-2b (3 million IU per day) was prescribed, and platelet count was controlled adequately. Headache and blurred vision gradually subsided within 2 months. Two months later, a repeat MRI showed partial recanalization of the right transverse-sigmoid sinus (Figures 1K,L), and the CSF opening pressure was 200 mm H2O. Repeated fundoscopic examination showed mild improvement of ODS; thus, acetazolamide (250 mg twice a day) was administered to control IH.
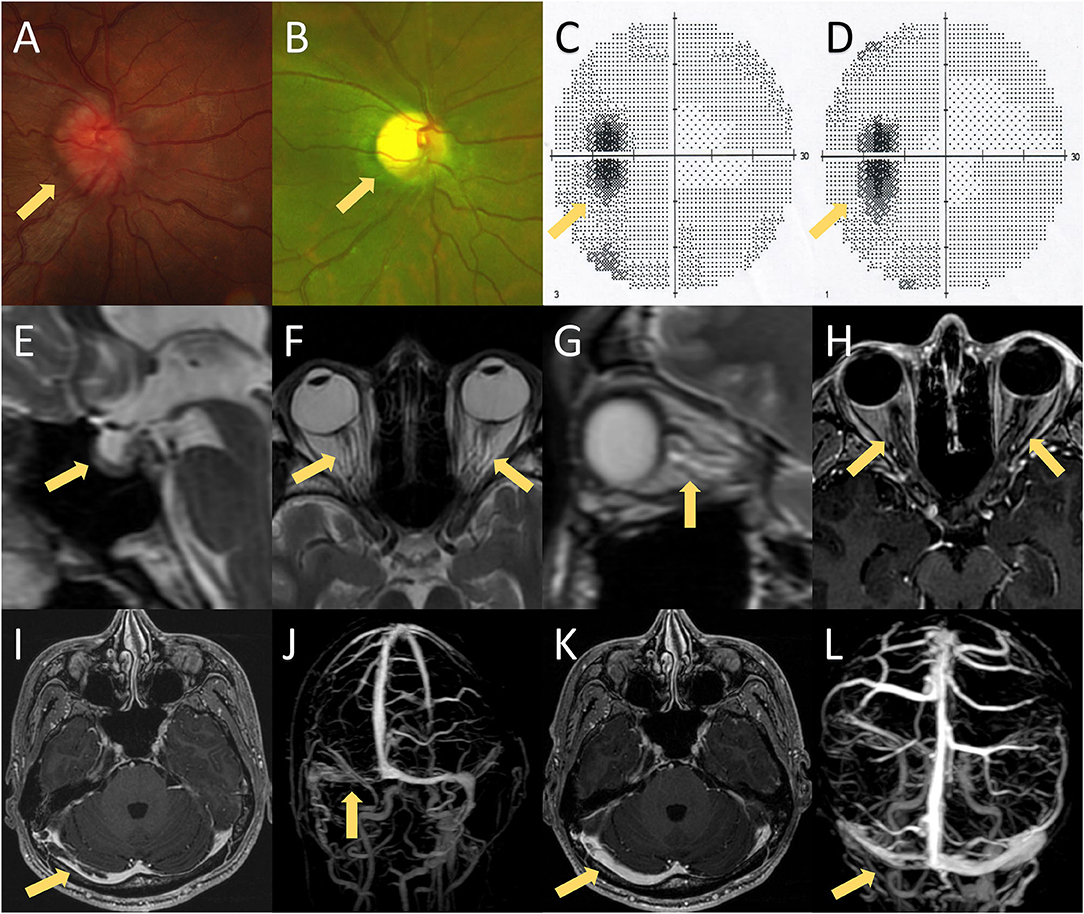
Figure 1. MRI findings, fundoscopic findings, and visual fields of the presented case. (A) Fundoscopic examination on the first hospitalization showed bilateral papilledema. (B) Fundoscopic examination on the second hospitalization revealed bilateral optic atrophy, which was probably caused by bilateral papilledema. (C,D) Visual fields on the first and second hospitalizations showed bilateral enlargement of the physiological blind spot without any other visual field defect. (E–G) Brain MRI exhibited several imaging signs of intracranial hypertension, including the empty-sella sign (E), perioptic subarachnoid space distension (F,G), optic nerve tortuosity (G), and posterior globe flattening (F,G). (H) Brain MRI with contrast showed no enhancement of the optic nerve. (I,J) Post-contrast 3D GRE T1-weighted imaging and contrast-enhanced magnetic resonance venography (CE-MRV) showed filling defects of the right transverse-sigmoid sinus. (K,L) Brain MRI and CE-MRV after 3 months of anticoagulation showed partial recanalization of the right transverse-sigmoid sinus.
