
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 25 February 2022
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.857279
This article is part of the Research TopicNeuromuscular Disorders and Peripheral Neuropathies - Case Report Collection 2021View all 13 articles
 Arianna Manini1
Arianna Manini1 Megi Meneri1,2
Megi Meneri1,2 Carmelo Rodolico3
Carmelo Rodolico3 Stefania Corti1,2
Stefania Corti1,2 Antonio Toscano3
Antonio Toscano3 Giacomo Pietro Comi1,4
Giacomo Pietro Comi1,4 Olimpia Musumeci3*
Olimpia Musumeci3* Dario Ronchi1*
Dario Ronchi1*The nuclear gene TK2 encodes the mitochondrial thymidine kinase, an enzyme involved in the phosphorylation of deoxycytidine and deoxythymidine nucleosides. Biallelic TK2 mutations are associated with a spectrum of clinical presentations mainly affecting skeletal muscle and featuring muscle mitochondrial DNA (mtDNA) instability. Current classification includes infantile- ( ≤ 1 year), childhood- (1–12 years), and late-onset (≥12 years) forms. In addition to age at onset, these forms differ for progression, life expectancy, and signs of mtDNA instability (mtDNA depletion vs. accumulation of multiple mtDNA deletions). Childhood-onset TK2 deficiency typically causes a rapidly progressive proximal myopathy, which leads to wheelchair-bound status within 10 years of disease onset, and severe respiratory impairment. Muscle biopsy usually reveals a combination of mitochondrial myopathy and dystrophic features with reduced mtDNA content. Here we report the case of an Italian patient presenting childhood-onset, slowly progressive mitochondrial myopathy, ptosis, hypoacusis, dysphonia, and dysphagia, harboring the TK2 variants c.278A>G and c.543del, the latter unreported so far. Compared to other childhood-onset TK2-patients, our case displays atypical features, including slowly progressive muscle weakness and absence of respiratory failure, which are usually observed in late-onset forms. This report extends the genetic background of TK2-related myopathy, highlighting the clinical overlap among different forms.
Mitochondrial DNA (mtDNA) maintenance defects are a heterogeneous group of clinical syndromes characterized by mtDNA deletions and/or depletion and derived from mutations in nuclear genes variably involved in mtDNA homeostasis (i.e., POLG1, POLG2, TWNK, DGUOK, TYMP) (1–5).
In 2001, Saada et al. (6) detected biallelic mutations in the thymidine kinase 2 (TK2) gene in four children presenting severe myopathy associated with muscle mtDNA depletion. TK2 is a nuclear-encoded mitochondrial enzyme involved in the phosphorylation of deoxycytidine and deoxythymidine nucleosides, which represents the first step of a salvage pathway aimed to provide deoxyribonucleotides (dNTPS) for mtDNA synthesis (7). Recessive TK2 mutations are now an established cause of mtDNA maintenance disorders, recently classified on the basis of clinical and biochemical features (8). According to age at onset, TK2 deficiency can lead either to an infantile-onset ( ≤ 1 year), childhood-onset (1–12 years) and late-onset (≥12 years) myopathy, which differ for rate of weakness progression, post-onset survival and predominance of mtDNA multiple deletions or depletion in muscle tissue, as summarized in Table 1 (8). Childhood-onset TK2 deficiency is typically associated with rapidly progressive, proximal myopathy, which leads to wheelchair-bound status within 10 years of disease onset in most patients, and post-onset survival longer than 13 years (8). More than half of childhood-onset TK2 patients require ventilatory support due to respiratory impairment (8). Ptosis, chronic external ophthalmoplegia (CPEO), facial weakness and dysphagia have been reported less frequently in this group of patients compared to late-onset cases (8–11), while cognitive decline, encephalopathy, seizures, and non-muscle manifestations are rare compared to the infantile-onset form (8–10). Needle electromyography (EMG) examination frequently evidences myopathic changes [i.e., polyphasic, short-duration, low-amplitude motor unit potentials (MUPs)] (8). Muscle histology reveals a combination of mitochondrial dysfunction [i.e., cytochrome C oxidase (COX)-negative fibers; ragged-red fibers (RRF)] and, mainly in pediatric cases, dystrophic features (i.e., atrophic fibers, fibrosis and increase of connective tissue) (8, 12, 13). Infantile and childhood-onset cases often showed mtDNA depletion in muscle tissue, whereas the late-onset ones are usually associated with mtDNA multiple deletions (8).
