
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 25 February 2022
Sec. Pediatric Neurology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.845877
This article is part of the Research TopicNeuronal Ceroid Lipofuscinosis: a Multidisciplinary UpdateView all 12 articles
 Marina Trivisano
Marina Trivisano Alessandro Ferretti
Alessandro Ferretti Costanza Calabrese
Costanza Calabrese Nicola PietrafusaLudovica PiscitelloGiusy Carfi' Pavia
Nicola PietrafusaLudovica PiscitelloGiusy Carfi' Pavia Federico Vigevano
Federico Vigevano Nicola Specchio*
Nicola Specchio*Neuronal ceroid lipofuscinoses (NCLs) are a heterogeneous group of neurodegenerative diseases, characterized by progressive cerebral atrophy due to lysosomal storage disorder. Common clinical features include epileptic seizures, progressive cognitive and motor decline, and visual failure, which occur over different time courses according to subtypes. During the latest years, many advances have been done in the field of targeted treatments, and in the next future, gene therapies and enzyme replacement treatments may be available for several NCL variants. Considering that there is rapid disease progression in NCLs, an early diagnosis is crucial, and neurophysiological features might have a key role for this purpose. Across the different subtypes of NCLs, electroencephalogram (EEG) is characterized by a progressive deterioration of cerebral activity with slowing of background activity and disappearance of spindles during sleep. Some types of heterogeneous abnormalities, diffuse or focal, prevalent over temporal and occipital regions, are described in many NCL variants. Photoparoxysmal response to low-frequency intermittent photic stimulation (IPS) is a typical EEG finding, mostly described in CLN2, CLN5, and CLN6 diseases. Visual evoked potentials (VEPs) allow to monitor the visual functions, and the lack of response at electroretinogram (ERG) reflects retinal neurodegeneration. Taken together, EEG, VEPs, and ERG may represent essential tools toward an early diagnosis of NCLs.
Neuronal ceroid lipofuscinoses (NCLs) are a heterogeneous group of autosomal recessive neurodegenerative disorders, characterized by progressive cerebral atrophy due to widespread accumulation of autofluorescent storage material within lysosomes (1–5). NCLs are caused by defective lysosomal processing enzymes or receptors (3–8). So far, 14 variants of NCLs are reported; they share some common clinical features including epileptic seizures, progressive cognitive and motor decline, and visual failure; all those symptoms occur over different time courses according to subtypes (9, 10).
Neuronal ceroid lipofuscinoses represent the most common cause of dementia in children (5, 8, 11), and the incidence varies worldwide from 1:12,500 to 1:100,000 (9).
Neuronal ceroid lipofuscinoses can be often misdiagnosed at the onset because of the appearance of non-specific presenting symptoms; therefore, the diagnosis may be delayed (5).
Clinical management is mostly palliative, although, in the recent years, many efforts have been done to identify targeted treatments, i.e., enzymatic replacement therapy (ERT) and genetic therapy for each of NCL variants (7, 12–16). Nevertheless, a targeted treatment with human recombinant enzyme is currently available only for CLN2 disease (12).
Considering the rapid disease progression (17) and the development of novel targeted treatments, early diagnosis remains crucial. Early diagnosis is also useful for genetic counseling to avoid family odyssey toward different hospitals looking for diagnosis and to avoid repetition of useless investigations (18). Next-generation sequencing techniques are successful in the identification of genetic childhood epilepsies and early detection of CLN2 disease (19). A recent study supported the value of target re-sequencing in patients with genetic childhood epilepsies, suggesting that this technique may be successful in the early detection of patients with CLN2 (19).
In this context, neurophysiological data may have a key role in the diagnosis of NCLs. Electroencephalogram (EEG) and evoked potentials may be of particular importance for this purpose, since they can provide critical information at a relatively early phase of the disease and are easy to carry out in a short time and without excessive costs.
This review aims to provide neurophysiological findings in NCLs, highlighting the neurophysiologic typical features of NCLs as well as what may allow differential diagnosis with developmental and epileptic encephalopathies (DEEs) due to other etiologies.
Two authors (MT and AF) performed a search in PubMed and EMBASE databases, from 1970 to 2021, using the following keywords: “ceroid lipofuscinosis” and “photoparoxysmal response,” “electroencephalogram”, “evoked potentials,” and “electroretinogram.” They looked for experimental and clinical studies and reviews. No meta-analyses were found.
Epilepsy is one of the cardinal symptoms of NCLs (5). The age at epilepsy onset may differ according to the NCLs variant, starting from the first months of life to adulthood; both focal and generalized seizures can be seen at onset (5). Epilepsy may represent the presenting symptom, as it happens in CLN2 and CLN6 diseases, or appear after visual loss or cognitive and motor decline, as in CLN3 and CLN5 diseases (20).
In the classic form of CLN1 disease, symptoms begin during infancy, and seizure onset is relatively early, typically between the age of 14 and 36 months. Additional phenotypes have been observed with late infantile, juvenile, and adult onset. In the latter cases, epilepsy typically begins several years after the initial symptom of visual impairment, following developmental regression and behavioral changes (20). Few data are available on EEG features in CLN1 disease (21, 22). It has been reported that a slowing of background activity and loss of sleep spindles is associated with high-voltage slow waves and spike and waves abnormalities (22). Between 5 and 12 years of age, a progressive flattening of cerebral activity consistent with the marked cortical atrophy due to massive neuronal death is evident (21). In summary, in CLN1 disease, changes of background activity have been distinguished in three stages: (1) decreasing of reactivity of the posterior rhythm to eye opening and closing; (2) decreasing of sleep spindles and subsequent disappearance; and (3) slowing and/or attenuation of EEG to inactivity, the so-called vanishing EEG pattern (22, 23).