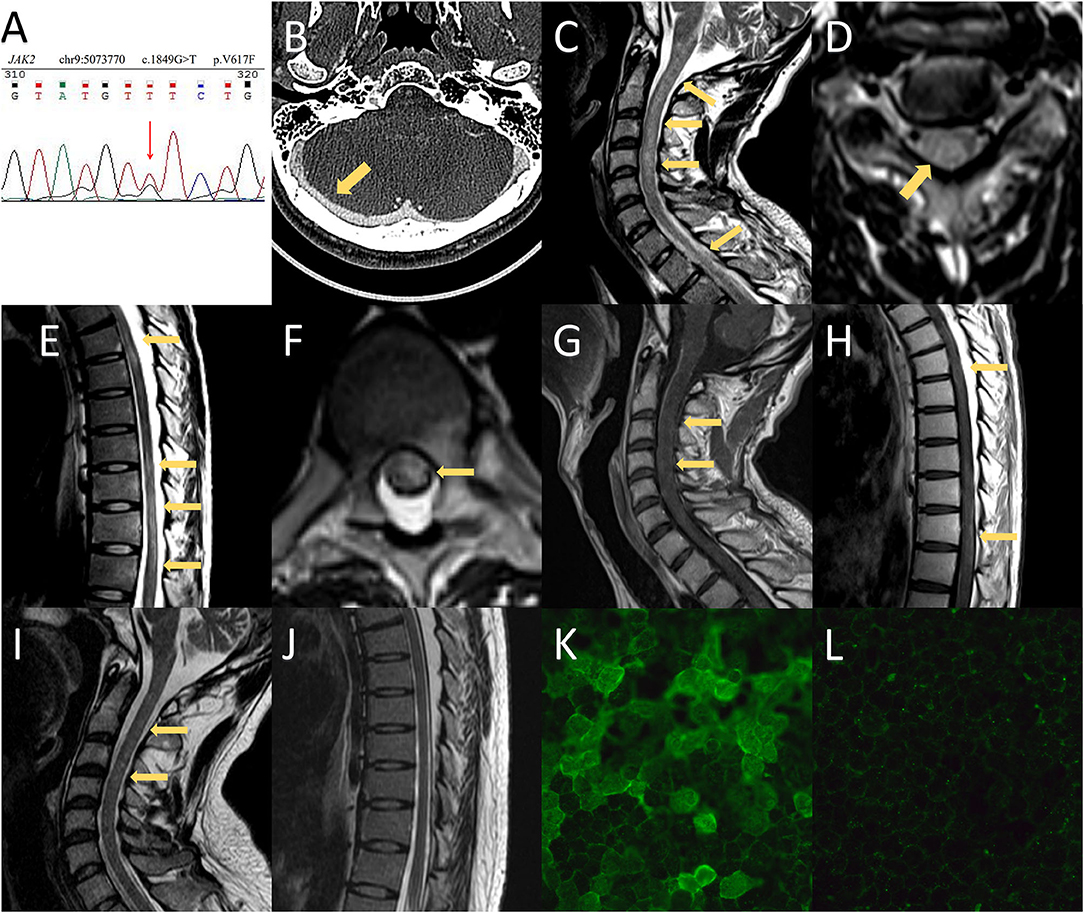
Figure 2. MRI and CT findings and anti-aquaporin-4 antibody (AQP4-IgG) array of the presented case. (A) Whole-exome sequencing revealed a mutation in JAK2-V617F. (B) CT venography of the second hospitalization showed no filling defects of the cerebral venous sinus. (C–H) Spinal MRI demonstrated T2-weighted imaging abnormalities in the medulla as well as extensive spinal cord involvement extending from C1 to the conus. Spinal MRI with contrast showed mild enhancement in the cervical and thoracic spinal cord. (I,J) Follow-up MRI showed prominent regression of the hyperintense lesion, which included the medulla and spinal cord. (K) The serum titer of AQP4-IgG using a cell-based assay was 1:1,000 at the time of neuromyelitis optica spectrum disorder diagnosis. (L) The patient was negative for AQP4-IgG at the 1-year follow-up.
The patient had no side effects of IFNα treatment, and his condition remained stable. However, 18 months later, he complained of persistent hiccups, nausea, and vomiting for 8 days and was admitted to the gastroenterology department on the 9th day for gastrointestinal endoscopy and abdominopelvic CT. However, no abnormalities were observed. The patient presented with acute urinary retention on day 12. Neurological consultation was requested following the lack of relief from antiemetic therapy. Given the high clinical suspicion of CVST recurrence, MRI, CT venography, lumbar puncture, and fundoscopic examinations were suggested. Fundoscopic and visual field examination on the 12th day showed bilateral optic atrophy and enlarged blind spot, which were considered a result of prolonged IH rather than acute IH (Figures 1B,D). However, the lack of filling defects of the cerebral venous system in CT venography ruled out the recurrence of CVST on the 13th day (Figure 2B). The patient developed numbness of the extremities and tingling around both calves on day 14 and was transferred to the neurology department. Neurological examination showed that he was conscious and oriented with horizontal nystagmus toward the left side. He had grade 5/5 strength and normal muscle tone; however, tendon hyperreflexia was observed in all four extremities. Sensory examination showed impaired proprioception sensations in both legs. Babinski and Hoffman's sign was positive bilaterally. Signs of meningeal irritation were absent. Repeated lumbar puncture was performed on the 15th day with a CSF opening pressure of 210 mm H2O. The CSF examination showed only mild mononuclear pleocytosis (80/ul) without abnormalities in proteins, immunoglobulin G (IgG) index, or oligoclonal bands. Spinal and brain MRI with contrast was undertaken on the 17th day, which showed an extensive signal abnormality that extended longitudinally from the medulla to the entire spinal cord, with slight edema and gadolinium enhancement (Figures 2C–H).
Serology for rheumatoid factor, anti-neutrophil cytoplasmic antibodies (ANCA), antiphospholipid antibodies, anti-Sjögren syndrome A/B antibodies, paraneoplastic panel, human leukocyte antigen (HLA) B5, and HLA-B51 were all negative. Serum and CSF infection panels were also negative. Antinuclear antibody (ANA) was positive at a titer of 1:100. Cytokines panel showed elevated interleukin-6 (IL-6) at 19 pg/ml (normal range 0–3 pg/ml). The cell-based AQP4-IgG assay of serum and CSF were positive at 1:1,000 and 1:100 (Figure 2K), respectively. Moreover, myelin-oligodendrocyte-glycoprotein IgG and glial-fibrillary-acidic-protein IgG were negative. Thus, the diagnosis of NMOSD was established according to the 2015 NMOSD criteria, and IFNα-2b treatment was suspended immediately. On the 18th day, the patient experienced weakness of both legs with an Expanded Disability Status Score (EDSS) of 3. However, neurological examination demonstrated only mild distal leg weakness (plantar flexion and ankle dorsiflexion, grade 4/5 strength). We administered a 12-day treatment plan with methylprednisolone (1,000 mg for 3 days, 500 mg for 3 days, 240 mg for 3 days, and 120 mg for 3 days). However, his clinical symptoms did not improve after 6 days of treatment. Intravenous immunoglobulin (0.4 g/kg/day for 5 days) was administered on the 23rd day, followed by mycophenolate mofetil (0.5 g twice a day) and oral prednisolone (60 mg per day, reduced to 20 mg per day step by step) as prophylaxis. In addition, hydroxyurea and aspirin were given instead of IFNα-2b to control ET.
