 Matthew Katz
Matthew Katz Fleur C. Garton
Fleur C. Garton Mark Davis3
Mark Davis3 Robert D. Henderson
Robert D. Henderson Pamela A. McCombe
Pamela A. McCombe
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 08 April 2022
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.868655
This article is part of the Research TopicNeuromuscular Disorders and Peripheral Neuropathies – Case Report Collection 2022View all 32 articles
Here we report on two unrelated adult patients presenting with Limb girdle muscular dystrophy who were found to have novel variants in ANO5. Both patients had prominent weakness of their proximal lower limbs with mild weakness of elbow flexion and markedly elevated creatine kinase. Next generation sequencing using a custom-designed neuromuscular panel was performed in both patients. In one patient, 336 genes were targeted for casual variants and in the other patient (using a later panel design), 464 genes were targeted. One patient was homozygous for a novel splice variant [c.294+5G>A; p.(Ala98Ins4*)] in ANO5. Another patient was compound heterozygous for two variants in ANO5; a common frameshift variant [c.191dupA; p.(Asn64fs)] and a novel missense variant [c.952G>C; p.(Ala318Pro)]. These findings support the utility of next generation sequencing in the diagnosis of patients presenting with a Limb girdle muscular dystrophy phenotype and extends the genotypic spectrum of ANO5 disease.
Anoctamin 5 (ANO5) is a protein that belongs to a family of calcium-activated chloride channels and phospholipid scramblases (1). The exact function of the ANO5 protein is not completely understood although it has been shown to play a role in sarcolemma repair (1). Recessive variants of ANO5 have been associated with a range of muscle diseases including limb girdle muscular dystrophy (LGMD R12), (2, 3) distal myopathy (Miyoshi-like muscular dystrophy type 3), (4) exercise induced myalgia, (5) recurrent rhabdomyolysis, (6) axial myopathy (7) and asymptomatic hyperCKemia (5).
Patients with LGMD R12 typically present in adulthood with slowly progressive proximal lower limb and bicep weakness that is often asymmetric. This is associated with markedly elevated creatine kinase (CK) levels (2, 3). For unknown reasons females homozygous for ANO5 variants tend to have a less severe phenotype than males (5). Dilated cardiomyopathy and cardiac conduction disease have been reported but are rare (8). Magnetic resonance imaging (MRI) of the lower limbs often shows selective atrophy and fatty infiltration of the posterior calf and thigh muscles (9).
Here we report the clinical, pathologic, and genetic findings of two unrelated patients presenting with LGMD who were found to have novel variants in ANO5.
This 35-year-old Iranian man presented with slowly progressive upper and lower limb weakness from the age of 20 years. At the time of presentation, the patient had difficulty standing from the seated position and washing his hair. There were no sensory symptoms. There was no significant medical history. The patient was borne to non-consanguineous parents. His brother had similar symptoms, but four other siblings were unaffected.
On examination he had a waddling gait. There was no muscle wasting. Tone was normal. There was MRC 4/5 weakness of hip flexion and extension, hip adduction and knee flexion and extension. There was MRC 4+/5 weakness of elbow flexion bilaterally. Distal upper and lower limb strength was normal (MRC 5/5). Facial, ocular and neck strength was normal (MRC 5/5). Reflexes were normal. The sensory examination was normal. The CK was 1990. An MRI of the upper limbs performed 3 years earlier showed increased T2 signal of teres major and long head of triceps bilaterally without atrophy or fatty infiltration (Figure 1). A muscle biopsy showed end-stage muscle disease consistent with a chronic muscular dystrophy.
