
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 12 April 2022
Sec. Experimental Therapeutics
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.870799
This article is part of the Research TopicViral Vector-based Gene Therapy in Neurological Disease: The Future is NowView all 7 articles
 Marika Marino1,2†
Marika Marino1,2† Matthew G. Holt1,2,3,4*†
Matthew G. Holt1,2,3,4*†In the last four decades, monoclonal antibodies and their derivatives have emerged as a powerful class of therapeutics, largely due to their exquisite targeting specificity. Several clinical areas, most notably oncology and autoimmune disorders, have seen the successful introduction of monoclonal-based therapeutics. However, their adoption for treatment of Central Nervous System diseases has been comparatively slow, largely due to issues of efficient delivery resulting from limited permeability of the Blood Brain Barrier. Nevertheless, CNS diseases are becoming increasingly prevalent as societies age, accounting for ~6.5 million fatalities worldwide per year. Therefore, harnessing the full therapeutic potential of monoclonal antibodies (and their derivatives) in this clinical area has become a priority. Adeno-associated virus-based vectors (AAVs) are a potential solution to this problem. Preclinical studies have shown that AAV vector-mediated antibody delivery provides protection against a broad range of peripheral diseases, such as the human immunodeficiency virus (HIV), influenza and malaria. The parallel identification and optimization of AAV vector platforms which cross the Blood Brain Barrier with high efficiency, widely transducing the Central Nervous System and allowing high levels of local transgene production, has now opened a number of interesting scenarios for the development of AAV vector-mediated antibody delivery strategies to target Central Nervous System proteinopathies.
Antibodies, or Immunoglobulins (Ig), are glycoproteins produced by the immune system, characterized by their ability to recognize and bind a specific region (epitope) of an antigen with high specificity and (generally) high affinity, neutralizing potential pathogens. The basic structure of an Ig, determined by X-ray crystallography, is a tetramer of ~150 kDa, formed by two identical pairs of heavy (50 kDa) and light polypeptide chains (25 kDa), joined by disulfide bonds. Heavy and light chains present highly variable complementarity determining regions (CDRs), which form the Fragment antigen binding (Fab) regions of the antibody, responsible for antigen recognition and binding. In contrast, the Fragment crystallizable (Fc) region binds to a variety of receptors on immune cells, to mediate antibody interaction with other components of the immune system and provide effector function (Figure 1).
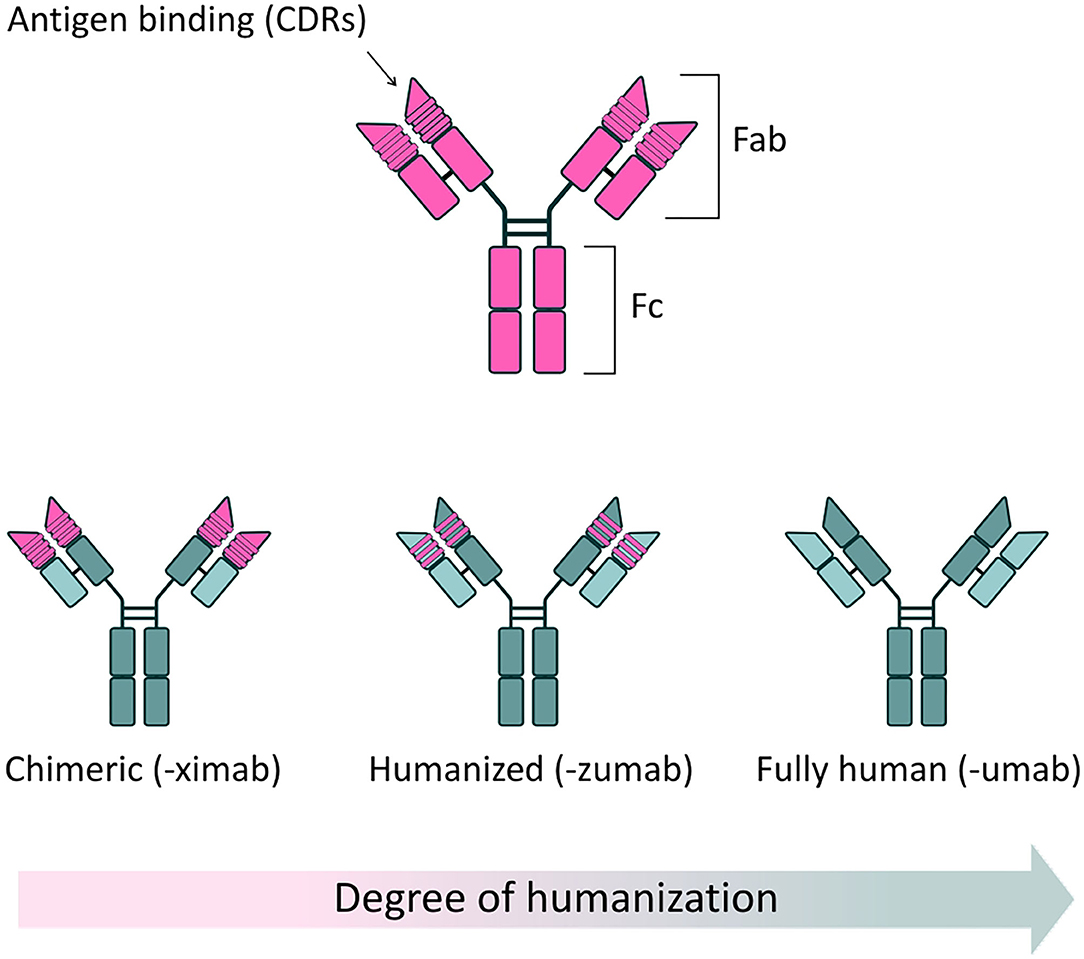
Figure 1. Full-length antibody structure. (Top) Schematic of a typical antibody structure, generated from a murine source. Two heavy chains and two light chains are connected in pairs by disulphide bonds. The fragment antigen binding (Fab) region contains highly variable complementarity determining regions (CDRs), responsible for the specific recognition and binding of antigens. The fragment crystallizable (Fc) region binds to specific receptors on immune cells, modulating the immune response, as well as some proteins of the complement system. (Bottom) Reduced immunogenicity of mAbs through protein engineering. The clinical success of mAbs has largely been driven by application of protein engineering techniques, which have allowed the development of safer, less immunogenic structures, when compared to mAbs of solely murine origin. Chimeric antibodies are characterized by approximately one third of their structure being derived from the original murine source. In these antibodies, murine-derived variable regions are linked to a human constant domain scaffold. By convention, chimeric mAbs are named using the “-ximab” suffix. In humanized mAbs, 90% of the structure is of human origin, with only the antigen binding sites deriving from the original mouse source. Humanized mAbs are named using the “-zumab” suffix. Fully human mAbs are entirely derived from human sources and thus display minimal immunogenicity in clinical use. Fully human antibodies are named using the “-umab” suffix.
The use of antibodies as therapeutic agents has increased considerably in the last 40 years, thanks to the generation of monoclonal antibodies (mAbs)—monovalent, laboratory-produced antibodies, derived from a single B-lymphocyte cell clone (1). Since the approval of the first therapeutic mAb, Muromonab-CD3 (OKT-3), by the US Food and Drug Administration (FDA) in 1985 (2), a further 80 mAb-based therapeutics have been approved for a range of clinical applications, with oncology, immunology and hematology being the predominant therapeutic areas (3, 4). This increased use of mAbs is no doubt partly fueled by progressive improvements in general safety, due to the development of less immunogenic chimeric (5, 6), humanized (7), and fully human structures (8) (Figure 1). mAbs have now achieved the status of best-selling pharmaceutical products: as of 2018, eight out of the ten best-selling drugs globally were mAbs; and in 2020 one single mAb, the anti-TNFα monoclonal Adalimumab (Humira®), generated an annual revenue of $19.9 billion (9). Interestingly, as of 5 years ago, ~570 mAbs were in evaluation in different phases of clinical trials and, based on previous completion rates, it is expected that a significant number of these novel mAbs therapeutics will come to market (10).
Proteinopathies are a large class of CNS diseases, characterized by the progressive accumulation of abnormal protein aggregates/inclusions, which are generally thought to confer a toxic gain-of-function (Table 1) (32). Generally, these toxic proteins accumulate intracellularly, although the toxic Aβ peptide classically associated with Alzheimer's disease accumulates extracellularly. Proteinopathies usually arise as adult onset, degenerative disorders, initially localized to specifically vulnerable regions of the CNS before spreading through the brain, possibly due to prion-like propagation mechanisms (33–36). Hence, drug delivery approaches that can widely target the CNS are required for the treatment of these severely debilitating conditions. In principle, mAbs represent an ideal therapeutic to treat these diseases, due to their high specificity for their protein targets [either directly targeting a toxic protein, or the processing enzymes responsible for its production, such as the β-site amyloid precursor protein cleaving enzyme 1 (BACE1) in Alzheimer's disease (AD)]. Importantly, the exquisite specificity of mAbs can be harnessed to target desired domains/functions of a protein while leaving the activity of other key domains intact (37). It can also be used to discriminate toxic protein variants from non-toxic forms, as shown by the ability of mAbs to distinguish different α-synuclein strains responsible for distinct synucleinopathies (38–40) and different structural variants of Aβ1−42 in Alzheimer's disease (AD) (41). This degree of targeting precision is superior to that achievable using alternative strategies, such as RNA interference (RNAi) and antisense oligonucleotides (ASOs), which lead to complete loss of protein function. RNAi and ASOs have also been reported to have additional problems linked to increased risks of toxicity (42–44) and off-target effects (45–47), and also appear unsuitable for depletion of long-lived proteins, as they operate at the transcriptional level, and so are dependent on protein depletion to have an effect (48).
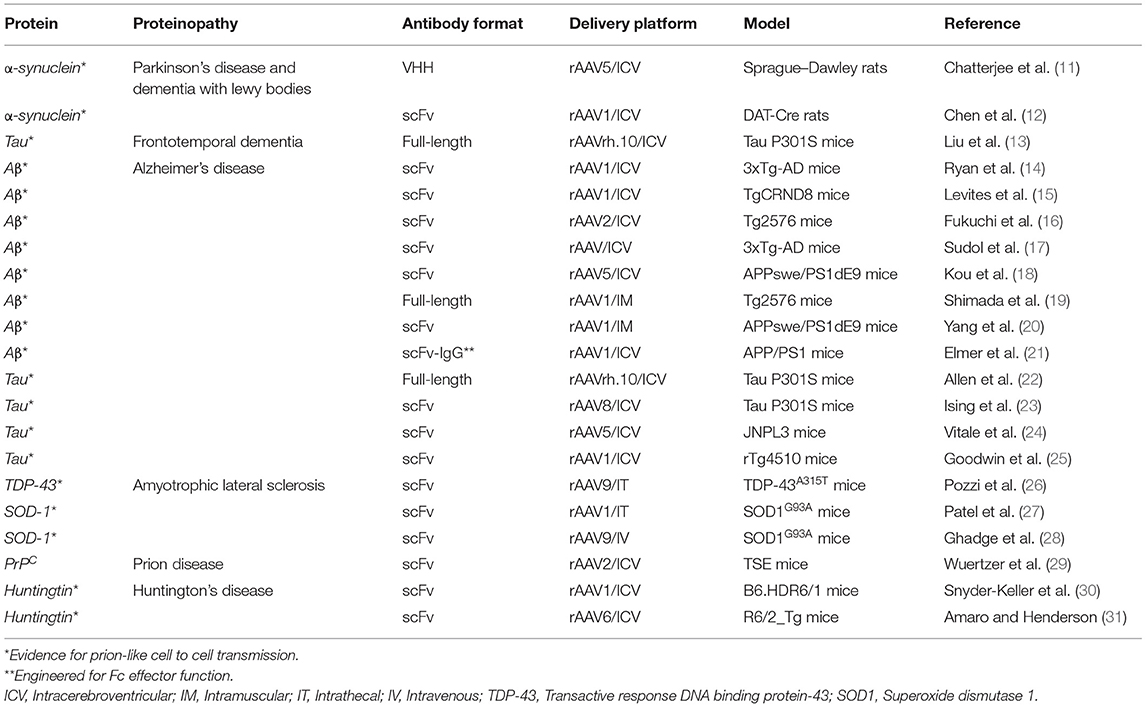
Table 1. List of A-MAD approaches in preclinical models of the most common CNS proteinopathies.
The use of mAbs as CNS therapeutics, however, is complicated by two main issues: (i) their large multimeric structure, limiting their ability to cross the Blood Brain Barrier (BBB) following intravenous administration (49) and (ii) their moderate half-life (~11–30 days in humans) (49). Combined, these issues mean that mAbs require repeated administration at high doses to maintain therapeutically relevant levels in the CNS, as shown by chAducanumab (Aduhelm®), which requires monthly intravenous infusions to achieve disease modifying effects in AD (50). Repeated administration does, however, have a number of disadvantages. First, it can negatively impact on patient compliance. Second, it will result in accumulating costs, particularly for long-term, adult-onset neurodegenerative diseases, which will require high levels of dosing over a sustained period.
As such, the vast majority of mAbs currently on the market are indicated for the treatment of acute peripheral conditions, with oncology, autoimmune diseases and dermatology being the most prevalent therapeutic areas. In contrast, the development of mAbs therapeutics for the treatment of CNS disorders is lagging behind, albeit with a few exceptions, such as anti-CGRP and anti-CGRP receptor mAbs for the treatment of migraine (forecast to become best-selling drugs by 2024) (51), and the aforementioned chAducanumab, regardless of the ongoing controversy surrounding its clinical efficacy.
Hence, to fully exploit the unique specificity properties of mAbs (and their derivatives) within the CNS, we believe it is essential to employ technologies that allow efficient BBB crossing and provide long-term, stable drug exposure in the CNS. In this respect, recombinant vectors based on Adeno-Associated Viruses (rAAV) have been identified as promising tools to unlock the potential of mAbs (and their derivatives) for the treatment of CNS proteinopathies. In this review, we will summarize the current status of the field and highlight areas of potential improvement that we believe will turn the prospect of rAAV vector-mediated antibody delivery (A-MAD) for the treatment of CNS proteinopathies into a reality in the near future.
“Blood brain barrier” is a working terminology that defines the microvasculature of the CNS, which possesses a unique structure and properties [extensively reviewed in (52)] (Figure 2A). The BBB allows the selective transport of molecules essential to maintain the CNS microenvironment and proper CNS function, while acting as a barrier against toxic compounds and dangerous microorganisms (Figure 2B). The shielding effect of the BBB, however, also severely limits the efficient penetration of therapeutics from the periphery into the CNS. In general, compounds which have a molecular weight >400 Da and are capable of forming more than 8 hydrogen bonds are efficiently excluded from the CNS by the BBB (53). This is especially problematic in the case of antibody-based therapies, due to their size. Typically, only 0.1–0.2% of systemically injected antibody accesses the CNS. Clearly, if therapeutic mAbs are not accessible to the brain, their utility for the treatment of proteinopathies will be limited. Hence, given the benefits of antibody-based therapies and the increasing unmet clinical need presented by CNS diseases, there is an ongoing search for technologies which can circumvent the CNS and efficiently deliver antibody-based therapeutics into the parenchyma (54) (see Section ‘Delivery of mAbs and their derivatives to the Central Nervous System’ below).