The patient gradually improved, and urinary retention and lower limb weakness diminished on the 27th day. Hiccups and nausea subsided on the 31st day, and the patient was discharged on the 32nd day. The patient only had slight limb numbness at discharge, with an EDSS score of 1. On the follow-up visit 1 year later, the patient remained symptom-free and relapse-free, and his EDSS score was 0. Spinal MRI revealed that most foci had recovered (Figures 2I,J). Serum AQP4-IgG (Figure 2L) and ANA tests were negative. Long-term administration of mycophenolate mofetil and low dosage prednisolone was maintained. The patient remained symptom-, relapse-, and side-effect-free until December 2021.
Our literature review identified six case reports and one small case series. Together with our patient, nine patients with IFNα-induced NMOSD, who met 2015 NMOSD criteria were included for the pooled analysis (1). The clinical summary is provided in Table 1. The median age of NMOSD occurrence in patients was 59 years (range: 24–65 years), and 55.6% (5/9) of patients were women. More than half of patients were characterized by relapsing NMOSD. Hepatitis C infection was the most common primary disease (six patients, 66.7%). The remaining two cases had malignant melanoma and systemic mastocytosis, respectively, whereas our patient had ET as the primary disease. Optic neuritis (ON) was the most common initial symptom (five patients, 55.6%), followed by myelitis in three patients and area postrema syndrome (APS) in one patient. Six patients presented with ON, six presented with transverse myelitis, and only one patient had APS. Four patients presented with brain lesions in MRI, of whom three were asymptomatic. Two patients were noted as having slight pleocytosis in the CSF. Correspondingly, these patients also had a slight-to-moderate increase in proteins in the CSF. Oligoclonal bands were not found in any patient. Serum AQP4-IgG was positively detected in all patients.

Table 1. Characteristics of the patients with Interferon-alpha-induced neuromyelitis optica spectrum disorder (NMOSD).
All patients received IFNα, and only two patients had transient exposure to IFNβ. The median time of IFNα exposure was 18 months (range 3–180 months). Notably, two patients developed NMOSD after discontinuation of IFNα (after 2 and 3 months, respectively). Autoimmune antibodies secondary to IFNα treatment were detected in two patients: ANA and an anti-glutamate decarboxylase (GAD) antibody. Less than half (4/9) of patients responded favorably to initial immunotherapy. Of the six patients who had available outcome data, only one patient showed no sequelae. After IFNα discontinuation and long-term immunotherapy, most (seven patients, 77.8%) patients remained relapse-free. Only two patients reported transverse myelitis relapse following immunotherapy.
We presented the first NMOSD case with ET, which is one of the myeloproliferative neoplasms (MPNs). Is MPN a potential cause of NMOSD? Immune dysregulation is an increasingly reported characteristic of MPNs (16). An association has already been reported between MPNs and several autoimmune diseases, including rheumatoid arthritis and systemic lupus erythematosus (SLE) (16). Galimberti and colleagues found that 7.8% of patients with an MPN presented with an overt autoimmune disease (16). Moreover, the unusual concurrence of MS and MPN has been reported in a Roskilde MPN population. However, all five patients developed MPNs after MS. The authors concluded that chronic inflammation in MS patients may contribute to the development of MPNs (17). To date, there are no reports describing the occurrence of NMOSD in patients with MPNs. These results suggest that the presence of MPNs is not sufficient to cause NMOSD.
In addition, we presented a specific case of NMOSD, which developed following the administration of IFNα with a significant diagnostic delay. Thus, several considerations are warranted. The ODS was the most prominent presence in the early stages of our case. Thus, we question whether ODS is an early manifestation of NMOSD. The most common causes of ODS include papilledema (resulting from intracranial hypertension), inflammatory ON, and anterior ischemic optic neuropathy (18). The ODS of different etiologies usually appear ophthalmoscopically similar. Binocular involvement relatively preserved visual function, and symptoms, such as headache and tinnitus, can frequently help physicians distinguish papilledema from other diseases with ODS (18, 19). However, binocular ODS can emerge in ischemic and inflammatory optic neuropathies (18). Indeed, there are several case reports, in which bilateral ODS of NMOSD was misinterpreted as IH, even with marked elevated intracranial pressure. Furthermore, there are some cases, in which ODS with CVST was mistaken for ON. It can sometimes be challenging to determine the underlying cause of ODS (20); however, there are several helpful techniques. The optical coherence tomography (OCT) and OCT angiography (OCTA) provide non-invasive imaging of the optic circulation and can be used to identify ischemic ODS. The most sensitive diagnostic modality for ON is MRI with fat-saturated sequences and contrast enhancement (19). The abnormal enhancement of the optic nerve is direct evidence of ON. In addition, MRI of the orbits can reveal signs suggestive of IH, which include the empty-sella sign, perioptic subarachnoid space distension, optic nerve tortuosity, and posterior globe flattening (21). Our case presented with headache, tinnitus, ODS, preserved visual function, and a CSF pressure of more than 400 mm H2O, which strongly supported the diagnosis of papilledema. We also carefully re-analyzed the patient's neuroradiological data. Characteristic MRI findings for papilledema were identified without optic nerve enhancement. Therefore, in our case, bilateral ODS and secondary optic atrophyoptic atrophy were considered a consequence of CVST rather than the initial presentation of NMOSD.
In addition, our patient, who presented with APS as the initial symptom of NMOSD, was first admitted to the gastroenterology department. Intractable hiccups and vomiting are commonly encountered problems in the gastroenterology department or general medicine service (22). More than 70% of patients with APS are reported to visit a gastroenterologist or internist initially, and neurologic evaluation is not commonly pursued (22). However, intractable hiccups and vomiting can be a common clinical presentation of IH; although the absence of headache and papilledema made it an unlikely diagnosis. Thus, clinicians need to be aware of NMOSD as a diagnosable central cause for patients presenting with unexplained intractable nausea and vomiting.