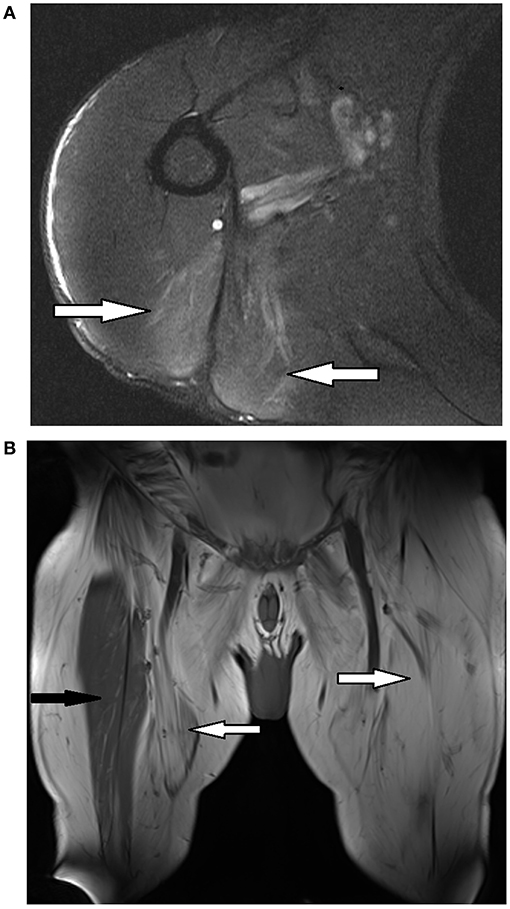
Figure 1. Magnetic resonance imaging (MRI) of selected muscles from patients 1 and 2. (A) Left shoulder of patient 1 - axial T2* weighted sequence showing increased T2 signal of teres major and long head triceps (white arrows). (B) Bilateral thighs of patient 2 - coronal T1∧ weighted sequence showing extensive fatty replacement of anterior and medial thigh muscles (white arrows) with relative preservation of the right rectus femoris (black arrow). *T2, transverse relaxation time; ∧T1, longitudinal relaxation time.
Previous genetic testing for Becker muscular dystrophy and facioscapulohumeral muscular dystrophy (FSHD) was negative. The patient had also undergone targeted sequencing of the fukutin related protein (FKRP) gene, which was normal. Next generation sequencing (NGS) using a custom-designed 336 gene neuromuscular panel (PathWest Laboratory, Australia) (10) revealed homozygosity for a novel splice variant [c.294+5G>A; p.(Ala98Ins4*)] in intron 5 of the ANO5 gene (ClinVar https://www.ncbi.nlm.nih.gov/clinvar/variation/1326848/?new_evidence=true).
While in silico analysis was not indicative of a likely splice effect, complementary DNA studies demonstrated intron inclusion and a complete absence of normal product (Figure 2). His brother was also found to be homozygous for the same variant on targeted sequencing of ANO5.
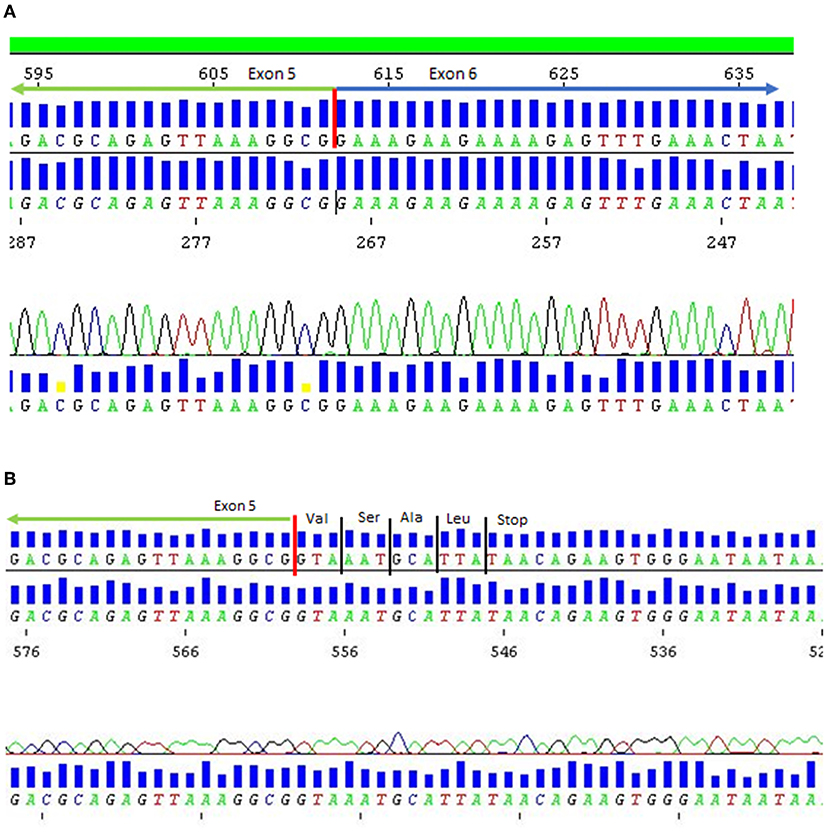
Figure 2. DNA sequence chromatogram for patient 1. (A) Normal control mRNA. (B) Patient 1 mRNA showing loss of exon 5 donor splice site resulting in intron inclusion and premature termination of protein due to presence of stop codon in variant mRNA.