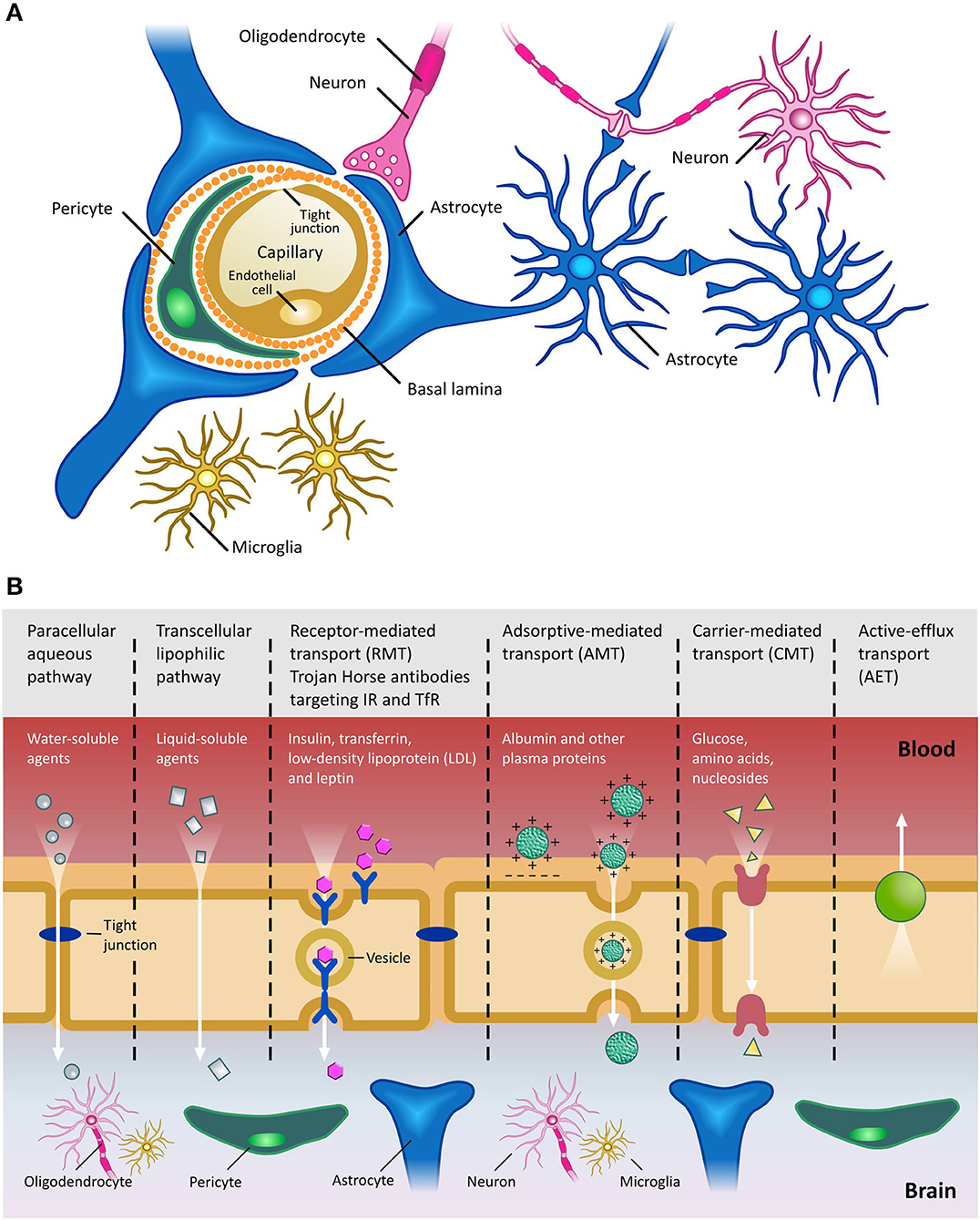
Figure 2. The Blood Brain Barrier. (A) Schematic of the BBB shown in cross-section. Main cellular components are shown. Endothelial cells are fused together by tight junctions, and surrounded by pericytes and astrocytic end feet. Figure adapted from (59). (B) Transport pathways across the Blood Brain Barrier. Ions, water and other small molecules necessary to maintain CNS homeostasis (such as glucose for metabolic support) can cross the BBB using a variety of mechanisms, including those based on passive diffusion, from high to low concentrations, and active transport, requiring ATP hydrolysis. Molecules can cross the BBB using either paracellular (between cells) or transcellular (across cells) pathways. Receptor-mediated transcytosis (RMT) allows selective uptake of macromolecules, including plasma proteins, enzymes, hormones and growth factors, via receptors expressed on the luminal side of the BBB. Trojan horse antibody strategies exploit RMT to enable delivery of conjugated drugs within the CNS. Adsorptive-mediated transport (AMT) relies on the electrostatic interaction between positively charged molecules (in general charged peptide moieties) and the negatively charged plasma membrane. Carrier-mediated transport (CMT) is an energy-dependent mechanism, allowing co-transport of molecules, either in the same direction (symport) or in the opposite direction (antiport). Active efflux transport (AET) is involved in the clearance of molecules, including drugs, from the brain. Due to the general impermeability of the BBB and the reliance on specialized and specific transport mechanisms for transport of molecules into the CNS, high molecular weight therapeutics, such as mAbs and their derivatives, are largely excluded from the CNS. Figure adapted from (60).
Disruption of the BBB has been reported at late stages of several proteinopathies, such as AD, Parkinson's disease (PD), Huntington's disease (HD) and Amyotrophic lateral sclerosis (ALS) (55, 56). In these conditions, in fact, multiple neurodegeneration pathways are driven by immune responses (associated with the influx of toxic blood-derived debris, cells and pathogenic microorganisms), as well as impaired oxygen transport and consequent hypoxia associated inflammation (56, 57). However, it is unclear whether such disruption would facilitate the entry of therapeutics into the CNS and, if it did, whether any benefits would offset the deleterious effects of barrier breakdown. Furthermore, the degree of BBB breakdown is likely variable and incomplete, suggesting that the use of BBB-crossing technologies to deliver antibodies into the CNS would still allow higher levels of therapeutic to be achieved (58), as shown in preclinical models of neurodegenerative disease with various degrees of BBB disruption. Finally, postponing drug administration until an advanced stage of disease, when extensive BBB disruption is potentially observed, would in all likelihood limit the therapeutic benefits, since significant (and currently irreparable) CNS damage would already have occurred. As such, developing novel technologies to circumvent the intact BBB, allowing the efficient delivery of mAbs into the CNS, is essential if we are to harness their full therapeutic potential for the treatment of CNS proteinopathies.
One way to overcome the shielding effect of the BBB is to inject therapeutics directly into the brain parenchyma. However, not only are direct injections highly invasive and damaging to the local tissue, but they are typically characterized by limited distribution of the therapeutic, which is retained near the injection site, limiting the use of this approach to treat multi-region CNS diseases (61–65). To some extent, these problems can be overcome by direct injection into the cerebrospinal fluid (CSF), but this method is also associated with potentially severe adverse effects, particularly if large boluses of drugs are rapidly administered (66). Drug entry into the CNS can also be achieved using administration of hyperosmotic solution (67) or by use of ultrasound to temporarily disrupt the BBB (68), although random opening of the barrier for undefined periods of time also involves the risk of potentially severe adverse effects (56). Ideally, the treatment of multi-region CNS diseases requires a drug delivery system which combines minimal invasiveness with widespread drug delivery.
To date, the most commonly exploited technology for the minimally invasive delivery of drugs to the CNS has been based on the naturally occurring process of receptor-mediated transcytosis (RMT), which serves to shuttle essential metabolites to the brain. This process can be subverted by so-called “Trojan Horses,” mAbs against endogenous BBB receptors, such as the insulin receptor (IR) or transferrin receptor (TfR). These antibodies bind their targets and are effectively carried across the BBB by RMT. By fusing the biologic of interest to the “Trojan Horse,” it is possible to obtain CNS delivery, following a single systemic bolus (69). This approach also underlies alternative technologies, such as the so-called “Brain Shuttle” (BS), which utilizes an antibody fragment against the TfR to transport biologics into the CNS (70). However, the “Trojan Horse” strategy is characterized by limited efficiency (CNS uptake of 1.1% and 3.5% of a systemically injected dose of TfR- or IR-targeting mAb in rhesus monkey, respectively) (71) and suffers from safety concerns linked to chronic administration of high antibody doses (71, 72). The widespread adoption of RMT-based technologies is impacted by a number of further issues. These include the fact that insulin and transferrin receptors are not uniquely expressed at the BBB site, raising safety concerns linked to non-site-specific distribution of the therapeutic (73). Potential solutions to overcome these concerns are currently being investigated, as shown by reports suggesting that a reduction in the antibody binding affinity to the TfR increases brain uptake, while mitigating adverse effects observed in the periphery (e.g., reduction in reticulocyte count) (73, 74). Additionally, receptor-specific “Trojan Horse” antibodies, such as anti-IR or -TfR are generally species-specific, requiring tuning of the binding profile in each species tested, which will negatively impact on the ease of their translation into humans (75, 76).
In contrast, rAAV vectors are emerging as ideal candidates for mAbs delivery to the CNS, as described below.
Naturally occurring AAV is a non-enveloped virus, in which a single stranded (ss) DNA genome of ~4.8 kb is contained within an icosahedral protein capsid. The genome encodes three genes which act in various processes relating to the viral life-cycle. The four proteins encoded by the rep gene (Rep78, Rep68, Rep52, and Rep40) are required for viral genome replication and packaging, while expression of the cap gene produces viral capsid proteins (VP1, VP2, and VP3 subunits). Finally, an alternate reading frame overlapping the cap gene encodes the assembly-activating protein (AAP), which promotes interaction between VPs for capsid formation and stability. These coding sequences are packaged between two inverted terminal repeats (ITRs), essential for genome replication and packaging (77) (Figure 3). The initial interaction of AAV with cells, prior to infection, is mediated by interactions with specific carbohydrates on the surface of target cells, such as sialic acid, galactose and heparin sulfate (78). The preferential binding for one or more of these molecules, determined by differences in the sequences of the different VP proteins, determines the tropism of specific AAV serotypes.
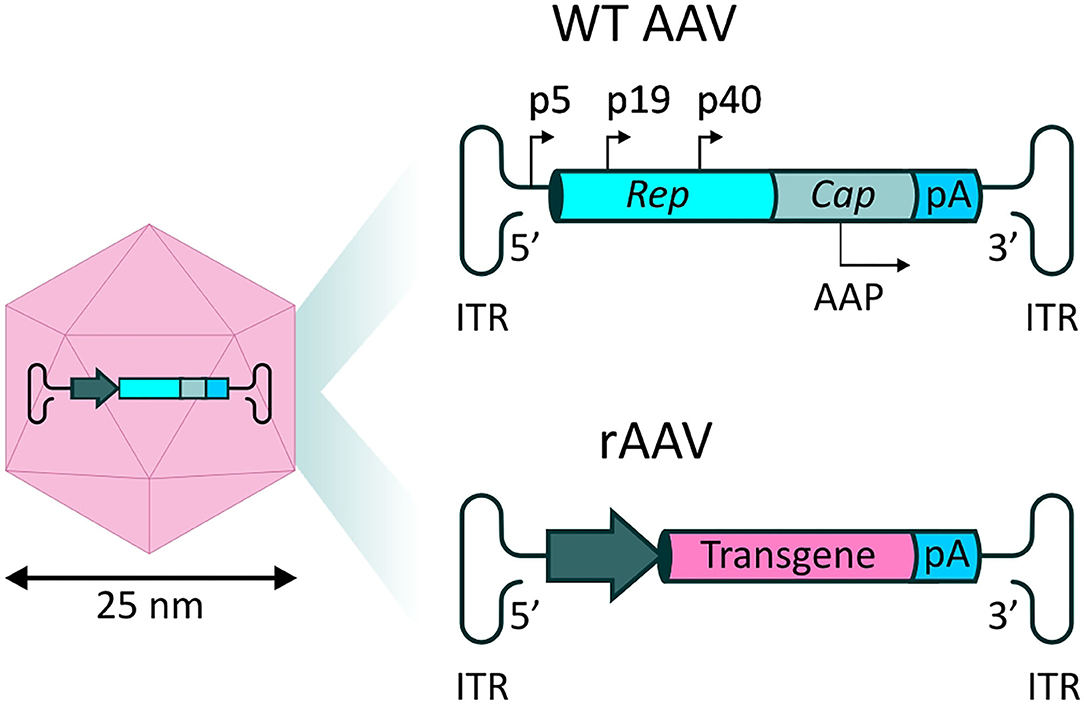
Figure 3. rAAV vectors for systemic delivery of mAbs. (Left) AAV capsid structure. The AAV capsid is composed of 60 protein monomers (VP1, VP2, and VP3 subunits with a stoichiometry of 1:1:10) arranged in icosahedral symmetry. (Top right) Representation of the single stranded (ss) DNA genome found in naturally occurring AAVs. Two open reading frames (ORFs) express genes necessary for replication (rep) and capsid structure (cap). An alternate reading frame overlapping the cap gene encodes the assembly-activating protein (AAP), promoting capsid formation and stability. (Bottom right) In recombinant AAV-based vectors (rAAV), the viral genes are substituted by a synthetic transgene cassette, in which a promoter drives expression of a transgene of interest, followed by a polyA tail.
Recombinant AAV (rAAV) vectors are formed by substitution of the viral genes with a synthetic expression cassette, in which a promoter element drives the expression of the therapeutic transgene of interest, followed by a polyadenylation (polyA) signal (Figure 3). In the typical rAAV vector production method, rep and cap are supplied in trans, alongside a helper plasmid containing essential genes (E4, E2a, and VA) which play a role in AAV replication (79). Standard ss rAAV have an optimal transgene capacity of ~4.8 kb, including regulatory sequences, such as promoters and regulatory elements, including the widely used polyadenylation sequence (polyA tail); packaging of coding sequences exceeding 5.2 kb is inefficient, and unfavorably impacts on transduction (80).
Over the past 30 years, rAAV vectors have been employed in a variety of clinical trials, both for peripheral (NCT03588299, NCT03001830, NCT02396342 for the treatment of hemophilia A and B) and CNS conditions (NCT01621581, NCT01973543, NCT03562494 for the treatment of Parkinson's disease; NCT00087789, NCT03634007 for the treatment of Alzheimer's disease) (81). These trials have shown rAAVs to have a largely safe clinical profile. One area of concern with systemic administration of high vector doses is potential hepatotoxicity, which has been reported in some clinical trials. However, it appears that this can be managed using corticosteroid administration (82). The FDA and the EMA approval of Zolgensma®, a pioneering AAV9-based medication for the treatment of Spinal Muscular Atrophy (SMA) in pediatric patients, is potentially the tip of the iceberg for AAV use in treating CNS diseases. In fact, as of January 2022, ~50 additional clinical studies employing rAAV vectors for the treatment of CNS disorders had been either completed or were ongoing (www.clinicaltrials.gov) (83).