In CLN2 disease, a late-infantile neuronal ceroid lipofuscinosis (LINCL), seizures often appear in an explosive fashion (24). Seizures are resistant to common anti-seizure medications (ASMs), and sodium channel blockers, such as carbamazepine, can even worsen seizure frequency and may increase myoclonus and ataxia (24).
The first EEG is usually performed within the fourth year of life and shows a normal background activity, associated with focal or diffuse abnormalities in 75% of the patients. High-voltage focal slow waves are prevalent over the temporal and occipital regions (25–28). Photoparoxysmal response (PPR) to intermittent photic stimulation (IPS) delivered at a low frequency of stimulation is typically seen in patients with CLN2 during the first stage of the disease (27). With disease progression, the background activity becomes slow and without reactivity to eye opening (29, 30). No spindles are present during sleep recordings. Generalized, focal and multifocal epileptiform discharges characterized by irregular spikes and poly-spikes and waves become common; however, a posterior predominance persists (24) (Figures 1A–E).
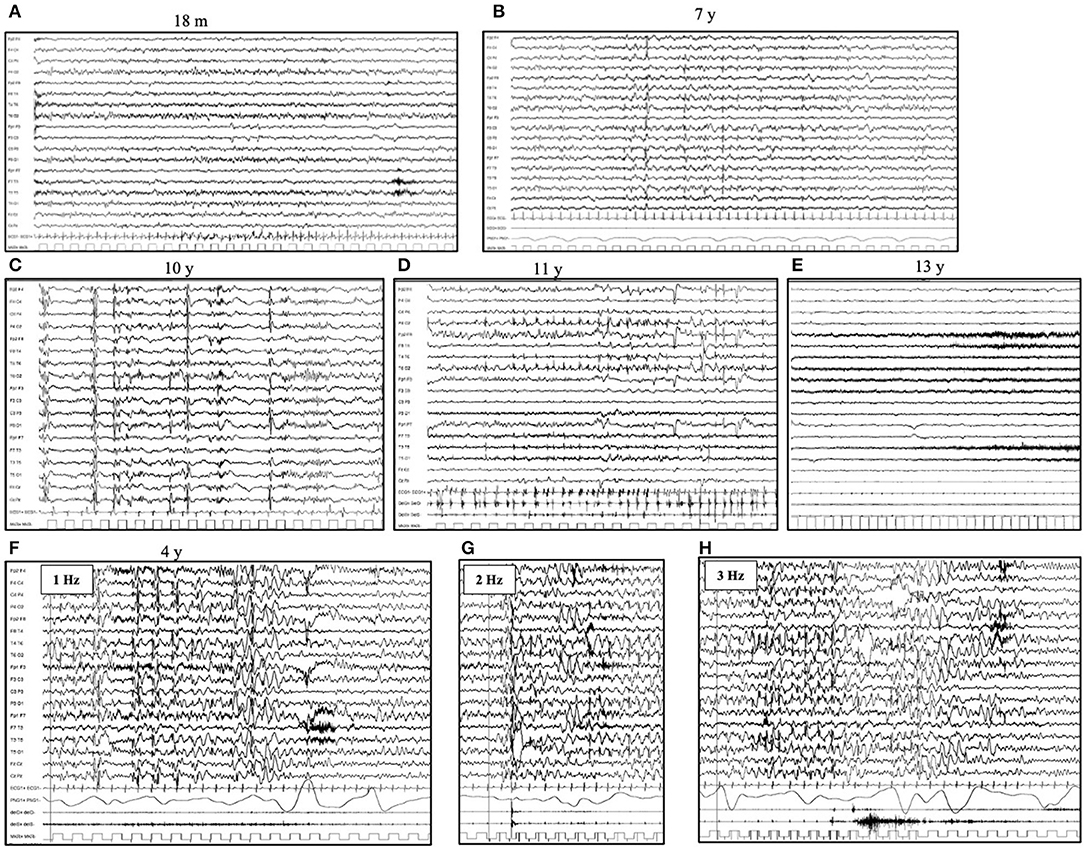
Figure 1. Electroencephalogram (EEG) evolution in CLN2 disease. Awake EEG (A) at the age of 18 months (pre-symptomatic phase) shows a normal background activity, with 8 Hz rhythm over bilateral posterior regions. No epileptiform abnormalities are evident. Awake EEG (B) at the age of 7 years shows a slow background activity and the lack of regional differentiation. There are diffuse and focal (mainly occipital) spikes and spike and waves abnormalities. During sleep (C), the EEG (age of 10 years) shows the lack of sleep spindles and bursts of rapid spikes and poly-spikes, alternated with the suppression of cerebral activity. (D) Myoclonic status can occur in the advanced phases of diseases (age of 11 years) and (E) A diffuse flattening of cerebral activity with an extremely slow and low-voltage cerebral activity is typical of the latter stage. (F–H) Photoparoxysmal response in a 4-year-old girl affected by CLN2 disease. The flash-per-flash response is evident at the lower frequency of intermittent photic stimulation, at 1, 2, and 3 Hz (respectively F–H).