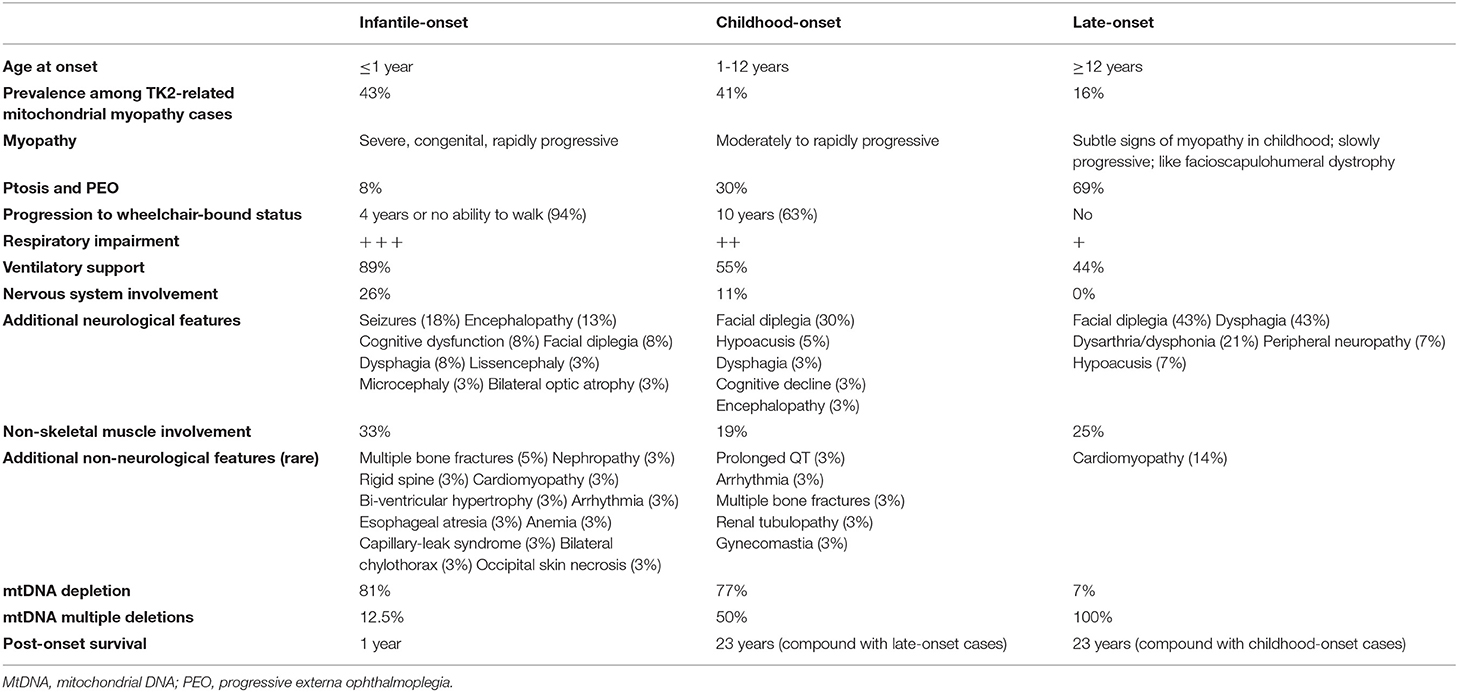
Table 1. Clinical and biochemical features associated with the three different phenotypes of TK2-related mitochondrial myopathy and mtDNA maintenance defects [data were obtained from Garone et al. (8)].