Convincing proof-of-principle has been obtained for rAAV vector-mediated for the treatment of peripheral conditions. In these studies, AAV vectors have been employed as gene transfer vehicles to deliver the coding sequence for specific antibodies into non-hematopoietic cells, driving antibody production and secretion for pre- or post-challenge treatment (84). This approach, known as Vectored Immunoprophylaxis (VIP), induced lifelong expression of significant doses of mAbs in multiple models of disease for the prevention of infection with various pathogens (85), such as Influenza A (86) and Hepatitis C (87) viruses, Plasmodium falciparum (88) and HIV (89–91). Such has been the success of this approach that VIP is currently being tested in phase 1 clinical trials (NCT01937455; NCT03374202) in HIV-infected adults (92).
Based on this accumulated weight of evidence, it is our opinion that the outstanding therapeutic potential of antibodies offers a unique opportunity to target CNS proteinopathies. Significant benefits of A-MAD strategies have emerged in preclinical models of CNS diseases (Table 1), and the translation of this technology into a therapeutic strategy for humans has now become a priority. We believe that combining mAbs derivatives and rAAV vectors truly has the potential to offer long-awaited therapeutic options against chronic and/or degenerative CNS proteinopathies, as (i) certain AAV serotypes have the ability to cross an intact BBB in multiple animal species, including non-human primates, resulting in widespread CNS transduction (93); (ii) rAAV vectors are able to infect the non-dividing (post-mitotic) cells of the CNS and are considered safe, due to their non-replicative and non-integrating nature; (iii) rAAV vectors provide stable, long-lasting production of the therapeutic of interest [up to 15 years in a non-human primate parkinsonian model (94)]; and (iv) due to the vector life-cycle, therapeutics are produced within the cell and, therefore, are able to access intracellular targets with ease (83).
Systemic administration of rAAV vectors with BBB crossing properties arguably represents the best option for treating multi-region CNS disease using mAbs (or derivatives). Unlike approaches relying on local vector administration, such as intraparenchymal (IP) (95), intrathecal (IT) (96, 97) and intracerebroventricular (ICV) (98) injections, in which transduction is largely limited to the area surrounding the injection site, systemic delivery results in widespread transduction of the CNS. However, systemic administration does require the use of much higher vector doses to achieve adequate levels of CNS transduction, raising concerns about potential immunotoxicity (99). Although systemic delivery of AAV vectors has proved to be largely safe in a number of clinical trials targeting peripheral conditions [e.g., Spinal Muscular Atrophy Type I (NCT02122952); Duchenne Muscular Dystrophy (NCT03368742); and Mucopolysaccharidosis (NCT03315182) (100)], potential concerns linked to off-target transduction of peripheral organs and immune responses to the AAV capsid and/or the transgene cannot be completely discounted, particularly when targeting adult CNS conditions, which will generally require higher rAAV vector doses than those administered to children.
Consequently, introduction of modifications to the rAAV vector capsid, responsible for vector tropism, and the transgene cassette, have become increasingly important to (i) minimalize safety concerns, limiting unwanted transduction of peripheral organs (ii) improve BBB crossing, and (iii) improve transgene expression levels in a cell-type specific manner. A major issue with systemic AAV administration is the presence of pre-existing neutralizing antibodies (NAbs) in approximately half of the human population, as a result of exposure to naturally occurring AAVs. NAbs are durable and display considerable cross-reactivity across serotypes, severely hindering transduction efficiency (101). Engineering the rAAV vector capsid to evade NAbs is, therefore, an important strategy to improve the efficiency of systemic delivery and A-MAD in the CNS (Figure 4). In the following sections, we will discuss recent innovations in the fields of capsid and transgene engineering, that we believe will help facilitate the use of A-MAD to treat CNS proteinopathies.
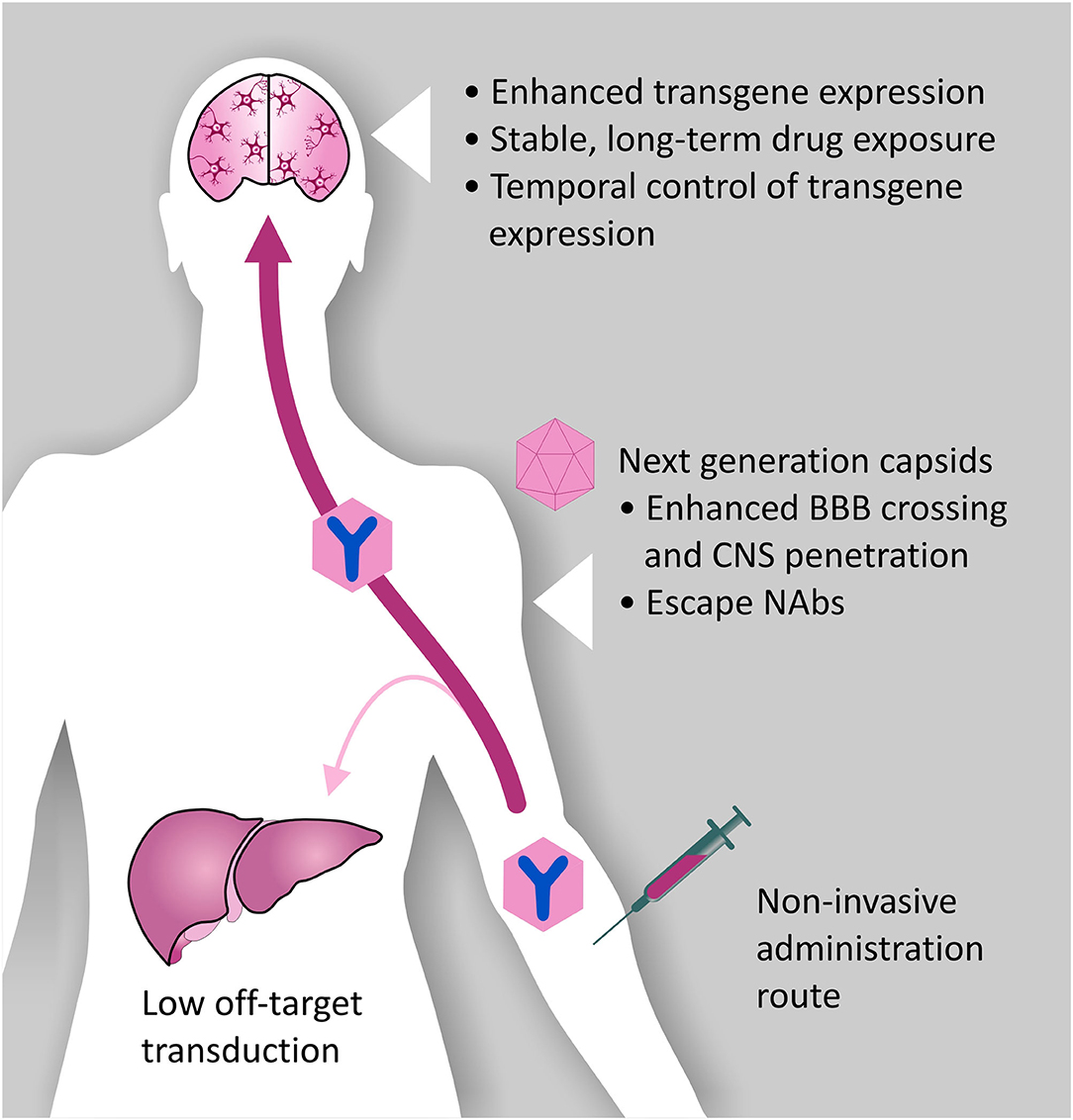
Figure 4. Desired characteristics of rAAV vectors for A-MAD. Certain rAAV vector serotypes show BBB crossing properties, thus allowing minimally invasive and long-term delivery of mAbs and their derivatives within the CNS. However, improvements to the rAAV vector platform are desirable to maximize the therapeutic potential of A-MAD strategies, and decrease the risk of potential side effects. In particular, capsid engineering can be used to enhance BBB penetration, and enable CNS cell-type specific tropism. These modifications would also have the benefit of reducing potential off-target toxicity which, in the case of systemic administration, is usually linked to liver transduction. Introduction of capsid modifications is also desirable to reduce the likelihood of vector capture by pre-existing neutralizing antibodies (NAbs), which have been found to severely limit the efficiency of the system. In parallel, engineering of the rAAV transgene cassette is desirable to maximize transgene expression efficiency, thus allowing high (therapeutically relevant) levels of mAbs (or derivatives) within the CNS. Moreover, following systemic administration, it will be essential to spatially restrict transgene expression, de-targeting the periphery. The ability to turn transgene expression “on” or “off” will serve to prevent, or limit, adverse reactions to the transgene. Incorporation of such modifications into basic vector design will, in all likelihood, allow remaining concerns over the safety of AAV systems for CNS use to be overcome, while also increasing their therapeutic potential.
Important advances in the clinical application of rAAV vectors have mostly been based on AAV9, as this serotype is characterized by higher transduction efficiency in the CNS, cardiac and skeletal muscle, pancreatic tissue and liver in comparison with other serotypes (102), and lacks the proinflammatory cytokine induction typically observed with lentiviral vectors. The ability of AAV9 to cross the BBB from the peripheral circulation and transduce non-dividing cells in the CNS has been behind the successful development of Zolgensma, with several other therapeutics currently being investigated for the treatment of CNS proteinopathies. An additional advantage of AAV9 is that the prevalence of anti-AAV9 NAbs in humans is lower than that observed for other serotypes, such as AAV1 and 2 (103). Given the useful properties of this serotype, we believe that further engineering of the AAV9 capsid could generate highly efficient BBB crossing capsids for use as minimally invasive delivery platforms for mAbs (and derivatives) for the treatment of CNS disorders.
Knowledge of AAV biology, including aspects of capsid structure, assembly and transduction mechanism, has allowed engineering of vectors with the aim of enhancing, or introducing, new and beneficial characteristics (104). This strategy is known as rational capsid design. Through rational design, novel AAV serotypes, showing enhanced specificity for CNS cell-types in comparison to peripheral organs, have been produced. Rational design generated AAV9.HR, a serotype able to preferentially transduce neurons upon systemic administration in neonatal mice, while de-targeting the periphery (105). AAV9.HR was derived from the parental serotype AAV9 by introducing the mutations His527Tyr and Arg533Ser, which influence galactose and LamR receptor binding (105, 106).
Rational capsid design has also allowed creation of entirely artificial AAV capsids, using phylogenic analyses to enable de novo gene synthesis of ancestral capsid proteins from the common ancestors of AAVs currently in circulation. These reconstructed ancestral capsids possess intriguing features, such as an enhanced thermostability and wide transduction patterns (107) that could open interesting scenarios for the delivery of mAbs (and derivatives) to a targeted CNS cell-type. Particularly relevant for CNS targeting is serotype Anc80L65, which has shown a 4-fold increase in the ability to transduce astrocytes, when compared to AAV9, following intravenous administration in adult mice (108). Preferential targeting of astrocytes via A-MAD could lead to significant benefits for the treatment of Transactive response DNA binding protein of 43 kDa (TDP-43)-related proteinopathies, such as ALS and frontotemporal dementia (FTD), where astrocyte activation and inflammation appear as strong effectors in disease progression (109). Of interest, a current gap in rAAV platform technology is the lack of serotypes able to efficiently transduce microglia, in our opinion one of the most valuable therapeutic targets in the CNS, due to their involvement in numerous proteinopathies (110–112). To date, modification of the AAV6 capsid through site-directed mutagenesis has been shown to increase transduction efficiency in monocyte-derived dendritic cells (113). However, microglia remain largely resistant to transduction (114). Further research efforts, directed toward the identification of microglia-targeting rAAVs, can open interesting scenarios for A-MAD strategies targeting the inflammatory component of CNS proteinopathies, although the potential impact of cell-transduction on the activation state of microglia may prove to be a significant challenge (115).
Modification of AAV capsids can also be achieved through methods that do not rely on a priori information, but on random events [capsid shuffling (116), peptide insertion (117), random mutagenesis (118)]. These approaches, known collectively as directed capsid evolution methodologies, have succeeded in generating rAAV capsids with remarkable BBB crossing properties and cell-type specific tropism following systemic administration (119).
Capsid shuffling generates hybrid serotypes such as AAV-Olig001, which was created by fusing together elements of different capsids originating from AAV serotypes 1, 2, 6, 8, and 9. AAV-Olig001 shows exceptional specificity for oligodendrocytes (95%) after intravenous administration in rats, with no reported transgene expression in astrocytes and microglia (120).
To date, perhaps the most successful strategy for AAV capsid evolution aimed at improving BBB crossing has been through modification via peptide insertion. This methodology forms the basis of the Cre recombination-based AAV directed evolution (CREATE) platform (121). In this method, a library of capsid variants was generated by introduction of a randomized sequence of 7 amino acids (aa) between aa 588 and 589 of the VP1 capsid subunit from AAV9. The capsid library was engineered around an expression cassette encoding a Cre-inducible fluorescent reporter, allowing recovery of capsid variants which successfully transduced cells in a Cre-expressing mouse (astrocyte-specific Gfap-promoter) following systemic injection. CREATE was used to generate the serotypes AAV-PHP.B, AAV-PHP.eB and AAV-PHP.S, which show enhanced BBB penetration with concomitant increases in brain (121) and spinal cord transduction (122), in comparison to the parental serotype. Unfortunately, the increased BBB crossing seen with AAV-PHP.B appears limited to C57Bl/6J mice, with other mouse strains and animal species (including non-human primates) showing limited CNS transduction following systemic administration, due to a lack of the obligate LY6A receptor on endothelial cells of the brain vasculature (123, 124). This limitation could be potentially overcome by the work of Hanlon et al. (125), who described a peptide insertion strategy, iTransduce, in which pseudorandom 21-base nucleotides were inserted between amino acids 588–589 of the AAV9 VP1 capsid subunit. Selection of BBB crossing capsids in iTransduce was again based on Cre-inducible marker expression, albeit in this case Cre was encoded by the AAV expression cassette and the vector was systemically applied to a fluorescent reporter mouse. This strategy was used to generate AAV-F, with a transgene expression efficiency in the murine brain comparable to that of AAV-PHP.B following intravenous administration. However, AAV-F does not rely on the LY6A receptor for BBB crossing, and it appears able to efficiently cross the BBB in non-C57Bl/6J genetic backgrounds (BALB/c) after systemic administration. To date, however, it is impossible to foresee whether AAV-F could be a clinically useful serotype, as it has not been systemically administered in non-human primates. In all likelihood, ongoing research efforts will provide us with a plethora of novel BBB crossing rAAV serotypes, allowing efficient targeting of CNS disorders in humans.