In CLN3 disease, the juvenile neuronal ceroid lipofuscinosis (Spielmeyer–Vogt disease or Batten disease), the presenting manifestation is a progressive visual loss starting around the age of 6–8 years. Epilepsy usually starts at the age of 10 years. Seizures at onset are usually bilateral tonic–clonic, whereas focal seizures increase during adolescence, and they are mostly characterized by clonic manifestations. Myoclonic seizures are rarely reported in CLN3 disease. EEG, in the early stages of the disease, shows focal epileptiform abnormalities (before 10 years of age), whereas bilateral and multifocal epileptiform discharges are significantly more prevalent in the later stages, along with a progressive slowing of the background activity (31). PPR has not been reported as a prominent feature in CLN3 disease (31).
The CLN6 disease may start both during infancy and adulthood. In both cases, epilepsy is one of the presenting symptoms (32). Background activity is poorly organized during awake and sleep since the onset, associated with irregular slow spike and waves discharges (at about 2.5 Hz). PPR to lower frequency is an early neurophysiological finding. During the advanced stage of disease, the cerebral activity became extremely slow, and low-voltage and spike and waves discharge became rare, often replaced by single spikes with multifocal distribution (32).
In adult patients affected by CLN6, the background activity is almost preserved in the early stage of the disease. Epileptiform abnormalities are the most prevalent over the posterior regions. Myoclonus is typically induced by active movements or provoked by IPS at a low frequency of stimulus. Abnormal PPR persists until the advanced stages of the disease (32).
The CLN5 disease, also known as the Finnish variant, starts between the age of 2 and 6 years with clumsiness and mental decline (33, 34). Seizure occurs relatively late (median age of 8 years), and epilepsy has its major expression with myoclonic seizures between the age of 7 and 11 years. In addition, for this LINCL, it has been reported that a progressive slowing of background activity is associated with multifocal epileptiform discharges (spikes, multiple spikes, and spike and wave complexes) (34) and the presence of posterior spikes triggered by low-frequency IPS (33, 34).
Regarding CLN7 disease, in few published patients, it has been reported that a progressive generalized slowing of background activity is associated with diffuse or multifocal abnormalities with occipital prevalence (Figures 2A–C). PPR has not been reported (35, 36).
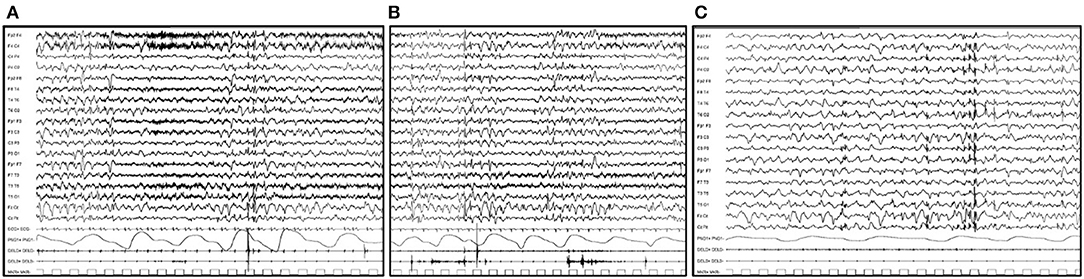
Figure 2. EEG features in a 6-year-old boy affected by CLN7 disease. (A) EEG trace shows a generalized slowing of background activity associated with diffuse or multifocal abnormalities. (B) Single myoclonic jerks involving the upper limbs with the EEG correlate of a diffuse single spike are evident. (C) During sleep, the cerebral activity is markedly slow and is associated with multifocal spike and spike and waves abnormalities, prevalent over bilateral posterior regions.
In patients with CLN8, it has been described as a progressively slowing of background activity with focal and/or generalized abnormalities since the onset. PPR has rarely been reported (37).
Few cases of CLN11 have been reported (38, 39). EEG is characterized by a quite preserved background activity associated with rare paroxysms of diffuse spikes and slow waves, prevalent over bilateral parietal regions. PPR has not been detected in the three cases reported so far (38, 39).
Limited data have been reported on EEG features for the other NCL variants, from CLN9 to CLN14, which are the more recently identified forms. More cases need to be reported to understand their typical neurophysiological findings.
Photoparoxysmal response in NCLs was firstly described in 1970 when it was reported in about 45% of patients (29). Later, other studies confirmed the presence of this peculiar neurophysiological finding (27, 32, 40, 41).
The susceptibility to PPR varies accordingly to the different diseases which are more prominent in the LINCLs, such as CLN2, CLN5, and CLN6 (Figures 1F–H). In CLN2 disease, PPR has been reported from 27 to 93% of patients (25, 27, 40, 41). This wide range may be explained by the lack of a universal approach of low frequency IPS in routine pediatric EEG recordings (24). In fact, in NCLs, as well as in other progressive myoclonic epilepsies, PPR is one of the earliest pieces of evidence of a neurodegenerative disorder, even before the onset of cognitive and motor regression (42). PPR to low-frequency stimulation has also been reported to occur in Lafora disease and mitochondrial diseases (43, 44).
In a recently reported series of 14 cases of CLN2, serial EEGs revealed a PPR in 93% of patients (27). PPR was evident since the first EEG, which was performed at 3.6 (3.1–4.0) years, in 43% of patients; it was documented at low (1–3 Hz) frequencies of stimulation in 69% of patients and has acquired the form of a flash-per-flash response in 69% of patients (27). Moreover, in the advanced stages of the disease, PPR was associated with massive myoclonic jerks (27). In about half of patients (54%), PPR disappears over time (25). PPR changes over time reflect the gray matter changes due to the progression of neurodegenerative disease (27).
