 Ruideng Wang
Ruideng Wang Rubing Zhou†
Rubing Zhou† Fang Zhou
Fang Zhou- Department of Orthopedics, Peking University Third Hospital, Beijing, China
It is been over 100 years since glial cells were discovered by Virchow. Since then, a great deal of research was carried out to specify these further roles and properties of glial cells in central nervous system (CNS). As it is well-known that glial cells, such as astrocytes, microglia, oligodendrocytes (OLs), and oligodendrocyte progenitor cells (OPCs) play an important role in supporting and enabling the effective nervous system function in CNS. After spinal cord injury (SCI), these glial cells play different roles in SCI and repair. In this review, we will discuss in detail about the role of glial cells in the healthy CNS and how they respond to SCI.
Introduction
Spinal cord injury (SCI) is a devastating and debilitating neurological and pathological condition with temporary or permanent major motor, sensory and autonomic dysfunctions. It is estimated that there are about 250,000~500,000 people suffering from SCI around the world every year. Besides, ~90% of these cases are caused by traumatic factors, despite the proportion of non-traumatic SCI appears to be growing (1). People with SCI are 2–5 times more likely to die prematurely than people without SCI. Meanwhile, these people with SCI have worse survival rates in low- and middle-income countries. In recent years, more and more studies have begun to reveal the pathophysiology, molecular mechanisms, and possible therapeutic strategies of spinal cord injury. Over the past 50 years, it is gradually realized that glial cells have critical roles in health and disease. Glial cells were first postulated by Virchow in the 19th century and called this unique tissue “Nervenkitt” (2). With time, scientists have been committed to specify these further roles and properties of glial cells in the central nervous system (CNS). The glial cells include four major groups: astrocytes, microglia, oligodendrocytes (OLs), and oligodendrocyte progenitor cells (OPCs). A large number of studies show that these glial cells play an important role in SCI. In this review, we will discuss how these glial cells function in the healthy CNS and respond to SCI.
Glial Cells Are Vital in Healthy CNS
Glial cells play a vital role in supporting and enabling effective nervous system function in the healthy CNS. During the development of the CNS, glial cells can constitute a cellular framework that contributes to the development of the nervous system, and induce the survival and differentiation of neuron. The main glial cells types include astrocytes, microglial, OLs, and OPCs. They cooperate with each other and perform different important functions in CNS (Table 1).
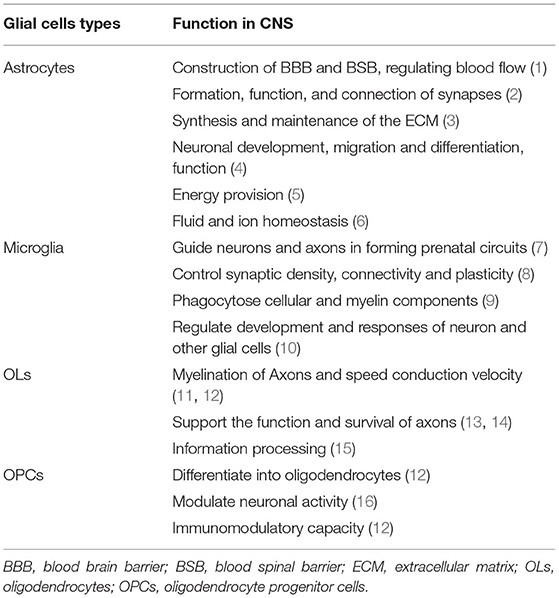
Table 1. Function of glial cells in the healthy CNS.
Astrocytes in The Healthy CNS
Astrocytes are the most abundant glial cells in CNS that have a large amount of complicated and fundamental functions in the healthy CNS. According to the differences in their cellular morphologies and anatomical locations, astrocytes are divided into two types: protoplasmic astrocytes with the morphological feature of several stem branches are found in gray matter while fibrous astrocytes with the morphological feature of many long fiber-like processes are found in white matter (3). In addition, both astrocyte subtypes have critical roles in health and disease. Astrocytes contribute to the construction of blood-brain barrier (BBB) and blood-spinal barrier (BSB) by combining with endothelial cells and perivascular pericytes through astrocytic endfeet (4–6). Previous studies have indicated that astrocytic endfeet show specialized feature characteristic as the astrocytic endfeet membrane expresses a large number of water channel aquaporin 4 (AQP4) and the Kir4.1 K+ channel, which is important for the properties of BBB (7–9). The Kir4.1 and AQP4 both bind to α-Syntrophin that could contribute to the inductive influence on BBB (10). Astrocytes are proved to produce a series of humoral agents, such as glial cell line-derived neurotrophic factor (GDNF), transforming growth factor-β (TGFβ), and angiopoetin-1 that can induce the aspects of BBB phenotypes (11–13). What's more, it is now recognized that the control of blood flow in brain is mediated by astrocytes. Neuronal activities may result in releasing potassium ions from astrocytic endfeet, extracellular K+ concentration can dilate the vessels through hyperpolarizing smooth muscle cells (14). The rise of Ca2+ concentration in astrocytic endfeet can also constrict vessels (15).
Astrocytes contribute to the formation, function, and connection of synapses as astrocytes have a close connection with synapses. The “tripartite synapse” concept was first described by Alfonso Araque, it includes the classic pre- and post-synaptic neuronal structures and astrocytes which should be viewed as integral modulatory elements of tripartite synapses (16). The role of astrocytes in synapses formation was first studied in 1995. Meyer-Franke et al. observed that retinal ganglion cells (RGCs) make very few synapses by purifying and culturing RGC neurons, however, RGCs can make many synapses if they are cultured in an astrocyte feeder layer or a culture medium that is previously conditioned by astrocytes (17). On the basis of the RGC culture system, subsequent studies identified that multiple factors secreted by astrocytes could control the formation of synapses. Thrombospondins (TSPs), the extracellular matrix (ECM) proteins secreted by astrocytes, have been proved to contribute to the formation of synapses (18, 19). By adding purified TSPs to cultured neurons greatly increased the number of synapses. In addition, Cagla Eroglu et al. showed that the von Willebrand factor A (VWF-A) domain of the calcium channel subunit α2δ1 interacts with the EGF-like receptors common to all TSPs which enhanced synaptogenesis both in vitro and in vivo (20). Hevin, another synaptogenic protein secreted by astrocytes, also induces an increase in the number of structural synapses by bridging presynaptic neurexin-1alpha (NRX1α) (21). Astrocytes can control the specific aspects of synapses function through many different signals, such as positive [cholesterol, glypican 4,6, ECM, tumor necrosis factor a (TNF-a)] and negative (SPARC, TSP) signals (22–27). For example, astrocyte-secreted cholesterol plays an important role in regulating the glutamatergic presynaptic function by complexing to apolipoprotein E-containing lipoproteins (27). Besides, astrocyte-secreted glypican 4/6 has an ability to upregulate the surface level of alpha-amino-3-hydroxy-5-methyl isoxazole propionic acid (AMPA) receptors (AMPARs) at synapses and increase the synaptic activity in neurons (26). As we all know, synapses can undergo rapid formation and elimination under certain conditions. Recent studies have identified some potential mechanisms, such as direct and indirect role of astrocytes in mediating synapses elimination. Microglia have been shown to recognize and phagocytose C1q/C3-coated synapses (28), and astrocytes would express TGF-β to induce the C1q expression which is critical for the phagocytic functions of microglia, and finally astrocytes mediate microglial-dependent synapses elimination (29). Meanwhile, astrocytes contribute to synapses elimination through MEGF10 and MERTK pathways (30).
Astrocytes are actively involved in the synthesis and maintenance of the ECM by secreting various substances in CNS. Tenascin-C, a glycoprotein, is expressed by astrocytes which can regulate cell growth, adhesion, and migration (31). Besides, astrocytes produce a large number of proteoglycans, such as chondroitin sulfate proteoglycans (CSPGs), which are suited for regulating neural development (32).
Other aspects of the role in CNS, such as astrocytes can store glycogen granules and make important contributions to the metabolism in CNS (33). The astrocyte-neuron lactate shuttle hypothesis which explains how astrocytes support neurons energy metabolism in detail. Glutamate released by neurons during the neuronal activity can bind to glutamate transporters (GLT-1), expressed by astrocytes, which mediate astrocytes taking up glucose from the blood circulation via glucose transporters (GLUT1). Then, glucose is subsequently metabolized to lactate and pyruvate. On the one hand, intracellular lactate can be shuttled to extracellular matrix via monocarboxylate transporter (MCT) 1 and MCT4 expressed by astrocytes and then could be absorbed by neurons through neuronal MCT2. Neuronal lactate can participate in the neuronal cell energy metabolism and promote ATP synthesis in the mitochondria directly or after conversion to pyruvate (34, 35). Similarly, ammonium () released by neurons increase lactate levels in astrocytes which can be shuttled to neurons (36). In summary, as astrocytes possess unique cellular properties, they play a vital role in the function and integrity of CNS (Table 1).