As introduced earlier, capsid modifications are also, to date, the most successful aid to AAV vector evasion of both capsid specific cytotoxic T-lymphocytes (CTL) and innate immune responses upon vector administration (126). For example, introduction of mutations in the epitope on the AAV2.5 capsid recognized by the monoclonal antibody A20 reduces levels of vector neutralization (127); while the chimera AAV-DJ shows the ability to evade neutralizing antibodies generated by exposure to other serotypes (128). In our opinion, the benefits of engineering new, immunosilent AAV serotypes exceed those of alternative strategies required to maximize the efficiency of systemic AAV administration and its use in A-MAD. Administration of empty “decoy” capsids, use of immunosuppressants in patients (129) and plasmapheresis have all been proposed as useful strategies but, in fact, all pose some concerns. To begin with, as NAbs recognize the same epitopes on both empty capsids and those carrying the therapeutic transgene, extremely high doses of “decoy” capsids (~10-fold higher) are needed to ensure that NAbs are effectively mopped up, leaving the therapeutic vector free to cross the BBB and transduce the CNS. In contrast, immunosuppressive agents and plasmapheresis are non-specific and effectively remove all circulating antibodies, leaving patients exposed to the risk of opportunistic infections (130). To overcome this issue, plasmapheresis methods based on AAV-specific immune absorption columns are being tested for their ability to exclusively deplete anti-AAV antibodies in non-human primates; the safety and efficacy of such an approach is yet to be determined in humans (131).
Upon successful cell transduction, a critical issue for successful A-MAD is the production and maintenance of a therapeutic threshold of biologics within the CNS. Production of the therapeutic transgene depends on conversion of the rAAV vector single stranded genome into double-stranded DNA (dsDNA), to enable subsequent mRNA transcription and translation. Synthesis of the second strand of DNA is considered a rate limiting step, delaying the onset of transgene expression and, in cells with particularly inefficient second strand synthesis, potentially limiting the steady-state level of therapeutic that can be produced (132, 133). This issue can be effectively overcome by creating a synthetic double-stranded genome, through mutating one of the ITRs so that it can form a short DNA hairpin—the so-called self-complementary (sc) configuration (134, 135). The sc configuration does, however, involve a 50% reduction in the transgene cassette capacity (~2.4 kb), thus limiting the choice of potential transgene candidates (136). As the cDNA of full-length mAbs occupies a minimum 2 kb of packaging space, A-MAD strategies using full length antibodies tend to accommodate a single copy of the antibody coding sequence in a single stranded genome (137, 138), which also includes standard regulatory elements, such as promoter sequences, the polyA signal and the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE, see Section ‘Enhancing VHH Expression’ below). On the contrary, mAbs derivatives, due to their small size, are optimal for incorporation into a scAAV system.
Insertion of full-length mAbs within a rAAV expression cassette often requires extensive reconfiguration, which can potentially alter antibody properties. mAbs derivatives of smaller size are therefore better suited for rAAV vector-mediated expression (139). In particular, camelid derived nanobodies, also known as VHH, are emerging as ideal candidates for incorporation into rAAV vectors, due to their single binding domain, exceptional stability and lack of an Fc effector function. This latter characteristic could contribute to a more favorable pharmacokinetic profile within the CNS, in comparison with full-length mAbs. In fact, it has been hypothesized that the binding of the Fc domain to Fc receptors in the BBB represents a potential route for the reverse transcytosis of antibodies across the BBB, facilitating their clearance from the brain (Figure 2B) (140–142).
VHH are the smallest naturally derived antigen-binding functional fragments (~15 kDa) (143), and as such are able to target epitopes inaccessible to full-length mAbs, such as the active site of enzymes (to modulate their catalytic activity) (144). The use of VHH largely overcomes the issues associated with the use of the single-chain fragment variable (scFv) format; to date, this is the most commonly employed antibody derivative used in A-MAD. However, scFv are notoriously difficult to engineer; scFv are formed from the variable regions of antibody heavy and light chains, joined by a short peptide linker, which requires extensive optimization to ensure correct protein folding and efficient antigen binding (145, 146), and they are frequently unstable in the highly reducing environment of the cell (147) (Figure 5).
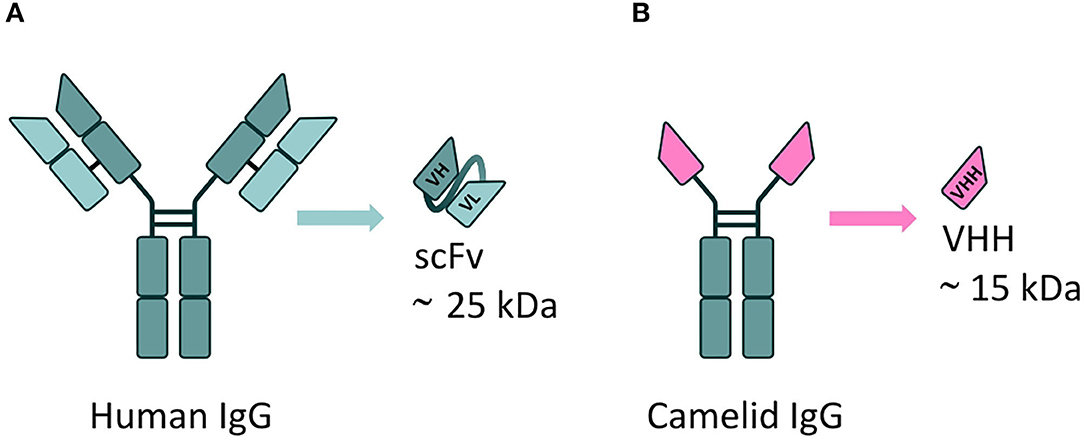
Figure 5. mAbs derivatives for A-MAD. (A) Representative scheme of a single-chain fragment variable (scFv) antibody fragment, derived from a human immunoglobulin. This class of engineered antibody is composed of the variable regions of the heavy (VH) and light chains (VL) fused by a linker. Their applicability is limited by potential folding and stability issues in the reducing environment of the cell. (B) Representative scheme of a VHH, the smallest antigen-binding functional fragment in nature, derived from a camelid immunoglobulin. The small size, high antigen specificity and exceptional stability of VHH (including in the reducing environment of the cell) make them an ideal cargo for rAAV vector-mediated delivery to target CNS proteinopathies.
One major advantage of the small size of VHH is the possibility to engineer multiple copies within a standard (sc) AAV expression cassette. This capacity can be used to introduce multiple copies of the same VHH to boost overall production levels, or to incorporate VHH raised against different targets. These could be targeting different domains within the same protein, to increase avidity, or completely independent proteins within a common signaling pathway. Employing such a strategy would significantly enhance the therapeutic potential of A-MAD in treating CNS proteinopathies, in a manner similar to that resulting from the use of multi-specific antibody fragments [bispecific T cell engagers (BiTEs) (148), diabodies (149) and dual affinity re-targeting antibodies (DARTs) (150)] for the treatment of peripheral conditions. Interestingly, VHH can be further engineered without loss of functionality, to (i) direct them into specific trafficking pathways and improve target engagement (151, 152), (ii) incorporate specific proteolysis-promoting sequences (PROTACs), to stimulate intracellular degradation of (toxic) proteins involved in CNS proteinopathies (153), or (iii) insert secretion signals allowing VHH to engage extracellular targets (which is desirable to prevent prion-type transmission), or allow so-called “cross-correction” in which VHH can be internalized into non-transduced cells to exert a therapeutic effect (152, 154, 155).
To date, the use of VHH in the clinics has not raised significant safety concerns (156). Importantly, VHH safety can be further enhanced through the process of humanization (157), and this increases their attractiveness for clinical applications (158). Indeed, Caplacizumab-yhdp (Cablivi®), a 28-kDa bivalent nanobody indicated for the treatment of thrombotic thrombocytopenic purpura (TTP), was recently granted EMA and FDA approval. Although the potential of using rAAV vectors for the long-term delivery of VHH into the CNS has yet to be fully explored, Marino et al. recently provided significant proof of concept for the feasibility of A-MAD strategies, based on VHH delivery, to stop or slow neurodegenerative disease progression, as shown by the disease modifying effects observed in a murine model of AD, following systemic administration of AAV-PHP.B encoding a highly-specific anti-BACE1 VHH (152).
In summary, it has been speculated for years that expression of VHH in vivo, with the aid of gene therapy vectors, could ultimately be their most powerful clinical application (159, 160). However, to take full advantage of A-MAD it will be necessary to optimize the transgene cassette to obtain robust and controllable expression of VHH in the CNS.
The use of strategies to maximize production of biologics within the desired cell-type is beneficial for two main reasons: i) systemic administration of lower vector doses would be sufficient to reach a therapeutic concentration of the biologics within the CNS, effectively minimizing the risks of potential side effects, as well as therapy costs; ii) targeted transduction of a limited number of cells could still suffice to achieve a disease modifying effect, particularly for biologics engineered for secretion, which could subsequently be taken up by surrounding cells.
Incorporation of the WPRE element within the rAAV transgene cassette is now a routine modification, which has been shown to enhance levels of transgene expression in the murine brain and in human retina (161, 162). An exciting recent development adopted to increase transgene expression in the periphery is the use of a core promoter in combination with a so-called cis-acting regulatory module (CRM). These short sequences comprise clusters of evolutionary conserved transcription factor binding sites that boost transcription in a cell-type specific manner (163, 164). To date, in silico approaches proved successful in discovering liver-(163), cardiac-(164) and skeletal muscle- (165) specific CRMs that, upon incorporation into the transgene cassette, enhanced transgene expression up to ~100-fold in comparison with the levels achieved by core promoters alone. The identification of CRMs for CNS would represent a large step forward as mAbs levels produced by A-MAD are likely critical for the successful treatment of proteinopathies (Figure 6). Nevertheless, although VHH show low immunogenicity, the possibility of adverse immune responses to robust VHH production cannot be overlooked in the clinic. Hence, systems to spatially and temporarily regulate VHH production with the CNS are desirable.
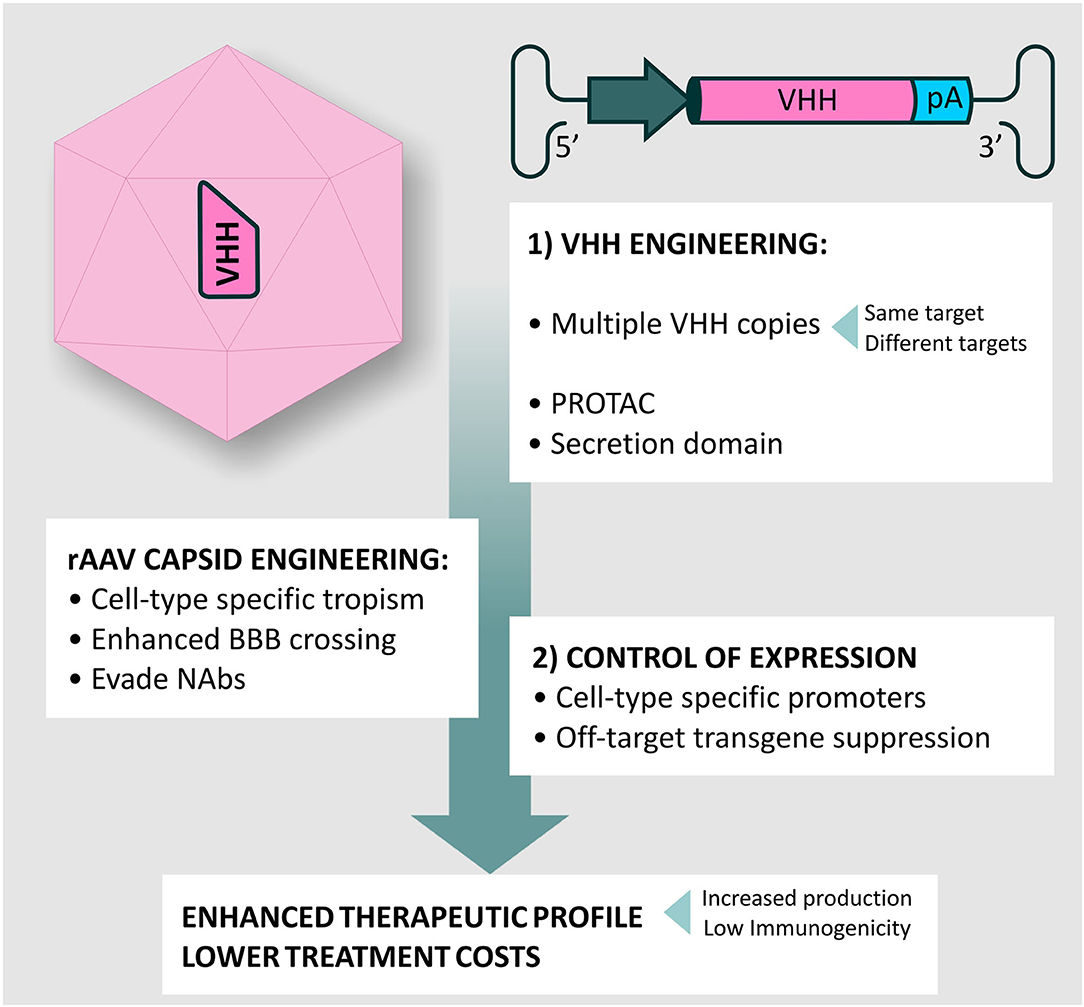
Figure 6. rAAV vectors and VHH engineering for A-MAD. Summary of potential modifications for A-MAD. Engineering the rAAV capsid and transgene cassette can maximize BBB crossing, NAbs evasion and spatially restrict VHH expression to the desired cell-type. More efficient production systems will lower production costs. The outstanding versatility of VHH allows easy engineering to generate multivalent drugs, or drive proteosomal degradation of toxic protein conformations via construction of Proteolysis targeting chimeras (PROTACs). Additionally, incorporation of secretion signals into the VHH will allow targeting of extracellular aggregates, preventing prion-type transmission. Together, the impact of improved rAAV vector technology and VHH engineering will produce more efficient A-MAD-based therapeutics, with concomitant reductions in undesired side-effects and lower treatment costs.
VHH expression can be driven within the cell-type of interest using specific promoters, overcoming issues such as (potential) toxicity resulting from off-target, multi-organ transgene overexpression, or deleterious immune responses resulting from the (undesired) transduction of antigen presenting cells (APCs) (166, 167).
Given the limited packaging capacity of rAAV vectors, short promoter sequences are beneficial, and the human synapsin 1 promoter [480 base pairs (bp) (168)] and truncated versions of the glial fibrillary acidic protein (GFAP) promoter [such as GfaABC1D, 694 bp (169)] have been well-characterized and are routinely used to restrict transgene expression to neurons and astrocytes, respectively (168–170). Far less characterized are short oligodendrocyte-specific promoters (171), albeit oligodendrocyte pathology seems to contribute significantly to proteinopathies, such as AD (172) and PD (173). Thus, we believe that increasing research efforts in this area should be prioritized to ensure that the full range of components driving CNS proteinopathies is effectively targeted. In this direction, ongoing research efforts are now aimed at refining patterns of transgene expression to target specific cell subtypes: for example, the mGAD65 promoter drives expression in GABAergic interneurons throughout the adult mouse brain, unlike the Dlx promoter, the previous gold standard for GABAergic interneuron targeting in the cortex, which shows mainly forebrain restricted expression (174). In addition, the mGAD65 promoter seems to preferentially drive expression in parvalbumin-positive interneurons and chandelier cells. Targeting (subsets of) GABAergic interneurons could provide significant therapeutic benefit in the treatment of tauopathies, where pathological tau accumulation has been implicated in memory deficits due to impaired GABAergic transmission (152, 175, 176).