This 55 year-old man presented with slowly progressive lower and upper limb weakness from the age of 35 years. At the time of presentation, he had difficulty rising from the seated position and walking up stairs. There was no significant medical history. There was no family history of muscle weakness.
On examination he had a waddling gait. There was wasting of the calf muscles bilaterally. The other limb muscles were of normal bulk. Tone was normal. There was MRC 4/5 weakness of hip flexion, hip extension, hip adduction, hip abduction, knee flexion and right knee extension. There was MRC 3/5 weakness of left knee extension. There was MRC 4+/5 weakness of elbow flexion and extension bilaterally. Distal upper and lower limb strength was normal (MRC 5/5). Facial, ocular and neck strength was normal (MRC 5/5). Reflexes were reduced in the upper and lower limbs. The sensory examination was normal. The CK was 4,910.
An EMG showed myopathic motor units without abnormal spontaneous activity in the right vastus medialis, vastus lateralis, biceps femoris and medial gastrocnemius. There were fibrillations with reduced recruitment of large motor units in the right tibialis anterior. MRI of the upper and lower limbs showed fatty infiltration and mild atrophy of the posterior calf and thigh muscles along with fatty infiltration and atrophy of the pectoral, latissimus dorsi and teres muscles (Figure 1). An echocardiogram did not show evidence of cardiomyopathy. The patient declined to undergo a muscle biopsy.
Next generation sequencing using a custom-designed 464 gene neuromuscular panel (PathWest Laboratory, Australia) (10) revealed compound heterozygosity for two variants in the ANO5 gene; a previously reported pathogenic frameshift variant [c.191dupA; p.(Asn64fs)] in exon 5 that results in a premature stop signal and a novel missense variant [c.952G>C; p.(Ala318Pro)] in exon 10 (ClinVar https://www.ncbi.nlm.nih.gov/clinvar/variation/1327991/?new_evidence=false#id_first). Segregation studies were not possible as the patient's first-degree relatives were not available for genetic testing.
We describe two patients presenting with LGMD who were found to have novel ANO5 variants after NGS using a custom-designed neuromuscular genetic panel. The use of multigene panels is especially helpful for the diagnosis of LGMD R12 as its phenotype can overlap with other types of LGMD (11) and muscle biopsies in these patients are often non-diagnostic and show non-specific myopathic or dystrophic changes (7, 12) although skeletal muscle amyloid deposits have been identified in some cases (13). The identification of disease causing variants with NGS also helps with genetic counseling in those with LGMD by allowing for targeted genetic testing in family members that could be carriers or at risk of inheriting the disease.
Patient 1 was a 35-year-old Iranian man who was homozygous for a novel splice variant [c.294+5G>A; p (Ala98Ins4*)]. This variant leads to intron inclusion resulting in premature truncation of the protein, is absent from controls (14–17) and segregates with disease in the affected sibling (13, 18) (Figure 2 and Table 1). Patient 1 has Iranian ancestry. Most reported cases of LGMD R12 have been in European patients (3). However, LGMD R12 has been reported in patients from the Middle East, including Saudi Arabia (18), Jordan (19) and Afghanistan (20).
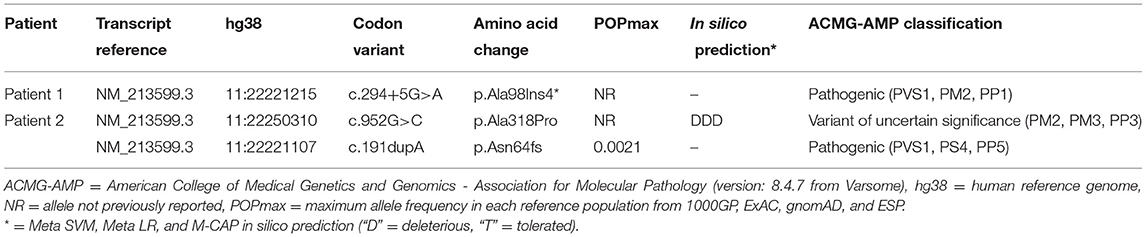
Table 1. Genetic summary for ANO5 variants.