The IFNα, a member of the IFNI family, plays a critical role in linking innate and adaptive immunity (23). The IFNIs exert their anti-proliferative, anti-tumor, anti-angiogenic, and immunomodulatory properties by activating the Janus kinase signal transducer and activator of transcription signals via the common type-I IFN receptor (IFNAR) (23, 24). The IFNα is still widely used in the treatment of chronic viral infections, hematological malignancies, and certain cancers (25), whereas IFNβ preparations are recommended for multiple isoforms of MS (5, 6). Opposing the beneficial actions of IFNI treatment, IFNI has gradually been recognized as a pro-inflammatory molecule that may not only unmask and aggravate underlying autoimmune processes, but also induce de novo autoimmune disorders, such as type-I diabetes, vitiligo, SLE, Sjögren syndrome, and autoimmune thyroid disease (26–28). Various neuroautoimmune diseases, including myasthenia gravis, inflammatory demyelinating polyneuropathy, and polymyositis, are also occasionally induced by IFNα therapy (29, 30).
Notwithstanding, NMOSD is still not formally regarded as a DID of treatment with IFNα. To date, few cases of IFNα-induced NMOSD have been reported, and this condition has likely been ignored by clinicians. Only eight cases with NMOSD secondary to IFNα therapy have been described to date. A few cases of IFNα-induced CNS demyelinating disease, including two cases with ON, two with ON and myelitis, and two with MS-like demyelinating disease, have been reported previously (15). However, AQP4-IgG was not detected in these cases, which hindered definite diagnoses. The first AQP4-IgG seropositive case of IFNα-induced NMOSD was reported in 2007 by Kajiyama et al. (14). The patient developed bilateral ON, transverse myelitis, and multiple periventricular white matter lesions after undergoing 13 months of IFNα treatment for chronic hepatitis C (14). Since then, nine patients have been reported with NMOSD with HCV infection as the primary disease, although three patients were not treated with IFNI (31). A possible association between NMOSD and HCV infection is likely; clinical data suggest that extrahepatic diseases are present in 40–74% of patients with hepatitis C as a result of complex interactions between HCV and B lymphocytes (31, 32). The HCV may contribute to immune system dysregulation, lymphocytes activation, and autoimmune antibody production, which include anti-ANA, anti-ANCA, and AQP4-IgG (32). Therefore, IFNα may form the bridge between NMOSD and HCV infection. Unfortunately, IFNα concentration was not measured in the NMOSD patients who did not undergo IFNα treatment.
In the remaining patients without HCV infection, IFNα was considered the independent trigger for the occurrence of NMOSD (9, 15). In addition, Williams and colleagues reported two cases in which NMOSD was secondary to exposure to significantly elevated endogenous IFNα, which was constitutively produced by the underlying interferonopathic disease, including genetic interferonopathy and SLE (9). The above evidence strongly supports the notion that NMOSD is a DID of IFNα treatment.
Interestingly, IFNβ treatment can exacerbate NMOSD but rarely induces newly onset NMOSD. In terms of mechanistic aspects, both IFNα and IFNβ may contribute to the development of NMOSD. The capacity to penetrate the blood-brain barrier (BBB) is an essential factor for DIDs of the CNS. A previous study showed that peripheral IFNα was able to exert its effects across the BBB (33). However, peripheral IFNβ was reported to have no direct access to an intact BBB (34).
The IFNα-induced NMOSD shares heterogeneous disease process patterns with AQP-IgG seropositive NMOSD (35), where clinical data, neuroradiological data, treatment response, and outcome features are largely similar across both groups. This is unsurprising because both conditions share the common pathogenetic AQP4-IgG pathway to NMOSD. Notably, the onset of NMOSD in some patients occurs several months after discontinuation of IFNα (12, 15). In an in vitro model, astrocytes showed markedly increased reactivity and dysregulation of the downstream gene and cytokines after exposure to IFNα for 3 weeks. Notably, these effects were not restored even 7 days after withdrawal of IFNα (36). Therefore, the prolonged exposure of IFNα may lead to persistent activation of the neuroautoimmune cascade even after the cessation of IFNα exposure.
Over the last decade, research progress has contributed to the substantial expansion of our understanding of the critical role of IFNI in the immunopathogenesis of NMOSD. Both central and peripheral mechanisms of the IFNI pathway may contribute to the development of NMOSD. We provide a detailed picture of the underlying mechanism in Figure 3.