The PPR seen in the NCLs is characterized by an occipital spike and waves response to the photic stimuli. However, it should also be highlighted that not in all NCL variants do patients have the same susceptibility to PPR; it has been reported more frequently in CLN2, CLN5, and CLN6 diseases (24, 27, 32, 33) and seems to not be a prominent feature in CLN1, CLN3, CLN8, and CLN11 diseases (21, 31, 37, 38). For the remaining NCLs, we do not have sufficient data about PPR to find out conclusive data.
On the other hand, the lack of the characteristic IPS response cannot rule out an NCL disease (45). The implementation of IPS, including IPS at low frequencies of stimulation as part of a standard EEG, may be useful as an early disease marker if associated with other clinical findings (24, 45, 46).
Few data are available in the literature regarding visual evoked potentials (VEPs) changes during the disease course in NCLs. What is known is that with disease progression, the so-called giant VEPs appear (Figures 3D,E). They are abnormally broad and of high amplitude, and their presence is a marker of cortical hyperexcitability, similar to PPR (32).
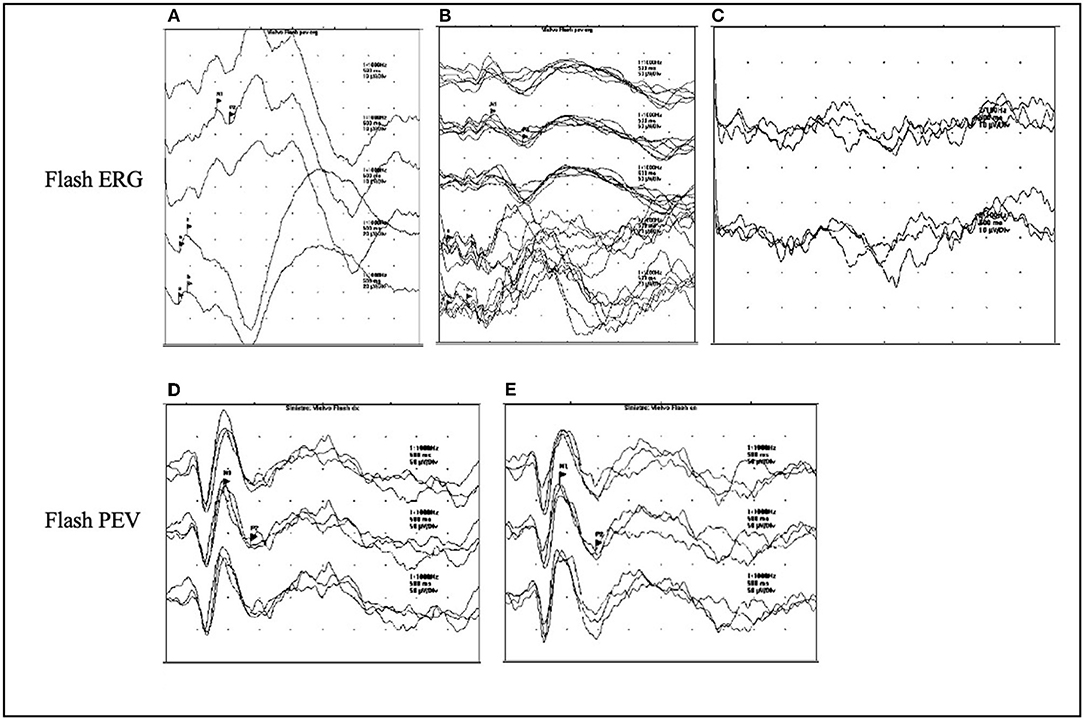
Figure 3. ERG and VEPs in CLN2 disease. (A–C) The ERG flash evolution in a patient affected with CLN2 at the ages of 3.5, 5.5, and 7 years. The response is normal at the onset of disease at the age of 3.5 years (A), at the age of 5.5 years, there is a reduction of the amplitude of ERG (B), which turns to be absent at the age of 7 years (C). (D,E) The presence of giant flash VEPs in the left and right eyes in a 4-year-old patient.
Abnormal VEPs have been found in about 75% of patients with CLN2 at a median age of 4.5 years (25). Furthermore, a more recent study on patients with CLN2 evidenced that most patients (89%) had an early and high-amplitude pattern reversal in VEPs, whereas few patients showed a bifid waveform, which is associated with a central scotoma, indicative of maculopathy or macular pathway dysfunction such as optic atrophy (47).
With further disease progression, the amplitude of VEP decreases, as it happens in other neurodegenerative disorders (48, 49).
The pattern-reversal VEP waveform is preferred to the flash VEP waveform, which shows a wide inter-individual variability. On the contrary, the pattern-reversal VEP has a relatively constant single positive peak throughout life and is a strong index of macular pathway function (47).
Variability of results may be due to the retrospective nature of the studies and the different ages and phases of the disease in which visual tests have been performed (25, 41, 47).
Vision-related problems are one of the cardinal signs of NCLs and, as in CLN3, are often an early sign, appearing prior to motor and mental deterioration (5). This is due to the accumulation of storage material into the retina, leading to its degeneration (50, 51).
Electroretinogram (ERG) represents the tool that allows to monitor the involvement of retina, and its use progressively disappears with disease progression (Figures 3A–C) at different ages according to the NCL variants (50). ERG is currently used to characterize the physiological changes in the degenerating retina in patients affected by NCLs. It allows to identify the retinopathy, which consists of symmetrical cone-rod dystrophy (47, 50).
In patients with CLN2, it has been demonstrated that changes in ERG appear even before the macular disruption on optical coherence tomography (OCT) from the age of 4 years and 10 months (47). Abnormal ERG has been reported in 66% of patients at a median age of 4.5 years (25). However, it is important to underlie that the flattening of ERG does not necessarily imply the total loss of retinal function, if some cortical VEP functions persist (29).