Although cell-specific promoters significantly restrict VHH expression to the target cell type, a residual amount of off-target transgene expression cannot be excluded following systemic vector administration. To prevent undesirable effects due to “leaky” transgene expression, vector targeting can be further improved at the post-transcriptional level, including the use of micro-RNA (miRNA)-based methods to suppress transgene expression (177). As an example, Xie et al. exploited a multi-tissue miRNA-dependent suppression mechanism to spatially limit transgene expression to the CNS of adult mice following systemic AAV9 administration (178).
Even though VHH display low immunogenicity, it is not possible to foresee whether permanent VHH expression in post-mitotic CNS cells, which display little or no turnover/renewal, could potentially increase the risk of immune responses, leading to irreversible cell damage/death. Hence, in our opinion, inclusion of a safety switch would be ideal to limit adverse reactions. One interesting approach to regulating transgene expression has been to take advantage of natural variations in promoter activity: upregulation of endogenous GFAP is a well-documented occurrence in several CNS pathologies in higher vertebrates (179), and proof of concept experiments with a lentiviral vector-based system for gene delivery suggest that GFAP promoters could even be used to enhance production of a therapeutic transgene in the event of significant astrogliosis (180). However, this approach is limited by a number of key issues, including that the pattern of GFAP expression across CNS areas is highly variable, and therefore, the response to injury is highly heterogeneous (181, 182).
In contrast, the use of small molecule inducible promoter systems appears a valid option, particularly as transient therapeutic expression may still produce a therapeutic effect, as previous studies on BACE1 suggest that transient inhibition of the enzyme is sufficient to produce sustained long-term reductions of amyloid-β (Aβ) accumulation in AD (183, 184). An ideal inducible system should be completely free of endogenous influence, and would be built around: (i) an inducible promoter dependent on a unique regulatory DNA sequence exogenous to the host genome; (ii) an effector protein that recognizes and binds specifically to this regulatory sequence, without interfering with any other sites within the host genome and iii) dose-dependent inducibility of the effector protein by a BBB penetrant drug that can be safely and repeatedly administered. Although reports of such systems in the literature are limited to date, the inducer/repressor tetracycline (Tet)-dependent system is an interesting example, and was successfully employed to modulate transgene expression for up to 5 years in the muscle of non-human primates, following locoregional intravenous AAV administration (185). However, delayed humoral and cellular immune responses directed against the bacterial component of the Tet-dependent system have been reported in some non-human primate studies (186, 187). Although these issues appear to have been overcome by fusing the reverse tetracycline-controlled transactivator (rtTA) with a glycine-alanine repeat (GARrtTA), which is known to enable the Epstein-Barr virus to evade host immune response (188, 189), caution is obviously needed moving forward. In the context of the CNS, the rtTA effector minocycline (190) appears particularly suited for the inducible expression of biologics, as it shows (i) high BBB permeability, (ii) low cytotoxicity [albeit minor side effects can be observed upon prolonged administration (191, 192)] and (iii) neuroprotective (anti-apoptotic and anti-inflammatory) effects, dependent on the inhibition of cytosolic cytochrome C translocation (193), caspase activity (194), microglial proliferation (195), and activation of inducible nitric oxide synthase and cytokine release in PD, HD, ALS, and cerebral ischemia (196, 197). These properties suggest minocycline-controlled systems (or equivalents) are likely to play an important role in the development of rAAV-mediated strategies (including A-MAD) for CNS use.
In the last four decades, the use of mAbs as therapeutics has rapidly expanded, with many now “blockbuster drugs,” which generate billions of dollars of annual revenue. As of November 2020, 88 antibody therapeutics were in stage 3 clinical studies for a variety of conditions (other than COVID-19) (198), and when combined with a relatively fast approval rate (currently twice as fast as for small molecules) their dominant position in the market place is likely to continue (198). In general, however, the approval of mAbs for CNS-related indications is lagging behind, albeit with a few noteworthy exceptions (50, 199–204). This is not surprising, as perhaps the biggest challenge in the development of non-invasive strategies to target CNS disorders is overcoming the restricted permeability of the BBB, which significantly limits mAbs access to the CNS from the systemic circulation. rAAV vector technology offers an interesting solution to this problem, with novel BBB-crossing serotypes allowing extended production of biologics within the CNS following a single systemic injection.
Interestingly, gene therapy-based methods are becoming increasingly popular for the treatment of previously incurable diseases. In fact, in 2019, the FDA authored a report predicting that by 2025 ~10–20 novel cell and gene therapy products will be approved per year (205). A comprehensive analysis of the 94 AAV-based clinical trials completed to date shows that the focus is directed towards four main therapeutic areas: liver, muscle, retinal and CNS diseases. The general trend towards optimism is in no small part due to the fact that in the fields of neurology and ophthalmology, areas with traditionally high rates of clinical trial failures, ~30% of the tested rAAV vector-based drugs successfully completed the path from investigational new drug (IND) to new drug application (NDA), a percentage exceeding the historical averages of any other drug type (206). The potential of AAVs as drug delivery platforms, particularly if supported by further successful outcomes in clinical trials, will undoubtedly drive research aimed at further improving and refining the technology, particularly for use in areas such as the CNS, where conventional small molecule therapies have traditionally had long developmental times and high failure rates (136). In fact, recent progress in both capsid engineering and manufacturing technology suggest that widespread gene delivery to the adult CNS via intravenous administration is now possible, while improvements in antibody engineering and expression cassette design point towards safe and effective A-MAD-based strategies for chronic, multi-region CNS diseases.
Given their unique properties, which are ideally suited for A-MAD, we anticipate use of VHH will play a key role in developing the technology, particularly as further research provides greater insights into disease causing mechanisms and identifies future targets for therapeutic intervention. The push towards using VHH in CNS disease is exemplified by the current efforts of major Biotech and Pharma companies, such as Ablynx (Sanofi) and Boehringer Ingelheim, who partnered to explore a potential VHH-based treatment for Alzheimer's disease; as of early 2022, the result of this partnership is an ongoing phase 1 clinical trial in AD patients for the Aβ-targeting biparatopic nanobody, NbBI.1031020 (207).
Nevertheless, some open concerns remain to be addressed before the clinical adoption of A-MAD in earnest. First and foremost, reports that the enhanced BBB crossing of AAV-PHP.B is restricted to BL6 mice and absent in non-human primates should be considered a cautionary tale in the rush to develop new AAV-based delivery platforms. In future, time and effort need to be invested in developing screening platforms which avoid issues arising out of cross-species differences in BBB composition and immune responses. Luckily, the growing opportunities to test vector performance in 3D cerebral organoids (208–210), which recapitulate aspects of the human brain, offer major advantages for the development of CNS relevant rAAV vectors. Second, and an issue specific to the use of VHH technology, is whether the lack of an Fc region is beneficial, reducing the risk of neuroinflammation and edema (211), or whether the effector function is actually needed. In all probability, this is likely dependent on the mechanism of VHH action; for example, inhibition of BACE1 activity in AD does not require an effector domain (152). In the event that effector function is required, however, the ease with which VHH can be engineered allows the straight-forward incorporation of the Fc region from human IgG, thus generating bivalency and effector function (212). Third, in the majority of cases, disease onset and the accumulation of irreversible cellular damage is thought to precede the appearance of symptoms by many years (213–216), raising the issue of when treatment should be initiated. Hence, it is likely that improvements in therapeutics and delivery systems will need to be mirrored by improvements in biomarker detection and cognitive testing.
Finally, methods for efficient, high-quality, large-scale manufacture of AAV vectors, exploiting easily scalable production systems [baculovirus–Sf9-systems (217); recombinant HSV-based systems (218); adenovirus–HeLa cell systems (219)] will ease the translation of A-MAD into clinical use, by significantly cutting production costs, which remain high at present. As an example, in early 2022, the cost of Zolgensma, indicated for children aged <2 years, is $2.48 million for a single treatment, and in all likelihood the higher doses required for adult therapeutics will be reflected in even higher costs. Nevertheless, it should be pointed out that A-MAD strategies would rely on a “single-dose” therapeutic approach, due to the ability of rAAV to effectively transform specific CNS cell-types into “biopharmacies,” capable of long-term biologic production. In contrast, high level dosing of standard, non-vector-based mAbs therapies, repeated over the course of decades, would itself be extremely expensive, allowing us to speculate that the final costs will in the end be potentially comparable.
To conclude, the growing toolbox available for A-MAD supports a bright future in which the full range of CNS proteinopathies can be effectively treated. Combining the cutting-edge power of rAAV vectors for CNS targeting with the unique properties of VHH promises to revolutionize our approach to CNS disease: the future is now!
Both authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
MM was supported by a pre-doctoral fellowship from the Fonds Wetenschappelijk Onderzoek (FWO) (SB/1S48018N). Work in the Holt lab has been supported by the Thierry Latran Foundation (SOD-VIP), FWO (Grant 1513616N), and European Research Council (ERC) (Starting Grant 281961 – AstroFunc; Proof of Concept Grant 713755 – AD-VIP). MGH is currently the ERANet Chair (NCBio) at i3S Porto funded by the European Commission (H2020-WIDESPREAD-2018-2020-6; NCBio; 951923).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Joost Schymkowitz, Frederic Rousseau, Maarten De Wilde, and members of the Holt lab for discussions and advice. Figures were produced by David Pennington (@Pennington Art).
1. Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. (1975) 256:495–7. doi: 10.1038/256495a0
2. Todd PA, Brogden RN. Muromonab CD3. Drugs. (1989) 37:871–99. doi: 10.2165/00003495-198937060-00004
3. Berger M, Shankar V, Vafai A. Therapeutic applications of monoclonal antibodies. Am J Med Sci. (2002) 324:18. doi: 10.1097/00000441-200207000-00004
4. Grilo AL, Mantalaris A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. (2019) 37:9–16. doi: 10.1016/j.tibtech.2018.05.014
5. Boulianne GL, Hozumi N, Shulman MJ. Production of functional chimaeric mouse/human antibody. Nature. (1984) 312:643–6. doi: 10.1038/312643a0
6. Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci. (1984) 81:6851–5. doi: 10.1073/pnas.81.21.6851
7. Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. (1986) 321:522–5. doi: 10.1038/321522a0
8. McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. (1990) 348:552–4. doi: 10.1038/348552a0
9. Kaplon H, Reichert JM. Antibodies to watch in 2019. mAbs. (2019) 11:219–38. doi: 10.1080/19420862.2018.1556465
10. Mullard A. FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov. (2021) 20:491–5. doi: 10.1038/d41573-021-00079-7
11. Chatterjee D, Bhatt M, Butler D, De Genst E, Dobson CM, Messer A, et al. Proteasome-targeted nanobodies alleviate pathology and functional decline in an α-synuclein-based Parkinson's disease model. npj Parkinson's Dis. (2018) 4:25. doi: 10.1038/s41531-018-0062-4
12. Chen Y-H, Wu K-J, Hsieh W, Harvey BK, Hoffer BJ, Wang Y, et al. Administration of AAV-alpha synuclein NAC antibody improves locomotor behavior in rats overexpressing alpha synuclein. Genes. (2021) 12:948. doi: 10.3390/genes12060948
13. Liu W, Zhao L, Blackman B, Parmar M, Wong MY, Woo T, et al. Vectored intracerebral immunization with the anti-Tau monoclonal antibody PHF1 markedly reduces Tau pathology in mutant Tau transgenic mice. J Neurosci. (2016) 36:12425–35. doi: 10.1523/JNEUROSCI.2016-16.2016
14. Ryan DA, Mastrangelo MA, Narrow WC, Sullivan MA, Federoff HJ, Bowers WJ. Aβ-directed single-chain antibody delivery via a serotype-1 AAV vector improves learning behavior and pathology in Alzheimer's disease mice. Mol Ther. (2010) 18:1471–81. doi: 10.1038/mt.2010.111
15. Levites Y, Jansen K, Smithson LA, Dakin R, Holloway VM, Das P, et al. Intracranial adeno-associated virus-mediated delivery of anti-pan amyloid beta, amyloid beta40, and amyloid beta42 single-chain variable fragments attenuates plaque pathology in amyloid precursor protein mice. J Neurosci. (2006) 26:11923–8. doi: 10.1523/JNEUROSCI.2795-06.2006
16. Fukuchi K, Tahara K, Kim H-D, Maxwell JA, Lewis TL, Accavitti-Loper MA, et al. Anti-Aβ single-chain antibody delivery via adeno-associated virus for treatment of Alzheimer's disease. Neurobiol Dis. (2006) 23:502–11. doi: 10.1016/j.nbd.2006.04.012
17. Sudol KL, Mastrangelo MA, Narrow WC, Frazer ME, Levites YR, Golde TE, et al. Generating differentially targeted amyloid-beta specific intrabodies as a passive vaccination strategy for Alzheimer's disease. Mol Ther. (2009) 17:2031–40. doi: 10.1038/mt.2009.174
18. Kou J, Kim H, Pattanayak A, Song M, Lim J-E, Taguchi H, et al. Anti-amyloid-β single-chain antibody brain delivery via AAV reduces amyloid load but may increase cerebral hemorrhages in an Alzheimer's disease mouse model. JAD. (2011) 27:23–38. doi: 10.3233/JAD-2011-110230
19. Shimada M, Abe S, Takahashi T, Shiozaki K, Okuda M, Mizukami H, et al. Prophylaxis and treatment of Alzheimer's disease by delivery of an adeno-associated virus encoding a monoclonal antibody targeting the amyloid beta protein. PLoS ONE. (2013) 8:e57606. doi: 10.1371/journal.pone.0057606
20. Yang J, Pattanayak A, Song M, Kou J, Taguchi H, Paul S, et al. Muscle-directed anti-Aβ single-chain antibody delivery via AAV1 reduces cerebral Aβ load in an Alzheimer's disease mouse model. J Mol Neurosci. (2013) 49:277–88. doi: 10.1007/s12031-012-9877-3
21. Elmer BM, Swanson KA, Bangari DS, Piepenhagen PA, Roberts E, Taksir T, et al. Gene delivery of a modified antibody to Aβ reduces progression of murine Alzheimer's disease. PLoS ONE. (2019) 14:e0226245. doi: 10.1371/journal.pone.0226245
22. Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, et al. Abundant Tau filaments and non-apoptotic neurodegeneration in transgenic mice expressing human P301S Tau protein. J Neurosci. (2002) 22:9340–51. doi: 10.1523/JNEUROSCI.22-21-09340.2002
23. Ising C, Gallardo G, Leyns CEG, Wong CH, Jiang H, Stewart F, et al. AAV-mediated expression of anti-Tau scFvs decreases Tau accumulation in a mouse model of Tauopathy. J Exp Med. (2017) 214:1227–38. doi: 10.1084/jem.20162125
24. Vitale F, Giliberto L, Ruiz S, Steslow K, Marambaud P, d'Abramo C. Anti-Tau conformational scFv MC1 antibody efficiently reduces pathological Tau species in adult JNPL3 mice. Acta Neuropathol Commun. (2018) 6:82. doi: 10.1186/s40478-018-0585-2
25. Goodwin MS, Sinyavskaya O, Burg F, O'Neal V, Ceballos-Diaz C, Cruz PE, et al. Anti-Tau scFvs targeted to the cytoplasm or secretory pathway variably modify pathology and neurodegenerative phenotypes. Mol Ther. (2021) 29:859–72. doi: 10.1016/j.ymthe.2020.10.007
26. Pozzi S, Thammisetty SS, Codron P, Rahimian R, Plourde KV, Soucy G, et al. Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. J Clin Invest. (2019) 129:1581–95. doi: 10.1172/JCI123931
27. Patel P, Kriz J, Gravel M, Soucy G, Bareil C, Gravel C, et al. Adeno-associated virus-mediated delivery of a recombinant single-chain antibody against misfolded superoxide dismutase for treatment of amyotrophic lateral sclerosis. Mol Ther. (2014) 22:498–510. doi: 10.1038/mt.2013.239
28. Ghadge GD, Kay BK, Drigotas C, Roos RP. Single chain variable fragment antibodies directed against SOD1 ameliorate disease in mutant SOD1 transgenic mice. Neurobiol Dis. (2019) 121:131–7. doi: 10.1016/j.nbd.2018.08.021
29. Wuertzer CA, Sullivan MA, Qiu X, Federoff HJ. CNS delivery of vectored prion-specific single-chain antibodies delays disease onset. Mol Ther. (2008) 16:481-6. doi: 10.1038/sj.mt.6300387
30. Snyder-Keller A, McLear JA, Hathorn T, Messer A. Early or late-stage anti-n-terminal huntingtin intrabody gene therapy reduces pathological features in b6.hdr6/1 mice. J Neuropathol Exp Neurol. (2010) 69:1078–85. doi: 10.1097/NEN.0b013e3181f530ec
31. Amaro IA, Henderson LA. An intrabody drug (rAAV6-INT41) reduces the binding of n-terminal huntingtin fragment(s) to dna to basal levels in pc12 cells and delays cognitive loss in the r6/2 animal model. J Neurodegen Dis. (2016) 2016:1–10. doi: 10.1155/2016/7120753
32. Bayer TA. Proteinopathies, a core concept for understanding and ultimately treating degenerative disorders? Eur Neuropsychopharmacol. (2015) 25:713–24. doi: 10.1016/j.euroneuro.2013.03.007
33. Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. (2010) 11:155–9. doi: 10.1038/nrn2786
34. Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. (2009) 64:783–90. doi: 10.1016/j.neuron.2009.12.016
35. Polymenidou M, Cleveland DW. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med. (2012) 209:889–93. doi: 10.1084/jem.20120741
36. Jucker M, Walker LC. Pathogenic protein seeding in alzheimer disease and other neurodegenerative disorders. Ann Neurol. (2011) 70:532–40. doi: 10.1002/ana.22615
37. Cao T, Heng BC. Commentary: intracellular antibodies (intrabodies) versus RNA interference for therapeutic applications. Ann Clin Lab Sci. (2005) 35:227–9.