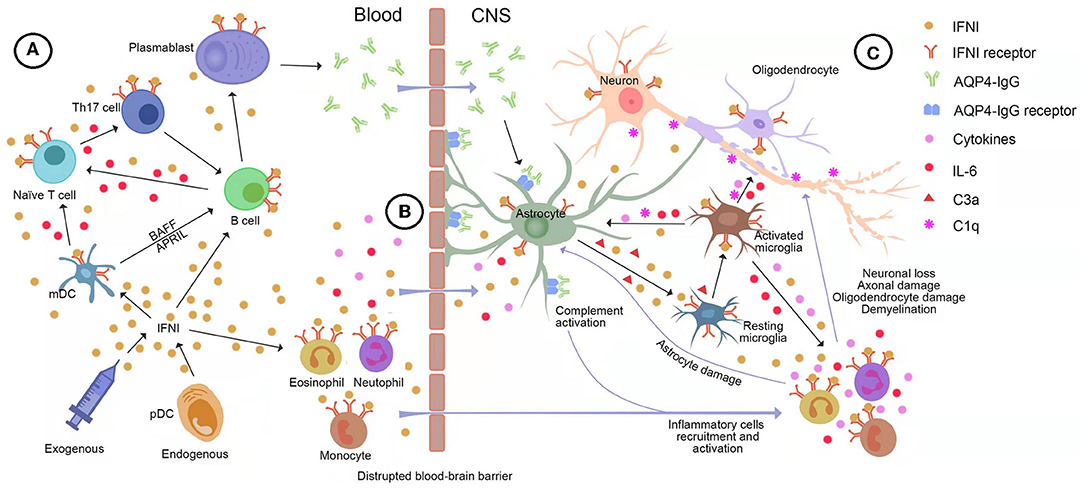
Figure 3. The potential role of type-I interferon (IFNI) signaling in the immunopathogenesis of neuromyelitis optica spectrum disorder (NMOSD). (A) Both endogenous and exogenous IFNI can drive B cells and myeloid dendritic cells (mDC) to produce large quantities of interleukin (IL)-6, which stimulates naive T cells to transform into inflammatory Th17 cells. Moreover, plasmacytoid dendritic cells (pDC) secretes IFNI into mDC, facilitating the generation of BAFF and APRIL, which are essential for the survival and maturation of B cells. In turn, inflammatory Th17 cells help B cells differentiate into AQP4-IgG-secreting plasma cells. (B) IFNI drives IL-6 and other pro-inflammatory molecules to disrupt and increase the permeability of the blood-brain barrier (BBB), allowing AQP4-IgG, pro-inflammatory cytokines, and immune cells to infiltrate the brain. (C) The IFNI-dependent astrocyte-microglia interaction drives the development of NMOSD pathology. Astrocytes are highly responsive to IFNI and the predominant source of IFNI in the central nervous system (CNS). The binding of AQP4-IgG to astrocytes induces massive production of IFNI and complement C3a, which results in the IFNI-dependent activation of microglia. Microglia respond to astroglial IFNI with the subsequent production of nitric oxide, inflammatory factors, complements, and downstream ISGs, which leads to a heightened activation state of microglia, immune cell recruitment, and complement-mediated CNS destruction. In turn, microglia secrete pro-inflammatory factors into astrocytes, especially IFNI, IL-1, IL-6, and TNF-α, leading to astrocyte activation, C1q production, and release of other pro-inflammatory factors feeding back to microglia. AQP4, aquaporin-4; APRIL, a proliferation-inducing ligand; BAFF, B-cell activating factor; BBB, brain blood barrier; C1q, Complement component 1 q; C3a, Complement component 3 a; IFN, interferon; IFNI, type-I interferon; IL-6, interleukin-6; IL-11, interleukin-11; mDC, myeloid dendritic cells; pDC, plasmacytoid dendritic cells; Th17, T helper type 17 cell; TNF-α, Tumor necrosis factor-α.
The AQP4-IgG is generally believed to form peripherally before entering the CNS (2). It is well-established that IFNI is the most important immune mediator of peripheral immunity. Prolonged exposure to IFNI may lead to the breakdown of immune tolerance and the initiation of an autoimmune response. The IFNI, predominantly IFNα, is either endogenously produced by plasmacytoid dendritic cells (pDCs) or administered exogenously and modulates the functions of key inflammatory cells in NMOSD in the periphery. The following are the most important immune effects of IFNI (37, 38): (1) IFNI promotes the expression of MHC-I molecules, which facilitate the processing and presentation of exogenous antigens, including AQP4; (2) IFNα increases the expression of MHC-II molecules and the production of cytokines, which stimulates monocytes differentiating into myeloid dendritic cells (mDCs). Subsequently, mDCs facilitate B-Cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL) generation, which are essential for the survival and maturation of B cells; (3) IFNI drives B cells and mDCs to produce large quantities of IL-6 and TGF-β, which are the key cytokines for the differentiation of Th17 cells and the suppression of Treg cell functions that lead to a protracted inflammatory response; (4) IFNI augments the differentiation of B cells into AQP4-IgG-secreting plasma cells via inflammatory Th17 cells and mDCs.
The IFNI also drives IL-6, CXCL10, and other pro-inflammatory molecules to disrupt and increase the permeability of the BBB, which allows AQP4-IgG, pro-inflammatory cytokines, and immune cells to infiltrate the brain.
The molecular and pathophysiological mechanisms involved in IFNI and NMOSD are being revealed via animal models of NMOSD. The most widely used animal models of autoinflammatory demyelinating disorders are referred to as experimental autoimmune encephalomyelitis (EAE) and can be divided into two clusters according to the passive transfer of myelin-specific TH1 or TH17 cells (TH1-EAE and TH17-EAE, respectively) (39, 40). Similar to NMOSD, TH17-EAE mice manifest ON and inflammatory demyelination in the CNS. Compared with effective treatment with IFNβ in TH1-EAE, IFNβ treatment of TH17-EAE shows significant deterioration of paralysis and increased spinal cord inflammation (39). Agasing et al. measured cytokines and inflammatory cells in TH17 mice treated with IFNβ and found that IFNI treatment of TH17-induced disease was associated with elevated serum IL-6 concentrations and TH17 cell numbers, but not with the number of neutrophils and inflammatory monocytes in the CNS. They also found that IFNIs could induce IL-6 from activated B cells to drive the pathogenic TH17 cells that play a crucial role in the occurrence and formation of NMOSD (39).
Khorooshi et al. established an animal model of NMOSD brain lesions, where typical astrocyte pathology characterized by the loss of AQP4 and GFAP was induced in mouse brains by intracerebral AQP4-IgG and human complement (huC). They observed that astrocyte pathology and associated granulocyte infiltration were reduced significantly more in IFNAR-deficient knockout (KO) mice than in wild-type mice. This result highlighted the role of IFNI signaling in the development of NMO-like pathology (41). The same team recently established a novel animal model of NMOSD-ON using intrathecal AQP4-IgG and huC. Typical astrocytopathy and NMOSD-like lesions in the optic nerve were observed in wild-type mice after intrathecal injection (42). However, NMOSD pathology was absent in IFNAR-KO mice (42). This result also strongly supported the notion that the presence of IFNI signaling is required for the development of NMOSD.