In CLN5 disease, retinal degeneration has been confirmed between the age of 6 and 10 years, when ERG has been found abolished in most of the patients (33, 34).
The use of ERG might be implemented in the future to evaluate the efficacy of experimental treatments with intravitreal therapies.
Somatosensory evoked potentials (SEPs) have been poorly investigated in NCL diseases. There are sporadic cases, across different forms of NCLs (21, 32, 33, 52), where it is highlighted that the presence of high-amplitude evoked potential, giant SEPs, which are the expressions of cortical hyperexcitability due to neuronal degeneration (32).
Giant SEPs are also a typical and specific marker of patients with cortical myoclonus, which is one of the main clinical features of NCLs (38).
Neurophysiological investigations can also be useful to differentiate NCLs from other DEEs. In detail, LINCLs should be differentiated from those DEEs with epilepsy onset between 2 and 6 years of age, mainly characterized by myoclonic seizures. Among these, it is important to consider epilepsy with myoclonic-atonic seizures (EMA) and Lennox–Gastaut syndrome (LGS), which are characterized by epilepsy with multiple types of seizures, intellectual disability, and drug resistance (24).
The EEG is particularly useful for this purpose because, in NCLs, there is a slowing of background activity with progressive loss of differentiation and disappearance of spindles during sleep, whereas in EMA, although with the presence of many epileptiform abnormalities, the background activity is preserved (53). Moreover, a poly-graphic study might highlight that, in EMA, myoclonic events mainly involve proximal, axial muscles, whereas, in NCLs, myoclonus involves mainly distal, segmental muscles (38, 53).
The LGS is otherwise characterized by specific EEG abnormalities, which are diffuse spike-and-slow wave complexes at 2.5 Hz during awake, and poly-spikes during sleep (54). In both syndromes, PPR is rarely reported and, when described, it is not induced by low-frequency IPS (53, 54).
On the other hand, PPR is reported in some epileptic syndromes of infancy, and among these, The Dravet syndrome (DS) is most frequent (55). Different from the majority of NCLs, in DS, epilepsy starts within the first year of life. In addition, in DS, PPR is not typically induced by low-frequency IPS and does not have the no characteristics of the flash-per-flash PPR (55).
During the last decades, technological advances have driven genetic discovery in epilepsy and increased the understanding of the molecular mechanisms of many epileptic disorders, in some cases providing targets for precision medicine (19, 56). Nevertheless, phenotyping and neurophysiological characterization are still critical for the diagnosis and better management of neurological symptoms.
An appropriate definition of neurophysiological features of NCLs is crucial for the possible role that they may have in the early diagnosis of such diseases. There are some features (Figure 4) that are common to different subtypes of NCLs, such as the progressive slowing of background activity, the disappearance of sleep spindles during sleep, and the presence of some type of heterogeneous abnormalities, such as bursts of diffuse or focal slow waves prevalent over temporal and occipital regions and diffuse spike and wave paroxysmal discharges (25–27).
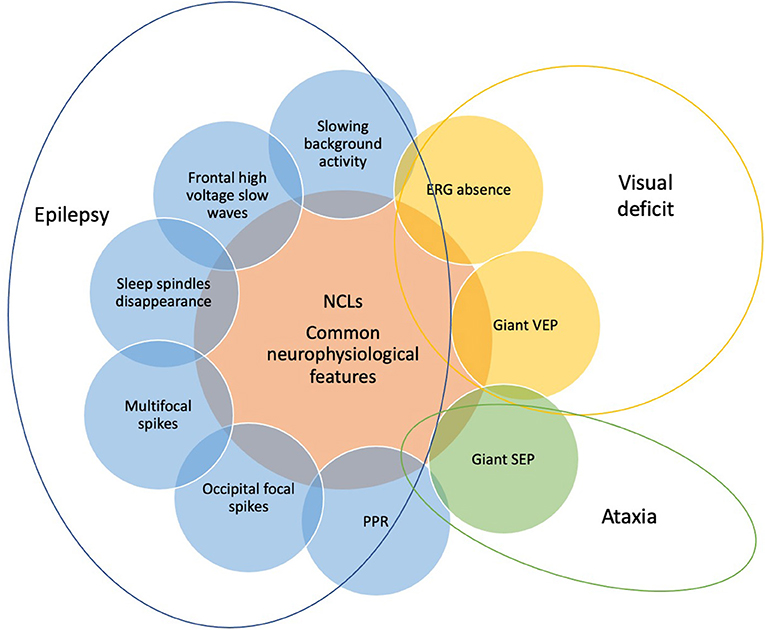
Figure 4. The common neurophysiological features of NCLs include EEG, visual evoked potentials (VEPs), somatosensory evoked potentials (SEPs), and electroretinogram findings. They can vary according to the different subtypes of NCLs and the age of patients. Each neurophysiological finding (slowing background activity, disappearance of sleep spindles, frontal high-voltage slow waves, occipital focal spikes, multifocal spikes, PPR, giant VEPs and SEP, and ERG absence) is the expression of one of the three main symptoms of NCLs, which are epilepsy, visual deficit, and ataxia and allows to monitor their progression.
Sensitivity to low-frequency IPS is a hallmark of neurodegenerative diseases such as NCLs and may be useful as an early marker if associated with other clinical findings (27). This peculiar EEG activation may be missed due to the lack of standardization of the test, which should be implemented with low-frequency stimulation, starting from 1 Hz (27).
The VEPs allow to monitor the visual pathway function, and the lack of visual response at ERG reflects retinal neurodegeneration.