Herein, we report the case of an Italian patient affected by a childhood-onset, slowly progressive mitochondrial myopathy with atypical clinical features, harboring two heterozygous TK2 variants, one of which has not been reported before.
The proband is a 55-year-old Italian woman from Sicily, born to non-consanguineous parents. Mild bilateral ptosis and dysphonia were noticed starting from 8 years of age. At 20 years of age, she started to complain of dysphagia of both solid food and liquids and began losing weight. Over the years she developed bilateral upper limb weakness and mild hypoacusis, with slow progression in the last 20 years. Cognitive abilities are normal.
Current clinical examination reveals bilateral ptosis and ophthalmoparesis, myopathic face, marked dysphonia, wasting, weakness of proximal upper limbs and of psoas muscles, more prominent on the right.
The complete timeline of relevant clinical signs and symptoms and of diagnostic assessments performed during disease progression is reported in Figure 1.
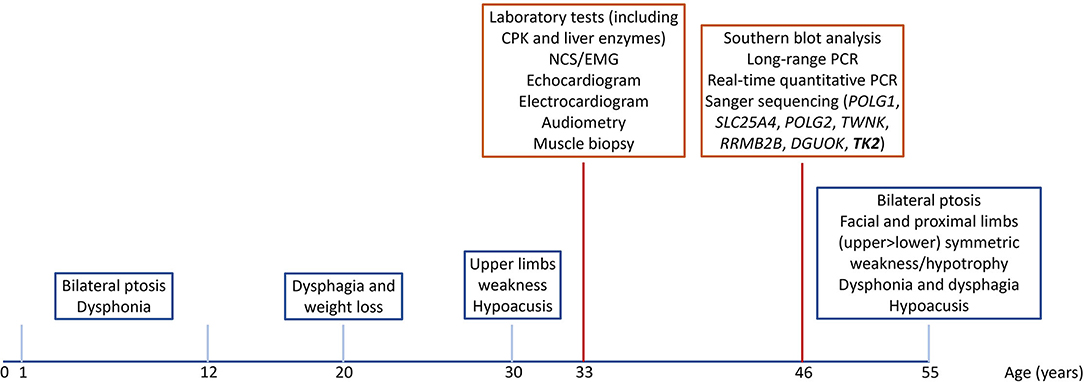
Figure 1. Timeline of relevant clinical signs and symptoms (blue squares), and of diagnostic assessments (red squares).
Creatine phosphokinase (CK) levels are currently 251 IU/L (normal value <200). During first hospitalization at 32 years of age, CK levels reached 3,880 IU/L, with concomitant increase of liver enzymes levels, including aspartate aminotransferase (AST) (243 IU/L; normal value <43 IU/L), alanine aminotransferase (ALT) (98 IU/L; normal value <45 IU/L) and lactate dehydrogenase (LDH) (1,459 IU/L; normal value <300 IU/L). Lactate levels were only mildly increased at baseline (2.6 mmol/L; normal value <1.5 mmol/L), but significantly raised under workload (4.6 mmol/L; normal value <2.3 mmol/L).
Needle EMG examination, performed at 33 years, showed rapid recruitment of short-duration, low-amplitude MUPs in bilateral biceps and first interosseous muscles. Audiometry, electrocardiogram, echocardiogram and spirometry were normal. At the same age, she underwent muscle biopsy of left biceps, which revealed increased fibers size, with coexistence of both atrophic and hypertrophic fibers in addition to mitochondrial dysfunction features, including 10% RRF, succinate dehydrogenase (SDH) hyperactive fibers and COX-negative fibers. The adenosine triphosphatase (ATPase) staining showed marked prevalence of type 1 fibers (almost 90%), and, to a lesser extent, type 2c fibers.