The IFNI-dependent astrocyte-microglia interaction is currently recognized as the driver of the development of NMOSD pathology (43, 44). Astrocytes are highly responsive to IFNI and the primary cells that produce IFNα in the CNS (45). The binding of AQP4-IgG to astrocytic AQP4 initiates astrocytic injury and stimulates the production and secretion of inflammatory cytokines and complement components, particularly IFNI, IL-6, and complement C3a by C3 cleavage (2). In turn, IFNI activates astrocytes to produce further inflammatory cytokines. Microglia, a critical mediator of the classical complement pathway in NMOSD pathology, can directly exacerbate neuroinflammation and promote neuroglial damage (46). Previous studies have shown that AQP4-IgG cannot directly activate microglia without astrocyte involvement. Indeed, the binding of astrocytic C3a to C3aR on resting microglia promotes microglial activation. Microglia-astrocyte crosstalk and motor impairment were shown to be absent in C3aR-deficient mice receiving AQP4-IgG, which highlights the need for the C3-C3aR axis in NMOSD pathology. Microglia respond to astroglial IFNI and C3a with subsequent production of nitric oxide, inflammatory factors, complements, and downstream ISGs, which leads to a heightened activation state of microglia, immune cell recruitment, complement cascades boost, and complement-mediated CNS destruction (43, 44). In turn, active microglia secrete abundant inflammatory factors and complements into astrocytes, which include IFNI, C1q, and IL-6, leading to a positive feedback loop. Particularly in the initial phase when there is no evident leukocyte infiltration, strong microgliosis, which is dependent on IFNIs from AQP4-IgG-binding astrocytes, corresponds to the facilitation of NMOSD-like pathology. Activated microglia significantly increases after intrastriatal injection of IFNβ in NMO mice, which subsequently exacerbates NMOSD-like pathology. In contrast, NMO-like pathology, microglia activation, and immunoreactivity markers are absent in IFNAR-KO mice that receive AQP4-IgG. In addition, the percentage of CD11c+ microglia was shown to be lower in IFNAR-KO mice than in control mice (46). Recent studies have found that microglia respond more strongly to IFNI than to other cell types in the CNS (33, 47). As mentioned earlier, IFNI drives the production of IL-6 and also exerts a pathogenic effect on NMOSD via the IL-6 pathway (39). The highly reactive microglia near the interface between the parenchyma and CSF has been identified to be closely associated with CSF IL-6 levels in patients with NMO. Correspondingly, the morphological characteristics of microglia in the IL-6 mice model are highly similar to those in patients with NMOSD (48). Therefore, it is reasonable to assume that IFNIs activate microglia via IL-6-driven pathology.
Taken together, IFNIs play a fundamental role in the immunopathogenesis and developmental process of NMOSD. The NMOSD is a severe neuroautoimmune disease of the CNS with high relapse and disability rates (1). Early diagnosis and rational immunotherapy strategies are crucial for improving the outcomes of patients with NMOSD. Insufficient awareness of NMOSD is a rare, yet life-threatening complication of IFNα therapy that may lead to misdiagnoses or delayed diagnoses associated with severe sequelae. Thus, screening for NMOSD should be performed as soon as possible, when patients present with early manifestations of NMOSD during IFNα treatment.
Our findings highlight the potential pathogenic risk of NMOSD of IFNα treatment. We present this case report and review of the published literature to alert physicians of this rare yet devastating consequence of IFNα. Monitoring early manifestations of NMOSD during or even after IFNα treatment is vital. Moreover, prompt suspension of IFNα treatment and early initiation of guideline-directed immunotherapy strategies are essential.
The original contributions presented in the study are included in the article. Deidentified data, including clinical manifestations, neuroimaging data, serum tests, and cerebrospinal fluid tests, are available upon appropriate request to the corresponding author.
FF and YL contributed to the design of the study, revised the manuscript, and are responsible for the integrity and accuracy of the data. JR and NX contributed to collecting clinical data, drafting the manuscript, and reviewing the published literature. JS was the attending doctor of the patient and contributed to the acquisition and analysis of the clinical data. All authors read and approved the final manuscript to be published.
Natural Science Foundation of Zhejiang Province of China (No. LY21H090015) and the Wenzhou Basic Scientific Research Project (No. Y20210900).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.872684/full#supplementary-material
Supplementary Figure 1. The timeline of the presented case.
ANA, antinuclear antibody; ANCA, anti-neutrophilcytoplasmic antibody; APRIL, a proliferation-inducing ligand; APS, area postrema syndrome; AQP4, aquaporin-4; BAFF, B-Cell activating factor; CE-MRV, contrast-enhanced magnetic resonance venography; CNS, central nervous system; CSF, cerebrospinal fluid; CT, computed tomography; CVST, cerebral venous sinus thrombosis; DID, drug-induced disease; EAE, experimental autoimmune encephalomyelitis; EDSS, Expanded disability status score; ET, essential thrombocythemia; GAD, glutamate decarboxylase; GFAP, glial fibrillary acidic protein; HCV, hepatitis C virus; HLA, human leukocyte antigen; huC, human complement; IFN, interferon; IFNI, type I interferon; IFNAR, type-I IFN receptor; IgG, immunoglobulin G; IL-6, interleukin-6; NMOSD, neuromyelitis optica spectrum disorder; MRI, magnetic resonance imaging; MS, multiple sclerosis; mDC, myeloid dendritic cells; OCT, optical coherence tomography; OCTA, optical coherence tomography angiography; ON, optic neuritis; pDCs, plasmacytoid dendritic cells; SLE, systemic lupus erythematosus.
1. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
2. Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. (2012) 11:535–44. doi: 10.1016/S1474-4422(12)70133-3
3. Guerau-de-Arellano M, Smith KM, Godlewski J, Liu Y, Winger R, Lawler SE, et al. Micro-Rna dysregulation in multiple sclerosis favours pro-inflammatory T-cell-mediated autoimmunity. Brain. (2011) 134(Pt 12):3578–89. doi: 10.1093/brain/awr262
4. Benveniste EN, Qin H. Type I interferons as anti-inflammatory mediators. Sci STKE. (2007) 2007:pe70. doi: 10.1126/stke.4162007pe70
5. Li H, Hu F, Zhang Y, Li K. Comparative efficacy and acceptability of disease-modifying therapies in patients with relapsing-remitting multiple sclerosis: a systematic review and network meta-analysis. J Neurol. (2020) 267:3489–98. doi: 10.1007/s00415-019-09395-w
6. Wiendl H, Gold R, Berger T, Derfuss T, Linker R, Maurer M, et al. Multiple sclerosis therapy consensus group (Mstcg): position statement on disease-modifying therapies for multiple sclerosis (white paper). Ther Adv Neurol Disord. (2021) 14:17562864211039648. doi: 10.1177/17562864211039648
7. Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult Scler. (2012) 18:1480–3. doi: 10.1177/1352458512439439
8. Palace J, Leite MI, Nairne A, Vincent A. Interferon beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol. (2010) 67:1016–7. doi: 10.1001/archneurol.2010.188
9. Williams J, McGlasson S, Irani S, Duffy D, Crow Y, Hunt D, et al. Neuromyelitis optica in patients with increased interferon alpha concentrations. Lancet Neurol. (2020) 19:31–3. doi: 10.1016/S1474-4422(19)30445-4
10. Kawazoe T, Araki M, Lin Y, Ogawa M, Okamoto T, Yamamura T, et al. New-onset type 1 diabetes mellitus and anti-aquaporin-4 antibody positive optic neuritis associated with type 1 interferon therapy for chronic hepatitis C. Intern Med. (2012) 51:2625–9. doi: 10.2169/internalmedicine.51.7771
11. Yamasaki M, Matsumoto K, Takahashi Y, Nakanishi H, Kawai Y, Miyamura M. [Case of Nmo (neuromyelitis optica) spectum disorder triggered by interferon alpha, which involved extensive pyramidal tract lesion of the brain]. Rinsho Shinkeigaku. (2012) 52:19–24. doi: 10.5692/clinicalneurol.52.19
12. Usmani N, McCarthy M, Rammohan KW, Ortega MR. Fulminant myelitis with Nmo igg antibody following treatment with interferon alpha. J Neurol. (2014) 261:240–1. doi: 10.1007/s00415-013-7202-x
13. Mangioni D, Soria A, Brighina L, Bandera A, Ferrarese C, Gori A. A case of classic neuromyelitis optica (devic's syndrome) triggered by pegylated-interferon alpha. BMC Pharmacol Toxicol. (2014) 15:56. doi: 10.1186/2050-6511-15-56
14. Kajiyama K, Tsuda K, Takeda M. Multiple sclerosis with positive anti-aquaporin-4 antibody, manifested after interferon-alpha-2b/ribavirin therapy for chronic hepatitis C. A case report. Neurol Med. (2007) 66:180–4.
15. Gao D, He M, Xu Q, Huang D, Wei S, Tian Y. Neuromyelitis optica spectrum disorder occurred after interferon alpha therapy in malignant melanoma. Mult Scler Relat Disord. (2019) 32:33–6. doi: 10.1016/j.msard.2019.04.023
16. Galimberti S, Baldini C, Barate C, Fornili M, Balducci S, Ricci F, et al. Myeloid neoplasms and autoimmune diseases: markers of association. Clin Exp Rheumatol. (2022) 40:49–55. doi: 10.55563/clinexprheumatol/ddxmp9
17. Thorsteinsdottir S, Bjerrum OW, Hasselbalch HC. Myeloproliferative neoplasms in five multiple sclerosis patients. Leuk Res Rep. (2013) 2:61–3. doi: 10.1016/j.lrr.2013.06.004
18. Jung JJ, Baek SH, Kim US. Analysis of the causes of optic disc swelling. Korean J Ophthalmol. (2011) 25:33–6. doi: 10.3341/kjo.2011.25.1.33
19. Trobe JD. Papilledema: the vexing issues. J Neuroophthalmol. (2011) 31:175–86. doi: 10.1097/WNO.0b013e31821a8b0b
20. Rougier MB. [Bilateral papilledema]. J Fr Ophtalmol. (2017) 40:422–9. doi: 10.1016/j.jfo.2017.02.002
21. Julayanont P, Karukote A, Ruthirago D, Panikkath D, Panikkath R. Idiopathic intracranial hypertension: ongoing clinical challenges and future prospects. J Pain Res. (2016) 9:87–99. doi: 10.2147/JPR.S60633
22. Iorio R, Lucchinetti CF, Lennon VA, Farrugia G, Pasricha PJ, Weinshenker BG, et al. Intractable nausea and vomiting from autoantibodies against a brain water channel. Clin Gastroenterol Hepatol. (2013) 11:240–5. doi: 10.1016/j.cgh.2012.11.021
23. Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. (2005) 23:307–36. doi: 10.1146/annurev.immunol.23.021704.115843
24. Kieseier BC. The mechanism of action of interferon-beta in relapsing multiple sclerosis. CNS Drugs. (2011) 25:491–502. doi: 10.2165/11591110-000000000-00000
25. Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. (2022) 19:237–53. doi: 10.1038/s41571-021-00588-9
26. Wahren-Herlenius M, Dorner T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet. (2013) 382:819–31. doi: 10.1016/S0140-6736(13)60954-X
27. Lombardi A, Tsomos E, Hammerstad SS, Tomer Y. Interferon alpha: the key trigger of Type 1 diabetes. J Autoimmun. (2018) 94:7–15. doi: 10.1016/j.jaut.2018.08.003