Taken together, EEG, VEPs, and ERG may represent essential tools that can address the clinicians toward an early diagnosis of NCL disease. Although treatment remains essentially symptomatic in NCLs, together with palliative, supportive, and rehabilitative measures, in the near future, ERT and gene therapies may be available and earlier diagnosis will be mandatory. In this context, the use of NGS-based approaches results important for the early identification of patients with NCL, allowing a timely adoption of the most accurate treatment strategies.
MT and AF prepared the first draft of the manuscript. NS and FV coordinated the job and supervised critically the manuscript and reviewed the final version of the manuscript. CC did the reference search and provided support for the discussion. LP and GC prepared the figures and contributed to the figure legends and reviewed critically the manuscript. NP reviewed critically the manuscript and contributed to the final draft. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Platt FM. Emptying the stores: lysosomal diseases and therapeutic strategies. Nat Rev Drug Discov. (2018) 17:133–50. doi: 10.1038/nrd.2017.214
2. Nelvagal HR, Lange J, Takahashi K, Tarczyluk-Wells MA, Cooper JD. Pathomechanisms in the neuronal ceroid lipofuscinoses. Biochim Biophys Acta Mol Basis Dis. (2019) 31:165570. doi: 10.1016/j.bbadis.2019.165570
3. Rakheja D, Bennett MJ. Neuronal ceroid-lipofuscinoses. Transl Sci Rare Dis. (2018) 3:83–95. doi: 10.3233/TRD-180024
4. Glees P, Hasan M. Lipofuscin in neuronal aging and diseases. Norm Pathol Anat (Stuttg). (1976) 32:1–68.
5. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases – clinical perspectives. Biochim Biophys Acta. (2013) 1832:1801–6. doi: 10.1016/j.bbadis.2013.04.008
6. Sondhi D, Crystal RG, Kaminsky SM. Gene Therapy for Inborn Errors of Metabolism: Batten Disease. In: Tuszynski MH, editor. Translational Neuroscience: Fundamental Approaches for Neurological Disorders. Boston, MA: Springer US (2016). p. II 1–129.
7. Mole SE, Anderson G, Band HA, Berkovic SF, Cooper JD, Kleine Holthaus SM, et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. (2019) 18:107–16. doi: 10.1016/S1474-4422(18)30368-5
8. Johnson TB, Cain JT, White KA, Ramirez-Montealegre D, Pearce DA, Weimer JM. Therapeutic landscape for Batten disease: current treatments and future prospects. Nat Rev Neurol. (2019) 15:161–78. doi: 10.1038/s41582-019-0138-8
9. Mole SE, Haltia M. Chapter 70 - The Neuronal Ceroid-Lipofuscinoses (Batten Disease). In: Rosenberg RN, Pascual JM, editors. Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease (Fifth Edition). Boston: Academic Press (2015). p. 793–808. doi: 10.1016/B978-0-12-410529-4.00070-X
10. Nita DA, Mole SE, Minassian BA. Neuronal ceroid lipofuscinoses. Epileptic Disord. (2016) 18:73–88. doi: 10.1684/epd.2016.0844
11. Donsante A, Boulis NM. Progress in gene and cell therapies for the neuronal ceroid lipofuscinoses. Expert Opin Biol Ther. (2018) 18:755–64. doi: 10.1080/14712598.2018.1492544
12. Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, et al. CLN2 Study Group. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N Engl J Med. (2018) 378:1898–907. doi: 10.1056/NEJMoa1712649
13. Specchio N, Ferretti A, Trivisano M, Pietrafusa N, Pepi C, Calabrese C, et al. Neuronal ceroid lipofuscinosis: potential for targeted therapy. Drugs. (2021) 81:101–23. doi: 10.1007/s40265-020-01440-7
14. Desnick RJ, Schuchman EH. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu Rev Genomics Hum Genet. (2012) 13:307–35. doi: 10.1146/annurev-genom-090711-163739
15. Solomon M, Muro S. Lysosomal enzyme replacement therapies: Historical development, clinical outcomes, and future perspectives. Adv Drug Deliv Rev. (2017) 118:109–34. doi: 10.1016/j.addr.2017.05.004
16. Kruer MC, Pearce DA, Orchard PJ, Steiner RD. Prospects for stem cell therapy in neuronal ceroid lipofuscinosis. Regen Med. (2013) 8:527–9. doi: 10.2217/rme.13.46
17. Nickel M, Simonati A, Jacoby D, Lezius S, Kilian D, Vande GraafB, et al. Disease characteristics and progression in patients with late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: an observational cohort study [published correction appears in Lancet Child Adolesc Health. 2018 2:e24]. Lancet Child Adolesc Health. (2018) 2:582–90. doi: 10.1016/S2352-4642(18)30179-2
18. Fietz M, AlSayed M, Burke D, Cohen-Pfeffer J, Cooper JD, Dvoráková L, et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): Expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab. (2016) 119:160–7. doi: 10.1016/j.ymgme.2016.07.011
19. Amadori E, Scala M, Cereda GS, Vari MS, Marchese F, Di Pisa V, et al. Targeted re-sequencing for early diagnosis of genetic causes of childhood epilepsy: the Italian experience from the 'beyond epilepsy' project. Ital J Pediatr. (2020) 46:92. doi: 10.1186/s13052-020-00860-1
20. Augustine EF, Adams HR, de Los Reyes E, Drago K, Frazier M, Guelbert N, et al. Management of CLN1 disease: international clinical consensus. Pediatr Neurol. (2021) 120:38–51. doi: 10.1016/j.pediatrneurol.2021.04.002