Southern blot analysis and long-range polymerase chain reaction (PCR) of muscle mtDNA showed multiple mtDNA deletions (Figures 2A,B). Quantitative PCR showed loss of mtDNA integrity (30% compared to age-matched controls; Figure 2C).
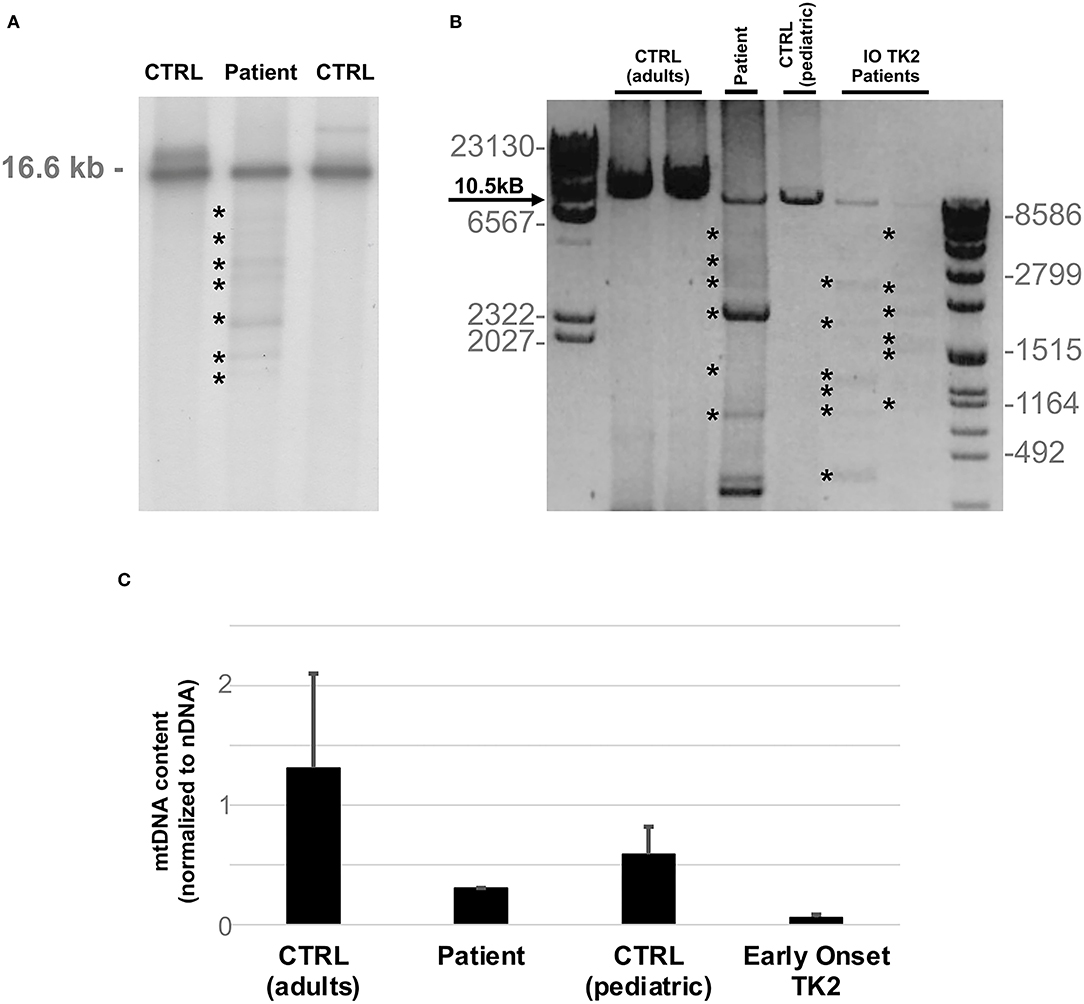
Figure 2. MtDNA studies. (A) Southern blot analysis of mtDNA obtained from patient's muscle biopsy and age matched controls. Asterisks indicate multiple bands corresponding to partially deleted mitochondrial genomes. The expected size of normal linearized mtDNA is indicated. (B) Long-range PCR analysis of mtDNA obtained from patient's muscle biopsy and two previously described infantile onset TK2 patients (IO TK2) compared to respective age matched controls. Asterisks indicate multiple bands corresponding to multiple mtDNA deletions. Black arrow indicates the expected size of a wild-type PCR amplicon (10.5 kB: FOR5635-RC16135). The sizes of the bands of the ladders (DNA Molecular Weight Marker II and VII, Roche) are indicated. (C) Histogram plot showing muscle mtDNA content in patient's muscle and two previously described infantile onset TK2 patients compared to respective age matched controls (n = 8, each group). Bars represent mean values. Error bars indicate standard deviation. MtDNA quantification, normalized to nuclear DNA (nDNA) content, was performed by quantitative PCR.