38. Covell DJ, Robinson JL, Akhtar RS, Grossman M, Weintraub D, Bucklin HM, et al. Novel conformation-selective alpha-synuclein antibodies raised against different in vitro fibril forms show distinct patterns of Lewy pathology in Parkinson's disease. Neuropathol Appl Neurobiol. (2017) 43:604–20. doi: 10.1111/nan.12402
39. Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. (2015) 522:340–4. doi: 10.1038/nature14547
40. Chavarría C, Rodríguez-Bottero S, Quijano C, Cassina P, Souza JM. Impact of monomeric, oligomeric and fibrillar alpha-synuclein on astrocyte reactivity and toxicity to neurons. Biochem J. (2018) 475:3153–69. doi: 10.1042/BCJ20180297
41. Hatami A, Albay R, Monjazeb S, Milton S, Glabe C. Monoclonal antibodies against Aβ42 fibrils distinguish multiple aggregation state polymorphisms in vitro and in Alzheimer disease brain. J Biol Chem. (2014) 289:32131–43. doi: 10.1074/jbc.M114.594846
42. Dias N, Stein CA. Antisense oligonucleotides: basic concepts and mechanisms. Mol Cancer Ther. (2002) 1:347–55.
43. Eder PS, DeVine RJ, Dagle JM, Walder JA. Substrate specificity and kinetics of degradation of antisense oligonucleotides by a 3′ exonuclease in plasma. Antisense Res Dev. (1991) 1:141–51. doi: 10.1089/ard.1991.1.141
44. Tamm I, Dörken B, Hartmann G. Antisense therapy in oncology: new hope for an old idea? Lancet. (2001) 358:489–97. doi: 10.1016/S0140-6736(01)05629-X
45. Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. (2003) 21:635–7. doi: 10.1038/nbt831
46. Jackson AL. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA. (2006) 12:1179–87. doi: 10.1261/rna.25706
47. Persengiev SP. Nonspecific, concentration-dependent stimulation and repression of mammalian gene expression by small interfering RNAs (siRNAs). RNA. (2004) 10:12–8. doi: 10.1261/rna5160904
48. Beghein E, Gettemans J. Nanobody technology: a versatile toolkit for microscopic imaging, protein-protein interaction analysis, and protein function exploration. Front Immunol. (2017) 8:771. doi: 10.3389/fimmu.2017.00771
49. Ovacik M, Lin K. Tutorial on monoclonal antibody pharmacokinetics and its considerations in early development. Clin Transl Sci. (2018) 11:540–52. doi: 10.1111/cts.12567
50. Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature. (2016) 537:50–6. doi: 10.1038/nature19323
51. Assas MB. Anti-migraine agents from an immunological point of view. J Transl Med. (2021) 19:23. doi: 10.1186/s12967-020-02681-6
52. Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. (2015) 7:a020412. doi: 10.1101/cshperspect.a020412
53. Pardridge WM. Drug Transport across the blood–brain barrier. J Cereb Blood Flow Metab. (2012) 32:1959–72. doi: 10.1038/jcbfm.2012.126
54. Atwal JK, Chen Y, Chiu C, Mortensen DL, Meilandt WJ, Liu Y, et al. A therapeutic antibody targeting BACE1 inhibits Amyloid-β production in vivo. Sci Transl Med. (2011) 3:84ra43. doi: 10.1126/scitranslmed.3002254
55. Hussain B, Fang C, Chang J. Blood–brain barrier breakdown: an emerging biomarker of cognitive impairment in normal aging and dementia. Front Neurosci. (2021) 15:688090. doi: 10.3389/fnins.2021.688090
56. Sweeney MD, Sagare AP, Zlokovic BV. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. (2018) 14:133–50. doi: 10.1038/nrneurol.2017.188
57. Elahy M, Jackaman C, Mamo JC, Lam V, Dhaliwal SS, Giles C, et al. Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun Ageing. (2015) 12:2. doi: 10.1186/s12979-015-0029-9
58. Bien-Ly N, Boswell CA, Jeet S, Beach TG, Hoyte K, Luk W, et al. Lack of widespread BBB disruption in Alzheimer's disease models: focus on therapeutic antibodies. Neuron. (2015) 88:289–97. doi: 10.1016/j.neuron.2015.09.036
59. Abbott NJ, Rönnbäck L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. (2006) 7:41–53. doi: 10.1038/nrn1824
60. Neves V, Aires-da-Silva F, Corte-Real S, Castanho MARB. Antibody approaches to treat brain diseases. Trends Biotechnol. (2016) 34:36–48. doi: 10.1016/j.tibtech.2015.10.005
61. Pardridge WM. Drug transport in brain via the cerebrospinal fluid. Fluids Barriers CNS. (2011) 8:7. doi: 10.1186/2045-8118-8-7
62. Xie H, Chung J-K, Mascelli MA, McCauley TG. Pharmacokinetics and bioavailability of a therapeutic enzyme (idursulfase) in cynomolgus monkeys after intrathecal and intravenous administration. PLoS ONE. (2015) 10:e0122453. doi: 10.1371/journal.pone.0122453
63. Papisov MI, Belov V, Fischman AJ, Belova E, Titus J, Gagne M, et al. Delivery of proteins to CNS as seen and measured by positron emission tomography. Drug Deliv Transl Res. (2012) 2:201–9. doi: 10.1007/s13346-012-0073-3
64. Cohen-Pfeffer JL, Gururangan S, Lester T, Lim DA, Shaywitz AJ, Westphal M, et al. Intracerebroventricular delivery as a safe, long-term route of drug administration. Pediatr Neurol. (2017) 67:23–35. doi: 10.1016/j.pediatrneurol.2016.10.022
65. Mörocz IÁ, Hynynen K, Gudbjartsson Hl, Peled S, Colucci V, Jólesz FA. Brain edema development after MRI-guided focused ultrasound treatment. J Magn Reson Imaging. (1998) 8:136–42. doi: 10.1002/jmri.1880080126
66. Belov V, Appleton J, Levin S, Giffenig P, Durcanova B, Papisov M. Large-volume intrathecal administrations: impact on CSF pressure and safety implications. Front Neurosci. (2021) 15:604197. doi: 10.3389/fnins.2021.604197
67. Dorovini-Zis K, Bowman PD, Lorris Betz A, Goldstein GW. Hyperosmotic arabinose solutions open the tight junctions between brain capillary endothelial cells in tissue culture. Brain Res. (1984) 302:383–6. doi: 10.1016/0006-8993(84)90254-3
68. Konofagou E, Tunga Y-S, Choia J, Deffieuxa T, Baseria B, Vlachosa F. Ultrasound-induced blood-brain barrier opening. Curr Pharm Biotechnol. (2012) 13:1332–45. doi: 10.2174/138920112800624364
69. Pardridge WM, Boado RJ. Pharmacokinetics and safety in rhesus monkeys of a monoclonal antibody-GDNF fusion protein for targeted blood-brain barrier delivery. Pharm Res. (2009) 26:2227–36. doi: 10.1007/s11095-009-9939-6
70. Weber F, Bohrmann B, Niewoehner J, Fischer JAA, Rueger P, Tiefenthaler G, et al. Brain shuttle antibody for Alzheimer's disease with attenuated peripheral effector function due to an inverted binding mode. Cell Rep. (2018) 22:149–62. doi: 10.1016/j.celrep.2017.12.019
71. Pardridge WM, Chou T. Mathematical models of blood-brain barrier transport of monoclonal antibodies targeting the transferrin receptor and the insulin receptor. Pharmaceuticals. (2021) 14:535. doi: 10.3390/ph14060535
72. Pardridge WM, Boado RJ, Patrick DJ, Ka-Wai Hui E, Lu JZ. Blood-brain barrier transport, plasma pharmacokinetics, and neuropathology following chronic treatment of the rhesus monkey with a brain penetrating humanized monoclonal antibody against the human transferrin receptor. Mol Pharm. (2018) 15:5207–16. doi: 10.1021/acs.molpharmaceut.8b00730
73. Couch JA, Yu YJ, Zhang Y, Tarrant JM, Fuji RN, Meilandt WJ, et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med. (2013) 5:183ra57. doi: 10.1126/scitranslmed.3005338
74. Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y, et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med. (2011) 3:84ra44. doi: 10.1126/scitranslmed.3002230
75. Zhou Q-H, Boado RJ, Pardridge WM. Selective plasma pharmacokinetics and brain uptake in the mouse of enzyme fusion proteins derived from species-specific receptor-targeted antibodies. J Drug Target. (2012) 20:715–9. doi: 10.3109/1061186X.2012.712132
76. Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C, et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med. (2014) 6:261ra154. doi: 10.1126/scitranslmed.3009835
77. Naso MF, Tomkowicz B, Perry WL, Strohl WR. Adeno-associated virus (AAV) as a vector for gene therapy. Biodrugs. (2017) 31:317–34. doi: 10.1007/s40259-017-0234-5
78. Wu Z, Asokan A, Samulski RJ. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther. (2006) 14:316–27. doi: 10.1016/j.ymthe.2006.05.009
79. Fripont S, Marneffe C, Marino M, Rincon MY, Holt MG. Production, purification, and quality control for adeno-associated virus-based vectors. J Vis Exp. (2019) 143:58960. doi: 10.3791/58960
80. Chamberlain K, Riyad JM, Weber T. Expressing transgenes that exceed the packaging capacity of adeno-associated virus capsids. Hum Gene Ther Methods. (2016) 27:1–12. doi: 10.1089/hgtb.2015.140
81. Cappella M, Ciotti C, Cohen-Tannoudji M, Biferi MG. Gene therapy for ALS—a perspective. Int J Mol Sci. (2019) 20:4388. doi: 10.3390/ijms20184388
82. Chand D, Mohr F, McMillan H, Tukov FF, Montgomery K, Kleyn A, et al. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J Hepatol. (2021) 74:560–6. doi: 10.1016/j.jhep.2020.11.001
83. Riyad JM, Weber T. Intracellular trafficking of adeno-associated virus (AAV) vectors: challenges and future directions. Gene Ther. (2021) 28:683–96. doi: 10.1038/s41434-021-00243-z
84. Sanders JW, Ponzio TA. Vectored immunoprophylaxis: an emerging adjunct to traditional vaccination. Trop Dis Travel Med Vaccines. (2017) 3:3. doi: 10.1186/s40794-017-0046-0
85. Deal CE, Balazs AB. Engineering humoral immunity as prophylaxis or therapy. Curr Opin Immunol. (2015) 35:113–22. doi: 10.1016/j.coi.2015.06.014
86. Balazs AB, Bloom JD, Hong CM, Rao DS, Baltimore D. Broad protection against influenza infection by vectored immunoprophylaxis in mice. Nat Biotechnol. (2013) 31:647–52. doi: 10.1038/nbt.2618
87. de Jong YP, Dorner M, Mommersteeg MC, Xiao JW, Balazs AB, Robbins JB, et al. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci Transl Med. (2014) 6:254ra129. doi: 10.1126/scitranslmed.3009512
88. Deal C, Balazs AB, Espinosa DA, Zavala F, Baltimore D, Ketner G. Vectored antibody gene delivery protects against Plasmodium falciparum sporozoite challenge in mice. Proc Natl Acad Sci USA. (2014) 111:12528–32. doi: 10.1073/pnas.1407362111
89. Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature. (2012) 481:81–4. doi: 10.1038/nature10660
90. Balazs AB, Ouyang Y, Hong CM, Chen J, Nguyen SM, Rao DS, et al. Vectored immunoprophylaxis protects humanized mice from mucosal HIV transmission. Nat Med. (2014) 20:296–300. doi: 10.1038/nm.3471
91. Priddy FH, Lewis DJM, Gelderblom HC, Hassanin H, Streatfield C, LaBranche C, et al. Adeno-associated virus vectored immunoprophylaxis to prevent HIV in healthy adults: a phase 1 randomised controlled trial. Lancet HIV. (2019) 6:e230–9. doi: 10.1016/S2352-3018(19)30003-7
92. Phelps M, Balazs AB. Contribution to HIV prevention and treatment by antibody-mediated effector function and advances in broadly neutralizing antibody delivery by vectored immunoprophylaxis. Front Immunol. (2021) 12:734304. doi: 10.3389/fimmu.2021.734304
93. Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. (2008) 16:1073–80. doi: 10.1038/mt.2008.76
94. Sehara Y, Fujimoto K, Ikeguchi K, Katakai Y, Ono F, Takino N, et al. Persistent expression of dopamine-synthesizing enzymes 15 years after gene transfer in a primate model of Parkinson's disease. Hum Gene Ther Clin Dev. (2017) 28:74–9. doi: 10.1089/humc.2017.010
95. Rosenberg JB, Chen A, De BP, Dyke JP, Ballon DJ, Monette S, et al. Safety of direct intraparenchymal AAVrh.10-mediated central nervous system gene therapy for metachromatic leukodystrophy. Hum Gene Ther. (2021) 32:563–80. doi: 10.1089/hum.2020.269
96. Federici T, Taub JS, Baum GR, Gray SJ, Grieger JC, Matthews KA, et al. Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther. (2012) 19:852–9. doi: 10.1038/gt.2011.130
97. Bey K, Deniaud J, Dubreil L, Joussemet B, Cristini J, Ciron C, et al. Intra-CSF AAV9 and AAVrh10 administration in nonhuman primates: promising routes and vectors for which neurological diseases? Mol Ther Methods Clin Dev. (2020) 17:771–84. doi: 10.1016/j.omtm.2020.04.001
98. Galvan A, Petkau TL, Hill AM, Korecki AJ, Lu G, Choi D, et al. Intracerebroventricular administration of AAV9-PHP.B SYN1-EmGFP induces widespread transgene expression in the mouse and monkey central nervous system. Hum Gene Ther. (2021) 32:599–615. doi: 10.1089/hum.2020.301
99. Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther. (2018) 29:285–98. doi: 10.1089/hum.2018.015
100. Perez BA, Shutterly A, Chan YK, Byrne BJ, Corti M. Management of neuroinflammatory responses to AAV-mediated gene therapies for neurodegenerative diseases. Brain Sci. (2020) 10:119. doi: 10.3390/brainsci10020119