21. Simonati A, Tessa A, Bernardina BD, Biancheri R, Veneselli E, Tozzi G, et al. Variant late infantile neuronal ceroid lipofuscinosis because of CLN1 mutations. Pediatr Neurol. (2009) 40:271–6. doi: 10.1016/j.pediatrneurol.2008.10.018
22. Vanhanen SL, Sainio K, Lappi M, Santavuori P. (1997) EEG and evoked potentials in infantile ceroid-lipofuscinosis. Dev Med Child Neurol 39:456-463Jalanko A, Braulke T Neuronal ceroid lipofuscinoses. Biochim Biophys Acta. (2009) 1793:697–709. doi: 10.1016/j.bbamcr.2008.11.004
23. Veneselli E, Biancheri R, Buoni S, Fois A. Clinical and EEG findings in 18 cases of late infantile neuronal ceroid lipofuscinosis. Brain Dev. (2001) 23:306–11. doi: 10.1016/S0387-7604(01)00231-5
24. Johnson AM, Mandelstam S, Andrews I, Boysen K, Yaplito-Lee J, Fietz M, et al. Neuronal ceroid lipofuscinosis type 2: an Australian case series. J Paediatr Child Health. (2020) 56:1210–8. doi: 10.1111/jpc.14890
25. Beltrán L, Valenzuela GR, Loos M, Vargas R, Lizama R, Spinsanti P, et al. Late-onset childhood neuronal ceroid lipofuscinosis: early clinical and electroencephalographic markers. Epilepsy Res. (2018) 144:49–52. doi: 10.1016/j.eplepsyres.2018.05.005
26. Binelli S, Canafoglia L, Panzica F, Pozzi A, Francescheti A. Electroencephalographic features in a series of patients with neuronal ceroid lipofuscinoses. Neurol Sci. (2000) 21:S83. doi: 10.1007/s100720070045
27. Specchio N, Bellusci M, Pietrafusa N, Trivisano M, de Palma L, Vigevano F. Photosensitivity is an early marker of neuronal ceroid lipofuscinosis type 2 disease. Epilepsia. (2017) 58:1380–8. doi: 10.1111/epi.13820
28. Veneselli E, Biancheri R, Perrone MV, Buoni S, Fois A. Neuronal ceroid lipofuscinoses: clinical and EEG findings in a large study of Italian cases. Neurol Sci. (2000) 21(3 Suppl):S75-81. doi: 10.1007/s100720070044
29. Pampiglione G, Harden A. So-called neuronal ceroid lipofuscinosis. J Neurol Neurosurg Psychiatry. (1977) 40:323–30. doi: 10.1136/jnnp.40.4.323
30. Aicardi J, Plouin P, GoutieÂres F. Ceroid-lipofuscinoses. Rev EEG Neurophysiol. (1978) 8:149±159. doi: 10.1016/S0370-4475(78)80130-0
31. Arntsen V, Strandheim J, Helland IB, Sand T, Brodtkorb E. Epileptological aspects of juvenile neuronal ceroid lipofuscinosis (CLN3 disease) through the lifespan. Epilepsy Behav. (2019) 94:59–64. doi: 10.1016/j.yebeh.2019.02.020
32. Canafoglia L, Gilioli I, Invernizzi F, Sofia V, Fugnanesi V, Morbin M, et al. Electroclinical spectrum of the neuronal ceroid lipofuscinoses associated with CLN6 mutations. Neurology. (2015) 85:316–24. doi: 10.1212/WNL.0000000000001784
33. Holmberg V, Lauronen L, Autti T, Santavuori P, Savukoski M, Uvebrant P, et al. Phenotype-genotype correlation in eight patients with Finnish variant late infantile NCL (CLN5). Neurology. (2000) 55:579–81. doi: 10.1212/WNL.55.4.579
34. Simonati A, Williams RE, Nardocci N, Laine M, Battini R, Schulz A, et al. Phenotype and natural history of variant late infantile ceroid-lipofuscinosis 5. Dev Med Child Neurol. (2017) 59:815–21. doi: 10.1111/dmcn.13473
35. Aiello C, Terracciano A, Simonati A, Discepoli G, Cannelli N, Claps D, et al. Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid lipofuscinosis. Hum Mutat. (2009) 30:E530–40. doi: 10.1002/humu.20975
36. Craiu D, Dragostin O, Dica A, Hoffman-Zacharska D, Gos M, Bastian AE, et al. Rett-like onset in late-infantile neuronal ceroid lipofuscinosis (CLN7) caused by compound heterozygous mutation in the MFSD8 gene and review of the literature data on clinical onset signs. Eur J Paediatr Neurol. (2015) 19:78–86. doi: 10.1016/j.ejpn.2014.07.008
37. Striano P, Specchio N, Biancheri R, Cannelli N, Simonati A, Cassandrini D, et al. Clinical and electrophysiological features of epilepsy in Italian patients with CLN8 mutations. Epilepsy Behav. (2007) 10:187–91. doi: 10.1016/j.yebeh.2006.10.009
38. Canafoglia L, Morbin M, Scaioli V, Pareyson D, D'Incerti L, Fugnanesi V, et al. Recurrent generalized seizures, visual loss, and palinopsia as phenotypic features of neuronal ceroid lipofuscinosis due to progranulin gene mutation. Epilepsia. (2014) 55:e56–9. doi: 10.1111/epi.12632
39. Kamate M, Detroja M, Hattiholi V. Neuronal ceroid lipofuscinosis type-11 in an adolescent. Brain Dev. (2019) 41:542–5. doi: 10.1016/j.braindev.2019.03.004
40. Albert DV, Yin H, De Los Reyes EC, Vidaurre J. Unique Characteristics of the Photoparoxysmal Response in Patients With Neuronal Ceroid Lipofuscinosis Type 2: Can EEG Be a Biomarker? J Child Neurol. (2016) 31:1475–82. doi: 10.1177/0883073816658659
41. Dozières-Puyravel B, Nasser H, Elmaleh-Bergès M, Lopez Hernandez E, Gelot A, Ilea A, et al. Paediatric-onset neuronal ceroid lipofuscinosis: first symptoms and presentation at diagnosis. Dev Med Child Neurol. (2020) 62:528–30. doi: 10.1111/dmcn.14346
42. Kasteleijn-Nolst Trenité D, Mastrangelo M, Spalice A, Parisi P. Photosensitivity as an early marker of epileptic and developmental encephalopathies. Epilepsia. (2018) 59:1086–7. doi: 10.1111/epi.14031
43. Roger J, Pellissier JF, Bureau M, Dravet C, Revol M, Tinuper P. Early diagnosis of Lafora disease: significance of paroxysmal visual manifestations and contribution of skin biopsy. Rev Neurol (Paris). (1983) 139:115–24.