Patient 2 was a 55-year-old Caucasian man who was compound heterozygous for the frameshift variant [c.191dupA; p.(Asn64fs)] and a novel missense variant [c.952G>C; p.(Ala318Pro)]. Through a founder effect, the frameshift variant [c.191dupA; p.(Asn64fs)] is the most common pathogenic ANO5 variant identified in European patients with LGMD R12 (3). The missense variant [c.952G>C; p.(Ala318Pro)] in patient 2 is absent from controls, is strongly conserved (21) and predicted to be damaging in silico (22, 23) (Table 1). Currently considered a variant of uncertain significance as per the American College of Medical Genetics and Genomics—Association for Molecular Pathology (ACMG-AMP) guidelines (24) additional functional evidence could help elucidate its impact. Detailed, mutation-specific assessments across ANO5 are lacking in the literature but loss of ANO5 has been shown to cause defective membrane repair (1). The novel missense variant we report in patient 2 is located within the first transmembrane domain of the protein (25) and could influence the effectiveness of membrane transport for repair (ie. coordination of annexins), particularly in muscle where it is highly expressed (26). The clinical presentation of patient 2 with adult onset, slowly progressive proximal lower limb and biceps weakness and significantly elevated CK is typical of other cases of LGMD R12 reported in the literature (2, 3).
ANO5 variants are not uncommon in LGMD but occur with a spectrum of LGMD severity. Our findings extend the genotypic spectrum of ANO5 disease and supports the utility of next generation sequencing in the diagnostic process. Further work on understanding disease mechanisms underlying ANO5 associated LGMD is needed to address relevant therapeutic avenues.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author/s.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
FG and MD assisted with the genetic data and review of paper. RH and PM assisted with overall review of the paper. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank Jonas Bjorkman (Pathology Queensland) for his contribution of genetic data to Figure 2.
1. Foltz SJ, Cui YY, Choo HJ, Hartzell HC. ANO5 ensures trafficking of annexins in wounded myofibers. J Cell Biol. (2021) 220:1–19. doi: 10.1083/jcb.202007059
2. Bolduc V, Marlow G, Boycott KM, Saleki K, Inoue H, Kroon J, et al. Recessive mutations in the putative calcium-activated chloride channel anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am J Hum Genet. (2010) 86:213–21. doi: 10.1016/j.ajhg.2009.12.013
3. Hicks P, Sarkozy A, Muelas N, Koehler K, Huebner A, Hudson G, et al. P63 a founder mutation in Anoctamin 5 is a major cause of limb girdle muscular dystrophy. Neuromuscul Disord. (2011) 21:S24–5. doi: 10.1016/S0960-8966(11)70082-7
4. Mahjneh I, Jaiswal J, Lamminen A, Somer M, Marlow G, Kiuru-Enari S, et al. A new distal myopathy with mutation in anoctamin 5. Neuromuscul Disord. (2010) 20:791–5. doi: 10.1016/j.nmd.2010.07.270
5. Penttilä S, Palmio J, Suominen T, Raheem O, Evilä A, Gomez NM, et al. Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology. (2012) 78:897–903. doi: 10.1212/WNL.0b013e31824c4682
6. Milone M, Liewluck T, Winder TL, Pianosi PT. Amyloidosis and exercise intolerance in ANO5 muscular dystrophy. Neuromuscul Disord. (2012) 22:13–5. doi: 10.1016/j.nmd.2011.07.005
7. Silva AMS, Coimbra-Neto AR, Souza PVS, Winckler PB, Gonçalves MVM, Cavalcanti EBU, et al. Clinical and molecular findings in a cohort of ANO5-related myopathy. Ann Clin Transl Neurol. (2019) 6:1225–38. doi: 10.1002/acn3.50801
8. Wahbi K, Béhin A, Bécane HM, Leturcq F, Cossée M, Laforêt P, et al. Dilated cardiomyopathy in patients with mutations in anoctamin 5. Int J Cardiol. (2013) 168:76–9. doi: 10.1016/j.ijcard.2012.09.070