Microglia Functions in The Healthy CNS
Debate on microglial origin still continues in this field, recent studies showed that microglia were derived from erythromyeloid precursors in the yolk sac through Pu.1- and Irf8-dependent pathways (37). Microglia are crucial for the development of CNS. They arise around the same time as neurons and critically contribute to the establishment of complex neuronal networks. During the early development of CNS, microglia act as guidepost cells to guide neurons and axons to form prenatal circuits (38). Moreover, microglia are involved in the regulation of surrounding cellular milieu by secreting trophic factors [brain-derived neurotrophic factor (BDNF) (39), insulin-like growth factor-1 (IGF-1) (40), and hepatocyte growth factor (HGF) (41)] which could promote the survival of neurons. For instance, the best-known trophic factors, IGF-1, can enhance the survival of cortical neurons. On contrary, inhibiting IGF-1 signaling (minocycline, CD11b-DTR, and Cx3cr1GFP/GFP) would result in the cell death in layer V (40). Besides, microglia are the sensors of damage as they can phagocytose apoptotic neuron driven by both TAM receptor ligands Gas6 and protein S (42). Additionally, they engulf excess new born neural progenitor cells via primary phagocytosis which is beneficial to the homeostasis during the development of CNS (43).
Microglia play an important role in the control of synaptic density, connectivity, and plasticity. Microglia can selectively remove synapses from injured neurons which is termed “synaptic stripping” (44, 45). This process is identified to be mediated through several mechanisms. C3 receptors (CR3) expressed by microglia can bind to C1q and C3, the complement proteins expressed by damaged cells, which could lead the microglia to be involved in the active removal or “stripping” of these synaptic contacts and finally contribute to synaptic elimination (46). Microglia can also activate “synaptic stripping” through the fractalkine/CX3CR1 signaling pathway (47). Except for the receptor binding mode, microglia can also shape the strength and plasticity of synapses by releasing reactive oxygen species (ROS) (48), nitric oxide (NO) (49), TNF-α (50) as well as neurotrophic factors [BDNF (51)]. For example, microglia-derived BDNF activates Trk in spinal neurons that could impact synapse activity (52). Above all, microglia are vital for neuronal health and survival during the development of CNS (Table 1).
OLs and OPCs Functions in The Healthy CNS
Another major glial cell type is OLs, generated from OPCs, are fundamental to the myelin formation in CNS. The newborn OPCs can express DM-20 during embryonic development, and first appear in a restricted region of the embryonic ventral neural tube at embryonic day 12.5 in mice (53). Then, they finally differentiate into OLs through a complicated process. Importantly, OPCs are observed to differentiate into OLs throughout development and adulthood. Except for differentiating into OLs, OPCs can tile throughout the entire CNS and constitute ~5% of all cells (54). The fate of OPCs to keep as precursor cells or differentiate into OLs is influenced by many factors, such as mechanical environment and extracellular matrix elasticity (55–57). OPCs continue to be precursor cells by self-renewal to achieve homeostasis in CNS. Besides, OPCs express GABA receptors, kainite glutamate receptors, and AMPA receptors to form neuron-OPC synapses which modulate the neuronal activity (58, 59).
Oligodendrocytes are crucial for maintaining the function and integrity of axons. The most important function of OLs is to generate myelin sheath, as we all know that myelin sheath is an extension structure of the OLs plasma membrane wrapping the nerve axons. Myelination is a complex and tightly regulated process: OLs in the growth zone of CNS undergo proliferation under certain factors, then contact and arrange along the axon, respectively. The inner and outer plasma membrane wrapping the nerve axons interact with each other through cytoplasmic channels which pushes the inner plasma membrane layer after layer to generate the compact myelin. Once the appropriate number of plasma membrane wrapping per axon is generated, this process is called myelination (60). Functionally, the myelin sheath enables fast and efficient nerve conduction in the nervous system and provides metabolic support to the axons (61).
Oligodendrocytes have a physiological role in supporting the function and survival of axons that is independent of myelination. In the absence of PLP and DM20, the membrane proteolipids of myelin sheath that are integral for myelinated axons, myelination is not disrupted but with subsequently widespread axonal dysfunction (62). Subsequent studies found that PLP/DM20 was important for OLs in supporting the axonal energy metabolism (63, 64). With the further study, it is now well-recognized that OLs are essential for supporting the axons energy metabolism (65). The mechanisms how OLs provide neuronal metabolic support are described in detail as following. OLs can express a large number of MCT1, which can mediate metabolic support to neurons by co-transporting lactate and pyruvate (66). OLs can take up glucose from the extracellular matrix via GLUT1 expressed by OLs and then convert glucose into lactate and pyruvate by glycolysis. Besides, glutamate released by neuron after neuronal activity can bind to NMDA receptors (NMDARs) expressed by OLs which subsequently result in an increased glucose uptake as well as more lactate and pyruvate production in OLs (67). Moreover, the gap junctions between astrocytes and OLs may contribute to OLs metabolic support as lactate and glucose derived from astrocytes could be shuttled into OLs through gap junctions, such as Cx32-Cx30, Cx32-Cx26, Cx47-Cx30, and Cx47-Cx43 (68–70). All the functions of OLs and OPCs in healthy CNS are shown in Table 1.
Glial Cells Respond To SCI
As discussed above, glial cells, such as astrocytes, microglia, OLs, and OPCs all are crucial for the development of CNS and maintaining homeostasis in healthy CNS. They have different and vital physiological functions for the CNS due to their cytological properties and cellular interactions. After SCI, the noxious mechanical forces cause tissue damage, such as cells death and disrupt the homeostasis of local CNS, as a result, these events trigger diverse multi-cellular responses and can lead either to the neural repair or secondary cellular injury. Glial cells exhibit various pathophysiological functions to repair the damage and maintain local microenvironment homeostasis due to various internal and external factors after SCI. Next, we will describe in detail the response of various glial cells to SCI.
Astrocytes: Reactive Astrocytes and Glial Scar Formation
Astrocytes, as discussed above, are essential to maintain the homeostasis in healthy CNS. Similarly, astrocytes also play an important role after SCI. After SCI, various intrinsic and extrinsic factors subsequently regulate astrocytes into reactive astrocytes with significant morphological, phenotypical, and functional changes, such changes are mainly based on different factors, such as the injury severity, the injury time, and the distance of astrocytes to the lesion. Reactive astrocytes have characteristics in morphology, such as cellular hypertrophy, thicker processes, and increased expression of intermediate filament proteins. Besides, the degree of changes are proportional to the stimulus intensity (71). On the basis of discrete gene-expression identifiers and functions, different types of reactive astrocytes have been recognized, such as A1, A2, and scar-forming astrocytes (72, 73). For example, complement component 3 is highly expressed by A1 astrocytes, and S100A10 is a specific hallmark for A2 astrocytes while type I collagen for scar-forming astrocytes (73, 74). Compared with the normal astrocytes, accumulating evidence suggests that reactive astrocytes show various abnormal functions, such as releasing proinflammatory chemokines and cytokines (71).
Molecules and Signaling Pathways Implicated in Formation of Reactive Astrocytes
Mechanical forces usually cause direct damage to the normal tissue and disrupt local homeostasis when patients or animals undergo SCI, which on the other hand triggers multitudinous multi-cellular responses. Although it is incompletely understood how mechanical forces and damaged tissues initially trigger the activation of astrocytes after SCI. The previous study has identified that astrocytes are susceptible to membrane distortions and debris (75). Traumatic membrane deformation could activate mechanosensitive ion channels and result in the rapid influx of extracellular calcium and sodium in astrocytes (76–78). Other studies show that plasma membrane stretching can rise the release of intracellular calcium and ATP via extracellular signal-regulated protein kinase (ERK) and PKB/Akt signaling pathways (79, 80). Besides, astrocytes may also release endothelin-1 (ET-1), isoprostanes, and matrix metalloproteinases 9 (MMP-9) after stretch-induced injury (81, 82). More studies are needed to have a deeper understanding of these.
Accumulating studies have identified that a lot of molecules, such as chemokines, cytokines, transcription factors, and growth factors are the mediators for the activation of astrocytes (factors are shown in Table 2). For example, proinflammatory cytokines, such as TNF-α, interleukin (IL)-6, and IL-1β initially trigger the reactivity of astrocytes during the acute phase after SCI while other molecules maintain astrocytes reactivity in the later stages (83–85). Additionally, it is worth mentioning that reactive astrocytes can release triggering molecules, such as TNF-α, IL-6, and MMP-9, which in turn activate more astrocytes (86). Besides, other glial cells, such as activated microglia, are identified to induce the activation of astrocytes by secreting various factors, such as Il-1α, TNF, and C1q (87). Other related molecules involved in the activation of astrocytes are shown in Table 2.

Table 2. The activation of astrocytes.
Many signaling pathways are closely involved in the activation of astrocytes, such as STAT3, TGF-β, NF-κB, JNK/c-Jun, and MAPK (more signaling ways are shown in Table 2). Here we will mainly introduce TGF-β and STAT3 signaling pathways. The STAT3 signaling pathway is one of the most important signaling pathways to mediate the formation of reactive astrocytes. Mice with STAT3 knock-out in astrocytes showed the attenuated upregulation of GFAP, unsuccessful cell hypertrophy, and failed scar formation after SCI (88). Other groups also identified that selective STAT3 deletion in mice could limit the migration of astrocytes and result in the widespread infiltration of inflammatory cells, degeneration of neurons, and demyelination of axons that can lead to severe motor deficits. However, by the activation of STAT3 signaling pathway, they observed that reactive astrocytes migrated rapidly around the lesion and secluded inflammatory cells that lead to a notable improvement in functional recovery (89). These results provided a potential intervention target of STAT3 signaling pathway in the treatment of SCI. TGF-β signaling pathway greatly contributes to the formation of reactive astrocytes. As discussed above, TGF-β is a key upstream trigger in the formation of reactive astrocytes. The previous study has shown that TGF-β could increase the expression of anti-regenerative molecules, such as CSPGs, laminin, and fibronectin by several-fold in reactive astrocytes (90). Interestingly, fibrinogen could act as a stimulating factor which can activate TGF-β signaling pathway, as a result, it could induce the activation of astrocytes and formation of CSPGs (91). In addition, it could induce astrogliosis by injecting fibrinogen into the mouse cortex (91). On contrary, with the genetical ablation of fibrinogen in mice, they found inhibited TGF-β activation and hampered glial scar formation (91). Other signaling pathways are shown in Table 2.
Reactive Astrocytes Expression Change and Their Functions
Recent years, numerous studies have identified that the activation of astrocytes could lead to the change of functions with releasing a range of molecules, such as cytokines [TNF-α, IL-6, IL-10, IL-1β, etc. (85, 92, 93)], chemokines [CCL2, CCL3, etc. (94, 95)], growth factors [BDNF, GDNF, etc. (96, 97)], toxic amino acids [GABA and glutamate (98, 99)], extracellular matrix [CSPGs, collagen I, fibronectin, MMP-9, etc. (100–102)], and intermediate filaments [Nestin, vimentin, and GFAP (100, 102)], which would have a significant influence on the spinal cord microenvironment after SCI (Table 2). The molecules released by reactive astrocytes can activate more normal astrocytes into reactive astrocytes and contribute to glial scar forming. On the other hand, they also affect other cells, such as neurons, OPCs, and microglia through a variety of complexed effects (71).
Over the past years, reactive astrocytes were thought to be detrimental for recovery after SCI. However, recent studies have identified that reactive astrocytes also contribute to SCI repair. Here, we will discuss the beneficial and detrimental effects of reactive astrocytes after SCI (Table 3).
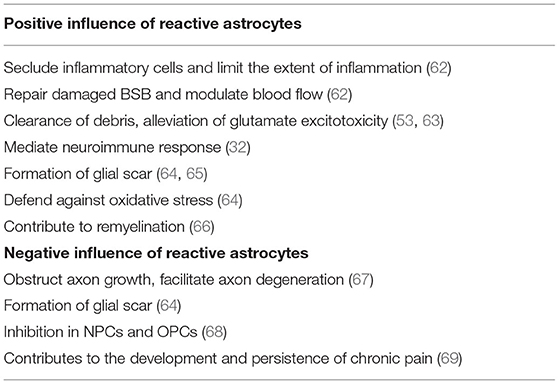
Table 3. Positive and negative influence of reactive astrogliosis.
Reactive astrocytes are considered to be a defense mechanism of astrocytes responding to SCI. After SCI, BBB breaks down and becomes leaky to endogenous and exogenous blood-borne macromolecules that can result in disastrous consequence. These changes will mediate reactive astrocytes to upregulate Sonic hedgehog (SHH) and activate signaling cascades to repair the tight junctions of the BBB (103). Interestingly, with the absence of reactive astrocytes, it was failure in repairing the damaged BBB (104). At acute stage after SCI, reactive astrocytes migrate rapidly around the lesion to seclude inflammatory cells and limit the extent of inflammation that has a notable improvement in functional recovery (89). Further, Jill et al. found significantly increased and prolonged infiltration of inflammatory cells around the lesion with selective and conditional reactive astrocytes ablation in mice (104). Various endogenous and exogenous factors result in the release and accumulation of cell debris and neurotoxic factors in the extracellular spaces after SCI. Recently, reactive astrocytes were identified to play a crucial role in removing these cell debris and neurotoxic factors. More importantly, reactive astrocytes have the ability to phagocytose dead cells in vitro and in vivo via the upregulation of ABCA1 (105, 106). Reactive astrocytes can also reduce the impact of glutamate excitotoxicity on neurons and OPCs by clearing excess glutamate from the blood or necrotic neuronal cell death (107). Besides, reactive astrocytes can affect immune cells through releasing various molecules, such as TNF-α, TGF-β, and proteoglycans. CSPGs have a close relationship with immune activity as they can recruit chemokines and growth factors that enhances the connection of immune cells (71).
Glial scar formation has been recognized for many years. After SCI, inflammatory cells (macrophages, neutrophils, and lymphocytes), fibrotic cells, and other cells, such as pericytes, fibroblasts, and OPCs migrate rapidly into the lesion, and subsequently newly proliferated, elongated reactive astrocytes come around the lesion to form a border which could separate necrotic tissue from healthy tissue (108–110). The border formed by reactive astrocytes can limit further expansion of the lesion and restrict inflammatory cells within damaged tissue that will protect the surrounding viable neural tissue from secondary damage (111). Further, selective inhibition of astrocyte reactivity results in the widespread propagation of inflammatory cells beyond the lesion.
In addition to the above protective effects, reactive astrocytes also have detrimental effects. As discussed above, reactive astrocytes can form a physical barrier to confine the lesion, however, it can also obstruct axonal growth. Besides, reactive astrocytes secrete inhibitory proteins, such as CSPGs, which are considered to be the major inhibitors of axonal regeneration. CSPGs derived from reactive astrocytes inhibit the growth of axons in vitro, and axonal regeneration is observed to stop at CSPG-rich regions in vivo. On the contrary, Chondroitinase ABC, by removing CSPG glycosaminoglycan (GAG) chains, attenuates the inhibitory activity of CSPGs, which is shown to facilitate axonal regeneration and functional recovery (112). Further, Hyunjung Lee et al. found that using thermostabilized Chondroitinase ABC through a hydrogel-microtube scaffold system could enhance the axonal regrowth, sprouting, and improve functional recovery after SCI (113). Additionally, other studies have shown that CSPGs inhibited axonal regeneration while the inhibition of CSPGs could improve functional recovery (114).
Reactive astrocytes play a modulatory role in NPCs and OPCs post-SCI. OPCs have extremely powerful ability in remyelination as they can proliferate and differentiate into OLs that will replenish a large number of lost OLs after SCI. Recently, Justin R Siebert et al. have discovered that astrocytes-derived CSPGs highly inhibited the migration and differentiation of OPCs in vitro, and the number of OPCs surrounding the lesion significantly increased when treated with the enzyme chondroitinase ABC (115). Other study also proved that CSPGs had a dampening effect on the outgrowth and differentiation of OPCs, and treated with chondroitinase ABC could completely eliminate this inhibition (116). In addition to CSPGs, other molecules, such as BMP and ET-1 released by reactive astrocytes can also inhibit the differentiation of OPCs and finally influence remyelination (117, 118). Besides, reactive astrocytes have a role in inhibiting the neuronal differentiation of NPCs by expressing insulin-like growth factor binding protein 6 (IGFBP6) and CSPGs (119).
Microglia/Macrophages: Neuroinflammation
Microglia/macrophages maybe the most potent modulators to launch the innate immune response after SCI. As discussed above, we know that microglia are resident in CNS while macrophages derive from the periphery. However, activated microglia and macrophages are difficult to distinguish through the morphology or antigenic markers following CNS injury, so they are referred as microglia/macrophages. Over the past years, the studies have revealed that microglia/macrophages had both the detrimental and beneficial effects on neurological recovery due to their different phenotypes at different stages after SCI (120).
Microglia/Macrophages Phenotypes
Microglia/macrophages phenotypes are mainly determined by the focal lesion and new stimuli can change the phenotypes. It is now well-acknowledged that microglia/macrophages are activated into different functional phenotypes after SCI. M1/M2 dichotomy is the earliest and simplest concept. M1 macrophages (or ‘classically' activated macrophages) are activated by the prototypical T helper 1 cytokine (TH1), interferon-γ (IFNγ), and lipopolysaccharide (LPS), which typically release inflammatory cytokines (IL-1, IL-6, TNFα, etc.), chemokines (CCL8, CCL 15, CXCL 10, CXCL 11, etc.), and the high levels of oxidative metabolites (ROS and NOS). On the contrast, M2 macrophages (or “alternatively” activated macrophages) are activated by the prototypic TH2 cytokine IL-4 and IL-13, which can produce numerous protective factors (TGFβ, IL-10, IL-1Ra, etc.) and clear cellular debris (120–122). However, the status and functional phenotypes of microglia/macrophages are much more complicated in vivo. Accumulating studies have identified the multiformity in M2 phenotype subpopulations, such as M2a, M2b, and M2c phenotypes, each phenotype is characterized by unique physiological features and distinct biological functions (121). Nowadays, microglia/macrophages in many other situations did not show a clear M1 or M2 phenotype or showed phenotypic plasticity during the disease progression. Single cell techniques and other new tools are, contributing to the understanding of polarization heterogeneity (123). By single-cell analysis, Lindsay M Milich at el. identified four microglial subtypes in the injured mouse spinal cord, which were labeled homeostatic, inflammatory, dividing, and migrating microglia. Homeostatic microglia were identified by several annotated markers of steady-state microglia, such as P2ry12, Siglech, and Tmem119. Inflammatory microglia were identified by the low expression of purinergic receptor P2ry12 and increased expression of Igf1. Dividing microglia expressed low levels of P2ry12, increased expression of Msr1, and high levels of cell cycle–related genes, such as Cdk1. Migrating microglia had the low levels of P2ry12, and the high levels of Msr1 and the growth factor Igf1 (124). Besides, two macrophage subtypes were named chemotaxis-inducing macrophages and inflammatory macrophages in addition to the border-associated macrophages based on their gene ontology terms. Both subtypes expressed the lysosomal gene Cd63, however, chemotaxis-inducing macrophages preferentially express heme oxygenase Hmox1 while inflammatory macrophages express Apoe (124, 125).
Microglia/Macrophages Respond to SCI
Activated microglia could release a large number of pro-inflammatory cytokines, chemokines, and other cytotoxic factors after SCI. They respond to SCI within minutes by producing pro-inflammatory molecules which can lead to the influx of multiple inflammatory cells from the circulation. Neutrophils are the first circulating leukocytes to infiltrate into the lesion and are prominently located in severely damaged site (126, 127). Besides, peripheral macrophages will infiltrate into the lesion and help clear apoptotic cells (127). However, these neutrophils and macrophages may be destructive to the lesion as they can produce various molecules, such as MMP-9 and disrupt the functions of the BSB (128). Besides, T and B lymphocytes are found to infiltrate into the injured lesion and cause a systemic autoimmune response (129). Here, we will mainly discuss the harmful and beneficial effects of neuroinflammation induced by activated microglia/macrophages after SCI.
Activated M1 microglia/macrophages induce neurons death and contribute to the secondary damage by releasing pro-inflammatory factors, such as IL-1β, IL-6, TNF-α, CCL5, and iNOS. Here we mainly elaborate IL-1β and TNF-α that play a detrimental role after SCI. IL-1β expressed by astrocytes and microglia was detected to reach peak at 12 h after SCI in rodents (130). IL-1β and TNF-α were proved to involve in the recruitment and activation of peripheral immune cells and the activation of astrocytes and microglia. In rats, the infusion of IL-1β markedly enhanced the cortical neuronal loss, on the contrast, it could significantly inhibit neuronal damage by IL-1 receptor antagonist (IL-1ra) (131). Other study also identified that IL-1β contributed to ischemic brain damage while IL-1ra markedly protected the focal cerebral from ischemia in the rat (132). TNF-α, another proinflammatory cytokine, expressed mainly by activated microglia/macrophages, contributes to neuronal cells death after SCI by binding to TNFRI and TNFRII (133). In addition, soluble TNFRI, which can compete with TNF-α by binding to TNFR, eventually reduces the neuronal cells death (133). Tiziana Genovese et al. indicated that the genetic inhibition of TNF-α significantly reduced the degree of inflammation, tissue injury, and apoptosis in an experimental model of spinal cord trauma (134). Besides, overexpressing TNF-α was shown to mediate OLs, OPCs death, and myelin vacuolization which could finally result in spontaneous demyelination (135).
Activated M2 microglia/macrophages have anti-inflammatory and neuroprotective effects by increasing the expression of anti-inflammatory molecules, such as IL-10, TGF-β, IGF-1, and BNDF. For example, IL-10 shows a wide range of regulatory activities in response to SCI. Tiziana Genovese et al. found that there was a significant augmentation of TNF-α, IL-1β and S100β which worsened the recovery of limb function in IL-10 KO mice (136). Recently, the group of Jessica Y Chen delivered IL-10 into mice SCI model by loading an implantable biomaterial scaffold. They observed that IL-10 could significantly reduce damage to tissue and improve subsequent motor deficits (137). IGF-I is a potent neurotrophic factor released by activated microglia/macrophages with anti-inflammatory response. The previous study showed that IGF-I gene transfer after SCI could inhibit the loss of neurons and significantly improve the neurological dysfunction (138). Besides, other study showed that BDNF and IGF-I could significantly enhance neuroprotective effects, such as repairing BSCB damage, alleviating edema, and cells injury by the downregulation of nNOS after SCI in rat model (139).
OLs and OPCs: Demyelination and Remyelination
OLs and Demyelination
In addition to the immediate trauma damage, there is a prolonged secondary damage after SCI. OLs are quite susceptible to changes in the surrounding microenvironment after SCI which can result in the necrosis, apoptosis, and autophagy of OLs (140–142). Acute OLs death has previously been investigated to occur within 15 min after injury and the number of OLs steadily declined by 7 days post-injury (143, 144). Previous studies have identified several aspects of subsequent damage that can lead to the death of OLs. Ischemia is an apparent reason to result in OLs death in the damaged areas of white matter (145). Ischemia and reperfusion contribute to the formation of free radical, such as reactive oxygen and nitrogen species, and OLs are particularly vulnerable to the oxidative stress. After SCI, ROS (hydroxyl radicals and superoxide) and RNS (nitric oxide, peroxynitrite, and nitrated protein) were detected to be at the increased levels (146–148). By oxidizing protein, lipids and nuclear material, ROS and NOS damage OLs which results in the necrosis and apoptosis of OLs. Besides, excitotoxicity is another major factor leading to the OLs death after SCI. The glutamate will reach a toxic level after SCI that can lead to the OLs death in vitro and in vivo. Glutamate binding to AMPA/kainate glutamate receptors expressed in OLs leads to OLs death via receptor overactivation and the specific inhibitors of AMPA receptors can block OLs death (149). As discussed above, extracellular ATP released by multiple cell types after SCI can also contribute to OLs death. ATP is proved to cause OLs death via an activation of calcium-permeable P2X(7) and treatment with P2X(7) antagonists reduces demyelination and improve neurological symptoms (150). In addition, recent studies reveal that proinflammatory cytokines potentially contribute to OLs cell death. An overexpression of TNF-α was observed to induce OLs apoptosis which could contribute to the degenerative change and demyelination via TNFR1 and TNFR2 expressed in OLs (151). Other cytokines, such as IL-2, IL-1, IFNγ, and proNGF, all contribute to OLs apoptosis (142). In addition to apoptosis and necrosis, autophagy is activated in SCI, and Beclin1, a promoter of autophagy, is highly expressed in OLs (152).
Oligodendrocytes are fundamental to myelin formation as described earlier. The injury or death of OLs results in the degeneration of myelin sheaths and the support of axons by OLs would be disrupted after SCI which eventually lead to the widespread demyelination of spared axons. As a matter of fact, accumulating studies have demonstrated that demyelination indeed occurred in animal models and human after SCI (153, 154). For example, demyelinated axons were seen within 2 weeks after injury in paraplegic domestic animals in previous study (153). More interestingly, the extent of demyelination mainly contingents on the type and severity of injury. The normal myelinated axons are characterized by the regular distribution of sodium and potassium channels, after demyelination, the distribution of sodium and potassium channels is disrupted that contributes to an axonal conduction block (155). Besides, demyelination is identified to increase voltage-gated Na+ channels, which may result in Na+ influx during action potential propagation. To eliminate the excess Na+, more ATP is required which can disrupt the axonal internal energy balance. Additionally, the excess Na+ may lead to axonal Ca2+ overload via the Na+/Ca2+ exchangers. These events eventually result in axonal degeneration (156, 157). Besides, the demyelinated axons are vulnerable to damage in the microenvironment after SCI and ultimately lead to axonal degeneration (158).
OPCs and Remyelination
After SCI, OPCs are multipotential stem cells which can differentiate into remyelinated cells to involve in axonal remyelination and contribute to the glial scar formation. McTigue et al. assessed the proliferation of NG2+ cells and OLs by bromodeoxyuridine incorporation and they found increased proliferation of NG2+ cells persisting throughout the first 4 weeks post-injury while the number of OLs continuously reduced by 7 days post-injury. However, they detected an increased number of OLs at 14 days post-injury. These results showed that proliferated NG2+ cells may differentiate into OLs after injury (159). Besides, the study using fate mapping confirmed that 30% of new OLs responsible for myelin regeneration were derived from OPCs while OPCs differentiate into the majority of myelinating Schwann cells (160). Other group also revealed that OPCs from the PDGFRα-expressing lineage could be transformed into functional myelinating Schwann cells after SCI (161). Moreover, by using genetic fate mapping, Hackett et al. found that ~25% of astrocytes were derived from NG2+ cells in the glial scar by 4 weeks after SCI (162). It is worth mentioning that the functions of OPCs are intricately modulated by a complex network including various factors (Table 4). We will not go into further discussion here. In addition to OPCs, the endogenous NPCs can also contribute to OLs replacement as they will be activated and migrate into the lesion after SCI (163, 164).
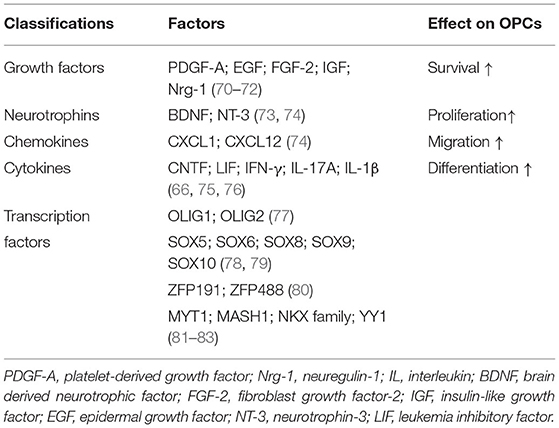
Table 4. Factors regulate remyelination via different effects on OPCs.
Remyelination occurs spontaneously on residual axons after SCI. Remyelination is difficult to detect until genetic fate mapping approaches are applied, the scientists can distinguish new myelin from preexisted myelin via labeling new myelination. Assinck et al. found that spontaneous remyelination was induced by OLs and myelinating Schwann cells in mice after SCI (160). Besides, other group detected remarkably clear visualization of spontaneously regenerated myelin in vivo (165). However, endogenous remyelination was limited due to multi-factors (166). Nashmi et al. found that the spontaneous remyelination in the injured white matter was non-optimal and incomplete because the newly formed myelin around the injured axons was thinner than normal myelinated axons (167). Recent studies have uncovered that multiple factors affected remyelination, such as (1) the myelinating OLs derived from OPCs are inadequate (168), (2) OLs maturation and myelination are limited (142), (3) axonal ensheathment and remyelination is influenced (169), and (4) OPCs, neural progenitor cells (NPCs) are affected by the unfriendly microenvironment (166). Therefore, more endogenous mechanisms of remyelination are needed to be explored.
Conclusions
Glial cells play a crucial role in maintaining the function and homeostasis of the CNS. Once the homeostasis of the CNS is disrupted, glial cells will respond to the different kinds of damage by multiplying, differentiating, activating, and so on. Nowadays, based on the animal models of SCI, we have gained a better understanding of the pathophysiological changes of glial cells after SCI. For example, after SCI, various factors lead to the activation of astrocytes, which can secrete various molecules, such as cytokines and chemokines in response to SCI. Besides, multicellular and multi-molecular components are involved in forming glial scar that has beneficial and detrimental effects in axonal regeneration and neuro-inflammation. Therefore, an in-depth exploration of the role of glial cells in SCI is conducive to the development of SCI repair strategies. Further studies should develop novel targets and strategies that contribute to the post-SCI reparative responses of glial cells.
Author Contributions
RW and RZ contributed to the writing of the manuscript. ZC and SG contributed to a systematic literature search. All authors discussed the results and contributed to the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81971160).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Quadri SA, Farooqui M, Ikram A, Zafar A, Khan MA, Suriya SS, et al. Recent update on basic mechanisms of spinal cord injury. Neurosur Rev. (2020) 43:425–41. doi: 10.1007/s10143-018-1008-3
2. Letterer E. [Virchow's contribution to modern pathology; on the 100th anniversary of cellular pathology, August 20, 1858]. Hippokrates. (1958) 29:505–11.
3. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. (2010) 119:7–35. doi: 10.1007/s00401-009-0619-8
4. Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. (2009) 335:75–96. doi: 10.1007/s00441-008-0658-9
5. Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. (2002) 200:629–38. doi: 10.1046/j.1469-7580.2002.00064.x
6. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. (2010) 37:13–25. doi: 10.1016/j.nbd.2009.07.030
7. Wolburg H. Orthogonal arrays of intramembranous particles: a review with special reference to astrocytes. J Hirnforsch. (1995) 36:239–58.
8. Verbavatz JM, Ma T, Gobin R, Verkman AS. Absence of orthogonal arrays in kidney, brain and muscle from transgenic knockout mice lacking water channel aquaporin-4. J Cell Sci. (1997) 110(Pt 22):2855–60. doi: 10.1242/jcs.110.22.2855
9. Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. (1997) 17:171–80. doi: 10.1523/JNEUROSCI.17-01-00171.1997
10. Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. (2006) 7:41–53. doi: 10.1038/nrn1824
11. Igarashi Y, Utsumi H, Chiba H, Yamada-Sasamori Y, Tobioka H, Kamimura Y, et al. Glial cell line-derived neurotrophic factor induces barrier function of endothelial cells forming the blood-brain barrier. Biochem Biophys Res Commun. (1999) 261:108–12. doi: 10.1006/bbrc.1999.0992
12. Haseloff RF, Blasig IE, Bauer HC, Bauer H. In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cell Mol Neurobiol. (2005) 25:25–39. doi: 10.1007/s10571-004-1375-x
13. Lee SW, Kim WJ, Choi YK, Song HS, Son MJ, Gelman IH, et al. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med. (2003) 9:900–6. doi: 10.1038/nm889
14. Paulson OB, Newman EA. Does the release of potassium from astrocyte endfeet regulate cerebral blood flow? Science. (1987) 237:896–8. doi: 10.1126/science.3616619
15. Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. (2004) 431:195–9. doi: 10.1038/nature02827
16. Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. (1999) 22:208–15. doi: 10.1016/S0166-2236(98)01349-6
17. Meyerfranke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion-cells in culture. Neuron. (1995) 15:805–19. doi: 10.1016/0896-6273(95)90172-8
18. Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. (2005) 120:421–33. doi: 10.1016/j.cell.2004.12.020
19. Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, et al. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cerebr Blood F Met. (2008) 28:1722–32. doi: 10.1038/jcbfm.2008.65
20. Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, et al. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. (2009) 139:380–92. doi: 10.1016/j.cell.2009.09.025
21. Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, et al. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci USA. (2011) 108:E440–9. doi: 10.1073/pnas.1104977108
22. Chung W-S, Allen NJ, Eroglu C. Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol. (2015) 7:a020370. doi: 10.1101/cshperspect.a020370
23. Goritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci. (2005) 29:190–201. doi: 10.1016/j.mcn.2005.02.006
24. Albrecht D, Lopez-Murcia FJ, Perez-Gonzalez AP, Lichtner G, Solsona C, Llobet A. SPARC prevents maturation of cholinergic presynaptic terminals. Mol Cell Neurosci. (2012) 49:364–74. doi: 10.1016/j.mcn.2012.01.005
25. Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, et al. Control of synaptic strength by glial TNFalpha. Science. (2002) 295:2282–5. doi: 10.1126/science.1067859
26. Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, et al. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature. (2012) 486:410–4. doi: 10.1038/nature11059
27. Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. (2001) 294:1354–7. doi: 10.1126/science.294.5545.1354
28. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
29. Bialas AR, Stevens B. TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci. (2013) 16:1773–82. doi: 10.1038/nn.3560
30. Chung W-S, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. (2013) 504:394–400. doi: 10.1038/nature12776
31. Joester A, Faissner A. The structure and function of tenascins in the nervous system. Matrix Biol. (2001) 20:13–22. doi: 10.1016/S0945-053X(00)00136-0
32. Wiese S, Karus M, Faissner A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front Pharmacol. (2012) 3:120. doi: 10.3389/fphar.2012.00120
33. Brown AM, Ransom BR. Astrocyte glycogen and brain energy metabolism. Glia. (2007) 55:1263–71. doi: 10.1002/glia.20557
34. Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. (1994) 91:10625–9. doi: 10.1073/pnas.91.22.10625
35. Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. (2004) 305:99–103. doi: 10.1126/science.1096485
36. Lerchundi R, Fernández-Moncada I, Contreras-Baeza Y, Sotelo-Hitschfeld T, Mächler P, Wyss MT, et al. NH4(+) triggers the release of astrocytic lactate via mitochondrial pyruvate shunting. Proc Natl Acad Sci USA. (2015) 112:11090–5. doi: 10.1073/pnas.1508259112
37. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. (2013) 16:273–80. doi: 10.1038/nn.3318
38. Squarzoni P, Thion MS, Garel S. Neuronal and microglial regulators of cortical wiring: usual and novel guideposts. Front Neurosci. (2015) 9:248. doi: 10.3389/fnins.2015.00248
39. Trang T, Beggs S, Salter MW. Brain-derived neurotrophic factor from microglia: a molecular substrate for neuropathic pain. Neuron Glia Biol. (2011) 7:99–108. doi: 10.1017/S1740925X12000087
40. Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. (2013) 16:543–51. doi: 10.1038/nn.3358
41. Yamagata T, Muroya K, Mukasa T, Igarashi H, Momoi M, Tsukahara T, et al. Hepatocyte growth factor specifically expressed in microglia activated Ras in the neurons, similar to the action of neurotrophic factors. Biochem Biophys Res Commun. (1995) 210:231–7. doi: 10.1006/bbrc.1995.1651
42. Fourgeaud L, Través PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, et al. TAM receptors regulate multiple features of microglial physiology. Nature. (2016) 532:240–4. doi: 10.1038/nature17630
43. Luo C, Koyama R, Ikegaya Y. Microglia engulf viable newborn cells in the epileptic dentate gyrus. Glia. (2016) 64:1508–17. doi: 10.1002/glia.23018
44. Blinzinger K, Kreutzberg G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z Zellforsch Mikrosk Anat. (1968) 85:145–57. doi: 10.1007/BF00325030
45. Perry VH, O'Connor V. The role of microglia in synaptic stripping and synaptic degeneration: a revised perspective. ASN Neuro. (2010) 2:e00047. doi: 10.1042/AN20100024
46. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
47. Hoshiko M, Arnoux I, Avignone E, Yamamoto N, Audinat E. Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J Neurosci. (2012) 32:15106–11. doi: 10.1523/JNEUROSCI.1167-12.2012
48. Zhang J, Malik A, Choi HB, Ko RWY, Dissing-Olesen L, MacVicar BA. Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron. (2014) 82:195–207. doi: 10.1016/j.neuron.2014.01.043
49. Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. (2015) 96:70–82. doi: 10.1016/j.neuropharm.2014.10.027
50. Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. (2006) 440:1054–9. doi: 10.1038/nature04671
51. Tong L, Prieto GA, Kramár EA, Smith ED, Cribbs DH, Lynch G, et al. Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1β via p38 mitogen-activated protein kinase. J Neurosci. (2012) 32:17714–24. doi: 10.1523/JNEUROSCI.1253-12.2012
52. Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. (2005) 438:1017–21. doi: 10.1038/nature04223
53. Timsit S, Martinez S, Allinquant B, Peyron F, Puelles L, Zalc B. Oligodendrocytes originate in a restricted zone of the embryonic ventral neural tube defined by DM-20 mRNA expression. J Neurosci. (1995) 15:1012–24. doi: 10.1523/JNEUROSCI.15-02-01012.1995
54. Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci. (2000) 20:6404–12. doi: 10.1523/JNEUROSCI.20-17-06404.2000
55. Jagielska A, Norman AL, Whyte G, Vliet KJV, Guck J, Franklin RJM. Mechanical environment modulates biological properties of oligodendrocyte progenitor cells. Stem Cells Dev. (2012) 21:2905–14. doi: 10.1089/scd.2012.0189
56. Lourenço T, Paes de Faria J, Bippes CA, Maia J, Lopes-da-Silva JA, Relvas JB, et al. Modulation of oligodendrocyte differentiation and maturation by combined biochemical and mechanical cues. Sci Rep. (2016) 6:21563. doi: 10.1038/srep21563
57. Urbanski MM, Kingsbury L, Moussouros D, Kassim I, Mehjabeen S, Paknejad N, et al. Myelinating glia differentiation is regulated by extracellular matrix elasticity. Sci Rep. (2016) 6:33751. doi: 10.1038/srep33751
58. Lin S-c, Bergles DE. Synaptic signaling between GABAergic interneurons and oligodendrocyte precursor cells in the hippocampus. Nat Neurosci. (2004) 7:24–32. doi: 10.1038/nn1162
59. Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. (2000) 405:187–91. doi: 10.1038/35012083
60. Bercury KK, Macklin WB. Dynamics and mechanisms of CNS myelination. Dev Cell. (2015) 32:447–58. doi: 10.1016/j.devcel.2015.01.016
61. Stadelmann C, Timmler S, Barrantes-Freer A, Simons M. Myelin in the central nervous system: structure, function, and pathology. Physiol Rev. (2019) 99:1381–431. doi: 10.1152/physrev.00031.2018
62. Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. (1998) 280:1610–3. doi: 10.1126/science.280.5369.1610
63. Ferreirinha F, Quattrini A, Pirozzi M, Valsecchi V, Dina G, Broccoli V, et al. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest. (2004) 113:231–42. doi: 10.1172/JCI200420138
64. Tarrade A, Fassier C, Charvin D, Charvin D, Vitte J, Peris L, et al. A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum Mol Genet. (2006) 15:3544–58. doi: 10.1093/hmg/ddl431
65. Philips T, Rothstein JD. Oligodendroglia: metabolic supporters of neurons. J Clin Invest. (2017) 127:3271–80. doi: 10.1172/JCI90610
66. Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. (2012) 487:443–8. doi: 10.1038/nature11314
67. Saab AS, Tzvetavona ID, Trevisiol A, Baltan S, Dibaj P, Kusch K, et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron. (2016) 91:119–32. doi: 10.1016/j.neuron.2016.05.016
68. Magnotti LM, Goodenough DA, Paul DL. Functional heterotypic interactions between astrocyte and oligodendrocyte connexins. Glia. (2011) 59:26–34. doi: 10.1002/glia.21073
69. Niu J, Li T, Yi C, Huang N, Koulakoff A, Weng C, et al. Connexin-based channels contribute to metabolic pathways in the oligodendroglial lineage. J Cell Sci. (2016) 129:1902–14. doi: 10.1242/jcs.178731
70. Orthmann-Murphy JL, Abrams CK, Scherer SS. Gap junctions couple astrocytes and oligodendrocytes. J Mol Neurosci. (2008) 35:101–16. doi: 10.1007/s12031-007-9027-5
71. Li X, Li M, Tian L, Chen J, Liu R, Ning B. Reactive astrogliosis: implications in spinal cord injury progression and therapy. Oxid Med Cell Longev. (2020) 2020:9494352. doi: 10.1155/2020/9494352
72. Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci. (2012) 32:6391–410. doi: 10.1523/JNEUROSCI.6221-11.2012
73. Hara M, Kobayakawa K, Ohkawa Y, Kumamaru H, Yokota K, Saito T, et al. Interaction of reactive astrocytes with type I collagen induces astrocytic scar formation through the integrin-N-cadherin pathway after spinal cord injury. Nat Med. (2017) 23:818–28. doi: 10.1038/nm.4354
74. Boghdadi AG, Teo L, Bourne JA. The neuroprotective role of reactive astrocytes after central nervous system injury. J Neurotrauma. (2020) 37:681–91. doi: 10.1089/neu.2019.6938
75. Cullen DK, Vernekar VN, LaPlaca MC. Trauma-induced plasmalemma disruptions in three-dimensional neural cultures are dependent on strain modality and rate. J Neurotrauma. (2011) 28:2219–33. doi: 10.1089/neu.2011.1841
76. Bowman CL, Ding JP, Sachs F, Sokabe M. Mechanotransducing ion channels in astrocytes. Brain Res. (1992) 584:272–86. doi: 10.1016/0006-8993(92)90906-P
77. Islas L, Pasantes-Morales H, Sanchez JA. Characterization of stretch-activated ion channels in cultured astrocytes. Glia. (1993) 8:87–96. doi: 10.1002/glia.440080204
78. Floyd CL, Gorin FA, Lyeth BG. Mechanical strain injury increases intracellular sodium and reverses Na+/Ca2+ exchange in cortical astrocytes. Glia. (2005) 51:35–46. doi: 10.1002/glia.20183
79. Neary JT, Kang Y, Willoughby KA, Ellis EF. Activation of extracellular signal-regulated kinase by stretch-induced injury in astrocytes involves extracellular ATP and P2 purinergic receptors. J Neurosci. (2003) 23:2348–56. doi: 10.1523/JNEUROSCI.23-06-02348.2003
80. Neary JT, Kang Y, Tran M, Feld J. Traumatic injury activates protein kinase B/Akt in cultured astrocytes: role of extracellular ATP and P2 purinergic receptors. J Neurotrauma. (2005) 22:491–500. doi: 10.1089/neu.2005.22.491
81. Hoffman SW, Rzigalinski BA, Willoughby KA, Ellis EF. Astrocytes generate isoprostanes in response to trauma or oxygen radicals. J Neurotrauma. (2000) 17:415–20. doi: 10.1089/neu.2000.17.415
82. Ostrow LW, Suchyna TM, Sachs F. Stretch induced endothelin-1 secretion by adult rat astrocytes involves calcium influx via stretch-activated ion channels (SACs). Biochem Biophys Res Commun. (2011) 410:81–6. doi: 10.1016/j.bbrc.2011.05.109
83. Giulian D, Lachman LB. Interleukin-1 stimulation of astroglial proliferation after brain injury. Science. (1985) 228:497–9. doi: 10.1126/science.3872478
84. Selmaj KW, Farooq M, Norton WT, Raine CS, Brosnan CF. Proliferation of astrocytes in vitro in response to cytokines. A primary role for tumor necrosis factor. J Immunol. (1990) 144:129–35.
85. Balasingam V, Tejada-Berges T, Wright E, Bouckova R, Yong VW. Reactive astrogliosis in the neonatal mouse brain and its modulation by cytokines. J Neurosci. (1994) 14:846–56. doi: 10.1523/JNEUROSCI.14-02-00846.1994
86. Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. (1997) 20:570–7. doi: 10.1016/S0166-2236(97)01139-9
87. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
88. Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosc. (2008) 28:7231–43. doi: 10.1523/JNEUROSCI.1709-08.2008
89. Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. (2006) 12:829–34. doi: 10.1038/nm1425
90. Gris P, Tighe A, Levin D, Sharma R, Brown A. Transcriptional regulation of scar gene expression in primary astrocytes. Glia. (2007) 55:1145–55. doi: 10.1002/glia.20537
91. Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, et al. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J Neurosci. (2010) 30:5843–54. doi: 10.1523/JNEUROSCI.0137-10.2010
92. Winter CG, Saotome Y, Levison SW, Hirsh D. A role for ciliary neurotrophic factor as an inducer of reactive gliosis, the glial response to central nervous system injury. Proc Natl Acad Sci USA. (1995) 92:5865–9. doi: 10.1073/pnas.92.13.5865
93. Rabchevsky AG, Weinitz JM, Coulpier M, Fages C, Tinel M, Junier MP. A role for transforming growth factor alpha as an inducer of astrogliosis. J Neurosci. (1998) 18:10541–52. doi: 10.1523/JNEUROSCI.18-24-10541.1998
94. Lo U, Selvaraj V, Plane JM, Chechneva OV, Otsu K, Deng W. p38α (MAPK14) critically regulates the immunological response and the production of specific cytokines and chemokines in astrocytes. Sci Rep. (2014) 4:7405. doi: 10.1038/srep07405
95. Ambrosini E, Remoli ME, Giacomini E, Rosicarelli B, Serafini B, Lande R, et al. Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. J Neuropathol Exp Neurol. (2005) 64:706–15. doi: 10.1097/01.jnen.0000173893.01929.fc
96. Saba J, Turati J, Ramírez D, Carniglia L, Durand D, Lasaga M, et al. Astrocyte truncated tropomyosin receptor kinase B mediates brain-derived neurotrophic factor anti-apoptotic effect leading to neuroprotection. J Neurochem. (2018) 146:686–702. doi: 10.1111/jnc.14476
97. Duarte Azevedo M, Sander S, Tenenbaum GDNF L. a neuron-derived factor upregulated in glial cells during disease. J Clin Med. (2020) 9:456. doi: 10.3390/jcm9020456
98. Oh S-J, Han K-S, Park H, Woo DH, Kim HY, Traynelis SF, et al. Protease activated receptor 1-induced glutamate release in cultured astrocytes is mediated by Bestrophin-1 channel but not by vesicular exocytosis. Mol Brain. (2012) 5:38. doi: 10.1186/1756-6606-5-38
99. Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease. Nat Med. (2014) 20:886–96. doi: 10.1038/nm.3639
100. McKeon RJ, Schreiber RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. (1991) 11:3398–411. doi: 10.1523/JNEUROSCI.11-11-03398.1991
101. Hsu JYC, Bourguignon LYW, Adams CM, Peyrollier K, Zhang H, Fandel T, et al. Matrix metalloproteinase-9 facilitates glial scar formation in the injured spinal cord. J Neurosci. (2008) 28:13467–77. doi: 10.1523/JNEUROSCI.2287-08.2008
102. Liddelow SA, Barres BA. Reactive astrocytes: production, function, therapeutic potential. Immunity. (2017) 46:957–67. doi: 10.1016/j.immuni.2017.06.006
103. Pitter KL, Tamagno I, Feng X, Ghosal K, Amankulor N, Holland EC, et al. The SHH/Gli pathway is reactivated in reactive glia and drives proliferation in response to neurodegeneration-induced lesions. Glia. (2014) 62:1595–607. doi: 10.1002/glia.22702
104. Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. (2004) 24:2143–55. doi: 10.1523/JNEUROSCI.3547-03.2004
105. Lööv C, Hillered L, Ebendal T, Erlandsson A. Engulfing astrocytes protect neurons from contact-induced apoptosis following injury. PLoS ONE. (2012) 7:e33090. doi: 10.1371/journal.pone.0033090
106. Morizawa YM, Hirayama Y, Ohno N, Shibata S, Shigetomi E, Sui Y, et al. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat Commun. (2017) 8:28. doi: 10.1038/s41467-017-00037-1
107. Liang J, Takeuchi H, Doi Y, Kawanokuchi J, Sonobe Y, Jin S, et al. Excitatory amino acid transporter expression by astrocytes is neuroprotective against microglial excitotoxicity. Brain Res. (2008) 1210:11–9. doi: 10.1016/j.brainres.2008.03.012
108. Anjum A, Yazid Di M, Fauzi Daud M, Idris J, Ng AMH, Selvi Naicker A, et al. Spinal cord injury: pathophysiology, multimolecular interactions, and underlying recovery mechanisms. Int J Mol Sci. (2020) 21:7533. doi: 10.3390/ijms21207533
109. Cooper JG, Jeong SJ, McGuire TL, Sharma S, Wang W, Bhattacharyya S, et al. Fibronectin EDA forms the chronic fibrotic scar after contusive spinal cord injury. Neurobiol Dis. (2018) 116:60–8. doi: 10.1016/j.nbd.2018.04.014
110. Hawkins LA, Devitt A. Current understanding of the mechanisms for clearance of apoptotic cells-a fine balance. J Cell Death. (2013) 6:57–68. doi: 10.4137/JCD.S11037
111. Yuan Y-M, He C. The glial scar in spinal cord injury and repair. Neurosci Bull. (2013) 29:421–35. doi: 10.1007/s12264-013-1358-3
112. Bradbury EJ, Moon LDF, Popat RJ, King VR, Bennett GS, Patel PN, et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. (2002) 416:636–40. doi: 10.1038/416636a
113. Lee H, McKeon RJ, Bellamkonda RV. Sustained delivery of thermostabilized chABC enhances axonal sprouting and functional recovery after spinal cord injury. Proc Natl Acad Sci USA. (2010) 107:3340–5. doi: 10.1073/pnas.0905437106
114. Smith-Thomas LC, Stevens J, Fok-Seang J, Faissner A, Rogers JH, Fawcett JW. Increased axon regeneration in astrocytes grown in the presence of proteoglycan synthesis inhibitors. J Cell Sci. (1995) 108(Pt 3):1307–15. doi: 10.1242/jcs.108.3.1307
115. Siebert JR, Stelzner DJ, Osterhout DJ. Chondroitinase treatment following spinal contusion injury increases migration of oligodendrocyte progenitor cells. Exp Neurol. (2011) 231:19–29. doi: 10.1016/j.expneurol.2011.05.002
116. Siebert JR, Osterhout DJ. The inhibitory effects of chondroitin sulfate proteoglycans on oligodendrocytes. J Neurochem. (2011) 119:176–88. doi: 10.1111/j.1471-4159.2011.07370.x
117. Wang Y, Cheng X, He Q, Zheng Y, Kim DH, Whittemore SR, et al. Astrocytes from the contused spinal cord inhibit oligodendrocyte differentiation of adult oligodendrocyte precursor cells by increasing the expression of bone morphogenetic proteins. J Neurosci. (2011) 31:6053–8. doi: 10.1523/JNEUROSCI.5524-09.2011
118. Hammond TR, Gadea A, Dupree J, Kerninon C, Nait-Oumesmar B, Aguirre A, et al. Astrocyte-derived endothelin-1 inhibits remyelination through notch activation. Neuron. (2014) 81:588–602. doi: 10.1016/j.neuron.2013.11.015
119. Barkho BZ, Song H, Aimone JB, Smrt RD, Kuwabara T, Nakashima K, et al. Identification of astrocyte-expressed factors that modulate neural stem/progenitor cell differentiation. Stem Cells Dev. (2006) 15:407–21. doi: 10.1089/scd.2006.15.407
120. David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. (2011) 12:388–99. doi: 10.1038/nrn3053
121. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
122. Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol. (2015) 11:56–64. doi: 10.1038/nrneurol.2014.207
123. Murray PJ. Macrophage polarization. Annu Rev Physiol. (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
124. Milich LM, Choi JS, Ryan C, Cerqueira SR, Benavides S, Yahn SL, et al. Single-cell analysis of the cellular heterogeneity and interactions in the injured mouse spinal cord. J Exp Med. (2021) 218:e20210040. doi: 10.1084/jem.20210040
125. Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. (2019) 22:1021–35. doi: 10.1038/s41593-019-0393-4
126. Carlson SL, Parrish ME, Springer JE, Doty K, Dossett L. Acute inflammatory response in spinal cord following impact injury. Exp Neurol. (1998) 151:77–88. doi: 10.1006/exnr.1998.6785
127. Stirling DP, Yong VW. Dynamics of the inflammatory response after murine spinal cord injury revealed by flow cytometry. J Neurosci Res. (2008) 86:1944–58. doi: 10.1002/jnr.21659
128. Scholz M, Cinatl J, Schädel-Höpfner M, Windolf J. Neutrophils and the blood-brain barrier dysfunction after trauma. Med Res Rev. (2007) 27:401–16. doi: 10.1002/med.20064
129. Ankeny DP, Lucin KM, Sanders VM, McGaughy VM, Popovich PG. Spinal cord injury triggers systemic autoimmunity: evidence for chronic B lymphocyte activation and lupus-like autoantibody synthesis. J Neurochem. (2006) 99:1073–87. doi: 10.1111/j.1471-4159.2006.04147.x
130. Pineau I, Lacroix S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol. (2007) 500:267–85. doi: 10.1002/cne.21149
131. Lawrence CB, Allan SM, Rothwell NJ. Interleukin-1beta and the interleukin-1 receptor antagonist act in the striatum to modify excitotoxic brain damage in the rat. Eur J Neurosci. (1998) 10:1188–95. doi: 10.1046/j.1460-9568.1998.00136.x
132. Loddick SA, Rothwell NJ. Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab. (1996) 16:932–40. doi: 10.1097/00004647-199609000-00017
133. Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, et al. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neurosci. (2008) 28:11391–400. doi: 10.1523/JNEUROSCI.3708-08.2008
134. Genovese T, Mazzon E, Crisafulli C, Di Paola R, Muià C, Esposito E, et al. TNF-alpha blockage in a mouse model of SCI: evidence for improved outcome. Shock. (2008) 29:32–41. doi: 10.1097/shk.0b013e318059053a
135. Probert L, Eugster HP, Akassoglou K, Bauer J, Frei K H, Lassmann, et al. TNFR1 signalling is critical for the development of demyelination and the limitation of T-cell responses during immune-mediated CNS disease. Brain. (2000) 123(Pt 10):2005–19. doi: 10.1093/brain/123.10.2005
136. Genovese T, Esposito E, Mazzon E, Di Paola R, Caminiti R, Bramanti P, et al. Absence of endogenous interleukin-10 enhances secondary inflammatory process after spinal cord compression injury in mice. J Neurochem. (2009) 108:1360–72. doi: 10.1111/j.1471-4159.2009.05899.x
137. Chen JY, Fu EJ, Patel PR, Hostetler AJ, Sawan HA, Moss KA, et al. Lentiviral interleukin-10 gene therapy preserves fine motor circuitry and function after a cervical spinal cord injury in male and female mice. Neurotherapeutics. (2021) 18:503–14. doi: 10.1007/s13311-020-00946-y
138. Hung K-S, Tsai S-H, Lee T-C, Lin J-W, Chang C-K, Chiu W-T. Gene transfer of insulin-like growth factor-I providing neuroprotection after spinal cord injury in rats. J Neurosurg Spine. (2007) 6:35–46. doi: 10.3171/spi.2007.6.1.35
139. Sharma HS, Nyberg F, Westman J, Alm P, Gordh T, Lindholm D. Brain derived neurotrophic factor and insulin like growth factor-1 attenuate upregulation of nitric oxide synthase and cell injury following trauma to the spinal cord. An immunohistochemical study in the rat. Amino Acids. (1998) 14:121–9. doi: 10.1007/BF01345252
140. Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. (1997) 3:73–6. doi: 10.1038/nm0197-73
141. Kanno H, Ozawa H, Sekiguchi A, Yamaya S, Itoi E. Induction of autophagy and autophagic cell death in damaged neural tissue after acute spinal cord injury in mice. Spine. (2011) 36:E1427–34. doi: 10.1097/BRS.0b013e3182028c3a
142. Almad A, Sahinkaya FR, McTigue DM. Oligodendrocyte fate after spinal cord injury. Neurotherapeutics. (2011) 8:262–73. doi: 10.1007/s13311-011-0033-5
143. Grossman SD, Rosenberg LJ, Wrathall JR. Temporal-spatial pattern of acute neuronal and glial loss after spinal cord contusion. Exp Neurol. (2001) 168:273–82. doi: 10.1006/exnr.2001.7628
144. Lytle JM, Wrathall JR. Glial cell loss, proliferation and replacement in the contused murine spinal cord. Eur J Neurosci. (2007) 25:1711–24. doi: 10.1111/j.1460-9568.2007.05390.x
145. Griffiths IR, McCulloch MC. Nerve fibres in spinal cord impact injuries. Part 1. Changes in the myelin sheath during the initial 5 weeks. J Neurol Sci. (1983) 58:335–49. doi: 10.1016/0022-510X(83)90093-X
146. Bao F, Liu D. Hydroxyl radicals generated in the rat spinal cord at the level produced by impact injury induce cell death by necrosis and apoptosis: protection by a metalloporphyrin. Neuroscience. (2004) 126:285–95. doi: 10.1016/j.neuroscience.2004.03.054
147. Liu D, Sybert TE, Qian H, Liu J. Superoxide production after spinal injury detected by microperfusion of cytochrome c. Free Radic Biol Med. (1998) 25:298–304. doi: 10.1016/S0891-5849(98)00055-0
148. Liu D, Ling X, Wen J, Liu J. The role of reactive nitrogen species in secondary spinal cord injury: formation of nitric oxide, peroxynitrite, nitrated protein. J Neurochem. (2000) 75:2144–54. doi: 10.1046/j.1471-4159.2000.0752144.x
149. McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. (1998) 4:291–7. doi: 10.1038/nm0398-291
150. Matute C, Torre I, Perez-Cerda F, Perez-Samartin A, Alberdi E, Etxebarria E, et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci. (2007) 27:9525–33. doi: 10.1523/JNEUROSCI.0579-07.2007
151. Inukai T, Uchida K, Nakajima H, Yayama T, Kobayashi S, Mwaka ES, et al. Tumor necrosis factor-alpha and its receptors contribute to apoptosis of oligodendrocytes in the spinal cord of spinal hyperostotic mouse (twy/twy) sustaining chronic mechanical compression. Spine. (2009) 34:2848–57. doi: 10.1097/BRS.0b013e3181b0d078
152. Kanno H, Ozawa H, Sekiguchi A, Itoi E. Spinal cord injury induces upregulation of Beclin 1 and promotes autophagic cell death. Neurobiol Dis. (2009) 33:143–8. doi: 10.1016/j.nbd.2008.09.009
153. Smith PM, Jeffery ND. Histological and ultrastructural analysis of white matter damage after naturally-occurring spinal cord injury. Brain Pathol. (2006) 16:99–109. doi: 10.1111/j.1750-3639.2006.00001.x
154. Guest JD, Hiester ED, Bunge RP. Demyelination and Schwann cell responses adjacent to injury epicenter cavities following chronic human spinal cord injury. Exp Neurol. (2005) 192:384–93. doi: 10.1016/j.expneurol.2004.11.033
155. Plemel JR, Keough MB, Duncan GJ, Sparling JS, Yong VW, Stys PK, et al. Remyelination after spinal cord injury: is it a target for repair? Prog Neurobiol. (2014) 117:54–72. doi: 10.1016/j.pneurobio.2014.02.006
156. Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. J Neurosci. (1992) 12:430–9. doi: 10.1523/JNEUROSCI.12-02-00430.1992
157. Li S, Stys PK. Na(+)-K(+)-ATPase inhibition and depolarization induce glutamate release via reverse Na(+)-dependent transport in spinal cord white matter. Neuroscience. (2001) 107:675–83. doi: 10.1016/S0306-4522(01)00385-2
158. Irvine KA, Blakemore WF. Remyelination protects axons from demyelination-associated axon degeneration. Brain. (2008) 131:1464–77. doi: 10.1093/brain/awn080
159. McTigue DM, Wei P, Stokes BT. Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J Neurosci. (2001) 21:3392–400. doi: 10.1523/JNEUROSCI.21-10-03392.2001
160. Assinck P, Duncan GJ, Plemel JR, Lee MJ, Stratton JA, Manesh SB, et al. Myelinogenic plasticity of oligodendrocyte precursor cells following spinal cord contusion injury. J Neurosci. (2017) 37:8635–54. doi: 10.1523/JNEUROSCI.2409-16.2017
161. Bartus K, Burnside ER, Galino J, James ND, Bennett DLH, Bradbury EJ. ErbB receptor signaling directly controls oligodendrocyte progenitor cell transformation and spontaneous remyelination after spinal cord injury. Glia. (2019) 67:1036–46. doi: 10.1002/glia.23586
162. Hackett AR, Yahn SL, Lyapichev K, Dajnoki A, Lee DH, Rodriguez M, et al. Injury type-dependent differentiation of NG2 glia into heterogeneous astrocytes. Exp Neurol. (2018) 308:72–9. doi: 10.1016/j.expneurol.2018.07.001
163. Horky LL, Galimi F, Gage FH, Horner PJ. Fate of endogenous stem/progenitor cells following spinal cord injury. J Comp Neurol. (2006) 498:525–38. doi: 10.1002/cne.21065
164. Karimi-Abdolrezaee S, Schut D, Wang J, Fehlings MG. Chondroitinase and growth factors enhance activation and oligodendrocyte differentiation of endogenous neural precursor cells after spinal cord injury. PLoS ONE. (2012) 7:e37589. doi: 10.1371/journal.pone.0037589
165. Powers BE, Sellers DL, Lovelett EA, Cheung W, Aalami SP, Zapertov N, et al. Remyelination reporter reveals prolonged refinement of spontaneously regenerated myelin. Proc Natl Acad Sci USA. (2013) 110:4075–80. doi: 10.1073/pnas.1210293110
166. Alizadeh A, Dyck SM, Karimi-Abdolrezaee S. Myelin damage and repair in pathologic CNS: challenges and prospects. Front Mol Neurosci. (2015) 8:35. doi: 10.3389/fnmol.2015.00035
167. Nashmi R, Fehlings MG. Changes in axonal physiology and morphology after chronic compressive injury of the rat thoracic spinal cord. Neuroscience. (2001) 104:235–51. doi: 10.1016/S0306-4522(01)00009-4
168. Mothe AJ, Tator CH. Proliferation, migration, and differentiation of endogenous ependymal region stem/progenitor cells following minimal spinal cord injury in the adult rat. Neuroscience. (2005) 131:177–87. doi: 10.1016/j.neuroscience.2004.10.011
Keywords: spinal cord injury, glial cells, reactive astrocytes, microglia, neuroinflammation, remyelination
Citation: Wang R, Zhou R, Chen Z, Gao S and Zhou F (2022) The Glial Cells Respond to Spinal Cord Injury. Front. Neurol. 13:844497. doi: 10.3389/fneur.2022.844497
Received: 28 December 2021; Accepted: 08 March 2022;
Published: 06 May 2022.
Edited by:
Mårten Risling, Karolinska Institutet (KI), SwedenReviewed by:
Andrew David Gaudet, University of Texas at Austin, United StatesAminata Coulibaly, University of Virginia, United States
Copyright © 2022 Wang, Zhou, Chen, Gao and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Zhou, emhvdWZAYmptdS5lZHUuY24=
†These authors have contributed equally to this work