28. Ramaswamy M, Tummala R, Streicher K, Nogueira da Costa A, Brohawn PZ. The pathogenesis, molecular mechanisms, and therapeutic potential of the interferon pathway in systemic lupus erythematosus and other autoimmune diseases. Int J Mol Sci. (2021) 22:11286. doi: 10.3390/ijms222011286
29. Owens T, Khorooshi R, Wlodarczyk A, Asgari N. Interferons in the central nervous system: a few instruments play many tunes. Glia. (2014) 62:339–55. doi: 10.1002/glia.22608
30. Supakornnumporn S, Katirji B. Autoimmune neuromuscular diseases induced by immunomodulating drugs. J Clin Neuromuscul Dis. (2018) 20:28–34. doi: 10.1097/CND.0000000000000214
31. Ferreira JD, Caldas AC, de Sa JC, Geraldes R. Longstanding spastic paraparesis in a patient infected with hepatitis C virus and seropositive for aquaporin-4 antibody - case report and review of the literature. Mult Scler Relat Disord. (2016) 8:120–3. doi: 10.1016/j.msard.2016.05.016
32. Younossi Z, Park H, Henry L, Adeyemi A, Stepanova M. Extrahepatic manifestations of hepatitis C: a meta-analysis of prevalence, quality of life, and economic burden. Gastroenterology. (2016) 150:1599–608. doi: 10.1053/j.gastro.2016.02.039
33. Aw E, Zhang Y, Carroll M. Microglial responses to peripheral Type 1 interferon. J Neuroinflammation. (2020) 17:340. doi: 10.1186/s12974-020-02003-z
34. Blank T, Detje CN, Spiess A, Hagemeyer N, Brendecke SM, Wolfart J, et al. Brain endothelial- and epithelial-specific interferon receptor chain 1 drives virus-induced sickness behavior and cognitive impairment. Immunity. (2016) 44:901–12. doi: 10.1016/j.immuni.2016.04.005
35. Nagireddy RBR, Kumar A, Singh VK, Prasad R, Pathak A, Chaurasia RN, et al. Clinicoradiological comparative study of aquaporin-4-Igg seropositive neuromyelitis optica spectrum disorder (Nmosd) and Mog antibody associated disease (Mogad): a prospective observational study and review of literature. J Neuroimmunol. (2021) 361:577742. doi: 10.1016/j.jneuroim.2021.577742
36. Cuadrado E, Jansen MH, Anink J, De Filippis L, Vescovi AL, Watts C, et al. Chronic exposure of astrocytes to interferon-alpha reveals molecular changes related to aicardi-goutieres syndrome. Brain. (2013) 136(Pt 1):245–58. doi: 10.1093/brain/aws321
37. Di Domizio J, Cao W. Fueling autoimmunity: Type I interferon in autoimmune diseases. Expert Rev Clin Immunol. (2013) 9:201–10. doi: 10.1586/eci.12.106
38. Eloranta ML, Ronnblom L. Cause and consequences of the activated Type I interferon system in Sle. J Mol Med (Berl). (2016) 94:1103–10. doi: 10.1007/s00109-016-1421-4
39. Agasing AM, Wu Q, Khatri B, Borisow N, Ruprecht K, Brandt AU, et al. Transcriptomics and proteomics reveal a cooperation between interferon and T-helper 17 cells in neuromyelitis optica. Nat Commun. (2020) 11:2856. doi: 10.1038/s41467-020-16625-7
40. Larabee CM, Hu Y, Desai S, Georgescu C, Wren JD, Axtell RC, et al. Myelin-specific Th17 cells induce severe relapsing optic neuritis with irreversible loss of retinal ganglion cells in C57bl/6 mice. Mol Vis. (2016) 22:332–41.
41. Khorooshi R, Wlodarczyk A, Asgari N, Owens T. Neuromyelitis optica-like pathology is dependent on Type I interferon response. Exp Neurol. (2013) 247:744–7. doi: 10.1016/j.expneurol.2013.02.005
42. Soerensen SF, Wirenfeldt M, Wlodarczyk A, Moerch MT, Khorooshi R, Arengoth DS, et al. An experimental model of neuromyelitis optica spectrum disorder-optic neuritis: insights into disease mechanisms. Front Neurol. (2021) 12:703249. doi: 10.3389/fneur.2021.703249
43. Chen T, Lennon VA, Liu YU, Bosco DB, Li Y, Yi MH, et al. Astrocyte-microglia interaction drives evolving neuromyelitis optica lesion. J Clin Invest. (2020) 130:4025–38. doi: 10.1172/JCI134816
44. Moinfar Z, Zamvil SS. Microglia complement astrocytes in neuromyelitis optica. J Clin Invest. (2020) 130:3961–4. doi: 10.1172/JCI138804
45. Blank T, Prinz M. Type I interferon pathway in cns homeostasis and neurological disorders. Glia. (2017) 65:1397–406. doi: 10.1002/glia.23154
46. Wlodarczyk A, Khorooshi R, Marczynska J, Holtman IR, Burton M, Jensen KN, et al. Type I interferon-activated microglia are critical for neuromyelitis optica pathology. Glia. (2021) 69:943–53. doi: 10.1002/glia.23938
47. Li W, Viengkhou B, Denyer G, West PK, Campbell IL, Hofer MJ. Microglia have a more extensive and divergent response to interferon-alpha compared with astrocytes. Glia. (2018) 66:2058–78. doi: 10.1002/glia.23460
Keywords: NMOSD, AQP4 antibody, interferon, drug-induced disease, autoimmune disease, case report
Citation: Rao J, Xu N, Sun J, Li Y and Fu F (2022) Case Report: Interferon-Alpha-Induced Neuromyelitis Optica Spectrum Disorder. Front. Neurol. 13:872684. doi: 10.3389/fneur.2022.872684
Received: 09 February 2022; Accepted: 14 March 2022;
Published: 25 April 2022.
Edited by:
Robert Weissert, University of Regensburg, GermanyReviewed by:
Weihe Zhang, China-Japan Friendship Hospital, ChinaCopyright © 2022 Rao, Xu, Sun, Li and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Li, eWFubGlAd3poZWFsdGguY29t; Fangwang Fu, d2FuZ3BhcGVyQDE2My5jb20=
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.