9. Sarkozy A, Deschauer M, Carlier RY, Schrank B, Seeger J, Walter MC, et al. Muscle MRI findings in limb girdle muscular dystrophy type 2L. Neuromuscul Disord. (2012) 22(Suppl. 2):122–9. doi: 10.1016/j.nmd.2012.05.012
10. Beecroft SJ, Yau KS, Allcock RJN, Mina K, Gooding R, Faiz F, et al. Targeted gene panel use in 2249 neuromuscular patients: the Australasian referral center experience. Ann Clin Transl Neurol. (2020) 7:353–62. doi: 10.1002/acn3.51002
11. Savarese M, Di Fruscio G, Tasca G, Ruggiero L, Janssens S, De Bleecker J, et al. Next generation sequencing on patients with LGMD and nonspecific myopathies: findings associated with ANO5 mutations. Neuromuscul Disord. (2015) 25:533–41. doi: 10.1016/j.nmd.2015.03.011
12. Seguí F, Gonzalez-Quereda L, Sanchez A, Matas-García A, Garrabou G, Rodriguez MJ, et al. Anoctamin 5 (ANO5) muscular dystrophy—three different phenotypes and a new histological pattern. Neurol Sci. (2020) 41:2967–71. doi: 10.1007/s10072-020-04453-y
13. Liewluck T, Winder TL, Dimberg EL, Crum BA, Heppelmann CJ, Wang Y, et al. ANO5-muscular dystrophy: clinical, pathological and molecular findings. Eur J Neurol. (2013) 20:1383–9. doi: 10.1111/ene.12191
14. Auton A, Abecasis GR, Altshuler DM, Durbin RM, Bentley DR, Chakravarti A, et al. A global reference for human genetic variation. Nature. (2015) 526:68–74. doi: 10.1038/nature15393
15. Lek M, Karczewski KJ, Minikel E V, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536:285–91. doi: 10.1038/nature19057
16. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv[Preprint]. (2019). bioRxiv: 531210. doi: 10.1101/531210
17. Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available online at: https://evs.gs.washington.edu/EVS/
18. Bohlega S, Monies DM, Abulaban AA, Murad HN, Alhindi HN, Meyer BF. Clinical and genetic features of anoctaminopathy in Saudi Arabia. Neurosciences. (2015) 20:173–7. doi: 10.17712/nsj.2015.2.20140547
19. Little AA, Mckeever PE, Gruis KL. Novel mutations in the anoctamin 5 gene (ANO5) associated with limb-girdle muscular dystrophy 2L. Muscle and Nerve. (2013) 47:287–91. doi: 10.1002/mus.23542
20. Schessl J, Kress W, Schoser B. Novel ANO5 mutations causing hyper-CK-emia, limb girdle muscular weakness and miyoshi type of muscular dystrophy. Muscle and Nerve. (2012) 45:740–2. doi: 10.1002/mus.23281
21. Ramani R, Krumholz K, Huang YF, Siepel A. Phastweb: a web interface for evolutionary conservation scoring of multiple sequence alignments using phastCons and phyloP. Bioinformatics. (2019) 35:2320–2. doi: 10.1093/bioinformatics/bty966
22. Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. (2015) 24:2125–37. doi: 10.1093/hmg/ddu733
23. Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. (2016) 48:1581–6. doi: 10.1038/ng.3703
24. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology sue. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
25. Bateman A, Martin MJ, Orchard S, Magrane M, Agivetova R, Ahmad S, et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. (2021) 49:D480–9.
Keywords: case report, limb girdle muscular dystrophy, ANO5, novel variant, next generation sequencing
Citation: Katz M, Garton FC, Davis M, Henderson RD and McCombe PA (2022) Novel Variants of ANO5 in Two Patients With Limb Girdle Muscular Dystrophy: Case Report. Front. Neurol. 13:868655. doi: 10.3389/fneur.2022.868655
Received: 03 February 2022; Accepted: 11 March 2022;
Published: 08 April 2022.
Edited by:
Giovanni Meola, University of Milan, ItalyReviewed by:
Matthew Wicklund, University of Colorado, United StatesCopyright © 2022 Katz, Garton, Davis, Henderson and McCombe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew Katz, bWF0dGhldy5rYXR6QGhlYWx0aC5xbGQuZ292LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.