After excluding mutations in the common genes associated with multiple mtDNA deletions (POLG1, SLC25A4, POLG2, TWNK, RRMB2B, DGUOK), direct sequencing of TK2 (NM_004614.4) revealed the heterozygous mutations c.278A>G and c.543del resulting in protein changes p.Asn93Ser and p.Leu182Phefs*11, respectively. The c.278A>G variant [rs142291440; Genome Aggregation Database (gnomAD) Minor Allele Frequency (MAF) 1.2 × 10−6] has been previously detected in an African American patient affected by childhood-onset mitochondrial myopathy (14). The microdeletion c.543del is absent in publicly available databases. DNA from relatives was not available for molecular studies.
We herein describe the case of an Italian patient from Sicily affected with a mild form of childhood-onset mitochondrial myopathy, with muscle mtDNA multiple deletions and depletion, associated with two heterozygous mutations in TK2, one of which is novel. To date, this is the second Italian case of TK2-related myopathy described so far (15). Intriguingly, both families are of Sicilian origin.
Both the p.Asn93Ser and p.Leu182Phefs*11 variants are located within the deoxynucleoside kinase domain, which spans amino acids 53–260. This large functional domain is involved in the phosphorylation of deoxypyrimidine nucleosides, the first step of the mtDNA synthesis cascade (7). The p.Asn93Ser has been already reported by Oskoui et al. (14). Both our proband and the patient described by Oskoui and colleagues display childhood-onset myopathy with progressive development of fatigue, proximal muscle weakness, and facial diplegia, and similar levels of mtDNA depletion assessed in muscle tissue. Main differences between the two cases are represented by the absence of ptosis and the presence of neuropathic changes at EMG reported by Oskoui and colleagues (14). The novel p.Leu182Phefs*11 variant is located in exon 8, which encodes the α8 helix involved in the catalysis of adenosine triphosphate (ATP) molecules due to its capacity of binding phosphate groups. Exon 8 is the second hot spot of TK2 pathogenic variants, following exon 5 (8). Small indels causing frameshift represent the second most prevalent type of TK2 variants in mtDNA maintenance defects (13%), following missense mutations (66%), and are distributed throughout TK2 length (16).
Consistently with classical cases of childhood-onset TK2 deficiency, analysis of mtDNA levels in muscle tissue of our patient demonstrated both depletion and multiple deletions, and CK levels were mildly elevated (8). Muscle histology findings included non-specific myopathic changes (i.e., fiber size variability, with type 1 predominance), and mitochondrial myopathy markers without dystrophic features. However, several clinical features are atypical compared to previous reports of childhood-onset TK2-related myopathy (Table 1). First, muscle weakness has remained stable over the last 20 years, and our patient is currently able to walk without support. Childhood-onset TK2-related myopathy is usually rapidly progressive, and patients become wheelchair-bound within 10 years of disease onset. In the retrospective analysis of natural history data of genetically confirmed TK2 deficient patients performed by Garone and colleagues, only 4 out of 30 childhood-onset patients were able to walk independently after 10 years from disease onset (8). Of the remaining childhood-onset cases, 19 (63%) became wheelchair-bound within 10 years of disease onset, and 8 (27%) were within 10 years of disease onset, so that their muscle impairment rate of progression could not be assessed. Second, respiratory failure with invasive or non-invasive ventilatory dependency is frequent, reaching about half childhood-onset cases (8). At 55 years of age, our patient has not developed respiratory impairment. Third, dysphagia, which has been described in our case, is rare in childhood-onset TK2-related myopathy, especially compared to late-onset forms (8).
As deoxynucleosides therapies for TK2 deficiency are under investigation, identifying patients carrying TK2 mutations affected by mtDNA maintenance defects becomes pivotal (17). Domínguez-Gonzàlez and colleagues have recently reported the results of an open-label study in which pyrimidine deoxynucleosides and deoxynucleotides were orally administered to 16 patients with mitochondrial myopathy due to TK2 deficiency (17). In early-onset patients affected with severe forms of myopathy, the treatment was effective in increasing survival and ameliorating muscle weakness, respiratory function, and dysphagia, with no major side effects (17). On the contrary, the beneficial effects of the administration of pyrimidine deoxynucleosides and deoxynucleotides in adult-onset cases were limited, and hepatic toxicity was suspected in two patients (17).
The application of NGS-based sequencing in a clinical setting is rapidly expanding the number of novel diagnoses in mitochondrial disorders. On the other hand, the identification of specific changes (such as the coexistence of mtDNA depletion and deletions) might restrict the number of genes to be investigated to those involved in mitochondrial dNTPs supply pathways. Timely or early molecular diagnosis for TK2 patients is crucial for the recruitment in the ongoing clinical trials and the access to rescue therapies in the near future.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the Comitato Etico Milano Area 2 Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico (Milan, Italy). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
AM and DR interpreted the results, conceived the idea, revised the literature, and wrote the manuscript. DR performed genetic analysis and mtDNA studies. OM made the clinical evaluation. OM, MM, CR, AT, GC, and SC performed a critical revision of the manuscript for important intellectual content. All the authors have read and approved the manuscript.
This study was funded by Italian Ministry Fundation IRCCS Ca' Granda Ospedale Maggiore Policlinico Ricerca Corrente 2020 to GC. This work was promoted within the European Reference Network (ERN) for Neuromuscular Diseases.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the Associazione Centro Dino Ferrari for its support. Muscle biopsy and DNA samples were provided by the Bank of muscle tissue, peripheral nerve, DNA, and Cell Culture, member of Telethon Network of Genetic biobanks, at Fondazione IRCCS Ca' Granda, Ospedale Maggiore Policlinico, Milano, Italy.
1. Van Goethem G, Dermaut B, Löfgren A, Martin J-J, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. (2001) 28:211–2. doi: 10.1038/90034
2. Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, Taylor RW, et al. Mutant POLG2 disrupts DNA polymerase γ subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. (2006) 78:1026–34. doi: 10.1086/504303
3. Spelbrink JN, Li F-Y, Tiranti V, Nikali K, Yuan Q-P, Tariq M, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. (2001) 28:223–31. doi: 10.1038/90058
4. Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. (2001) 29:337–41. doi: 10.1038/ng746
5. Ronchi D, Liu C, Caporali L, Piga D, Li H, Tagliavini F, et al. Novel mutations in DNA2 associated with myopathy and mtDNA instability. Ann Clin Transl Neurol. (2019) 6:1893–9. doi: 10.1002/acn3.50888
6. Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. (2001) 29:342–4. doi: 10.1038/ng751
7. Fasullo M, Endres L. Nucleotide salvage deficiencies, DNA damage and neurodegeneration. Int J Mol Sci. (2015) 16:9431–49. doi: 10.3390/ijms16059431
8. Garone C, Taylor RW, Nascimento A, Poulton J, Fratter C, Domínguez-González C, et al. Retrospective natural history of thymidine kinase 2 deficiency. J Med Genet. (2018) 55:515–21. doi: 10.1136/jmedgenet-2017-105012
9. Domínguez-González C, Hernández-Laín A, Rivas E, Hernández-Voth A, Sayas Catalán J, Fernández-Torrón R, et al. Late-onset thymidine kinase 2 deficiency: a review of 18 cases. Orphanet J Rare Dis. (2019) 14:100. doi: 10.1186/s13023-019-1071-z
10. Tyynismaa H, Sun R, Ahola-Erkkila S, Almusa H, Poyhonen R, Korpela M, et al. Thymidine kinase 2 mutations in autosomal recessive progressive external ophthalmoplegia with multiple mitochondrial DNA deletions. Hum Mol Genet. (2012) 21:66–75. doi: 10.1093/hmg/ddr438
11. Alston CL, Schaefer AM, Raman P, Solaroli N, Krishnan KJ, Blakely EL, et al. Late-onset respiratory failure due to TK2 mutations causing multiple mtDNA deletions. Neurology. (2013) 81:2051–53. doi: 10.1212/01.wnl.0000436931.94291.e6
12. Collins J, Bove KE, Dimmock D, Morehart P, Wong L-J, Wong B. Progressive myofiber loss with extensive fibro-fatty replacement in a child with mitochondrial DNA depletion syndrome and novel thymidine kinase 2 gene mutations. Neuromuscul Disord. (2009) 19:784–7. doi: 10.1016/j.nmd.2009.08.002
13. Vila MR, Segovia-Silvestre T, Gamez J, Marina A, Naini AB, Meseguer A, et al. Reversion of mtDNA depletion in a patient with TK2 deficiency. Neurology. (2003) 60:1203–5. doi: 10.1212/01.WNL.0000055928.58122.47
14. Oskoui M, Davidzon G, Pascual J, Erazo R, Gurgel-Giannetti J, Krishna S, et al. Clinical spectrum of mitochondrial DNA depletion due to mutations in the thymidine kinase 2 gene. Arch Neurol. (2006) 63:1122. doi: 10.1001/archneur.63.8.1122
15. Galbiati S, Bordoni A, Papadimitriou D, Toscano A, Rodolico C, Katsarou E, et al. New mutations in TK2 gene associated with mitochondrial DNA depletion. Pediatr Neurol. (2006) 34:177–85. doi: 10.1016/j.pediatrneurol.2005.07.013
16. Wang J, Kim E, Dai H, Stefans V, Vogel H, Al Jasmi F, et al. Clinical and molecular spectrum of thymidine kinase 2-related mtDNA maintenance defect. Mol Genet Metab. (2018) 124:124–30. doi: 10.1016/j.ymgme.2018.04.012
Keywords: thymidine kinase 2, TK2, mitochondrial DNA, mtDNA maintenance defects, myopathy, deoxynucleosides
Citation: Manini A, Meneri M, Rodolico C, Corti S, Toscano A, Comi GP, Musumeci O and Ronchi D (2022) Case Report: Thymidine Kinase 2 (TK2) Deficiency: A Novel Mutation Associated With Childhood-Onset Mitochondrial Myopathy and Atypical Progression. Front. Neurol. 13:857279. doi: 10.3389/fneur.2022.857279
Received: 18 January 2022; Accepted: 31 January 2022;
Published: 25 February 2022.
Edited by:
Edoardo Malfatti, Hôpitaux Universitaires Henri Mondor, FranceReviewed by:
Catarina M. Quinzii, Columbia University, United StatesCopyright © 2022 Manini, Meneri, Rodolico, Corti, Toscano, Comi, Musumeci and Ronchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dario Ronchi, ZGFyaW8ucm9uY2hpQHVuaW1pLml0; Olimpia Musumeci, b2xpbXBpYS5tdXN1bWVjaUB1bmltZS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.