 De-Tian Liu1†
De-Tian Liu1† Xue-Qing Tang1†
Xue-Qing Tang1† Rui-Ping Wan2†Sheng Luo1Bao-Zhu Guan1
Rui-Ping Wan2†Sheng Luo1Bao-Zhu Guan1 Bin Li1Li-Hong Liu1
Bin Li1Li-Hong Liu1 Bing-Mei Li1
Bing-Mei Li1 Zhi-Gang Liu2Long-Shan Xie3*
Zhi-Gang Liu2Long-Shan Xie3* Yong-Hong Yi1*
Yong-Hong Yi1*- 1Key Laboratory of Neurogenetics and Channelopathies of Guangdong Province and the Ministry of Education of China, Institute of Neuroscience and Department of Neurology of the Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
- 2Department of Pediatrics, Foshan Women and Children Hospital Affiliated to Southern Medical University, Foshan, China
- 3First People's Hospital of Foshan, Foshan, China
Introduction: PRRT2 is a major causative gene for self-limited familial neonatal-infantile epilepsy, paroxysmal kinesigenic dyskinesia, and paroxysmal kinesigenic dyskinesia with infantile convulsions. Voluntary movement trigger is prominent in adolescence and adulthood, but the triggers are unknown in infants.
Methods: A gene panel designed for targeted next-generation sequencing (NGS) was used to screen genetic abnormalities in a cohort of 45 cases with infantile convulsions. The copy number variation was detected by a computational method based on the normalized depth of coverage and validated by a quantitative real-time polymerase chain reaction (RT-qPCR) method. The genotype-phenotype correlation of the PRRT2 mutation gene was analyzed.
Results: A de novo heterozygous PRRT2 deletion was identified in a child who had infantile convulsions induced by vigorous sucking. Seizures happened during the change of feeding behavior from breast to formula, which led to hungry and vigorous sucking. Ictal electroencephalograms recorded seizures with focal origination, which provided direct evidence of epileptic seizures in infants with PRRT2 mutations. Seizures stopped soon after the feeding behavior was changed by reducing feeding interval time and extending feeding duration. Data reanalysis on our previously reported cases with PRRT2 mutations showed that six of 18 (33.3%) patients had infantile convulsions or infantile non-convulsion seizures during feeding. The mutations included two truncating mutations (c.579dupA/p.Glu194Argfs*6, and c.649dupC/p.Arg217Profs*8) that were identified in each of the three affected individuals.
Conclusions: This study suggests that feeding, especially vigorous sucking, is potentially a trigger and highlights the significance of feeding behavior in preventing seizures in infants with PRRT2 mutations. Identification of PRRT2 haploinsufficiency mutations in the patients with infantile convulsions induced by sucking suggested a potential genotype-phenotype correlation.
Introduction
The PRRT2 gene (OMIM: 614386) encodes a transmembrane protein containing a proline-rich domain in the N-terminal half and being involved in synaptic transmission (1). PRRT2-associated diseases are mainly paroxysmal kinesigenic dyskinesia (PKD) and paroxysmal hypnogenic dyskinesia (PHD) in adults and self-limited familial neonatal-infantile epilepsy or infantile convulsion and choreoathetosis (ICCA) in infants. Infantile convulsion (IC) is the most common feature in early life among the wide spectrum of PRRT2 phenotypes (2). A previous study demonstrated that self-limited familial neonatal-infantile epilepsy accounted for 41.7%, PKD accounted for 38.7%, and ICCA accounted for 14.3% of the PRRT2-associated phenotypes (3). The PRRT2 mutations previously reported included missense, non-sense, splicing, deletions, insertions, indels, and complex rearrangement; and the majority are truncating mutations that lead to haploinsufficiency. Voluntary movement triggering is a prominent feature in patients of adolescence and adulthood, which is highlighted in the diagnostic criteria for PKD (2). However, the triggers of attacks in early childhood were unknown. We previously observed several patients with PRRT2 mutations that had attacks during feeding, suggesting sucking as a possible kinesigenic trigger (4). In this study, we screened a cohort of 45 cases with infant seizures and identified a PRRT2 heterozygous deletion mutation in a child with frequent seizures induced by vigorous sucking and became seizure-free after changing feeding behavior. We analyzed the clinical and genetic features of the cases who had attacks during feeding and found that their PRRT2 mutations were all truncating or deletion mutations that would lead to haploinsufficiency, which suggested a potential genotype-phenotype correlation and clinical significance.
Materials and methods
Patients
A total of 45 patients with infant convulsion were recruited from the Epilepsy Center of the Second Affiliated Hospital of Guangzhou Medical University and the First People's Hospital of Foshan. Epileptic seizures and epilepsy syndromes were diagnosed and classified according to the criteria of the Commission on Classification and Terminology of the International League Against Epilepsy (1981, 1989, 2001, 2010, and 2017). The collected clinical data included detailed semiology, onset age, seizure type, the evolution of disorder and frequency, family history, response to anti-seizure medications, general and neurological examination, and brain magnetic resonance imaging (MRI). Long-term video-EEG monitoring records that included open-close eyes test, hyperventilation, intermittent photic stimulation, and sleep recording were obtained.
This study complied with the principles of the International Committee of Medical Journal Editors about patient consent for research or participation. Written informed consent was obtained from the individuals or legal guardians. Ethical approval had been obtained from the Ethics Committee of the Second Affiliated Hospital of Guangzhou Medical University.
Targeted sequencing and filtering
Peripheral blood samples were collected from the probands, their parents, and other family members if available. Genomic DNA (gDNA) was extracted using the Qiagen Flexi Gene DNA Kit (Qiagen, Hilden, Germany). The PRRT2 mutations in the early six cases in our previous study were identified by direct Sanger sequencing, targeting the coding regions (4). A custom-targeted NGS gene panel was performed in recent years to screen gene abnormalities associated with episodic disorders (5), including PRRT2, CHRNA4, KCNA1, KCNQ2, KCNMA1, PNKD, SCN2A, and SLC2A1 genes that are associated with BFIS and PKD. Trios-based exome paired-end sequencing was performed on the Illumina Nextseq 500 platform by capture probe NeuMet_V1. The mean read depth for each sample was 208-fold. The raw data were aligned to the reference genome (GRCh37/hg19). Single nucleotide variants (SNVs) and insertions/deletions (indels) filtering were performed by using the Genome Analysis Toolkit (6). Stepwise filtering was applied to the derived candidate causative variants as we previously described (7). The copy number variations (CNVs) were performed by a computational method (CNVkit) based on the normalized depth of coverage, which used both the targeted reads and the non-specifically captured off-target reads to infer copy number (8). Sanger sequencing was applied to confirm the candidate variants.
Quantitative real-time PCR (QPCR) analysis
The qPCR was used to verify the heterozygous PRRT2 deletion at the gDNA level. The parents were regarded as healthy controls. Primers F1 (5′-GGATGCAGAGGGAGTGGAATG-3′) and R1 (5′-CCCAGCACCCCAAAATCAAC-3′) were designed to amplify the fore part of PRRT2, primers F2 (5′-GAAGGCACCCAGAAACCTCG-3′) and R2 (5′-ACAGCATAAGCGAAGGCCAC-3′) were designed to amplify the middle part of PRRT2, and primers F3 (5′-AACACCTCCCCAACTCTGCG-3′) and R3 (5′-CCAGGGTGGAGGTCCAGAGAAT-3′) were designed to amplify the whole length of PRRT2. The total volume of each sample reaction was 10 μl, including 1.5 μl (15 ng gDNA), 1 μl forward primer (2 μM), 1 μl reverse primer (2 μM),.2 μl ROX Reference Dye (50×), 5 μl SYBR Premix Ex Taq (2×), and 1.3 μl double distilled water. Each sample was run in triplicate on a 7300 Realtime PCR System (Applied Biosystems) by using the SYBR® Premix Ex Taq™ Reagent (Takara). The expression of POLR2A as a reference gene was used for data normalization. Relative ratios of the gene expression were calculated by formula: r = 2−ΔΔCt with ΔΔCt = (CtPRRT2 – CtPOLR2A) ind tested - (CtPRRT2 – CtPOLR2A) ind ref. Values were calculated about the father and arbitrarily attributed to the value of 1.0. SPSS 19.0 software (Chicago, USA) was used for statistical analysis.
Results
One de novo PRRT2 mutation was identified in a child with infant seizures. The child was a full-term boy who was born to healthy non-consanguineous parents. His early development was normal. The seizures began at the age of 4 months when breastfeeding was switched to formula feeding. He presented his first convulsion when he started sucking eagerly and vigorously. The seizure began with a deviation of head and eyes to the right, then cyanosis, and followed by the loss of consciousness and generalized hypertonic lasting about 1 min. The attacks repeated 2–10 times per day and mostly occurred during vigorous sucking. The ictal EEGs recorded a seizure that started with spikes from the left frontal lobe and gradually generalized, lasting for 75 s, followed by electrical suppression (Figure 1). Interictal EEGs revealed low voltage spikes and waves in the left frontal lobe with a normal background. Brain MRI was normal. Blood routine and biochemical tests were normal. Valproate (16 mg/kg/d) was applied on the third day after onset but did not stop the seizures. Vigorous sucking was subsequently avoided by shortening interval time, extending the duration of feeding, and adding the rice paste. The child was seizure-free since then (followed-up for 2 years until now). EEG became normal in the third month after being seizure-free. Valproate was discontinued after a year. The neurodevelopment was normal at 3 years of follow-up. Using the gene panel, we failed to find any pathogenic point mutations in this child. Heterozygous PRRT2 gene deletion was identified by the computational method of CNVs (Figure 2A) and validated by the qPCR method (Figure 2B). The mutation was a heterozygous whole deletion of one allele of the PRRT2 gene, which led to haploinsufficiency. Co-segregation analysis showed that the mutation was a de novo mutation (Figure 2C).
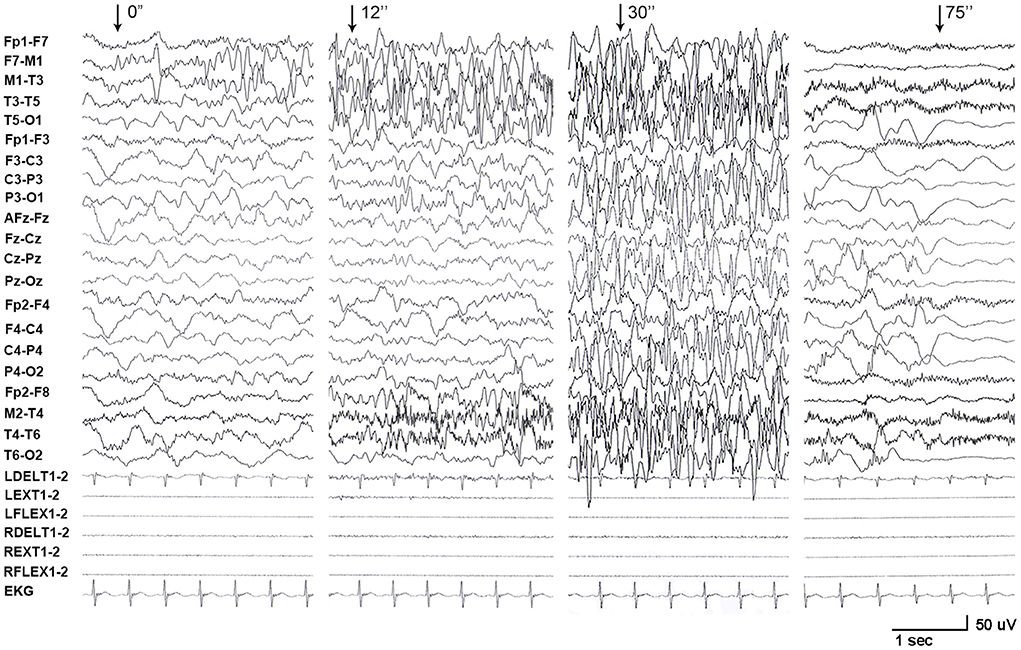
Figure 1. EEG recordings from patient with PRRT2 variants. A long-term video electroencephalogram recorded a seizure that started with spikes originating from the left frontal lobe and gradually generalized, lasting for 75 s, followed by electrical suppression.
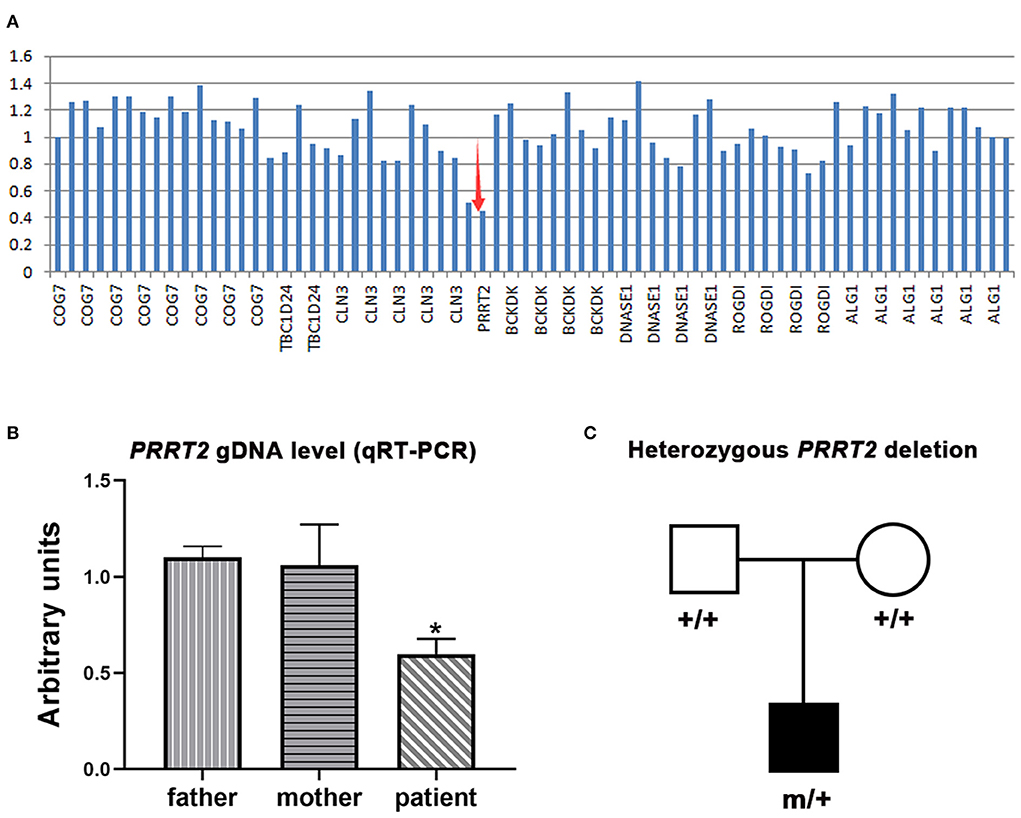
Figure 2. Genetic data of the cases with PRRT2 variants. (A) Copy number variations analysis showed a heterozygous PRRT2 deletion (red arrows). (B) Relative quantification of genomic DNA (gDNA) confirmed the deletion. The patient presented a reduced PRRT2 gDNA level (half of the level of his parents). Error bar, mean ± SEM, n = 3; *p < 0.05, vs. his father and mother. (C) Pedigree of the family, showing the heterozygous PRRT2 deletion was a de novo mutation.
We reanalyzed the data from the cohort of 34 individuals with PRRT2 mutations (Figure 2 in that article) (4). Six patients were found to have experienced feeding-associated infantile convulsions or infantile non-convulsion seizures. The clinical and genetic features of the patients are summarized in Table 1. All patients had heterozygous deleterious mutations, including two truncating mutations, c.579dupA/p.Glu194Argfs*6, and c.649dupC/p.Arg217Profs*8, that were identified in each of the three affected individuals. Mutation c.649dupC/p.Arg217Profs*8 has previously been reported as a hot-spot mutation (3). Except for the deletion mutation that was de novo, the others were of familial inheritance. The average age at onset was 6.1 ± 2.1 months (range 4–10 months). Five patients exhibited classical convulsions. Two patients presented non-convulsive seizures that manifested as a stop of suction, loss of responses, and then cyanosis without a generalized tonic seizure. All patients had normal EEG backgrounds. Three of them had focal (frontal/ temporal) spikes and sharp or slow waves. Six were in remission before 12 months and one was in remission at age of 3 years old. Five patients developed paroxysmal disorders or paroxysmal dyskinesias later in adolescence.
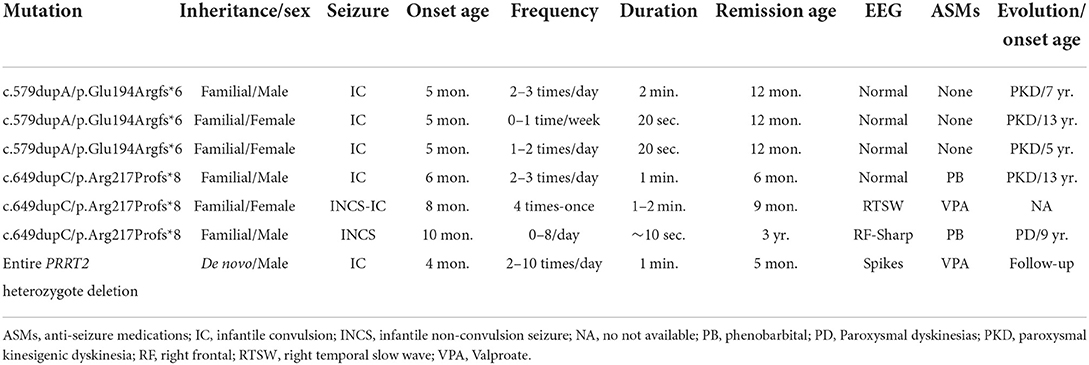
Table 1. Genetic and clinical characteristics of patients with feeding-induced infantile convulsion/non-convulsion seizure caused by PRRT2 mutations.
Discussion and conclusions
The PRRT2 has been identified as the most common causative gene of PKD (9) and other paroxysmal disorders. Loss of function of the gene results in a delay of neuronal migration and a marked decrease in synaptic density, which were suggested to be the mechanism underlying PRRT2-associated disease (1, 10). Paroxysmal movement attack induced by triggering is a prominent feature of the disorders associated with PRRT2 mutations. In this study, we reported seven cases with PRRT2 mutations who presented feeding-induced infantile seizures. Ictal EEGs recorded a seizure with focal origination in the case induced by vigorous sucking. This study suggests feeding, especially sucking, as a potential trigger factor. The seven cases had deletion or truncating mutations that resulted in haploinsufficiency of the PRRT2 gene, suggesting a potential genotype-phenotype correlation. To date, 155 PRRT2 mutations have been identified in patients with episodic disorders (Human Gene Mutation Database, www.hgmd.cf.ac.uk/ac/index.php). Mutations of haploinsufficiency accounted for 60.6%, including truncating, deletion, insertion, and splice-site mutations. The other mutations were missenses. The hot spot mutation c.649dupC/p.Arg217Profs*8 alone even contributed to 73.8% of PKD/IC etiology in a previous study (3). In our early cohort, six of the 18 (33.3%) patients with PRRT2 variant had IC and infantile no-convulsion seizures during feeding (4). Therefore, feeding potentially triggers movement in infants with PRRT2 mutations, especially in those with mutations of haploinsufficiency.
Previously, it was not determined whether the infant convulsion in patients with PRRT2 mutations was epileptic seizures in some cases. Marini et al. (11) reported a recorded focal seizure in a child with PRRT2 mutation. The reported case and the case presented in this study provided direct evidence of epileptic seizures in infants with PRRT2 mutations. Therefore, attention should be paid to triggers in infants with convulsions or seizures. The child in this study got seizure-free since the change of feeding behavior, highlighting the significance of feeding behavior in the early prevention of epileptic seizures and the possible secondary brain injury due to frequent seizures. Vigorous sucking behavior, such as feeding in hunger, should be avoided by maneuvers like reducing feeding interval time and extending feeding duration in infants with PRRT2 mutations. This case also suggests sucking-induced seizures as a reminder of PRRT2 mutations and further genetic tests. Although some patients do not have seizures after a year or two, frequent seizures are harmful. It is crucial to identify the trigger and use appropriate antiepileptic drugs, such as carbamazepine, to stop the seizures.
In conclusion, we identified a de novo heterozygous PRRT2 deletion in a patient who presented infantile seizures induced by sucking. Data reanalysis on our previously reported cases with PRRT2 mutations showed that 33.3% of patients had infantile convulsions or infantile no-convulsion seizures during feeding. This study suggests sucking as a potential trigger movement in infants with PRRT2 mutations, highlighting the significance of feeding behavior in the early prevention of epileptic seizures. All seven patients had PRRT2 mutations of haploinsufficiency, suggesting a potential genotype-phenotype correlation.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: NCBI - PRJNA833568.
Ethics statement
All procedures performed were in accordance with the ethical standards of the institutional committee. The present study was approved by the Ethics Committee of the Second Affiliated Hospital of Guangzhou Medical University.
Author contributions
Substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data were made by SL, B-ZG, BL, L-HL, B-ML, and Z-GL. Drafting the article or revising it critically for important intellectual content were made by Y-HY, L-SX, D-TL, X-QT, and R-PW. Final approval of the version to be published was done by all authors.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 81870903). The funders had no role in study design, data collection, and analysis or in the decision to publish and neither in the preparation of the manuscript.
Acknowledgments
We thank the patient and his parents for participating in this study. Our sincere thanks to Professor Wei-Ping Liao for editing this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
PKD, paroxysmal kinesigenic dyskinesia; PHD, paroxysmal hypnogenic dyskinesia; ICCA, infantile convulsion and choreoathetosis; IC, infantile convulsion; EEG, electroencephalogram; MRI, brain magnetic resonance imaging; CNVs, copy number variations; qPCR, quantitative real-time-polymerase chain reaction.
References
1. Liu YT, Nian FS, Chou WJ, Tai CY, Kwan SY, Chen C, et al. PRRT2 mutations lead to neuronal dysfunction and neurodevelopmental defects. Oncotarget. (2016) 7:39184–96. doi: 10.18632/oncotarget.9258
2. Bruno MK, Hallett M, Gwinn-Hardy K, Sorensen B, Considine E, Tucker S, et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology. (2004) 63:2280–7. doi: 10.1212/01.WNL.0000147298.05983.50
3. Ebrahimi-Fakhari D, Saffari A, Westenberger A, Klein C. The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain. (2015) 138(Pt. 12):3476–95. doi: 10.1093/brain/awv317
4. Liu XR, Wu M, He N, Meng H, Wen L, Wang JL, et al. Novel PRRT2 mutations in paroxysmal dyskinesia patients with variant inheritance and phenotypes. Genes Brain Behav. (2013) 12:234–40. doi: 10.1111/gbb.12008
5. Zhou P, He N, Zhang JW, Lin ZJ, Wang J, Yan LM, et al. Novel mutations and phenotypes of epilepsy-associated genes in epileptic encephalopathies. Genes Brain Behav. (2018) 17:e12456. doi: 10.1111/gbb.12456
6. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. (2011) 43:491–8. doi: 10.1038/ng.806
7. Chen ZR, Liu DT, Meng H, Liu L, Bian WJ, Liu XR, et al. Homozygous missense TPP1 mutation associated with mild late infantile neuronal ceroid lipofuscinosis and the genotype-phenotype correlation. Seizure. (2019) 69:180–5. doi: 10.1016/j.seizure.2018.08.027
8. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. (2016) 12:e1004873. doi: 10.1371/journal.pcbi.1004873
9. Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan GH, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet. (2011) 43:1252–5. doi: 10.1038/ng.1008
10. Tan G-H, Liu Y-Y, Wang L, Li K, Zhang Z-Q, Li H-F, et al. PRRT2 deficiency induces paroxysmal kinesigenic dyskinesia by regulating synaptic transmission in cerebellum. Cell Res. (2017) 28:90–110. doi: 10.1038/cr.2017.128
Keywords: PRRT2, infantile convulsion, seizures, sucking-induced, genotype-phenotype correlation
Citation: Liu D-T, Tang X-Q, Wan R-P, Luo S, Guan B-Z, Li B, Liu L-H, Li B-M, Liu Z-G, Xie L-S and Yi Y-H (2022) PRRT2 gene mutations associated with infantile convulsions induced by sucking and the genotype-phenotype correlation. Front. Neurol. 13:836048. doi: 10.3389/fneur.2022.836048
Received: 11 January 2022; Accepted: 29 June 2022;
Published: 26 July 2022.
Edited by:
Carl E. