 Shaina L. Rosenblum
Shaina L. Rosenblum Daniel J. Kosman
Daniel J. Kosman
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 15 March 2022
Sec. Neuroepidemiology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.855751
This article is part of the Research Topic Metals and Cognitive Decline: Pathophysiology, Treatment, and Prevention View all 8 articles
The redox properties that make iron an essential nutrient also make iron an efficient pro-oxidant. Given this nascent cytotoxicity, iron homeostasis relies on a combination of iron transporters, chaperones, and redox buffers to manage the non-physiologic aqueous chemistry of this first-row transition metal. Although a mechanistic understanding of the link between brain iron accumulation (BIA) and neurodegenerative diseases is lacking, BIA is co-morbid with the majority of cognitive and motor function disorders. The most prevalent neurodegenerative disorders, including Alzheimer's Disease (AD), Parkinson's Disease (PD), Multiple System Atrophy (MSA), and Multiple Sclerosis (MS), often present with increased deposition of iron into the brain. In addition, ataxias that are linked to mutations in mitochondrial-localized proteins (Friedreich's Ataxia, Spinocerebellar Ataxias) result in mitochondrial iron accumulation and degradation of proton-coupled ATP production leading to neuronal degeneration. A comorbidity common in the elderly is a chronic systemic inflammation mediated by primary cytokines released by macrophages, and acute phase proteins (APPs) released subsequently from the liver. Abluminal inflammation in the brain is found downstream as a result of activation of astrocytes and microglia. Reasonably, the iron that accumulates in the brain comes from the cerebral vasculature via the microvascular capillary endothelial cells whose tight junctions represent the blood-brain barrier. A premise amenable to experimental interrogation is that inflammatory stress alters both the trans- and para-cellular flux of iron at this barrier resulting in a net accumulation of abluminal iron over time. This review will summarize the evidence that lends support to this premise; indicate the mechanisms that merit delineation; and highlight possible therapeutic interventions based on this model.
A strong correlation between cognitive decline and systemic pathology underlying chronic inflammation has been long recognized (1–4). Chronic inflammation common as one ages can be linked to arthritis, diabetes, cancer and/or chronic bacterial infection including periodontal disease (5). Another metabolic trajectory common to the elderly is brain iron accumulation as noted by both post-mortem quantitative analyses (6) and fMRI (7). This brain iron accumulation occurs despite the commonly co-morbid anemia linked also to chronic inflammation (8). Here we review the molecular cell mechanisms that underlie these clinical patterns. This synthesis provides support for the premise that a combination of therapeutic management of the brain iron and oxidative stress exacerbated by chronic inflammation is a mechanistically rational pharmacologic approach in the suppression of cognitive decline.
Iron is an essential trace element that is necessary for normal cell functioning, but can be harmful in excess. Due to Fenton chemistry, excessive amounts of iron can produce free radicals that can result in oxidative stress and ultimately, cell death (9). Therefore, iron metabolism must be highly regulated within tissues and cells. Iron functions as a co-factor for several enzymatic processes, energy metabolism, myelination, and DNA synthesis (10–12). The brain has critical need for iron due to this organ's high metabolic activity. In this context, maintenance of iron homeostasis is essential so as to avoid iron-dependent oxidative stress leading to neuronal degeneration and death. Healthy, normal brains accumulate iron specifically within the globus pallidus, caudate nucleus, putamen, dentate nucleus, and substantia nigra as shown by postmortem analysis and MRI (13–15). Iron in the brain can be found in several different forms. Ferrous (Fe2+) and ferric (Fe3+) iron is chelated by small molecules such and citrate and glutathione; Fe2+ is bound to the iron chaperones, PCBP1 and 2; and both redox forms are prosthetic groups of a multitude of proteins, both catalytic and structural. As a polyphosphate complex, ferric iron is stored in ferritin. When ferritin is degraded in the lysosome, hemosiderin is produced, which contains both ferrous and ferric iron. Within neurons, neuromelanin forms complexes with metals, including iron, and is found in autolysosomal organelles mostly in neurons of the substantia nigra and locus coeruleus, but in other regions of the human brain as well (16, 17). Cells of the brain do not have direct access to nutrients, including iron, from the systemic circulation because the blood-brain barrier (BBB) separates the brain from the systemic circulation, preventing toxic material transport. Iron transport into the brain occurs at the BBB, which is composed of brain microvascular endothelial cells (BMVECs) and is supported by astrocytes, neurons and microglia (18).
Several proteins on the luminal, or systemic blood membrane, and on the abluminal, or membrane closest to the brain interstitium, of the BBB mediate iron uptake and efflux into the brain. Iron uptake occurs through either transferrin-bound iron (TBI) or non-transferrin bound iron (NTBI) mechanisms. The TBI uptake pathway involves the binding of iron to the transferrin (Tf) protein, the binding of transferrin to the transferrin receptor (TfR), endocytosis of this complex, and finally, iron reduction and release into the cytosol through the divalent metal transporter 1 (DMT1) (19). NTBI uptake can occur by first either, reduction of extracellular free ferric iron (Fe3+), or reduction of TBI by a ferrireductase, Steap2/3. Once reduced, NTBI is brought into the cell by either ZIP8 (gene SLC39A8) or ZIP14 (SLC39A14), which are divalent metal ioin transporters that also transport Zn2+ and Mn2+ (20). Once in the cytoplasm of the BMVEC, ferrous iron can be chaperoned by the iron chaperones, PCBP1/2 for storage in ferritin (as Fe3+) or metalation of cytosolic iron-dependent enzymes; or delivered to the mitochondria for assembly of Fe,S clusters and heme. BMVEC-accumulated iron also can be exported apically back into circulation or basolaterally released into the abluminal space. Lysosomal degradation of ferritin releases ferrous iron into the cytosol when needed, with the help of the ferrireductases Steap2/3 and Lcytb (21). Ferrous iron efflux is supported by the sole iron efflux transporter, ferroportin (FPN). FPN-dependent efflux requires the subsequent ferrous iron oxidation—ferroxidation—by a ferroxidase, either ceruloplasmin (CP) or hephaestin (HP) (22). Efflux is regulated by the abundance of FPN in the plasma membrane. The peptide hormone, hepcidin, binds to FPN and triggers its internalization and degradation (23–25). Systemically, hepcidin is released from hepatocytes; in the brain, it is released from glial cells (26, 27). These pathways are displayed in Figure 1 and the proteins of interest are presented in Table 1. In BMVECs, endosomal-independent NTBI and TBI incorporation pathways are likely to be the main mode of iron transport into the brain. Evidence for this comes from many veins, showing that removing ferrireductases, chelating extracellular iron, and knocking down the ZIP proteins all inhibit TBI uptake (20, 28). These results implicate ZIP-mediated iron transport as the most prominent method of iron accumulation in BMVEC, the first step in the accumulation of iron in the abluminal space and the glia and neurons therein. Much is understood about the basic cellular processes involved with iron metabolism, but how these processes are affected by chronic inflammation is an underrepresented area of investigation.
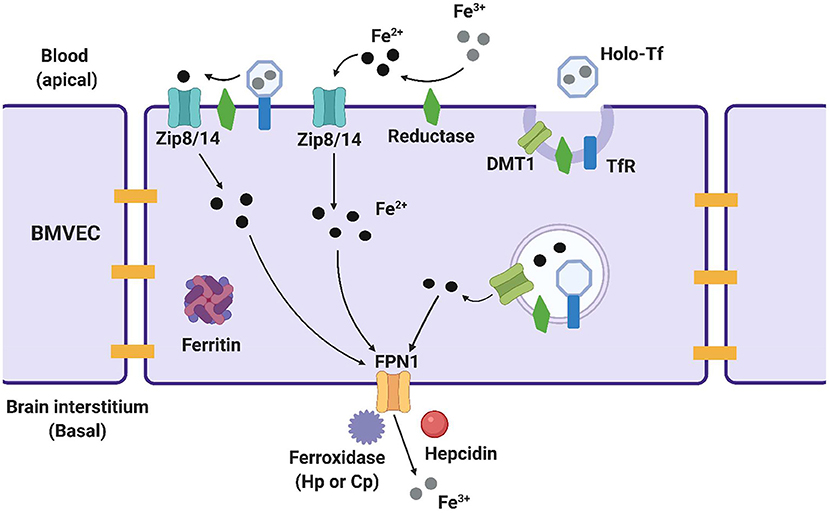
Figure 1. Methods of iron trafficking in BMVECs. This image highlights the uptake and efflux pathways used for iron transport in BMVECs. The yellow bars represent tight junctions which will be discussed in a later section. ZIP8/14 can transport transferrin bound iron (TBI) and non-transferrin bound iron (NTBI), with the help of a reductase which reduces the iron and allows it to enter the cell. Ferric (Fe3+) iron binds to transferrin, creating holo-Tf which binds to TfR and is endocytosed into an endolysosome in the cell. Here, the iron is reduced and leaves the endolysosome through DMT1. Within the cell, ferrous iron (Fe2+) can be stored in ferritin for later use. When the iron is ready to exit the cell, it is first oxidized back to ferric iron by a ferroxidase, and exits through ferroportin (FPN). Hepcidin is an effector hormone known to induce degradation of ferroportin and prevent cellular iron efflux. This image was created with Biorender.com.
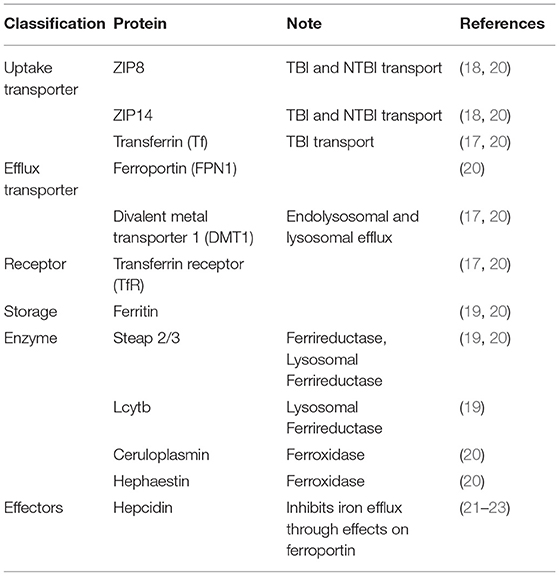
Table 1. Iron metabolism proteins in BMVEC.
The focus of this review is broadly on the relationship between inflammation and iron accumulation in the brain. More specifically, it is to describe knowledge about the mechanisms behind brain iron accumulation that presents in chronic inflammatory disorders. To understand chronic inflammation, acute inflammation must be discussed. Acute inflammation can be activated by changes in body homeostasis such as bacterial infection, viral infection, body injury, or immunological disorders. These occurrences can induce the acute phase response (APR), which is characterized by changes in the concentration of plasma proteins known as acute phase proteins (APPs) (29). Following an immunological trigger, monocytes become activated and release cytokines (IL-6, TNFα, IL-1, IFN-γ, TGF-β). These cytokines induce the synthesis and release of acute phase proteins from hepatocytes in the liver (Figure 2). Of note, IL-6 is known as the most potent cytokine that induces the APR (30, 31). An acute phase protein is any plasma protein that increases or decreases by 25% during inflammation (29). Interestingly, there are several APPs that are associated with iron homeostasis. For example, concentrations of the iron storage protein ferritin and the ferroxidase ceruloplasmin increase during acute phase inflammation. Also, the iron uptake protein transferrin is decreased during this response. Once released, APPs can travel to various tissues and initiate downstream effects relating to an inflammatory response, such as fever, leukocytosis, and muscle breakdown (29). One relevant downstream effect of the APR is a decrease in the serum concentration of iron, or hypoferremia, suggesting that iron is being sequestered within cells and tissues (32). Of high importance, another APP, hepcidin, is a key regulator of this effect due to its ability to prevent iron efflux from cells (33). Indeed, a significant feature of the inflammatory response due to this hepcidin release is iron sequestration in macrophages (34–36), duodenal enterocytes affecting dietary iron absorption (37–39), and in hepatocytes (40), but the effect of inflammation on brain iron sequestration is not well understood (Figure 2). Overall, acute phase inflammation and its relationship to iron homeostasis is relevant to understanding the connection between chronic inflammation and brain iron accumulation.
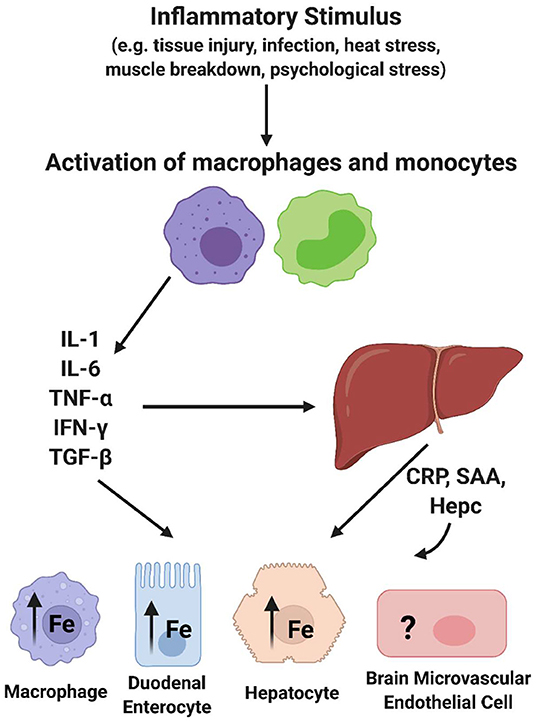
Figure 2. Acute phase response effect on cellular iron trafficking. Essentially all of the cytokines released upon activation of macrophages and monocytes trigger changes in most if not all tissues with impact on their iron trafficking. In addition, several of the acute phase proteins released from hepatocytes (APP) have a sustaining effect on these processes. Hepcidin (Hepc) has the specific role of modulating the steady-state level of the sole iron efflux transporter, Ferroportin (FPN), in a cell's plasma membrane, thus regulating cell iron efflux. This image was created with Biorender.com.
Chronic inflammation is characterized as slow, long-term inflammation that lasts for several months to years. This long-term inflammation can be due to failing to remove the agent causing acute inflammation, such as a virus or bacterial infection, autoimmune disorders, such as rheumatoid arthritis, or chronic diseases that involve an inflammatory response (41). Features of acute phase inflammation continue as the inflammation becomes chronic, specifically vasodilation, increased barrier permeability, and activation of monocytes and resulting release of pro-inflammatory cytokines. This helps contribute to the goal of chronic inflammation, which is infiltration of the primary inflammatory cells into the tissue site, production of inflammatory cytokines and growth factors, and resulting tissue damage and/or repair (42). Chronic inflammatory diseases are the leading cause of mortality worldwide and therefore the biggest threat to human health (43). Highly prevalent chronic inflammatory disorders include diabetes, stroke, asthma, coronary heart disease, and allergies. Neurodegenerative disorders often present with chronic inflammation, and therefore are also considered chronic inflammatory disorders (41). Several studies have shown increased expression of pro-inflammatory cytokines in neurodegenerative disorders including Alzheimer's Disease (AD), Multiple Sclerosis (MS), and Parkinson's disease (PD) (44–48). In the brain, chronic inflammation is mediated by microglia and astrocytes, which respond to signals from the systemic circulation and subsequently produce inflammatory stimuli themselves (Table 2). Specifically, microglia, which are the macrophages of the brain, become activated when signals from systemic serum are released and cross the BBB (55). Activated microglia release inflammatory signals that activate astrocytes to a reactive phenotype, in which they release several cytokines and factors affecting the brain microvascular endothelial cells and circulating through the brain interstitium (56). This chronic inflammatory activation can perpetuate neuronal damage, neurodegeneration, and subsequent cognitive decline, of which the mechanisms are not yet fully understood. Altogether, inflammation leads to a cascade of events and the inflammatory stimuli involved, when chronic, lead to alterations in iron metabolism in the periphery, and possibly in the brain.

Table 2. Glial cell responses to inflammation in normal aging and neurodegenerative disorders.
Consequences of chronic inflammatory mediators have mainly been studied on the endothelial cells of other known barriers, especially those of the macrovascular endothelium. For example, CRP has been shown to affect macrovascular endothelial cells by inducing cytokine release and upregulating the expression of adhesion molecules, promoting a proinflammatory response and atherosclerosis (57–59). IL-6 also activates macrovascular endothelial cells and increases adhesion molecule expression and chemokine release (60–62). Lastly, TNF cytokine family members increase adhesion molecule expression and chemokine production in macrovascular endothelial cells (63–65). Therefore, it is known that inflammatory signals associated with a chronic inflammatory response alter macrovascular endothelial cells and promote inflammation, but the function of these signals with respect to BIA mediated by BMVECs has not been elucidated.
With normal aging, there is physiological and cognitive deterioration. One theory to explain this has been described as the “free radical theory of aging,” basically stating that with age, there is greater production of reactive oxygen and nitrogen species, activating transcription factors and leading to the generation of pro-inflammatory molecules and a chronic inflammatory state. Developing on this theory, the chronic inflammatory state seen in aging is characterized by local infiltration of macrophages into tissue sites of inflammation, and increased levels of circulating pro-inflammatory cytokines, complement components, and adhesion molecules (66, 67). This chronic inflammatory state is especially displayed in the aged brain, which has been shown in both mouse and human studies. For example, older mice injected with LPS had an exaggerated inflammatory response indicating amplified neuroinflammation compared to young mice (68). Also, the brains of aged mice have greater levels of IL-1, IL-6, and reduced expression of the anti-inflammatory cytokines IL-10 and IL-4 (49, 69–71). Of importance, when primed with LPS, old mice had an increased inflammatory response in the hippocampus compared to younger cohorts which was accompanied by neurobehavioral complications. The same group found that hippocampal processing in these old mice was more easily disrupted than in younger ones when the peripheral innate immune system was stimulated (50). This indicates that inflammation may have negative downstream effects on cognition in aged brains. In the aging brain, glial cells respond in a pro-inflammatory manner. Microglia in the aged brain become primed to respond to inflammation (Table 2). They have greater MHC II expression, complement proteins, integrins and toll-like receptors, all allowing for a stronger pro-inflammatory reaction (72, 73). Also, microglia morphology changes with aging, as the cells become smaller with less process ramifications and less motility, allowing them to stay at inflammatory sites longer (74). Controversial questions still remain about whether aged, primed microglia have a better ability to respond to neuroinflammation or if their change in morphology is more reflective of senescence and dystrophy (75–77). Aged microglia release factors that can activate astrocytes to a reactive A1 phenotype, including TNFα, C1q, and IL1α (56). The aging induced A1-like reactive astrocyte phenotype is characterized by loss of normal astrocyte functions such as promoting neuron survival and synapse formation. Most importantly, A1 astrocytes become strongly neurotoxic and have the ability to rapidly kill neurons in the aged brain (78). These inflammatory alterations in the brain likely contribute to the neurodegeneration that takes place with normal aging.
Aging-induced inflammation contributes to another important phenomenon, anemia of inflammation. Anemia of inflammation (AI) can occur in patients with infection, sepsis, chronic inflammatory disorders, or in normal aging, in which it is called anemia of aging. As mentioned, prevalence of anemia increases with age, and is accompanied by rises in expression of pro-inflammatory markers, including CRP and IL-6 (79, 80). AI is characterized by decreased serum iron concentrations despite normal amounts of the iron storage protein ferritin, representing iron stores. Alterations in the expression of acute phase proteins is a defining feature of AI. First, the acute phase protein hepcidin plays a highly important role in this anemia. As mentioned previously, in an inflammatory state, there are greater hepcidin levels, which inhibits iron efflux by effects on the iron efflux protein ferroportin. A hallmark of AI is hepcidin-mediated hypoferremia that is induced by increased expression of cytokines. The concentration of serum transferrin, another acute phase protein, decreases in AI (81). How this affects brain iron accumulation is unknown, but a lack of transferrin would likely induce an increase in ZIP-mediated iron uptake, although more research is needed to confirm this. Overall, much is understood about systemic changes that occur with anemia of inflammation and aging, but little is known about what happens with respect to iron uptake and accumulation in the brain.
Unbalanced brain iron concentrations can result in neuronal death and subsequently, a loss of cognitive and motor function. Iron concentrations increase during the aging process in different human brain regions. As determined with MRI methodology, this iron is most strongly concentrated in areas of the basal ganglia, specifically in the substantia nigra and the striatum (82) (Table 3). In the substantia nigra, increases in different molecular forms of iron are found with aging, including total iron, heavy and light chain ferritin, and the neuromelanin-iron complex (16, 83, 103). In mice, similarly to humans, age-associated increases in iron particularly in the white matter tracts of basal ganglia regions were found. This was accompanied by glia dystrophy, removing the ability of the astrocytes and microglia to sequester iron and prevent neurotoxicity (104).
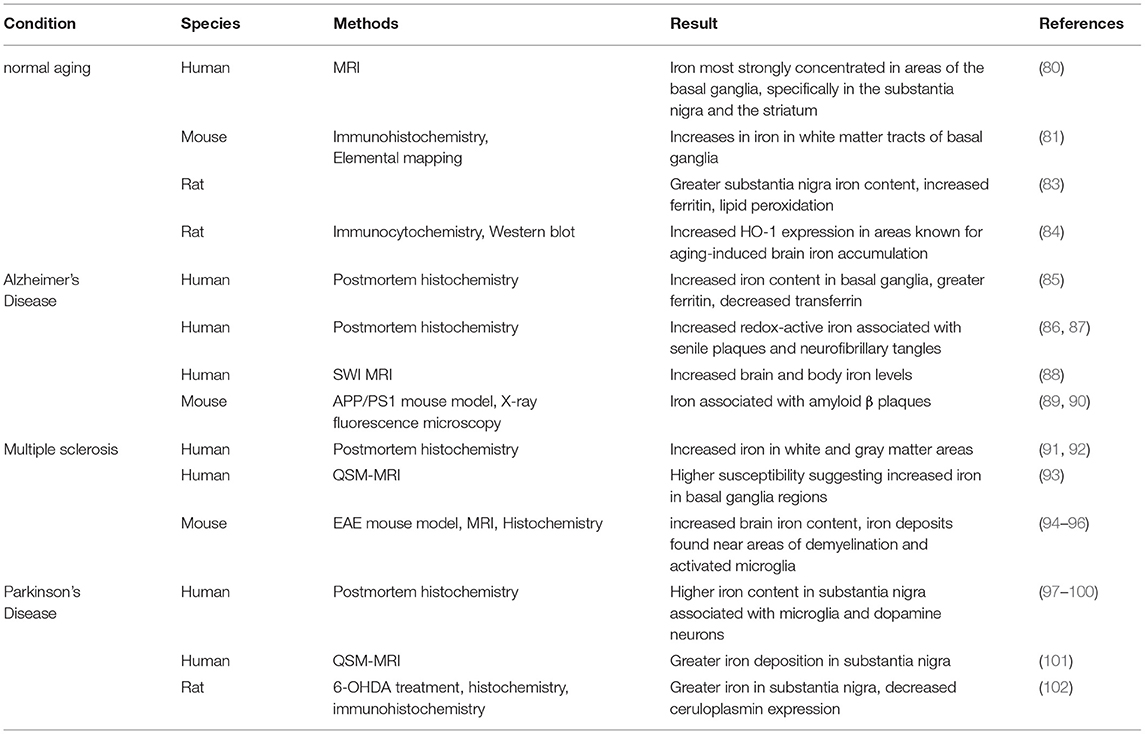
Table 3. Evidence of brain iron accumulation in aging and neurodegenerative disorders.
Studies focused on the accumulation of iron in neuromelanin-containing organelles have provided needed analytical evidence of the co-morbidity of brain iron accumulation in normal aging and associated with neurodegeneration. Using analytical transmission electron microscopy and nano-secondary ion mass spectroscopy, Biesemeier et al. demonstrated that iron was present at ~0.15 mol percent in these organelles, three-fold greater than for copper, for example (84). Neuromelanin has promise as a bio-marker of Parkinson's disease (105) and given the association of iron with this autophagic marker of catecholamine oxidation, it can be captured by MRI. However, one difficulty moving forward is to distinguish between the normal increase in neuromelanin with age and a co-morbid change associated with disease on-set (106). While not useful for the detection or treatment of disease, there are two new approaches that can provide higher resolution in the effort to localize the sites of brain iron accumulation. One of these is Scanning Transmission X-ray Microscopy (STXM). In this technique, at a given energy, the absorbed photon knocks an element's 1s electron into the continuum; for iron, this is known as the K-edge, and it differs for ferrous (Fe2+) in comparison to ferric (Fe3+) (107). Furthermore, given the plane polarized nature of light—including X-rays—the ‘optical activity' in the iron's electronic structure can be exposed. In this way, Telling et al. could dissect the redox state and differential coordination chemistry of iron in amyloid plaques in a mouse model of Alzheimer's Disease (108) and in post-mortem tissue from Alzheimer's Disease patients (107). Another developing technique for the high-resolution (~1–2 μm) quantification of metal ions is microParticle Induced X-ray Emission (μPIXE) (109). Rather than an X-photon as in STXM, μPIXE uses a proton (the “particle”) to couple with an energy transition in the metal ion. Using this approach, Friedrich et al. showed that in mid-brain (substantia nigra) sections from controls, oligodendroglial and astroglial cells were iron-rich, whereas in sections from Parkinson's disease patients, iron was increased throughout except for these two cell types (110). In addition, this neuronal iron increase appeared to be localized in the cytosol indicating an increase in the labile iron pool. An obvious extension of these experiments is the comparison between the quantitative neuronal iron increase revealed by the μPIXE approach and delineation of the redox state and coordination derived from the STXM measurement.
A question of how increased iron accumulates in the aging brain still persists. In aged mice, increased expression of the iron transporter DMT1 have been found (51, 111). Interestingly, in aged mice, expression of the transferrin receptor decreases (51). Decreases in the proteins of the transferrin uptake pathway take place in both aging and inflammation, suggesting again that ZIP-mediated transport may be more important for iron accumulation in these states. One noteworthy study injected young and aged rats with LPS and measured the amount of iron in the substantia nigra, the brain region most affected by Parkinson's Disease. Aged mice had greater substantia nigra iron content, along with increased ferritin expression, microglia activation, and lipid peroxidation (52). This study shows that inflammation may play a role in the brain iron accumulation that is seen in aging. Expression of heme oxygenase 1 (HO-1) increases with age. HO-1 is an enzyme that degrades heme, releasing several molecules including carbon monoxide and ferrous iron (Fe2+) (53, 54). Higher levels of systemic inflammation in aging can increase the expression of HO-1 in the brain, an increase that is co-morbid with cognitive impairment (112). Therefore, upregulation of HO-1 may be a component of brain iron accumulation with inflammation that is seen in aged populations. Another factor that can contribute to brain iron accumulation with aging is increased expression of abluminal hepcidin. In aged mice, hepcidin increases in the cortex. This results in decreased surface expression of ferroportin, and an increased cytosolic non-heme iron that would result from the expected knock down of ferrous iron efflux (113). This may result in iron accumulation in any of the various cell types encompassing the cortex. Therefore, increased expression of hepcidin may contribute to a greater brain iron retention associated with aging. This retention would predict a decrease in efflux of iron from the brain, into either the cerebral-spinal fluid, or by an abluminal to systemic flux of iron via the microcapillary endothelium of the blood brain barrier. Thus, the mechanisms for age-related increases of iron content are not clear, but clearly relevant to the accumulation of dysregulated iron in the aging brain.
Unhealthy aging, as in neurodegenerative disorder presentation, is accompanied by elements of an inflammatory response. As stated earlier, cytokine expression is upregulated in neurodegenerative disorders. For example, CSF collected from MS patients showed greater expression of IL-6 and TNFα compared to patients with non-inflammatory neurological diseases (114). Also, evidence of an abundance of macrophages and CD3+ T cells in MS brain tissue samples, including in patients with progressive disease, indicates continuous activation of the innate and adaptive immune system throughout prolongation of the disease (115). Systematic meta-analysis of several AD patient CSF cytokine studies revealed heightened expression of TGF-β and MCP-1 in AD patients compared to controls, suggesting that these cytokines could be used as biomarkers for this disease (85). Studies have found increased levels of pro-inflammatory cytokines, such as TNFα, IL-1β, and IL-6, in the nigrostriatal region of postmortem brains and in the CSF of patients with PD (86, 87). Serum samples taken from PD patients display amplified levels of monocytes, neutrophils, leukocytes, and CRP expression, suggesting the presentation of a heightened inflammatory response (116). Inflammatory mediator expression is a well known aspect of neurodegenerative disease and may be an upstream explanation for neurodegeneration and cognitive decline.
Glial cell activation and perpetuation of inflammatory response is prominent in neurodegenerative disorders. In the postmortem substantia nigra of PD patients, significant microglial activation has been found, correlating to increased MHC-II expression (91) (Table 2). MHC-II expressing microglia were discovered in the substantia nigra of a rat model of PD, along with CD4+ and CD8+ T cells, suggesting these microglia are in an environment where they are primed to present Parkinson's related antigens for T cell recognition, furthering disease pathogenesis (92). Microglia activation, release of cytokines, and induction of neuron death is associated with AD pathology. Aβ production and failure to clear this protein are key components in the development of AD. As this disease progresses, persistent microglia activation and release of cytokines and ROS increases Aβ generation and decreases clearance of this harmful protein (97, 117). These examples of glial cell mediated inflammation advocate for the idea that glial cells may play a prominent role describing mechanisms of inflammation induced brain iron accumulation.
Several studies with mice have made clear that inflammation can have an important effect on neurodegenerative disease pathogenesis. In a study examining AD, treating wild type ICR mice with LPS resulted in memory impairment as shown by the passive avoidance and the water maze tests. These cognitive deficits were accompanied by Aβ generation in the cortex and hippocampus and greater neuron death when treating with LPS (98). Similar results were seen when giving wild type C57BL/6J mice LPS for a longer period of time, confirming that long-term chronic inflammation can perpetuate AD pathophysiology and cognitive impairment (99). Another study investigating how systemic inflammation can result in neurodegeneration found that one systemic injection of LPS into adult wild type mice can produce increased levels of brain TNFα for 10 months. Researchers in this group found that this increase in brain TNFα was accompanied by activated microglia in the cortex, hippocampus, and substantia nigra. Lastly, they discovered a loss of dopamine neurons that did not become apparent until 7 months after the initial LPS treatment (100). This study illustrates that sustained neuroinflammation induced by a systemic stimulus can be a source of harm to neurons and can attenuate PD disease progression. Overall, several lines of evidence state that neurodegenerative disorders present with systemic and neuroinflammation, but mechanisms describing the effects of inflammation on disease progression are still lacking and need continued research.
One possible pathway to the neurodegeneration seen in neurodegenerative disorders is increased brain iron accumulation (Table 3). Increased disordered iron in brain tissue can result in greater production of ROS and neuronal death. Research in this area has been conducted using human postmortem tissue samples, human brain MRI, and animal models. Elevated iron levels in postmortem AD brains was first discovered in 1953 (88). Several studies following confirmed this, and added that iron colocalizes with the senile Aβ plaques and neurofibrillary tangles associated with this disorder (93, 118–120). Histopathological studies on MS patients have also been completed, finding increased brain iron accumulation in both gray and white matter regions (101, 121). One specific study found iron in normal controls near oligodendrocytes and myelin fibers, but when demyelination in MS patient lesions was analyzed, iron was more closely associated with microglia, suggesting dysregulation of iron homeostasis in this disease (94). In postmortem PD tissue, increased iron levels are found in the substantia nigra. Specifically, this iron has been detected associating with microglia and dopaminergic neurons, signifying iron deposition in these areas as a contributor to the neuronal death seen in this disease (95, 96, 122, 123). Excess reactive iron becomes associated with neuromelanin within neurons and extracellularly. When these neurons die, neuromelanin remains in the extra neuronal space and stimulates microglial release of neurotoxic factors including TNF-a, IL-6, and nitric oxide. This microglial activation is also stimulated by Aβ-iron complexes, suggesting a pattern of protein aggregation, metal complexing, and induction of inflammation (124). This release of inflammatory factors may function to increase brain iron accumulation through effects on iron transporters and BBB permeability, ultimately aggravating the neuropathology.
fMRI methods have been widely used to confirm alterations in iron concentrations of patients with neurodegenerative disorders. Iron, in general, is the most abundant paramagnetic ion in the brain. Different forms of iron in the brain have varying levels of paramagnetism, altering their magnetic susceptibility, or response to an applied magnetic field. For example, ferric iron stored in ferritin, a main source of iron in the brain, is superparamagnetic. In contrast, although oligodendrocyte-dependent myelin biosynthesis is strongly iron-dependent, much of this iron is the ferrous form and is diamagnetic. Thus, white matter areas of the brain display both dia- and para-magnetism. One important fact to consider is that MRI methods do not yet have a way to distinguish different molecular forms of iron, an area needing further development (102). Another approach is susceptibility weighted imaging. SWI is an MRI technique that uses the paramagnetic property of the iron storage protein ferritin to measure a phase shift relating to the amount of iron in the tissue, has been used to investigate iron in the brains of AD, PD, and MS patients. In all disorders, SWI revealed increased iron content in several brain regions compared to healthy patients (89, 90, 125, 126). QSM-MRI, a more advanced and quantitative method used to measure brain iron in vivo, measures the susceptibility of tissues to paramagnetic species. As noted, many forms of iron are paramagnetic and therefore this MRI method has been accepted as a quantitative method to assess tissue iron levels including iron content in neurodegenerative disordered brains. For example, QSM-MRI has demonstrated increased iron content in MS, PD, and AD brains (127–129).
Animal models of neurodegenerative disorders have been utilized to determine the involvement of altered brain iron metabolism in disease progression. Similar to humans, a mouse model of MS called experimental autoimmune encephalomyelitis (EAE), displays increased brain iron content, measured by both MRI and histochemical techniques. These iron deposits were found near areas of demyelination and activated microglia (130–132). This highlights a possible mechanism for iron accumulation and neurodegeneration in MS, which is that oligodendrocyte death leads to myelin destruction, iron release from dying oligodendrocytes, subsequent oxidative damage and neuron death. Injection with 6-hydroxydopamine (6-OHDA) is a method commonly used to create a mouse model of PD. Using this model, techniques such as elemental bio-imaging, immunohistochemistry, and MRI have been implemented to measure iron levels. Several pieces of evidence from these studies reveals increased iron content specifically in the substantia Nigra in the brains of animals modeling PD (133–135). In this same model, decreases in the iron efflux protein ferroportin and the ferroxidases hephaestin and ceruloplasmin were discovered, suggesting iron sequestration in CNS cells as a mechanism for dysregulated iron homeostasis in PD (136, 137). Lastly, the APP/PS1 mouse model of AD, which displays amyloid plaques closely resembling those of human AD, has been utilized to measure iron accumulation with neurodegeneration. Using this model, increases in microglial-iron retention and iron surrounding Aβ plaques was determined (138, 139). Altogether, there is ample research providing evidence for a relationship between brain iron accumulation and neurodegenerative disorders. Mechanisms for what triggers this iron accumulation need ongoing study to fully understand and prevent neurodegenerative disease propagation.
Much is known separately about the processes of inflammation and iron metabolism in healthy aging and neurodegenerative disease progression. What is still unclear is whether there is a cause and effect relationship between these two phenomena that can explain the cognitive decline that is present in both healthy and diseased aging brains. Considering the information presented above, two mechanisms explaining how brain iron accumulation occurs in states of inflammation will be discussed.
A likely mechanism for increased BIA in an inflammatory state is functional changes in iron uptake and efflux proteins (Figure 3). Specific inflammatory mediators are known to upregulate expression of iron metabolism related proteins. Also, a noteworthy study revealed that treating hippocampal neurons, cortical astrocytes, and cortical microglia with IL-6, TNFα, and LPS increases expression of DMT1 and decreases expression of Fpn1. The changes in these proteins was correlated to greater iron uptake into the hippocampal neurons and cortical microglia (140). Transgenic mice expressing IL-6 in astrocytes present with increased iron deposition in the cerebellum, greater ferritin expression, and decreased transferrin expression (141). Lastly, microglia are known to take up NTBI in response to LPS, which occurs through increased DMT1 expression (142). This research demonstrates that inflammatory mediators have the ability to alter iron-related protein synthesis and resulting iron accumulation, although the mechanisms behind this are not known. The decrease in FPN expression in response to inflammation begs the question of how would abluminal iron efflux from BMVECs occur. Stated before, inflammation induced hepcidin would likely decrease expression of FPN in BMVECs. One study found that mouse astrocytes overexpressing hepcidin reduced BMVEC expression of TfR1 and FPN, suggesting reduced iron flux across the BBB (143). Although this study lacks functional determination of iron flux across BMVEC and determination of non-canonical iron accumulation pathways, it highlights the ability of hepcidin to affect proteins involved with iron accumulation. Another report provided further insight, showing that glia grown in contact with BMVEC released hepcidin that knocked down BMVEC FPN-dependent iron efflux (144). Certainly, as inflammation is known to increase hepcidin, more research is needed to determine if inflammation increases or decreases iron flux across the BBB. While this information is provided in these cell types, research into how IL-6, and other significant inflammatory signals, affect iron uptake transporters in BMVECs is lacking. As mentioned previously, brain iron accumulation must occur at the BMVECs of the BBB, therefore it is extremely essential to understand how iron transporter trafficking is affected in this cell type.
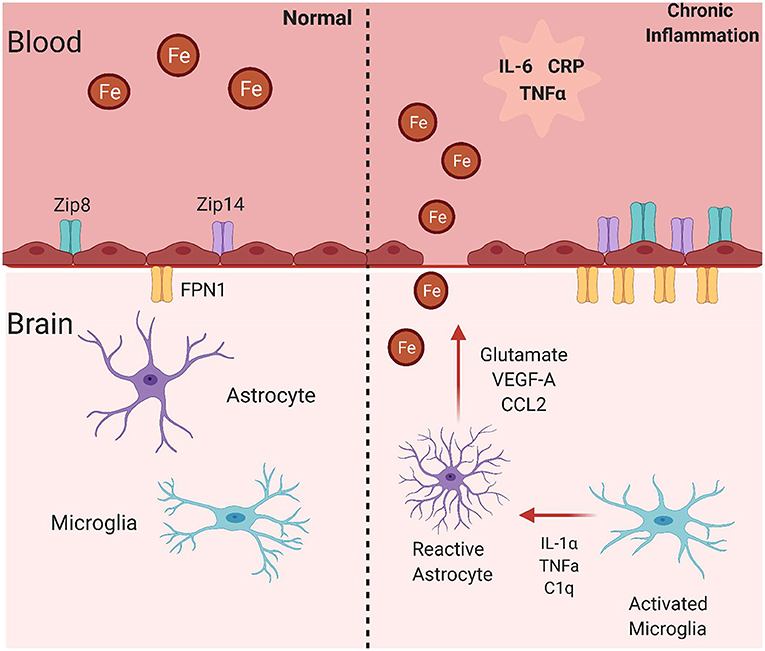
Figure 3. Mechanisms for brain iron accumulation in a state of chronic inflammation. In a state of chronic inflammation, systemic inflammatory factors increase plasma membrane iron transporter occupancy. These include the ferrous iron uptake transporters, ZIP8 and ZIP14, and the sole iron efflux transporter Fpn. Note that these changes occur at both the apical (blood) and basolateral (brain) side of the endothelial cell blood-brain barrier in response to systemic and abluminal signals. This image was created with Biorender.com.
As noted throughout this article, iron uptake into BMVECs is mostly mediated through the ZIP proteins, which stands for zinc-regulated, iron-regulated transport proteins (Figure 3). The ZIP protein family is very large, but ZIP8 and ZIP14 are the most homologous with 50% amino acid similarity (145). Structurally, ZIP proteins have 8 transmembrane domains, extracellular N- and C-termini, and 4 intracellular loops. The metal binding site is within transmembrane domain 5 and is conserved as HEXPHEXGD, except for in ZIP8 and ZIP14 in which the first histidine is changed to a glutamic acid. This is predicted to broaden the metal ion substrate range to include iron, zinc, manganese, and cadmium, unlike other ZIP proteins (145). Both of these proteins are expressed (145, 146). Both ZIP8 and ZIP14 are expressed in BMVEC, as shown by qPCR and immunologic analyses. Data from the same group demonstrated that LPS induces localization of both ZIP8 and ZIP14 to the BMVEC cell surface plasma membrane, showing that inflammation can alter localization of iron transporters (147). As LPS induces downstream cytokine release, it is likely that cytokines such as IL-6 and TNFα have similar effects on these proteins in BMVECs, but this hypothesis has not been tested. Research from multiple groups has revealed that IL-6 can increase expression of the both uptake proteins in liver hepatocytes and SH-SY5Y cells, a neuronal cell line (148, 149). Also, ZIP14 is known as the primary transporter facilitating zinc uptake in hepatocytes, which is regulated by the cytokine IL-1β (150). It has also been discovered that LPS treatment increases ZIP14 mRNA and protein expression, which was correlated to decreased serum zinc and increased liver zinc levels (151). As serum hypozincemia and hypoferremia both occur with inflammation, it is possible that inflammation is leading to ZIP14-mediated brain iron accumulation, as in the liver. Most research on inflammation and the ZIP proteins focuses on ZIP14 because of its direct relationship to IL-6, but ZIP8 can also be affected by inflammatory stimuli. ZIP8 transcript upregulation and amplified zinc uptake occurs in response to TNFα treatment in primary human lung epithelia and human immortalized lung epithelial cells (152). Treating synovial fibroblasts with TNFα and IL-1β induced higher zinc concentrations and greater expression of both ZIP8 and ZIP14, which was determined to be dependent on NF-κB signaling downstream of HIF-1α activation (153). ZIP8 expression is induced by the cytokine, IFN-γ, in intestinal epithelial cells, further propagating the changes in metal homeostasis seen in Crohn's Disease (154). This accumulation of evidence relates inflammation and the ZIP proteins to processes in numerous cell types, tissues, and diseases. This suggests that inflammation may regulate the ZIP proteins in the context of the brain, allowing for greater brain iron acquisition, oxidative stress, neuronal damage, and ultimately cognitive decline.
Changes in BBB permeability in response to inflammatory stimuli may be a significant mechanism describing BIA with inflammation (Figure 3). Permeability of the BBB refers to how well materials are able to cross, with increased permeability meaning that the barrier is less intact and more accessible to substance transport. Reviewed earlier, the BBB is composed of a neurovascular unit encompassing BMVECs, astrocytes, microglia, and neurons. Barrier permeability is controlled by tight junctions (TJ) and adherens junctions (AJ) which form between the BMVECs of the BBB. When these junctions are strong and intact, the brain's protection from systemic toxins is increased, but when they are broken down the brain is exposed to materials from the rest of the body. TJ and AJ of the cellular cytoskeleton are composed of multiple proteins converging to control permeability. TJ proteins highly expressed in BMVECs comprise claudin-3, claudin-5, occludin, and zona occludens 1 (ZO-1). AJ proteins expressed in BMVECs include VE-cadherin and β-catenin (155). Organization of these junctions in BMVECs is shown in Figure 4. Junctional proteins in many different cell types can be greatly affected by inflammation and various disease states. IL-6 receptor trans-signaling in human umbilical vein endothelial cells (HUVECs) results in a loss of ZO-1 and VE-cadherin localization, determined to be induced by STAT phosphorylation of these proteins (156). IL-6 incubated cerebral capillary endothelial cells (cEND) express less claudin-5, occludin, and VE-cadherin, and have reduced barrier functionality (157). Evidence demonstrates that the acute phase protein CRP plays a role in barrier breakdown. CRP disrupted TJ rearrangement and function of a co culture BBB model by activating a kinase that phosphorylates the myosin-light chain (158) (Table 4). Also, the blood retinal barrier (BRB) was disrupted by CRP, due to reduced expression of ZO-1 and occludin in retinal pigment epithelium (159). Oxidative stress and ROS can lead to BBB breakdown, as actin is glutathionylated in this cellular state. Actin glutathionylation limits the rate and extent of actin polymerization, in turn affecting TJ protein arrangement (160, 161). BMVECs treated with TNFα express less VE-Cadherin, occludin, and claudin-5. This correlated to increased expression of NADPH-oxidase subunits, which was shown to be responsible for the loss of TJ proteins and highlights the effect of oxidative stress on BBB integrity (162). Lastly, synergistic effects of both IL-6 and TNFα can affect BBB through decreased expression of ZO-1, claudin-5, occludin, and VE-Cadherin (163). Evidence presented above establishes that systemic inflammatory factors and oxidative stress can induce endothelial cell barrier breakdown, but factors from within the brain can have a similar effect.
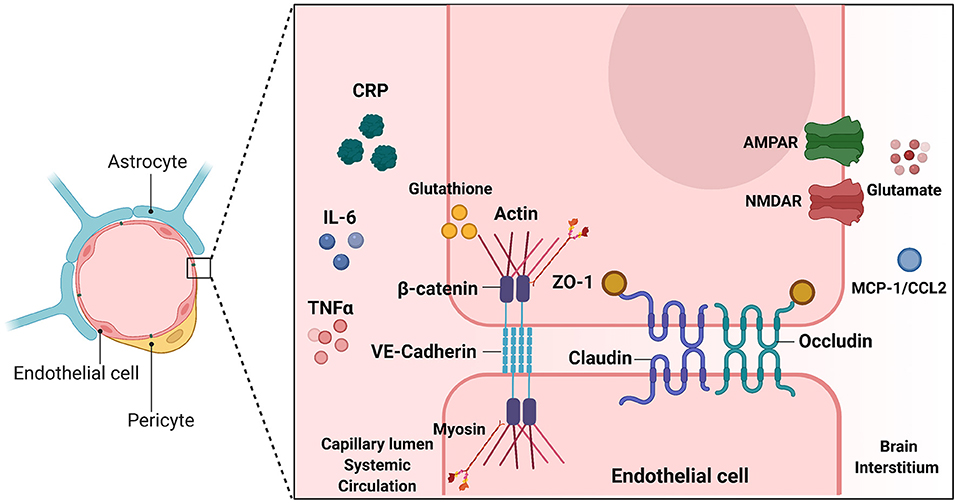
Figure 4. Endothelial junctions at the Blood Brain Barrier. Illustration depicting tight junctions and adherens junctions in brain microvascular endothelial cells of the BBB. Inducers of BBB breakdown are shown both in the systemic circulation and brain interstitium. TJ and AJ proteins are depicted between the two endothelial cells. Inducers shown on either side of the BBB make modifications such as phosphorylation, or glutathionylation, that have downstream effects on TJ/AJ localization and expression. This alters BBB integrity and barrier leakiness. This image was created with Biorender.com.
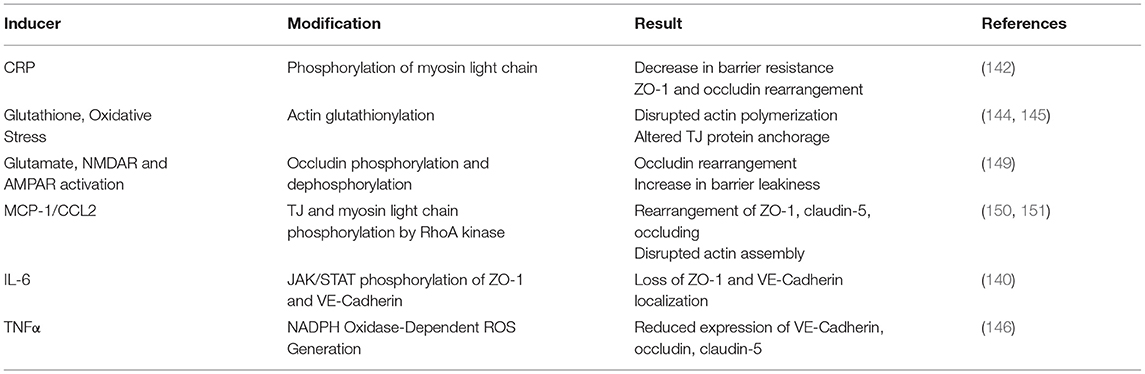
Table 4. Cytoskeletal modifications leading to changes in TJ/AJ function and barrier permeability.
Reactive astrocytes release factors that can have harmful effects on the BBB. In normal conditions, astrocytes take up glutamate from BMVECs, but in an inflammatory state, this function is disrupted and instead they release glutamate (164). Activation of both the NMDA and AMPA receptors is involved in glutamate-induced BBB breakdown. When glutamate binds to the NMDA receptor on BMVECs, Src kinases are activated, which phosphorylate tyrosine residues of occludin. When glutamate binds to the AMPA receptor, a PKCη phosphatase is activated which dephosphorylates serine and threonine residues of occludin. In both cases, occludin is removed from normal positioning, allowing for greater barrier leakiness (165). CCL2, or MCP-1, a chemokine released by astrocytes in inflammatory conditions, facilitates breakdown of TJ and migration of leukocytes across the BBB through changes in expression and localization of ZO-1, claudin-5, and occludin (166, 167). Astrocyte-derived vascular endothelial growth factor A (VEGF-A) breaks down the BBB by targeting claudin-5 and occludin, discovered using VEGF-A KO astrocytes (168). Overall, signals coming from both the systemic circulation and the brain interstitium can alter BBB integrity (Table 4).
The BBB is compromised in many diseases of neuroinflammation. In inflammatory diseases, a more permeable barrier would be harmful because macrophages and T cells harboring antigens to the disease would more easily pass through into the brain. This, in turn, activates glial cells, induces cytokine release, and propagates neuroinflammation. In HIV encephalitis, a complication of HIV associated with cognitive dysfunction, BBB integrity is lost. In this disease, this allows for accumulation of activated, HIV-infected macrophages and induction of reactive astrocytes, the mechanism behind this being significant loss of the TJ proteins occludin and ZO-1 (169). Systemic inflammatory pain rat models, including injection of formalin, carrageenan, and complete Freund's adjuvant (CFA) display increased BBB permeability. In both the carrageenan and CFA injected rats, this was due to decreased expression of ZO-1. Carrageenan and CFA inflammatory pain models are considered longer-term than the formalin model, suggesting that BBB breakdown is induced with chronic inflammation (170). Loss of BBB integrity is an integral aspect of neurodegenerative disorders. In EAE mice, BBB breakdown increases with disease severity, encompassing disruption of ZO-1 and actin in the spinal cord and BMVECs of the BBB (171). Results of this study correlate to what has been discovered in humans with MS, demonstrating significant BBB defects in this disease (172–174). BBB abnormalities have also been discovered in AD cases. Leakage of plasma proteins into the brain that associate with senile plaques was found in postmortem AD brains, indicating increased BBB permeability (175, 176). Also, an MRI technique called dynamic contrast-enhanced MRI (DCE-MRI), found increased extravasation of a gadolinium bolus into the CSF of AD patients compared to controls, again indicating BBB breakdown in this disease (177). Overall, many lines of evidence confirm alterations in the BBB protein architecture in inflammatory conditions and suggest a relationship between inflammation, BBB integrity, and cognitive decline. What has not been examined is if these changes in BBB permeability can lead to increased brain iron accumulation. This finding would link the inflammation presented in neurodegenerative disorders to brain iron accumulation, downstream oxidative stress, and cognitive decline, an important connection to be made.
Several different approaches can be taken to treat BIA induced by inflammation. Targets include oxidative stress, iron itself, and inflammatory mediators. Antioxidants have shown promise as therapeutics for the oxidative stress presented in healthy aging and neurodegenerative disease. For example, N-acetyl cysteine (NAC), a precursor to cysteine known to amplify glutathione levels, increased the levels of antioxidants, decreased the amount of prooxidants, and decreased expression of IL-6, TNFα, and IL1β in aging rat brains (178). Long term oral administration of NAC markedly reduced dopaminergic neuronal loss, oxidative stress, and motor abnormalities in a mouse model of PD (179, 180). N-acetyl cysteine had similar effects in human patients with PD, increasing dopamine transporter binding as a biomarker for PD pathology (181). This result demonstrates that antioxidants have therapeutic qualities to prevent the oxidative stress seen with neurodegeneration. N-acetyl cysteine was also discovered to prevent manganese toxicity in SH-SY5Y cells, a dopaminergic cell line, indicating a direct effect of antioxidants on metal toxicity (182). Non-steroidal anti-inflammatory drugs (NSAID) are another possible therapeutic intervention for inflammation-induced BIA. Ibuprofen treatment led to a reduction of plaque-associated microglia and a corresponding attenuation in proinflammatory cytokine levels in brains of Tg2576 mice overexpressing human amyloid precursor protein (APP), a mouse model of AD (183). A conjugate of glutathione and Ibuprofen attenuated AD characteristics, specifically loss of Aβ, in a rat model of AD, suggesting that antioxidant and NSAID together can be used to increase efficacy in neuroprotection (184).
In addition to these off-the-shelf small molecule anti-oxidants are ones designed based on them, eg, idebenone, a congener of ubiquinone or co-enzyme Q (CoQ) (185–187). These are quinone-based molecules with redox properties comparable to ascorbic acid and vitamin C. Although CoQ and idebenone have exhibited some therapeutic promise, eg., as in a Phase III clinical trial of the latter in the treatment of Duchenne muscular dystrophy (188), there are no approvals for use although in some European countries it is available in special cases (189). With respect to the use of idebenone in the treatment of cognitive decline, the results are unclear; an earlier study failed to find any benefit in the treatment of Alzheimer's disease (AD) (190) while a more recent one, employing an idebenone nanorod formulation taken orally, demonstrated efficacy in a mouse model of AD (191). This result indicates that as with any potential therapeutic, efficacy is often more a matter of delivery than function at the cell level.
Iron chelation could be useful to prevent the downstream BIA caused by inflammation. Deferiprone has shown promise in treating PD, reducing iron content in the caudate and dentate nuclei (192). Deferiprone shows efficiency at reducing tau phosphorylation and Aβ accumulation in a rabbit model (193). An issue with iron chelators is that because they must be delivered systemically, they are likely to affect iron levels in the systemic circulation. This would be damaging to elderly populations and those with chronic disease due to the anemia and iron deficiency seen in aging and inflammatory disorders. Treating with an iron chelator would be tackling the secondary harmful event, the iron accumulation, rather than the inflammation itself. A primary limitation of iron chelators such as deferiprone and deferasirox (Exjade@) is their affinity for iron. The stability constants of their complexes with iron exceed 1025, a value that indicates either would sequester essentially all of the iron in a cell or organism. Thus, while useful in the treatment of iron overload as in hemochromatosis (194), they are contraindicated for the management of the progressive iron accumulation downstream of chronic inflammation (195). A cautionary example is in the use of deferiprone for treatment of Friedreich's Ataxia, an autosomal recessive disorder resulting in the reduction of frataxin essential to the function of all iron-containing enzymes in the mitochondria. In this disease, iron accumulates in the mitochondria, a pathology logically addressed by iron chelation. Not surprisingly, deferiprone treatment exacerbates the iron enzyme deficiency, out-competing the cell's iron metabolism machinery for this essential co-factor (196).
Clearly, iron chelation and anti-oxidant approaches have proven frustrating in that the results have been variable, and in some cases, contra-indicating. On the other hand, there is accumulating evidence that a combination therapy may hold promise. For example, combination of deferiprone and N-acetyl cysteine was efficacious in suppression of the brain iron accumulation, mitochondrial dysfunction and reduced dendritic spine density in mice with chronic brain iron overload (197). An excellent argument has been given as to why managing neuronal iron and redox biology is likely to be a productive pharmacologic approach (198). Indeed, recent reports of the efficacy of a quinazolinone derivative, PBT434, in a Phase I trial involving Multiple System Atrophy indicate the potential of such chemical platforms (199). PBT434 suppresses α-synuclein toxicity in models of Parkinson's disease (200) and MSA (201). PBT434 has a moderate affinity for both ferrous and ferric iron (stability constants ~107); has a redox potential that supports cell anti-oxidant activity; and exhibits facile equilibration across the BBB without disrupting iron homeostasis in BMVEC (200, 202). In short, PBT434 reflects the chemical properties that appear most desirable in a rationally developed pharmaceutical approach to managing the cell dysfunction associated with BIA.
Ferroptosis is a more recently identified form of iron-dependent cell death. This type of cell death is induced by RAS-selective lethal compounds (RSLs), which were found to be selectively lethal to oncogenic RAS-mutant cell lines. Ferroptosis is divergent from other apoptotic pathways because it is associated with increased levels of ROS and is prevented by iron chelation, suggesting dependence on iron and oxidative stress. Erastin and RSL3, two important RSLs, can induce oxidative, iron dependent cell death (203). Glutathione can prevent this form of cell death, as depletion of a glutathione peroxidase and subsequently glutathione is a mechanism behind ferroptosis induction (204). Deferoxamine, another high-affinity iron chelator, is also efficient at preventing ferroptosis (205). As inflammation can result in oxidative stress, it is possible that inflammation-induced BIA can result in cell death by ferroptosis, but more research is needed to confirm this. If ferroptosis is induced by inflammation, glutathione and iron chelation may work as therapeutics for the resulting BIA.
Of course, all of the above pharmacologic strategies address the consequences of inflammation-dependent BIA and not the inflammatory process nor the mechanism(s) underlying the BIA itself. In regard to the latter, the fact is that the brain's iron content does increase. This can result from an increase in iron trafficking from the circulation at the BBB; a decrease in the rate of diffusive iron efflux at the cerebrospinal fluid barrier; or a combination of these changes in metabolite flux. This review has focused primarily on the latter reflecting the fact that accumulating evidence supports the premise that both transcellular and paracellular, apical to basolateral brain iron uptake is increased in an inflammatory state (Figure 3). This model suggests alternatives to the iron chelation and anti-oxidant approaches, namely, modulation of the activity of the iron efflux protein, FPN, and maintenance of the tight junctions that suppress paracellular blood-to-brain leakage.
A rational way of modulating the functional activity of Fpn is via managing its residence time in the plasma membrane. As noted, FPN membrane occupancy is regulated by its interaction with hepcidin. Systemic hepcidin regulates the function of FPN found at the apical, blood membrane of all cells, including BMVEC; glial cell-secreted hepcidin regulates FPN basolateral localization (Figure 3). Thus, conceptually, a hepcidin antagonist functions to up-regulate apical FPN-dependent BMVEC iron efflux while a basolateral hepcidin agonist would suppress iron efflux into the abluminal space. Both such compounds are in development. For example, a Phase I trial of an antagonist in the treatment of patients with an anemia caused by elevated circulating hepcidin has shown considerable clinical benefit (206). Such treatment increases the flux of iron into circulation from cells, eg, apical iron efflux from BMVEC. With respect to hepcidin “agonists” the most promising are not agonists, per se, but hepcidin-mimics, “mini-hepcidins” (MH) (207, 208); MH are poly-peptides including 7–10 residues of the hepcidin sequence. Several have been used in pre-clinical studies (209, 210) including a recent report of improvement in a mouse model of β-thalassemia major (211). The objective of targeting a hepcidin agonist to the basolateral-localized FPN—indeed, the FPN localized to any membrane facing the brain's interstitium—is its delivery across the BBB. The molecular bases for pursuing this rational pharmaceutic are in hand given the extensive study of both MH and methodologies for accessing the abluminal space.
Clearly, the mechanisms underlying inflammation-induced BIA and its link to neurodegenerative disorders remain a critical area of research in two areas: molecular cell and correlated animal model studies to interrogate the mechanisms underlying the increase in brain iron specifically; and use of these experimental paradigms to develop a mechanistic understanding underlying the efficacy of small molecules that support a remediation of the cell misfunction revealed in these model studies. One objective of this review was to provide likely targets for this future drug discovery. The bottom line is that, at present, there are no governmentally-approved pharmacologic approaches for the treatment of BIA or the associated neuronal pathology.
SR: conceptualization, methodology, validation, investigation, and writing-review and editing. SR and DK: writing-original draft preparation and supervision. DK: project administration, funding acquisition, chronic inflammation, iron trafficking, brain iron, neurodegeneration, aging, and blood-brain barrier. All authors contributed to the article and approved the submitted version.
This work was supported by an award from the National Institutes of Health of the United States of America (Award NS102337) to DK.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
BIA, Brain Iron Accumulation; AD, Alzheimer's Disease; PD, Parkinson's Disease; MSA, Multiple System Atrophy; MS, Multiple Sclerosis; APPs, Acute Phase Proteins; BBB, Blood Brain Barrier; BMVECs, Brain Microvascular Endothelial Cells; TBI, Transferrin Bound Iron; NTBI, non-transferrin bound iron; Tf, Transferrin; TfR, Transferrin Receptor; DMT1, Divalent Metal Transporter 1; FPN, Ferroportin; APR, Acute Phase Response; CRP, C-Reactive Protein; AI, Anemia of Inflammation; HO-1, Heme Oxygenase-1; ROS, Reactive Oxygen Species; SWI, Susceptibility Weighted Imaging; QSM-MRI, Quantitative Susceptibility Mapping MRI; EAE, Experimental Autoimmune Encephalomyelitis; 6-OHDA, 6-hydroxydopamine; HIF-1α, Hypoxia Inducible Factor 1α; TJs, Tight Junctions; AJs, Adherens Junctions; ZO-1, Zona-occludens 1; HUVECs, Human Umbilical Vein Endothelial Cells; BRB, Blood Retinal Barrier; VEGF-A, Vascular Endothelial Growth Factor, A; CFA, Complete Freund's Adjuvant; DCE-MRI, Dynamic Contrast Enhanced MRI; NAC, N-acetyl cysteine; NSAIDS, non-steroidal anti-inflammatory drugs; APP, Amyloid Precursor Protein; RSLs, RAS-selective lethal compounds; MH, Mini Hepcidins.
1. Sartori AC, Vance DE, Slater LZ, Crowe M. The impact of inflammation on cognitive function in older adults: implications for healthcare practice and research. J Neurosci Nurs. (2012) 44:206–17. doi: 10.1097/JNN.0b013e3182527690
2. Walker KA, Gottesman RF, Wu A, Knopman DS, Gross AL, Mosley TH, et al. Systemic inflammation during midlife and cognitive change over 20 years: The ARIC Study. Neurology. (2019) 92:e1256–67. doi: 10.1212/WNL.0000000000007094
3. Bibi F, Yasir M, Sohrab SS, Azhar EI, Al-Qahtani MH, Abuzenadah AM, et al. Link between chronic bacterial inflammation and Alzheimer disease. CNS Neurol Disord Drug Targets. (2014) 13:1140–7. doi: 10.2174/1871527313666140917115741
4. Paouri E, Georgopoulos S. Systemic and CNS Inflammation Crosstalk: Implications for Alzheimer's Disease. Curr Alzheimer Res. (2019) 16:559–574. doi: 10.2174/1567205016666190321154618
5. Cardoso EM, Reis C, Manzanares-Céspedes MC. Chronic periodontitis, inflammatory cytokines, and interrelationship with other chronic diseases. Postgrad Med. (2018) 130:98–104. doi: 10.1080/00325481.2018.1396876
6. Ramos P, Santos A, Pinto NR, Mendes R, Magalhães T, Almeida A. Iron levels in the human brain: a post-mortem study of anatomical region differences and age-related changes. J Trace Elem Med Biol. (2014) 28:13–7. doi: 10.1016/j.jtemb.2013.08.001
7. Daugherty AM, Raz N. Appraising the role of iron in brain aging and cognition: promises and limitations of MRI Methods. Neuropsychol Rev. (2015) 25:272–87. doi: 10.1007/s11065-015-9292-y
8. Weiss G, Ganz T, Goodnough LT. Anemia of inflammation. Blood. (2019) 133:40–50. doi: 10.1182/blood-2018-06-856500
9. Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. (1995) 82:969–74. doi: 10.1016/0378-4274(95)03532-X
10. Levi S, Rovida E. The role of iron in mitochondrial function. Biochim Biophys Acta. (2009) 1790:629–36. doi: 10.1016/j.bbagen.2008.09.008
11. Todorich B, Pasquini JM, Garcia CI, Paez PM, Connor JR. Oligodendrocytes and myelination: the role of iron. Glia. (2009) 57:467–78. doi: 10.1002/glia.20784
12. Puig S, Ramos-Alonso L, Romero AM, Martínez-Pastor MT. The elemental role of iron in DNA synthesis and repair. Metallomics. (2017) 9:1483–500. doi: 10.1039/C7MT00116A
13. Hallgren B, Sourander P. The effect of age on the non-haemin iron in the human brain. J Neurochem. (1958) 3:41–51. doi: 10.1111/j.1471-4159.1958.tb12607.x
14. Drayer B, Burger P, Darwin R, Riederer S, Herfkens R, Johnson GA. MRI of brain iron. AJR Am J Roentgenol. (1986) 147:103–10. doi: 10.2214/ajr.147.1.103
15. Vymazal J, Righini A, Brooks RA, Canesi M, Mariani C, Leonardi M, et al. T1 and T2 in the brain of healthy subjects, patients with Parkinson disease, and patients with multiple system atrophy: relation to iron content. Radiology. (1999) 211:489–95. doi: 10.1148/radiology.211.2.r99ma53489
16. Zecca L, Bellei C, Costi P, Albertini A, Monzani E, Casella L, et al. New melanic pigments in the human brain that accumulate in aging and block environmental toxic metals. Proc Natl Acad Sci U S A. (2008) 105:17567–72. doi: 10.1073/pnas.0808768105
17. Engelen M, Vanna R, Bellei C, Zucca FA, Wakamatsu K, Monzani E, et al. Neuromelanins of human brain have soluble and insoluble components with dolichols attached to the melanic structure. PLoS One. (2012) 7:e48490. doi: 10.1371/journal.pone.0048490
18. Keaney J, Campbell M. The dynamic blood-brain barrier. Febs j. (2015) 282:4067–79. doi: 10.1111/febs.13412
19. Rouault TA. Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat Rev Neurosci. (2013) 14:551–64. doi: 10.1038/nrn3453
20. Kosman DJ. A holistic view of mammalian (vertebrate) cellular iron uptake. Metallomics. (2020) 12:1323–34. doi: 10.1039/d0mt00065e
21. Meng F, Fleming BA, Jia X, Rousek AA, Mulvey MA, Ward DM. Lysosomal iron recycling in mouse macrophages is dependent upon both reductases LcytB and Steap3. Blood Adv. (2022) 22:5609. doi: 10.1182/bloodadvances.2021005609
22. McCarthy RC, Kosman DJ. Mechanisms and regulation of iron trafficking across the capillary endothelial cells of the blood-brain barrier. Front Mol Neurosci. (2015) 8:31. doi: 10.3389/fnmol.2015.00031
23. Sangkhae V, Nemeth E. Regulation of the iron homeostatic hormone hepcidin. Adv Nutr. (2017) 8:126–136. doi: 10.3945/an.116.013961
24. Wang CY, Babitt JL. Hepcidin regulation in the anemia of inflammation. Curr Opin Hematol. (2016) 23:189–97. doi: 10.1097/MOH.0000000000000236
25. Xu Y, Alfaro-Magallanes VM, Babitt JL. Physiological and pathophysiological mechanisms of hepcidin regulation: clinical implications for iron disorders. Br J Haematol. (2021) 193:882–93. doi: 10.1111/bjh.17252
26. Vela D. Hepcidin an emerging and important player in brain iron homeostasis. J Transl Med. (2018) 16:25. doi: 10.1186/s12967-018-1399-5
27. Qian ZM, Ke Y. Hepcidin and its therapeutic potential in neurodegenerative disorders. Med Res Rev. (2020) 40:633–53. doi: 10.1002/med.21631
28. McCarthy RC, Kosman DJ. Mechanistic analysis of iron accumulation by endothelial cells of the BB. Biometals B. (2012) 25:665–75. doi: 10.1007/s10534-012-9538-6
29. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. (1999) 340:448–54. doi: 10.1056/NEJM199902113400607
30. Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. (1998) 101:311–20. doi: 10.1172/JCI1368
31. Mackiewicz A, Schooltink H, Heinrich PC, Rose-John S. Complex of soluble human IL-6-receptor/IL-6 up-regulates expression of acute-phase proteins. J Immunol. (1992) 149:2021–7.
32. Weiss G. Iron metabolism in the anemia of chronic disease. Biochim Biophys Acta. (2009) 1790:682–93. doi: 10.1016/j.bbagen.2008.08.006
33. Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. (2003) 101:2461–3. doi: 10.1182/blood-2002-10-3235
34. Weiss G, Bogdan C, Hentze MW. Pathways for the regulation of macrophage iron metabolism by the anti-inflammatory cytokines IL-4 and IL-13. J Immunol. (1997) 158:420–5.
35. Noyes WD, Bothwell TH, Finch CA. The role of the reticulo-endothelial cell in iron metabolism. Br J Haematol. (1960) 6:43–55. doi: 10.1111/j.1365-2141.1960.tb06216.x
36. Fillet G, Cook JD, Finch CA. Storage iron kinetics. VII. A biologic model for reticuloendothelial iron transport. J Clin Invest. (1974) 53:1527–33. doi: 10.1172/JCI107703
37. Ganz T Hepcidin–a Hepcidin–a regulator of intestinal iron absorption and iron recycling by macrophages. Best Pract Res Clin Haematol. (2005) 18:171–82. doi: 10.1016/j.beha.2004.08.020
38. Lynch S. Influence of infection/inflammation, thalassemia and nutritional status on iron absorption. Int J Vitam Nutr Res. (2007) 77:217–23. doi: 10.1024/0300-9831.77.3.217
39. Cortell S, Conrad ME. Effect of endotoxin on iron absorption. Am J Physiol. (1967) 213:43–7. doi: 10.1152/ajplegacy.1967.213.1.43
40. Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. (2012) 1823:1434–43. doi: 10.1016/j.bbamcr.2012.01.014
42. Feghali CA, Wright TM. Cytokines in acute and chronic inflammation. Front Biosci. (1997) 2:d12–26. doi: 10.2741/A171
43. Global regional and national. Age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. (2018) 392:1736–88. doi: 10.1016/S0140-6736(18)32203-7
44. Hu WT, Howell JC, Ozturk T, Gangishetti U, Kollhoff AL, Hatcher-Martin JM, et al. CSF cytokines in aging, multiple sclerosis, and dementia. Front Immunol. (2019) 10:480. doi: 10.3389/fimmu.2019.00480
45. Reale M, Iarlori C, Gambi F, Feliciani C, Salone A, Toma L, et al. Treatment with an acetylcholinesterase inhibitor in Alzheimer patients modulates the expression and production of the pro-inflammatory and anti-inflammatory cytokines. J Neuroimmunol. (2004) 148:162–71. doi: 10.1016/j.jneuroim.2003.11.003
46. De Lella Ezcurra AL, Chertoff M, Ferrari C, Graciarena M, Pitossi F. Chronic expression of low levels of tumor necrosis factor-alpha in the substantia nigra elicits progressive neurodegeneration, delayed motor symptoms and microglia/macrophage activation. Neurobiol Dis. (2010) 37:630–40. doi: 10.1016/j.nbd.2009.11.018
47. Karpenko MN, Vasilishina AA, Gromova EA, Muruzheva ZM, Miliukhina IV, Bernadotte A. Interleukin-1β, interleukin-1 receptor antagonist, interleukin-6, interleukin-10, and tumor necrosis factor-α levels in CSF and serum in relation to the clinical diversity of Parkinson's disease. Cell Immunol. (2018) 327:77–82. doi: 10.1016/j.cellimm.2018.02.011
48. Alvarez A, Cacabelos R, Sanpedro C, García-Fantini M, Aleixandre M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol Aging. (2007) 28:533–6. doi: 10.1016/j.neurobiolaging.2006.02.012
49. Maher FO, Nolan Y, Lynch MA. Downregulation of IL-4-induced signalling in hippocampus contributes to deficits in LTP in the aged rat. Neurobiol Aging. (2005) 26:717–28. doi: 10.1016/j.neurobiolaging.2004.07.002
50. Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun. (2008) 22:301–11. doi: 10.1016/j.bbi.2007.08.014
51. Lu LN, Qian ZM, Wu KC, Yung WH, Ke Y. Expression of iron transporters and pathological hallmarks of parkinson's and Alzheimer's diseases in the brain of young, adult, aged rats. Mol Neurobiol. (2017) 54:5213–24. doi: 10.1007/s12035-016-0067-0
52. Hunter RL, Liu M, Choi DY, Cass WA, Bing G. Inflammation and age-related iron accumulation in F344 rats. Curr Aging Sci. (2008) 1:112–21. doi: 10.2174/1874609810801020112
53. Fernández-Mendívil C, Luengo E, Trigo-Alonso P, García-Magro N, Negredo P, López MG. Protective role of microglial HO-1 blockade in aging: implication of iron metabolism. Redox Biol. (2021) 38:101789. doi: 10.1016/j.redox.2020.101789
54. Bergeron M, Ferriero DM, Sharp FR. Developmental expression of heme oxygenase-1 (HSP32) in rat brain: an immunocytochemical study. Brain Res Dev Brain Res. (1998) 105:181–94. doi: 10.1016/S0165-3806(97)00169-7
55. Lyman M, Lloyd DG, Ji X, Vizcaychipi MP, Ma D. Neuroinflammation: the role and consequences. Neurosci Res. (2014) 79:1–12. doi: 10.1016/j.neures.2013.10.004
56. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
57. Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation. (2000) 102:2165–8. doi: 10.1161/01.CIR.102.18.2165
58. Verma S, Li SH, Badiwala MV, Weisel RD, Fedak PW, Li RK, et al. Endothelin antagonism and interleukin-6 inhibition attenuate the proatherogenic effects of C-reactive protein. Circulation. (2002) 105:1890–6. doi: 10.1161/01.CIR.0000015126.83143.B4
59. Fichtlscherer S, Rosenberger G, Walter DH, Breuer S, Dimmeler S, Zeiher AM. Elevated C-reactive protein levels and impaired endothelial vasoreactivity in patients with coronary artery disease. Circulation. (2000) 102:1000–6. doi: 10.1161/01.CIR.102.9.1000
60. Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. (1997) 6:315–25. doi: 10.1016/S1074-7613(00)80334-9
61. Watson C, Whittaker S, Smith N, Vora AJ, Dumonde DC, Brown KA. IL-6 acts on endothelial cells to preferentially increase their adherence for lymphocytes. Clin Exp Immunol. (1996) 105:112–9. doi: 10.1046/j.1365-2249.1996.d01-717.x
62. Suzuki M, Hashizume M, Yoshida H, Mihara M. Anti-inflammatory mechanism of tocilizumab, a humanized anti-IL-6R antibody: effect on the expression of chemokine and adhesion molecule. Rheumatol Int. (2010) 30:309–15. doi: 10.1007/s00296-009-0953-0
63. Bradley JR. TNF-mediated inflammatory disease. J Pathol. (2008) 214:149–60. doi: 10.1002/path.2287
64. Munro JM, Pober JS, Cotran RS. Tumor necrosis factor and interferon-gamma induce distinct patterns of endothelial activation and associated leukocyte accumulation in skin of Papio anubis. Am J Pathol. (1989) 135:121–33.
65. Rollins BJ, Yoshimura T, Leonard EJ, Pober JS. Cytokine-activated human endothelial cells synthesize and secrete a monocyte chemoattractant, MCP-1/JE. Am J Pathol. (1990) 136:1229–33.
66. Sarkar D, Fisher PB. Molecular mechanisms of aging-associated inflammation. Cancer Lett. (2006) 236:13–23. doi: 10.1016/j.canlet.2005.04.009
67. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. (1956) 11:298–300. doi: 10.1093/geronj/11.3.298
68. Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, et al. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. Faseb J. (2005) 19:1329–31. doi: 10.1096/fj.05-3776fje
69. Maher FO, Martin DS, Lynch MA. Increased IL-1beta in cortex of aged rats is accompanied by downregulation of ERK and PI-3 kinase. Neurobiol Aging. (2004) 25:795–806. doi: 10.1016/j.neurobiolaging.2003.08.007
70. Ye SM, Johnson RW. Increased interleukin-6 expression by microglia from brain of aged mice. J Neuroimmunol. (1999) 93:139–48. doi: 10.1016/S0165-5728(98)00217-3
71. Ye SM, Johnson RW. An age-related decline in interleukin-10 may contribute to the increased expression of interleukin-6 in brain of aged mice. Neuroimmunomodulation. (2001) 9:183–92. doi: 10.1159/000049025
72. Perry VH, Matyszak MK, Fearn S. Altered antigen expression of microglia in the aged rodent. CNGlia S. (1993) 7:60–7. doi: 10.1002/glia.440070111
73. Sheffield LG, Berman NE. Microglial expression of MHC class II increases in normal aging of nonhuman primates. Neurobiol Aging. (1998) 19:47–55. doi: 10.1016/S0197-4580(97)00168-1
74. Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT. Age-related alterations in the dynamic behavior of microglia. Aging Cell. (2011) 10:263–76. doi: 10.1111/j.1474-9726.2010.00660.x
75. Sheng JG, Mrak RE, Griffin WS. Enlarged and phagocytic, but not primed, interleukin-1 alpha-immunoreactive microglia increase with age in normal human brain. Acta Neuropathol. (1998) 95:229–34. doi: 10.1007/s004010050792
76. Miller KR, Streit WJ. The effects of aging, injury and disease on microglial function: a case for cellular senescence. Neuron Glia Biol. (2007) 3:245–53. doi: 10.1017/S1740925X08000136
77. Harry GJ. Microglia during development and aging. Pharmacol Ther. (2013) 139:313–26. doi: 10.1016/j.pharmthera.2013.04.013
78. Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A. (2018) 115:E1896–905. doi: 10.1073/pnas.1800165115
79. Ershler WB, Keller ET. Age-associated increased interleukin-6 gene expression, late-life diseases, and frailty. Annu Rev Med. (2000) 51:245–70. doi: 10.1146/annurev.med.51.1.245
80. Beghé C, Wilson A, Ershler WB. Prevalence and outcomes of anemia in geriatrics: a systematic review of the literature. Am J Med. (2004) 116:3s−10s. doi: 10.1016/j.amjmed.2003.12.009
81. Nemeth E, Ganz T. Anemia of inflammation. Hematol Oncol Clin North Am. (2014) 28:671–81, vi. doi: 10.1016/j.hoc.2014.04.005
82. Daugherty A, Raz N. Age-related differences in iron content of subcortical nuclei observed in vivo: a meta-analysis. Neuroimage. (2013) 70:113–21. doi: 10.1016/j.neuroimage.2012.12.040
83. Connor JR, Snyder BS, Arosio P, Loeffler DA, LeWitt P. A quantitative analysis of isoferritins in select regions of aged, parkinsonian, and Alzheimer's diseased brains. J Neurochem. (1995) 65:717–24. doi: 10.1046/j.1471-4159.1995.65020717.x
84. Biesemeier A, Eibl O, Eswara S, Audinot JN, Wirtz T, Pezzoli G, et al. Elemental mapping of Neuromelanin organelles of human Substantia Nigra: correlative ultrastructural and chemical analysis by analytical transmission electron microscopy and nano-secondary ion mass spectrometry. J Neurochem. (2016) 138:339–53. doi: 10.1111/jnc.13648
85. Chen X, Hu Y, Cao Z, Liu Q, Cheng Y. Cerebrospinal fluid inflammatory cytokine aberrations in alzheimer's disease, parkinson's disease and amyotrophic lateral sclerosis: a systematic review and meta-analysis. Front Immunol. (2018) 9:2122. doi: 10.3389/fimmu.2018.02122
86. Nagatsu T, Sawada M. Inflammatory process in Parkinson's disease: role for cytokines. Curr Pharm Des. (2005) 11:999–1016. doi: 10.2174/1381612053381620
87. Imamura K, Hishikawa N, Ono K, Suzuki H, Sawada M, Nagatsu T, et al. Cytokine production of activated microglia and decrease in neurotrophic factors of neurons in the hippocampus of Lewy body disease brains. Acta Neuropathol. (2005) 109:141–50. doi: 10.1007/s00401-004-0919-y
88. Goodman L. Alzheimer's disease; a clinico-pathologic analysis of twenty-three cases with a theory on pathogenesis. J Nerv Ment Dis. (1953) 118:97–130. doi: 10.1097/00005053-195308000-00001
89. Zhang J, Zhang Y, Wang J, Cai P, Luo C, Qian Z, et al. Characterizing iron deposition in Parkinson's disease using susceptibility-weighted imaging: an in vivo MR study. Brain Res. (2010) 1330:124–30. doi: 10.1016/j.brainres.2010.03.036
90. Haacke EM, Garbern J, Miao Y, Habib C, Liu M. Iron stores and cerebral veins in MS studied by susceptibility weighted imaging. Int Angiol. (2010) 29:149–57.
91. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. (1988) 38:1285–91. doi: 10.1212/WNL.38.8.1285
92. Cebrián C, Loike JD, Sulzer D. Neuronal MHC-I expression and its implications in synaptic function, axonal regeneration and Parkinson's and other brain diseases. Front Neuroanat. (2014) 8:114. doi: 10.3389/fnana.2014.00114
93. Yamamoto A, Shin RW, Hasegawa K, Naiki H, Sato H, Yoshimasu F, et al. Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer's disease. J Neurochem. (2002) 82:1137–47. doi: 10.1046/j.1471-4159.2002.t01-1-01061.x
94. Haider L, Simeonidou C, Steinberger G, Hametner S, Grigoriadis N, Deretzi G, et al. Multiple sclerosis deep grey matter: the relation between demyelination, neurodegeneration, inflammation and iron. J Neurol Neurosurg Psychiatry. (2014) 85:1386–95. doi: 10.1136/jnnp-2014-307712
95. Morris CM, Edwardson JA. Iron histochemistry of the substantia nigra in Parkinson's disease. Neurodegeneration. (1994) 3:277–82.
96. Sofic E, Paulus W, Jellinger K, Riederer P, Youdim MB. Selective increase of iron in substantia nigra zona compacta of parkinsonian brains. J Neurochem. (1991) 56:978–82. doi: 10.1111/j.1471-4159.1991.tb02017.x
97. Cai Z, Hussain MD, Yan LJ. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer's disease. Int J Neurosci. (2014) 124:307–21. doi: 10.3109/00207454.2013.833510
98. Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. (2008) 5:37. doi: 10.1186/1742-2094-5-37
99. Kahn MS, Kranjac D, Alonzo CA, Haase JH, Cedillos RO, McLinden KA, et al. Prolonged elevation in hippocampal Aβ and cognitive deficits following repeated endotoxin exposure in the mouse. Behav Brain Res. (2012) 229:176–84. doi: 10.1016/j.bbr.2012.01.010
100. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. (2007) 55:453–62. doi: 10.1002/glia.20467
101. Williams R, Buchheit CL, Berman NE, LeVine SM. Pathogenic implications of iron accumulation in multiple sclerosis. J Neurochem. (2012) 120:7–25. doi: 10.1111/j.1471-4159.2011.07536.x
102. Möller HE, Bossoni L, Connor JR, Crichton RR, Does MD, Ward RJ, et al. Iron, myelin, and the brain: neuroimaging meets neurobiology. Trends Neurosci. (2019) 42:384–401. doi: 10.1016/j.tins.2019.03.009
103. Zecca L, Stroppolo A, Gatti A, Tampellini D, Toscani M, Gallorini M, et al. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc Natl Acad Sci U S A. (2004) 101:9843–8. doi: 10.1073/pnas.0403495101
104. Ashraf A, Michaelides C, Walker TA, Ekonomou A, Suessmilch M, Sriskanthanathan A, et al. Regional distributions of iron, copper and zinc and their relationships with glia in a normal aging mouse model. Front Aging Neurosci. (2019) 11:351. doi: 10.3389/fnagi.2019.00351
105. Zucca FA, Vanna R, Cupaioli FA, Bellei C, De Palma A, Di Silvestre D, et al. Neuromelanin organelles are specialized autolysosomes that accumulate undegraded proteins and lipids in aging human brain and are likely involved in Parkinson's disease. NPJ Parkinsons Dis. (2018) 4:17. doi: 10.1038/s41531-018-0050-8
106. Sulzer D, Cassidy C, Horga G, Kang UJ, Fahn S, Casella L, et al. Neuromelanin detection by magnetic resonance imaging (MRI) and its promise as a biomarker for Parkinson's disease. NPJ Parkinsons Dis. (2018) 4:11. doi: 10.1038/s41531-018-0047-3
107. Everett J, Collingwood JF, Tjendana-Tjhin V, Brooks J, Lermyte F, Plascencia-Villa G, et al. Nanoscale synchrotron X-ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer's disease subjects. Nanoscale. (2018) 10:11782–796. doi: 10.1039/C7NR06794A
108. Telling ND, Everett J, Collingwood JF, Dobson J, van der Laan G, Gallagher JJ, et al. Iron biochemistry is correlated with amyloid plaque morphology in an established mouse model of alzheimer's disease. Cell Chem Biol. (2017) 24:1205–15. doi: 10.1016/j.chembiol.2017.07.014
109. Pallon J, De La Rosa N, Elfman M, Kristiansson P, Nilsson EJC, Ros L. A new quantitative X-ray system for micro-PIXE analysis. X-Ray Spectrometry. (2017) 46:319–24. doi: 10.1002/xrs.2779
110. Friedrich I, Reimann K, Jankuhn S, Kirilina E, Stieler J, Sonntag M, et al. Cell specific quantitative iron mapping on brain slices by immuno-microPIXE in healthy elderly and Parkinson's disease. Acta Neuropathol Commun. (2021) 9:47. doi: 10.1186/s40478-021-01145-2
111. Jeong SY, David S. Age-related changes in iron homeostasis and cell death in the cerebellum of ceruloplasmin-deficient mice. J Neurosci. (2006) 26:9810–9. doi: 10.1523/JNEUROSCI.2922-06.2006
112. Schipper HM, Bennett DA, Liberman A, Bienias JL, Schneider JA, Kelly J, et al. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol Aging. (2006) 27:252–61. doi: 10.1016/j.neurobiolaging.2005.01.016
113. Sato T, Shapiro JS, Chang HC, Miller RA, Ardehali H. Aging is associated with increased brain iron through cortex-derived hepcidin expression. Elife. (2022) 11. doi: 10.7554/eLife.73456
114. Maimone D, Gregory S, Arnason BG, Reder AT. Cytokine levels in the cerebrospinal fluid and serum of patients with multiple sclerosis. J Neuroimmunol. (1991) 32:67–74. doi: 10.1016/0165-5728(91)90073-G
115. Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. (2000) 47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
116. Jin H, Gu HY, Mao CJ, Chen J, Liu CF. Association of inflammatory factors and aging in Parkinson's disease. Neurosci Lett. (2020) 736:135259. doi: 10.1016/j.neulet.2020.135259
117. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. (2008) 28:8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008
118. Connor JR, Snyder BS, Beard JL, Fine RE, Mufson EJ. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer's disease. J Neurosci Res. (1992) 31:327–35. doi: 10.1002/jnr.490310214
119. Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. (1997) 94:9866–8. doi: 10.1073/pnas.94.18.9866
120. Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci. (1998) 158:47–52. doi: 10.1016/S0022-510X(98)00092-6
121. Craelius W, Migdal MW, Luessenhop CP, Sugar A, Mihalakis I. Iron deposits surrounding multiple sclerosis plaques. Arch Pathol Lab Med. (1982) 106:397–9.
122. Berg D, Hochstrasser H. Iron metabolism in Parkinsonian syndromes. Mov Disord. (2006) 21:1299–310. doi: 10.1002/mds.21020
123. Lhermitte J, Kraus WM, McAlpine D. Original papers: on the occurrence of abnormal deposits of iron in the brain in parkinsonism with special reference to its localisation. J Neurol Psychopathol. (1924) 5:195–208. doi: 10.1136/jnnp.s1-5.19.195
124. Rao KS, Hegde ML, Anitha S, Musicco M, Zucca FA, Turro NJ, et al. Amyloid beta and neuromelanin–toxic or protective molecules? the cellular context makes the difference. Prog Neurobiol. (2006) 78:364–73. doi: 10.1016/j.pneurobio.2006.03.004
125. Gao L, Jiang Z, Cai Z, Cai M, Zhang Q, Ma Y, et al. Brain iron deposition analysis using susceptibility weighted imaging and its association with body iron level in patients with mild cognitive impairment. Mol Med Rep. (2017) 16:8209–15. doi: 10.3892/mmr.2017.7668
126. Modica CM, Zivadinov R, Dwyer MG, Bergsland N, Weeks AR, Benedict RH. Iron and volume in the deep gray matter: association with cognitive impairment in multiple sclerosis. AJNR Am J Neuroradiol. (2015) 36:57–62. doi: 10.3174/ajnr.A3998
127. Zivadinov R, Tavazzi E, Bergsland N, Hagemeier J, Lin F, Dwyer MG, et al. Brain iron at quantitative MRI is associated with disability in multiple sclerosis. Radiology. (2018) 289:487–96. doi: 10.1148/radiol.2018180136
128. Tiepolt S, Rullmann M, Jochimsen TH, Gertz HJ, Schroeter ML, Patt M, et al. Quantitative susceptibility mapping in β-Amyloid PET-stratified patients with dementia and healthy controls—A hybrid PET/MRI study. Eur J Radiol. (2020) 131:109243. doi: 10.1016/j.ejrad.2020.109243
129. Chen Q, Chen Y, Zhang Y, Wang F, Yu H, Zhang C, et al. Iron deposition in Parkinson's disease by quantitative susceptibility mapping. BMC Neurosci. (2019) 20:23. doi: 10.1186/s12868-019-0505-9
130. Nathoo N, Agrawal S, Wu Y, Haylock-Jacobs S, Yong VW, Foniok T, et al. Susceptibility-weighted imaging in the experimental autoimmune encephalomyelitis model of multiple sclerosis indicates elevated deoxyhemoglobin, iron deposition and demyelination. Mult Scler. (2013) 19:721–31. doi: 10.1177/1352458512460602
131. Forge JK, Pedchenko TV, LeVine SM. Iron deposits in the central nervous system of SJL mice with experimental allergic encephalomyelitis. Life Sci. (1998) 63:2271–84. doi: 10.1016/S0024-3205(98)00512-8
132. LeVine SM. Iron deposits in multiple sclerosis and Alzheimer's disease brains. Brain Res. (1997) 760:298–303. doi: 10.1016/S0006-8993(97)00470-8
133. Hare DJ, George JL, Grimm R, Wilkins S, Adlard PA, Cherny RA, et al. Three-dimensional elemental bio-imaging of Fe, Zn, Cu, Mn and P in a 6-hydroxydopamine lesioned mouse brain. Metallomics. (2010) 2:745–53. doi: 10.1039/c0mt00039f
134. Wang J, Jiang H, Xie JX. [Correlation between iron levels and degeneration of dopaminergic neurons in rat nigrostriatal system during the early 6-OHDA lesions in medial forebrain bundle]. Sheng Li Xue Bao. (2003) 55:422–7.
135. Virel A, Faergemann E, Orädd G, Strömberg I. Magnetic resonance imaging (MRI) to study striatal iron accumulation in a rat model of Parkinson's disease. PLoS One. (2014) 9:e112941. doi: 10.1371/journal.pone.0112941
136. Wang J, Jiang H, Xie JX. Ferroportin1 and hephaestin are involved in the nigral iron accumulation of 6-OHDA-lesioned rats. Eur J Neurosci. (2007) 25:2766–72. doi: 10.1111/j.1460-9568.2007.05515.x
137. Wang J, Bi M, Xie J. Ceruloplasmin is involved in the nigral iron accumulation of 6-OHDA-lesioned rats. Cell Mol Neurobiol. (2015) 35:661–8. doi: 10.1007/s10571-015-0161-2
138. Holland R, McIntosh AL, Finucane OM, Mela V, Rubio-Araiz A, Timmons G, et al. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav Immun. (2018) 68:183–96. doi: 10.1016/j.bbi.2017.10.017
139. James SA, Churches QI, de Jonge MD, Birchall IE, Streltsov V, McColl G, et al. Iron, copper, and zinc concentration in Aβ plaques in the APP/PS1 mouse model of alzheimer's disease correlates with metal levels in the surrounding neuropil. ACS Chem Neurosci. (2017) 8:629–37. doi: 10.1021/acschemneuro.6b00362
140. Urrutia P, Aguirre P, Esparza A, Tapia V, Mena NP, Arredondo M, et al. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J Neurochem. (2013) 126:541–9. doi: 10.1111/jnc.12244
141. Castelnau PA, Garrett RS, Palinski W, Witztum JL, Campbell IL, Powell HC. Abnormal iron deposition associated with lipid peroxidation in transgenic mice expressing interleukin-6 in the brain. J Neuropathol Exp Neurol. (1998) 57:268–82. doi: 10.1097/00005072-199803000-00008
142. McCarthy RC, Sosa JC, Gardeck AM, Baez AS, Lee CH, Wessling-Resnick M. Inflammation-induced iron transport and metabolism by brain microglia. J Biol Chem. (2018) 293:7853–63. doi: 10.1074/jbc.RA118.001949
143. Zhang X, Gou YJ, Zhang Y, Li J, Han K, Xu Y, et al. Hepcidin overexpression in astrocytes alters brain iron metabolism and protects against amyloid-β induced brain damage in mice. Cell Death Discov. (2020) 6:113. doi: 10.1038/s41420-020-00346-3
144. McCarthy RC, Kosman DJ. Glial cell ceruloplasmin and hepcidin differentially regulate iron efflux from brain microvascular endothelial cells. PLoS One. (2014) 9:e89003. doi: 10.1371/journal.pone.0089003
145. Jenkitkasemwong S, Wang CY, Mackenzie B, Knutson MD. Physiologic implications of metal-ion transport by ZIP14 and ZIP8. Biometals. (2012) 25:643–55. doi: 10.1007/s10534-012-9526-x
146. Wang CY, Jenkitkasemwong S, Duarte S, Sparkman BK, Shawki A, Mackenzie B, et al. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J Biol Chem. (2012) 287:34032–43. doi: 10.1074/jbc.M112.367284
147. Steimle BL, Smith FM, Kosman DJ. The solute carriers ZIP8 and ZIP14 regulate manganese accumulation in brain microvascular endothelial cells and control brain manganese levels. J Biol Chem. (2019) 294:19197–208. doi: 10.1074/jbc.RA119.009371
148. Liuzzi JP, Lichten LA, Rivera S, Blanchard RK, Aydemir TB, Knutson MD, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc Natl Acad Sci U S A. (2005) 102:6843–8. doi: 10.1073/pnas.0502257102
149. Fujishiro H, Yoshida M, Nakano Y, Himeno S. Interleukin-6 enhances manganese accumulation in SH-SY5Y cells: implications of the up-regulation of ZIP14 and the down-regulation of ZnT10. Metallomics. (2014) 6:944–9. doi: 10.1039/C3MT00362K
150. Lichten LA, Liuzzi JP, Cousins RJ. Interleukin-1beta contributes via nitric oxide to the upregulation and functional activity of the zinc transporter Zip14 (Slc39a14) in murine hepatocytes. Am J Physiol Gastrointest Liver Physiol. (2009) 296:G860–7. doi: 10.1152/ajpgi.90676.2008
151. Aydemir TB, Chang SM, Guthrie GJ, Maki AB, Ryu MS, Karabiyik A, et al. Zinc transporter ZIP14 functions in hepatic zinc, iron and glucose homeostasis during the innate immune response (endotoxemia). PLoS One. (2012) 7:e48679. doi: 10.1371/journal.pone.0048679
152. Besecker B, Bao S, Bohacova B, Papp A, Sadee W, Knoell DL. The human zinc transporter SLC39A8 (Zip8) is critical in zinc-mediated cytoprotection in lung epithelia. Am J Physiol Lung Cell Mol Physiol. (2008) 294:L1127–36. doi: 10.1152/ajplung.00057.2008
153. Hatano N, Matsubara M, Suzuki H, Muraki Y, Muraki K. HIF-1α Dependent Upregulation of ZIP8, ZIP14, and TRPA1 Modify Intracellular Zn(2+) accumulation in inflammatory synoviocytes. Int J Mol Sci. (2021) 22:12. doi: 10.3390/ijms22126349
154. Melia JMP, Lin R, Xavier RJ, Thompson RB, Fu D, Wan F, et al. Induction of the metal transporter ZIP8 by interferon gamma in intestinal epithelial cells: potential role of metal dyshomeostasis in Crohn's disease. Biochem Biophys Res Commun. (2019) 515:325–31. doi: 10.1016/j.bbrc.2019.05.137
155. Cong X Kong W> Endothelial tight junctions and their regulatory signaling pathways in vascular homeostasis and disease. Cell Signal. (2020) 66:109485. doi: 10.1016/j.cellsig.2019.109485
156. Alsaffar H, Martino N, Garrett JP, Adam AP. Interleukin-6 promotes a sustained loss of endothelial barrier function via Janus kinase-mediated STAT3 phosphorylation and de novo protein synthesis. Am J Physiol Cell Physiol. (2018) 314:C589–c602. doi: 10.1152/ajpcell.00235.2017
157. Blecharz-Lang KG, Wagner J, Fries A, Nieminen-Kelhä M, Rösner J, Schneider UC, et al. Interleukin 6-mediated endothelial barrier disturbances can be attenuated by blockade of the il6 receptor expressed in brain microvascular endothelial cells. Transl Stroke Res. (2018) 9:631–42. doi: 10.1007/s12975-018-0614-2
158. Kuhlmann CR, Librizzi L, Closhen D, Pflanzner T, Lessmann V, Pietrzik CU, et al. Mechanisms of C-reactive protein-induced blood-brain barrier disruption. Stroke. (2009) 40:1458–66. doi: 10.1161/STROKEAHA.108.535930
159. Molins B, Pascual A, Méndez, Llorenç V, Zarranz-Ventura J, Mesquida M, et al. C-reactive protein isoforms differentially affect outer blood-retinal barrier integrity and function. Am J Physiol Cell Physiol. (2017) 312:C244–53. doi: 10.1152/ajpcell.00057.2016
160. Dalle-Donne I, Giustarini D, Rossi R, Colombo R, Milzani A. Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med. (2003) 34:23–32. doi: 10.1016/S0891-5849(02)01182-6
161. Smith FM, Kosman DJ. Molecular defects in friedreich's ataxia: convergence of oxidative stress and cytoskeletal abnormalities. Front Mol Biosci. (2020) 7:569293. doi: 10.3389/fmolb.2020.569293
162. Rochfort KD, Collins LE, Murphy RP, Cummins PM. Downregulation of blood-brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: consequences for interendothelial adherens and tight junctions. PLoS ONE. (2014) 9:e101815. doi: 10.1371/journal.pone.0101815
163. Rochfort KD, Collins LE, McLoughlin A, Cummins PM. Tumour necrosis factor-α-mediated disruption of cerebrovascular endothelial barrier integrity in vitro involves the production of proinflammatory interleukin-6. J Neurochem. (2016) 136:564–72. doi: 10.1111/jnc.13408
164. Vesce S, Rossi D, Brambilla L, Volterra A. Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int Rev Neurobiol. (2007) 82:57–71. doi: 10.1016/S0074-7742(07)82003-4
165. Andras IE, Deli MA, Veszelka S, Hayashi K, Hennig B, Toborek M. The NMDA and AMPA/KA receptors are involved in glutamate-induced alterations of occludin expression and phosphorylation in brain endothelial cells. J Cereb Blood Flow Metab. (2007) 27:1431–43. doi: 10.1038/sj.jcbfm.9600445
166. Song L, Pachter JS. Monocyte chemoattractant protein-1 alters expression of tight junction-associated proteins in brain microvascular endothelial cells. Microvasc Res. (2004) 67:78–89. doi: 10.1016/j.mvr.2003.07.001
167. Paul D, Ge S, Lemire Y, Jellison ER, Serwanski DR, Ruddle NH, et al. Cell-selective knockout and 3D confocal image analysis reveals separate roles for astrocyte-and endothelial-derived CCL2 in neuroinflammation. J Neuroinflammation. (2014) 11:10. doi: 10.1186/1742-2094-11-10
168. Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. (2012) 122:2454–68. doi: 10.1172/JCI60842
169. Dallasta LM, Pisarov LA, Esplen JE, Werley JV, Moses AV, Nelson JA, et al. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am J Pathol. (1999) 155:1915–27. doi: 10.1016/S0002-9440(10)65511-3
170. Huber JD, Witt KA, Hom S, Egleton RD, Mark KS, Davis TP. Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol. (2001) 280:H1241–8. doi: 10.1152/ajpheart.2001.280.3.H1241
171. Bennett J, Basivireddy J, Kollar A, Biron KE, Reickmann P, Jefferies WA, et al. Blood-brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. J Neuroimmunol. (2010) 229:180–91. doi: 10.1016/j.jneuroim.2010.08.011
172. Kirk J, Plumb J, Mirakhur M, McQuaid S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J Pathol. (2003) 201:319–27. doi: 10.1002/path.1434
173. Leech S, Kirk J, Plumb J, McQuaid S. Persistent endothelial abnormalities and blood-brain barrier leak in primary and secondary progressive multiple sclerosis. Neuropathol Appl Neurobiol. (2007) 33:86–98. doi: 10.1111/j.1365-2990.2006.00781.x
174. Plumb J, McQuaid S, Mirakhur M, Kirk J. Abnormal endothelial tight junctions in active lesions and normal-appearing white matter in multiple sclerosis. Brain Pathol. (2002) 12:154–69. doi: 10.1111/j.1750-3639.2002.tb00430.x
175. Wisniewski H.M., Kozlowski PB Evidence for blood-brain barrier changes in senile dementia of the Alzheimer type (SDAT). Ann N Y Acad Sci. (1982) 396:119–29. doi: 10.1111/j.1749-6632.1982.tb26848.x
176. Wisniewski HM, Vorbrodt AW, Wegiel J. Amyloid angiopathy and blood-brain barrier changes in Alzheimer's disease. Ann N Y Acad Sci. (1997) 826:161–72. doi: 10.1111/j.1749-6632.1997.tb48468.x
177. Starr JM, Farrall AJ, Armitage P, McGurn B, Wardlaw J. Blood-brain barrier permeability in Alzheimer's disease: a case-control MRI study. Psychiatry Res. (2009) 171:232–41. doi: 10.1016/j.pscychresns.2008.04.003
178. Garg G, Singh S, Singh AK, Rizvi SI. N-acetyl-l-cysteine attenuates oxidative damage and neurodegeneration in rat brain during aging. Can J Physiol Pharmacol. (2018) 96:1189–96. doi: 10.1139/cjpp-2018-0209
179. Clark J, Clore EL, Zheng K, Adame A, Masliah E, Simon DK. Oral N-acetyl-cysteine attenuates loss of dopaminergic terminals in alpha-synuclein overexpressing mice. PLoS One. (2010) 5:e12333. doi: 10.1371/journal.pone.0012333
180. Berman AE, Chan WY, Brennan AM, Reyes RC, Adler BL, Suh SW, et al. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1-/- mouse. Ann Neurol. (2011) 69:509–20. doi: 10.1002/ana.22162
181. Monti DA, Zabrecky G, Kremens D, Liang TW, Wintering NA, Cai J, et al. N-acetyl cysteine may support dopamine neurons in parkinson's disease: preliminary clinical and cell line data. PLoS One. (2016) 11:e0157602. doi: 10.1371/journal.pone.0157602
182. Maddirala Y, Tobwala S, Ercal N. N-acetylcysteineamide protects against manganese-induced toxicity in SHSY5Y cell line. Brain Res. (2015) 1608:157–66. doi: 10.1016/j.brainres.2015.02.006
183. Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, et al. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci. (2003) 23:7504–9. doi: 10.1523/JNEUROSCI.23-20-07504.2003
184. Pinnen F, Sozio P, Cacciatore I, Cornacchia C, Mollica A, Iannitelli A, et al. Ibuprofen and glutathione conjugate as a potential therapeutic agent for treating Alzheimer's disease. Arch Pharm (Weinheim). (2011) 344:139–48. doi: 10.1002/ardp.201000209
185. Jaber S, Polster BM. Idebenone and neuroprotection: antioxidant, pro-oxidant, or electron carrier? J Bioenerg Biomembr. (2015) 47:111–8. doi: 10.1007/s10863-014-9571-y
186. Suárez-Rivero JM, Pastor-Maldonado CJ, Povea-Cabello S, Álvarez-Córdoba M, Villalón-García I, Munuera-Cabeza M, et al. Coenzyme Q(10) analogues: benefits and challenges for therapeutics. Antioxidants (Basel). (2021) 10:2. doi: 10.3390/antiox10020236
187. Yan A, Liu Z, Song L, Wang X, Zhang Y, Wu N, et al. Idebenone alleviates neuroinflammation and modulates microglial polarization in LPS-Stimulated BV2 Cells and MPTP-induced parkinson's disease mice. Front Cell Neurosci. (2018) 12:529. doi: 10.3389/fncel.2018.00529
188. Buyse GM, Voit T, Schara U, Straathof CSM, D'Angelo MG, Bernert G, et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): a double-blind randomised placebo-controlled phase 3 trial. Lancet. (2015) 385:1748–1757. doi: 10.1016/S0140-6736(15)60025-3
190. Thal LJ, Grundman M, Berg J, Ernstrom K, Margolin R, Pfeiffer E, et al. Idebenone treatment fails to slow cognitive decline in Alzheimer's disease. Neurology. (2003) 61:1498–502. doi: 10.1212/01.WNL.0000096376.03678.C1
191. Huang Y, Ma M, Zhu X, Li M, Guo M, Liu P, et al. Effectiveness of idebenone nanorod formulations in the treatment of Alzheimer's disease. J Control Release. (2021) 336:169–80. doi: 10.1016/j.jconrel.2021.06.024
192. Martin-Bastida A, Ward RJ, Newbould R, Piccini P, Sharp D, Kabba C, et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson's disease. Sci Rep. (2017) 7:1398. doi: 10.1038/s41598-017-01402-2
193. Prasanthi JR, Schrag M, Dasari B, Marwarha G, Dickson A, Kirsch WM, et al. Deferiprone reduces amyloid-β and tau phosphorylation levels but not reactive oxygen species generation in hippocampus of rabbits fed a cholesterol-enriched diet. J Alzheimers Dis. (2012) 30:167–82. doi: 10.3233/JAD-2012-111346
194. Kontoghiorghes GJ, Kontoghiorghe CN. Iron and chelation in biochemistry and medicine: new approaches to controlling iron metabolism and treating related diseases. Cells. (2020) 9:6. doi: 10.3390/cells9061456
195. Nuñez MT, Chana-Cuevas P. New perspectives in iron chelation therapy for the treatment of neurodegenerative diseases. Pharmaceuticals (Basel). (2018) 11:4. doi: 10.3390/ph11040109
196. Goncalves S, Paupe V, Dassa EP, Rustin P. Deferiprone targets aconitase: implication for Friedreich's ataxia treatment. BMC Neurol. (2008) 8:20. doi: 10.1186/1471-2377-8-20
197. Sripetchwandee J, Wongjaikam S, Krintratun W, Chattipakorn N, Chattipakorn SC. A combination of an iron chelator with an antioxidant effectively diminishes the dendritic loss, tau-hyperphosphorylation, amyloids-β accumulation and brain mitochondrial dynamic disruption in rats with chronic iron-overload. Neuroscience. (2016) 332:191–2. doi: 10.1016/j.neuroscience.2016.07.003
198. Ma L, Gholam Azad M, Dharmasivam M, Richardson V, Quinn RJ, Feng Y, et al. Parkinson's disease: alterations in iron and redox biology as a key to unlock therapeutic strategies. Redox Biol. (2021) 41:101896. doi: 10.1016/j.redox.2021.101896
199. Stamler D, Bradbury M, Wong C, Offman E. A first in human study of PBT434, a novel small molecule inhibitor of α-synuclein aggregation (S4.001). Neurology. (2019) 92:S4.
200. Finkelstein DI, Billings JL, Adlard PA, Ayton S, Sedjahtera A, Masters CL, et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson's disease. Acta Neuropathol Commun. (2017) 5:53. doi: 10.1186/s40478-017-0456-2
201. Finkelstein DI, Shukla JJ, Cherny RA, Billings JL, Saleh E,N, Stefanova, et al. The compound ATH434 prevents alpha-synuclein toxicity in a murine model of multiple system atrophy. J Parkinsons Dis. (2021) 21:2877. doi: 10.3233/JPD-212877
202. Bailey DK, Clark W, Kosman DJ. The iron chelator, PBT434, modulates transcellular iron trafficking in brain microvascular endothelial cells. PLoS One. (2021) 16:e0254794. doi: 10.1371/journal.pone.0254794
203. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
204. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
205. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. (2008) 15:234–45. doi: 10.1016/j.chembiol.2008.02.010
206. Renders L, Budde K, Rosenberger C, van Swelm R, Swinkels D. First-in-human Phase I studies of PRS-080#22, a hepcidin antagonist, in healthy volunteers and patients with chronic kidney disease undergoing hemodialysis. PLoS One. (2019) 14:e0212023. doi: 10.1371/journal.pone.0212023
207. Preza GC, Ruchala P, Pinon R, Ramos E, Qiao B, Peralta MA, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest. (2011) 121:4880–8. doi: 10.1172/JCI57693
208. Casu C, Nemeth E, Rivella S. Hepcidin agonists as therapeutic tools. Blood. (2018) 131:1790–794. doi: 10.1182/blood-2017-11-737411
209. Casu C, Oikonomidou PR, Chen H, Nandi V, Ginzburg Y, Prasad P, et al. Minihepcidin peptides as disease modifiers in mice affected by β-thalassemia and polycythemia vera. Blood. (2016) 128:265–76. doi: 10.1182/blood-2015-10-676742
210. Ramos E, Ruchala P, Goodnough JB, Kautz L, Preza GC, Nemeth E, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. (2012) 120:3829–36. doi: 10.1182/blood-2012-07-440743
Keywords: chronic inflammation, iron trafficking, brain iron, neurodegeneration, aging, blood-brain barrier
Citation: Rosenblum SL and Kosman DJ (2022) Aberrant Cerebral Iron Trafficking Co-morbid With Chronic Inflammation: Molecular Mechanisms and Pharmacologic Intervention. Front. Neurol. 13:855751. doi: 10.3389/fneur.2022.855751
Received: 15 January 2022; Accepted: 14 February 2022;
Published: 15 March 2022.
Edited by:
Emel Koseoglu, Erciyes University, TurkeyReviewed by:
Luigi Zecca, National Research Council (CNR), ItalyCopyright © 2022 Rosenblum and Kosman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel J. Kosman, Y2Fta29zQGJ1ZmZhbG8uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.