101. Govindasamy L, Padron E, McKenna R, Muzyczka N, Kaludov N, Chiorini JA, et al. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J Virol. (2006) 80:11556–70. doi: 10.1128/JVI.01536-06
102. DiMattia MA, Nam H-J, Van Vliet K, Mitchell M, Bennett A, Gurda BL, et al. Structural insight into the unique properties of adeno-associated virus serotype 9. J Virol. (2012) 86:6947–58. doi: 10.1128/JVI.07232-11
103. Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. (2010) 21:704–12. doi: 10.1089/hum.2009.182
104. Brown HC, Zakas PM, George SN, Parker ET, Spencer HT, Doering CB. Target-cell-directed bioengineering approaches for gene therapy of hemophilia A. Mol Ther Methods Clin Dev. (2018) 9:57–69. doi: 10.1016/j.omtm.2018.01.004
105. Wang D, Li S, Gessler DJ, Xie J, Zhong L, Li J, et al. A rationally engineered capsid variant of AAV9 for systemic CNS-directed and peripheral tissue-detargeted gene delivery in neonates. Mol Ther Methods Clin Dev. (2018) 9:234–46. doi: 10.1016/j.omtm.2018.03.004
106. Kotchey NM, Adachi K, Zahid M, Inagaki K, Charan R, Parker RS, et al. A potential role of distinctively delayed blood clearance of recombinant adeno-associated virus serotype 9 in robust cardiac transduction. Mol Ther. (2011) 19:1079–89. doi: 10.1038/mt.2011.3
107. Zinn E, Pacouret S, Khaychuk V, Turunen HT, Carvalho LS, Andres-Mateos E, et al. In silico reconstruction of the viral evolutionary lineage yields a potent gene therapy vector. Cell Rep. (2015) 12:1056–68. doi: 10.1016/j.celrep.2015.07.019
108. Hudry E, Andres-Mateos E, Lerner EP, Volak A, Cohen O, Hyman BT, et al. Efficient gene transfer to the central nervous system by single-stranded Anc80L65. Mol Ther Methods Clin Dev. (2018) 10:197–209. doi: 10.1016/j.omtm.2018.07.006
109. Lee S, Kim S, Kang H-Y, Lim HR, Kwon Y, Jo M, et al. The overexpression of TDP-43 in astrocytes causes neurodegeneration via a PTP1B-mediated inflammatory response. J Neuroinflamm. (2020) 17:299. doi: 10.1186/s12974-020-01963-6
110. Xu Y, Jin M-Z, Yang Z-Y, Jin W-L. Microglia in neurodegenerative diseases. Neural Regen Res. (2021) 16:270. doi: 10.4103/1673-5374.290881
111. d'Errico P, Ziegler-Waldkirch S, Aires V, Hoffmann P, Mezö C, Erny D, et al. Microglia contribute to the propagation of Aβ into unaffected brain tissue. Nat Neurosci. (2022) 25:20–5. doi: 10.1038/s41593-021-00951-0
112. Xie M, Liu YU, Zhao S, Zhang L, Bosco DB, Pang Y-P, et al. TREM2 interacts with TDP-43 and mediates microglial neuroprotection against TDP-43-related neurodegeneration. Nat Neurosci. (2022) 25:26–38. doi: 10.1038/s41593-021-00975-6
113. Pandya J, Ortiz L, Ling C, Rivers AE, Aslanidi G. Rationally designed capsid and transgene cassette of AAV6 vectors for dendritic cell-based cancer immunotherapy. Immunol Cell Biol. (2014) 92:116–23. doi: 10.1038/icb.2013.74
114. Maes ME, Colombo G, Schulz R, Siegert S. Targeting microglia with lentivirus and AAV: recent advances and remaining challenges. Neurosci Lett. (2019) 707:134310. doi: 10.1016/j.neulet.2019.134310
115. O'Carroll SJ, Cook WH, Young D. AAV targeting of glial cell types in the central and peripheral nervous system and relevance to human gene therapy. Front Mol Neurosci. (2021) 13:618020. doi: 10.3389/fnmol.2020.618020
116. Li W, Asokan A, Wu Z, Van Dyke T, DiPrimio N, Johnson JS, et al. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol Ther. (2008) 16:1252–60. doi: 10.1038/mt.2008.100
117. Girod A, Ried M, Wobus C, Lahm H, Leike K, Kleinschmidt J, et al. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat Med. (1999) 5:1052–6. doi: 10.1038/12491
118. Rabinowitz JE, Xiao W, Samulski RJ. Insertional mutagenesis of AAV2 capsid and the production of recombinant virus. Virology. (1999) 265:274–85. doi: 10.1006/viro.1999.0045
119. Zhu D, Schieferecke AJ, Lopez PA, Schaffer DV. Adeno-associated virus vector for central nervous system gene therapy. Trends Mol Med. (2021) 27:524–37. doi: 10.1016/j.molmed.2021.03.010
120. Powell SK, Khan N, Parker CL, Samulski RJ, Matsushima G, Gray SJ, et al. Characterization of a novel adeno-associated viral vector with preferential oligodendrocyte tropism. Gene Ther. (2016) 23:807–14. doi: 10.1038/gt.2016.62
121. Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol. (2016) 34:204–9. doi: 10.1038/nbt.3440
122. Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu W-L, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. (2017) 20:1172–9. doi: 10.1038/nn.4593
123. Hordeaux J, Wang Q, Katz N, Buza EL, Bell P, Wilson JM. The neurotropic properties of AAV-PHP.B are limited to C57BL/6J mice. Mol Ther. (2018) 26:664–8. doi: 10.1016/j.ymthe.2018.01.018
124. Liguore WA, Domire JS, Button D, Wang Y, Dufour BD, Srinivasan S, et al. AAV-PHP.B administration results in a differential pattern of CNS biodistribution in non-human primates compared with mice. Mol Ther. (2019) 27:2018–37. doi: 10.1016/j.ymthe.2019.07.017
125. Hanlon KS, Meltzer JC, Buzhdygan T, Cheng MJ, Sena-Esteves M, Bennett RE, et al. Selection of an efficient AAV vector for robust CNS transgene expression. Mol Ther Methods Clin Dev. (2019) 15:320–32. doi: 10.1016/j.omtm.2019.10.007
126. Shao W, Earley LF, Chai Z, Chen X, Sun J, He T, et al. Double-stranded RNA innate immune response activation from long-term adeno-associated virus vector transduction. JCI Insight. (2018) 3:e120474. doi: 10.1172/jci.insight.120474
127. Li C, Diprimio N, Bowles DE, Hirsch ML, Monahan PE, Asokan A, et al. Single amino acid modification of adeno-associated virus capsid changes transduction and humoral immune profiles. J Virol. (2012) 86:7752–9. doi: 10.1128/JVI.00675-12
128. Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA, et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol. (2008) 82:5887–911. doi: 10.1128/JVI.00254-08
129. Manning WC, Zhou S, Bland MP, Escobedo JA, Dwarki V. Transient immunosuppression allows transgene expression following readministration of adeno-associated viral vectors. Hum Gene Ther. (1998) 9:477–85. doi: 10.1089/hum.1998.9.4-477
130. Kaplan A. Complications of apheresis: complications of apheresis. Semin Dialy. (2012) 25:152–8. doi: 10.1111/j.1525-139X.2011.01026.x
131. Bertin B, Veron P, Leborgne C, Deschamps J-Y, Moullec S, Fromes Y, et al. Capsid-specific removal of circulating antibodies to adeno-associated virus vectors. Sci Rep. (2020) 10:864. doi: 10.1038/s41598-020-57893-z
132. Ferrari FK, Samulski T, Shenk T, Samulski RJ. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virol. (1996) 70:3227–34. doi: 10.1128/JVI.70.5.3227-3234.1996
133. Fisher KJ, Gao GP, Weitzman MD, DeMatteo R, Burda JF, Wilson JM. Transduction with recombinant adeno-associated virus for gene therapy is limited by leading-strand synthesis. J Virol. (1996) 70:520–32. doi: 10.1128/JVI.70.1.520-532.1996
134. Wu Z, Sun J, Zhang T, Yin C, Yin F, Van Dyke T, et al. Optimization of self-complementary AAV vectors for liver-directed expression results in sustained correction of hemophilia B at low vector dose. Mol Ther. (2008) 16:280–9. doi: 10.1038/sj.mt.6300355
135. Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther. (2011) 19:1058–69. doi: 10.1038/mt.2011.72
136. Deverman BE, Ravina BM, Bankiewicz KS, Paul SM, Sah DWY. Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov. (2018) 17:641–59. doi: 10.1038/nrd.2018.110
137. Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. (2010) 18:80–6. doi: 10.1038/mt.2009.255
138. Zhan W, Muhuri M, Tai PWL, Gao G. Vectored immunotherapeutics for infectious diseases: can rAAVs be the game changers for fighting transmissible pathogens? Front Immunol. (2021) 12:673699. doi: 10.3389/fimmu.2021.673699
139. Zhao A, Tohidkia MR, Siegel DL, Coukos G, Omidi Y. Phage antibody display libraries: a powerful antibody discovery platform for immunotherapy. Crit Rev Biotechnol. (2016) 36:276–89. doi: 10.3109/07388551.2014.958978
140. Zhang Y, Pardridge WM. Mediated efflux of IgG molecules from brain to blood across the blood-brain barrier. J Neuroimmunol. (2001) 114:168–72. doi: 10.1016/s0165-5728(01)00242-9
141. Zhang Y, Pardridge WM. Rapid transferrin efflux from brain to blood across the blood-brain barrier. J Neurochem. (2001) 76:1597–600. doi: 10.1046/j.1471-4159.2001.00222.x
142. Cooper PR, Ciambrone GJ, Kliwinski CM, Maze E, Johnson L, Li Q, et al. Efflux of monoclonal antibodies from rat brain by neonatal Fc receptor, FcRn. Brain Res. (2013) 1534:13–21. doi: 10.1016/j.brainres.2013.08.035
143. Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. (2013) 82:775–97. doi: 10.1146/annurev-biochem-063011-092449
144. Jobling SA, Jarman C, Teh M-M, Holmberg N, Blake C, Verhoeyen ME. Immunomodulation of enzyme function in plants by single-domain antibody fragments. Nat Biotechnol. (2003) 21:77–80. doi: 10.1038/nbt772
145. Ewert S, Honegger A, Plückthun A. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods. (2004) 34:184–99. doi: 10.1016/j.ymeth.2004.04.007
146. Ahmad ZA, Yeap SK, Ali AM, Ho WY, Alitheen NBM, Hamid M. scFv antibody: principles and clinical application. Clin Dev Immunol. (2012) 2012:1–15. doi: 10.1155/2012/980250
147. Soetens E, Ballegeer M, Saelens X. An inside job: applications of intracellular single domain antibodies. Biomolecules. (2020) 10:E1663. doi: 10.3390/biom10121663
148. Wolf E, Hofmeister R, Kufer P, Schlereth B, Baeuerle PA. BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov Today. (2005) 10:1237–44. doi: 10.1016/S1359-6446(05)03554-3
149. Bates A, Power CA. David vs. Goliath: the structure, function, and clinical prospects of antibody fragments. Antibodies. (2019) 8:E28. doi: 10.3390/antib8020028
150. Johnson S, Burke S, Huang L, Gorlatov S, Li H, Wang W, et al. Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J Mol Biol. (2010) 399:436–49. doi: 10.1016/j.jmb.2010.04.001
151. Dmitriev OY, Lutsenko S, Muyldermans S. Nanobodies as probes for protein dynamics in vitro and in cells. J Biol Chem. (2016) 291:3767–75. doi: 10.1074/jbc.R115.679811
152. Marino M, Zhou L, Rincon MY, Callaerts-Vegh Z, Verhaert J, Wahis J et al. AAV-mediated delivery of an anti-BACE1 VHH alleviates pathology in an Alzheimer's disease model. EMBO Mol Med. (2022). doi: 10.15252/emmm.201809824
153. Baudisch B, Pfort I, Sorge E, Conrad U. Nanobody-directed specific degradation of proteins by the 26S-proteasome in plants. Front Plant Sci. (2018) 9:130. doi: 10.3389/fpls.2018.00130
154. Cheloha RW, Harmand TJ, Wijne C, Schwartz TU, Ploegh HL. Exploring cellular biochemistry with nanobodies. J Biol Chem. (2020) 295:15307–27. doi: 10.1074/jbc.REV120.012960
155. Verhelle A, Nair N, Everaert I, Van Overbeke W, Supply L, Zwaenepoel O, et al. AAV9 delivered bispecific nanobody attenuates amyloid burden in the gelsolin amyloidosis mouse model. Hum Mol Genet. (2017) 26:3030. doi: 10.1093/hmg/ddx207
156. Ackaert C, Smiejkowska N, Xavier C, Sterckx YGJ, Denies S, Stijlemans B, et al. Immunogenicity risk profile of nanobodies. Front Immunol. (2021) 12:632687. doi: 10.3389/fimmu.2021.632687
157. Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. (2009) 284:3273–84. doi: 10.1074/jbc.M806889200
158. Jovčevska I, Muyldermans S. The therapeutic potential of nanobodies. Biodrugs. (2020) 34:11–26. doi: 10.1007/s40259-019-00392-z
160. Wagner TR, Rothbauer U. Nanobodies right in the middle: intrabodies as toolbox to visualize and modulate antigens in the living cell. Biomolecules. (2020) 10:1701. doi: 10.3390/biom10121701
161. Wang L, Wang Z, Zhang F, Zhu R, Bi J, Wu J, et al. Enhancing transgene expression from recombinant AAV8 vectors in different tissues using woodchuck hepatitis virus post-transcriptional regulatory element. Int J Med Sci. (2016) 13:286–91. doi: 10.7150/ijms.14152
162. Patrício MI, Barnard AR, Orlans HO, McClements ME, MacLaren RE. Inclusion of the woodchuck hepatitis virus posttranscriptional regulatory element enhances AAV2-driven transduction of mouse and human retina. Mol Ther Nucleic Acids. (2017) 6:198–208. doi: 10.1016/j.omtn.2016.12.006
163. Chuah MK, Petrus I, De Bleser P, Le Guiner C, Gernoux G, Adjali O, et al. Liver-specific transcriptional modules identified by genome-wide in silico analysis enable efficient gene therapy in mice and non-human primates. Mol Ther. (2014) 22:1605–13. doi: 10.1038/mt.2014.114
164. Rincon MY, Sarcar S, Danso-Abeam D, Keyaerts M, Matrai J, Samara-Kuko E, et al. Genome-wide computational analysis reveals cardiomyocyte-specific transcriptional cis-regulatory motifs that enable efficient cardiac gene therapy. Mol Ther. (2015) 23:43–52. doi: 10.1038/mt.2014.178
165. Sarcar S, Tulalamba W, Rincon MY, Tipanee J, Pham HQ, Evens H, et al. Next-generation muscle-directed gene therapy by in silico vector design. Nat Commun. (2019) 10:492. doi: 10.1038/s41467-018-08283-7
166. Majowicz A, Maczuga P, Kwikkers KL, van der Marel S, van Logtenstein R, Petry H, et al. Mir-142-3p target sequences reduce transgene-directed immunogenicity following intramuscular adeno-associated virus 1 vector-mediated gene delivery: reduction of transgene directed immune responses in AAV1 gene delivery. J Gene Med. (2013) 15:219–32. doi: 10.1002/jgm.2712
167. Muhuri M, Zhan W, Maeda Y, Li J, Lotun A, Chen J, et al. Novel combinatorial microRNA-binding sites in AAV vectors synergistically diminish antigen presentation and transgene immunity for efficient and stable transduction. Front Immunol. (2021) 12:674242. doi: 10.3389/fimmu.2021.674242
168. Kügler S, Lingor P, Schöll U, Zolotukhin S, Bähr M. Differential transgene expression in brain cells in vivo and in vitro from AAV-2 vectors with small transcriptional control units. Virology. (2003) 311:89–95. doi: 10.1016/S0042-6822(03)00162-4
169. Griffin JM, Fackelmeier B, Fong DM, Mouravlev A, Young D, O'Carroll SJ. Astrocyte-selective AAV gene therapy through the endogenous GFAP promoter results in robust transduction in the rat spinal cord following injury. Gene Ther. (2019) 26:198–210. doi: 10.1038/s41434-019-0075-6
170. Finneran DJ, Njoku IP, Flores-Pazarin D, Ranabothu MR, Nash KR, Morgan D, et al. Toward development of neuron specific transduction after systemic delivery of viral vectors. Front Neurol. (2021) 12:685802. doi: 10.3389/fneur.2021.685802
171. von Jonquieres G, Fröhlich D, Klugmann CB, Wen X, Harasta AE, Ramkumar R, et al. Recombinant human myelin-associated glycoprotein promoter drives selective AAV-mediated transgene expression in oligodendrocytes. Front Mol Neurosci. (2016) 9:13. doi: 10.3389/fnmol.2016.00013
172. Nasrabady SE, Rizvi B, Goldman JE, Brickman AM. White matter changes in Alzheimer's disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun. (2018) 6:22. doi: 10.1186/s40478-018-0515-3
173. Bohnen NI, Albin RL. White matter lesions in Parkinson disease. Nat Rev Neurol. (2011) 7:229–36. doi: 10.1038/nrneurol.2011.21
174. Hoshino C, Konno A, Hosoi N, Kaneko R, Mukai R, Nakai J, et al. GABAergic neuron-specific whole-brain transduction by AAV-PHP.B incorporated with a new GAD65 promoter. Mol Brain. (2021) 14:33. doi: 10.1186/s13041-021-00746-1
175. Levenga J, Krishnamurthy P, Rajamohamedsait H, Wong H, Franke TF, Cain P, et al. Tau pathology induces loss of GABAergic interneurons leading to altered synaptic plasticity and behavioral impairments. Acta Neuropathol Commun. (2013) 1:34. doi: 10.1186/2051-5960-1-34
176. Ruan Z, Pathak D, Venkatesan Kalavai S, Yoshii-Kitahara A, Muraoka S, Bhatt N, et al. Alzheimer's disease brain-derived extracellular vesicles spread tau pathology in interneurons. Brain. (2021) 144:288–309. doi: 10.1093/brain/awaa376
177. Geisler A, Fechner H. MicroRNA-regulated viral vectors for gene therapy. World J Exp Med. (2016) 6:37. doi: 10.5493/wjem.v6.i2.37
178. Xie J, Xie Q, Zhang H, Ameres SL, Hung J-H, Su Q, et al. MicroRNA-regulated, systemically delivered rAAV9: a step closer to CNS-restricted transgene expression. Mol Ther. (2011) 19:526–35. doi: 10.1038/mt.2010.279
179. Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000). Neurochem Res. (2000) 25:1439–51. doi: 10.1023/a:1007677003387
180. Jakobsson J, Georgievska B, Ericson C, Lundberg C. Lesion-dependent regulation of transgene expression in the rat brain using a human glial fibrillary acidic protein-lentiviral vector. Eur J Neurosci. (2004) 19:761–5. doi: 10.1111/j.0953-816x.2003.03147.x
181. Batiuk MY, Martirosyan A, Wahis J, de Vin F, Marneffe C, Kusserow C, et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat Commun. (2020) 11:1220. doi: 10.1038/s41467-019-14198-8
182. Pestana F, Edwards-Faret G, Belgard TG, Martirosyan A, Holt MG. No longer underappreciated: the emerging concept of astrocyte heterogeneity in neuroscience. Brain Sci. (2020) 10:168. doi: 10.3390/brainsci10030168
183. Das P, Verbeeck C, Minter L, Chakrabarty P, Felsenstein K, Kukar T, et al. Transient pharmacologic lowering of Aβ production prior to deposition results in sustained reduction of amyloid plaque pathology. Mol Neurodegener. (2012) 7:39. doi: 10.1186/1750-1326-7-39
184. Karlnoski RA, Rosenthal A, Kobayashi D, Pons J, Alamed J, Mercer M, et al. Suppression of amyloid deposition leads to long-term reductions in Alzheimer's pathologies in Tg2576 mice. J Neurosci. (2009) 29:4964–71. doi: 10.1523/JNEUROSCI.4560-08.2009
185. Guilbaud M, Devaux M, Couzinié C, Le Duff J, Toromanoff A, Vandamme C, et al. Five years of successful inducible transgene expression following locoregional adeno-associated virus delivery in nonhuman primates with no detectable immunity. Hum Gene Ther. (2019) 30:802–13. doi: 10.1089/hum.2018.234
186. Latta-Mahieu M, Rolland M, Caillet C, Wang M, Kennel P, Mahfouz I, et al. Gene transfer of a chimeric trans-activator is immunogenic and results in short-lived transgene expression. Hum Gene Ther. (2002) 13:1611–20. doi: 10.1089/10430340260201707
187. Favre D, Blouin V, Provost N, Spisek R, Porrot F, Bohl D, et al. Lack of an immune response against the tetracycline-dependent transactivator correlates with long-term doxycycline-regulated transgene expression in nonhuman primates after intramuscular injection of recombinant adeno-associated virus. J Virol. (2002) 76:11605–11. doi: 10.1128/JVI.76.22.11605-11611.2002
188. Hoyng SA, Gnavi S, de Winter F, Eggers R, Ozawa T, Zaldumbide A, et al. Developing a potentially immunologically inert tetracycline-regulatable viral vector for gene therapy in the peripheral nerve. Gene Ther. (2014) 21:549–57. doi: 10.1038/gt.2014.22
189. Burnside ER, De Winter F, Didangelos A, James ND, Andreica E-C, Layard-Horsfall H, et al. Immune-evasive gene switch enables regulated delivery of chondroitinase after spinal cord injury. Brain. (2018) 141:2362–81. doi: 10.1093/brain/awy158
190. Chtarto A. Minocycline-induced activation of tetracycline-responsive promoter. Neurosci Lett. (2003) 352:155–8. doi: 10.1016/S0304-3940(03)01077-2
191. Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. (1996) 134:693–5. doi: 10.1111/j.1365-2133.1996.tb06972.x
192. Seaman HE, Lawrenson RA, Williams TJ, MacRae KD, Farmer RDT. The risk of liver damage associated with minocycline: a comparative study. J Clin Pharmacol. (2001) 41:852–60. doi: 10.1177/00912700122010753
193. Zhu S, Stavrovskaya IG, Drozda M, Kim BYS, Ona V, Li M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. (2002) 417:74–8. doi: 10.1038/417074a
194. Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. (2000) 6:797–801. doi: 10.1038/77528
195. Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. (2002) 22:1763–71. doi: 10.1523/JNEUROSCI.22-05-01763.2002
196. Thomas M, Le WD, Jankovic J. Minocycline and other tetracycline derivatives: a neuroprotective strategy in Parkinson's disease and Huntington's disease. Clin Neuropharmacol. (2003) 26:18–23. doi: 10.1097/00002826-200301000-00005
197. Arvin KL, Han BH, Du Y, Lin S, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol. (2002) 52:54–61. doi: 10.1002/ana.10242
198. Kaplon H, Reichert JM. Antibodies to watch in 2021. mAbs. (2021) 13:1860476. doi: 10.1080/19420862.2020.1860476
199. Hersh CM, Cohen JA. Alemtuzumab for the treatment of relapsing-remitting multiple sclerosis. Immunotherapy. (2014) 6:249–59. doi: 10.2217/imt.14.7
200. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung H-P, Hemmer B, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. (2017) 376:221–34. doi: 10.1056/NEJMoa1601277
201. Goadsby PJ, Reuter U, Hallström Y, Broessner G, Bonner JH, Zhang F, et al. A controlled trial of erenumab for episodic migraine. N Engl J Med. (2017) 377:2123–32. doi: 10.1056/NEJMoa1705848
202. Silberstein SD, Dodick DW, Bigal ME, Yeung PP, Goadsby PJ, Blankenbiller T, et al. Fremanezumab for the preventive treatment of chronic migraine. N Engl J Med. (2017) 377:2113–22. doi: 10.1056/NEJMoa1709038
203. Dhillon S. Eculizumab: a review in generalized myasthenia gravis. Drugs. (2018) 78:367–76. doi: 10.1007/s40265-018-0875-9
204. Frampton JE. Eculizumab: a review in neuromyelitis optica spectrum disorder. Drugs. (2020) 80:719–27. doi: 10.1007/s40265-020-01297-w
205. Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet. (2020) 21:255–72. doi: 10.1038/s41576-019-0205-4
206. Kuzmin DA, Shutova MV, Johnston NR, Smith OP, Fedorin VV, Kukushkin YS, et al. The clinical landscape for AAV gene therapies. Nat Rev Drug Discov. (2021) 20:173–4. doi: 10.1038/d41573-021-00017-7
207. Muyldermans S. Applications of nanobodies. Annu Rev Anim Biosci. (2021) 9:401–21. doi: 10.1146/annurev-animal-021419-083831
208. Phan DT, Bender RHF, Andrejecsk JW, Sobrino A, Hachey SJ, George SC, et al. Blood-brain barrier-on-a-chip: Microphysiological systems that capture the complexity of the blood-central nervous system interface. Exp Biol Med. (2017) 242:1669–78. doi: 10.1177/1535370217694100
209. Paşca SP. The rise of three-dimensional human brain cultures. Nature. (2018) 553:437–45. doi: 10.1038/nature25032
210. Williams-Medina A, Deblock M, Janigro D. In vitro models of the blood-brain barrier: tools in translational medicine. Front Med Technol. (2020) 2:623950. doi: 10.3389/fmedt.2020.623950
211. Fuller JP, Stavenhagen JB, Teeling JL. New roles for Fc receptors in neurodegeneration-the impact on Immunotherapy for Alzheimer's Disease. Front Neurosci. (2014) 8:235. doi: 10.3389/fnins.2014.00235
212. Vincke C, Muyldermans S. Introduction to heavy chain antibodies and derived Nanobodies. Methods Mol Biol. (2012) 911:15–26. doi: 10.1007/978-1-61779-968-6_2
213. Carrillo MC, Dean RA, Nicolas F, Miller DS, Berman R, Khachaturian Z, et al. Revisiting the framework of the National Institute on Aging-Alzheimer's Association diagnostic criteria. Alzheimers Dement. (2013) 9:594–601. doi: 10.1016/j.jalz.2013.05.1762
214. Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. (2014) 13:614–29. doi: 10.1016/S1474-4422(14)70090-0
215. Rodriguez-Vieitez E, Saint-Aubert L, Carter SF, Almkvist O, Farid K, Schöll M, et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer's disease. Brain. (2016) 139:922–36. doi: 10.1093/brain/awv404
216. De Strooper B, Karran E. The cellular phase of Alzheimer's disease. Cell. (2016) 164:603–15. doi: 10.1016/j.cell.2015.12.056
217. Kotin RM, Snyder RO. Manufacturing clinical grade recombinant adeno-associated virus using invertebrate cell lines. Hum Gene Ther. (2017) 28:350–60. doi: 10.1089/hum.2017.042
218. Adamson-Small L, Potter M, Falk DJ, Cleaver B, Byrne BJ, Clément N. A scalable method for the production of high-titer and high-quality adeno-associated type 9 vectors using the HSV platform. Mol Ther Methods Clin Dev. (2016) 3:16031. doi: 10.1038/mtm.2016.31
Keywords: AAV vectors, AAV vector-mediated antibody delivery (A-MAD), Monoclonal antibodies, Nanobodies (VHH), Central Nervous System, Blood Brain Barrier (BBB)
Citation: Marino M and Holt MG (2022) AAV Vector-Mediated Antibody Delivery (A-MAD) in the Central Nervous System. Front. Neurol. 13:870799. doi: 10.3389/fneur.2022.870799
Received: 07 February 2022; Accepted: 21 February 2022;
Published: 12 April 2022.
Edited by:
Jared Brent Smith, Regenxbio Inc., United StatesReviewed by:
Simon O'Carroll, The University of Auckland, New ZealandCopyright © 2022 Marino and Holt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew G. Holt, bWhvbHRAaTNzLnVwLnB0
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.