44. Canafoglia L, Franceschetti S, Antozzi C, et al. Epileptic phenotypes associated with mitochondrial disorders. Neurology. (2001) 56:1340–6. doi: 10.1212/WNL.56.10.1340
45. Trivisano M, Specchio N. Red flags for neuronal ceroid lipofuscinosis type 2 disease. Dev Med Child Neurol. (2020) 62:414. doi: 10.1111/dmcn.14389
46. Nickel M, Jacoby D, Lezius S, Down M, Simonati A, Genter F, et al. Natural history of CLN2 disease: quantitative assessment of disease characteristics and rate of progression. Neuropediatrics. (2016) 47 (FV04-03). doi: 10.1055/s-0036-1583730
47. Thompson DA, Handley SE, Henderson RH, Marmoy OR, Gissen P. An ERG and OCT study of neuronal ceroid lipofuscinosis CLN2 Battens retinopathy. Eye (Lond). (2021) 35:2438–48. doi: 10.1038/s41433-021-01594-y
48. Sainio K. Neurophysiological findings in neuronal ceroid lipofuscinoses. Neuropediatrics. (1997) 8:70. doi: 10.1055/s-2007-973673
49. Kobayashi K, Hitomi T, Matsumoto R, Kondo T, Kawamata J, Matsuhashi M, et al. Long-term follow-up of cortical hyperexcitability in Japanese Unverricht-Lundborg disease. Seizure. (2014) 23:746–50. doi: 10.1016/j.seizure.2014.06.002
50. Quagliato EMAB, Rocha DM, Sacai PY, Watanabe SS, Salom~ao SR, Berezovsky A. Retinal function in patients with the neuronal ceroid lipofuscinosis phenotype. Arq Bras Oftalmol. (2017) 80:215–9. doi: 10.5935/0004-2749.20170053
51. Goebel HH, Klein H, Santavuori P, Sainio K. Ultrastructural studies of the retina in infantile neuronal ceroid-lipofuscinosis. Retina. (1988) 8:59–66. doi: 10.1097/00006982-198808010-00011
52. Jadav RH, Sinha S, Yasha TC, Aravinda H, Gayathri N, Rao S, et al. Clinical, electrophysiological, imaging, and ultrastructural description in 68 patients with neuronal ceroid lipofuscinoses and its subtypes. Pediatr Neurol. (2014) 50:85–95. doi: 10.1016/j.pediatrneurol.2013.08.008
53. Trivisano M, Specchio N, Cappelletti S, Di Ciommo V, Claps D, Specchio LM, et al. Myoclonic astatic epilepsy: an age-dependent epileptic syndrome with favorable seizure outcome but variable cognitive evolution. Epilepsy Res. (2011) 97:133–41. doi: 10.1016/j.eplepsyres.2011.07.021
54. Berg AT, Levy SR, Testa FM. Evolution and course of early life developmental encephalopathic epilepsies: Focus on Lennox-Gastaut syndrome. Epilepsia. (2018) 59:2096–105. doi: 10.1111/epi.14569
55. Specchio N, Pontrelli G, Serino D, Trivisano M, Cappelletti S, Terracciano A, et al. Occipital seizures induced by intermittent photic stimulation in Dravet syndrome. Seizure. (2014) 23:309–13. doi: 10.1016/j.seizure.2013.12.009
Keywords: neuronal ceroid lipofuscinoses, EEG, photoparoxysmal response, intermittent photic stimulation, visual evoked potentials, electroretinogram, neurophysiological findings
Citation: Trivisano M, Ferretti A, Calabrese C, Pietrafusa N, Piscitello L, Carfi' Pavia G, Vigevano F and Specchio N (2022) Neurophysiological Findings in Neuronal Ceroid Lipofuscinoses. Front. Neurol. 13:845877. doi: 10.3389/fneur.2022.845877
Received: 30 December 2021; Accepted: 20 January 2022;
Published: 25 February 2022.
Edited by:
Alessandro Simonati, University of Verona, ItalyReviewed by:
Ruth Elizabeth Williams, Evelina London Children's Hospital, United KingdomCopyright © 2022 Trivisano, Ferretti, Calabrese, Pietrafusa, Piscitello, Carfi' Pavia, Vigevano and Specchio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Specchio, bmljb2xhLnNwZWNjaGlvQG9wYmcubmV0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.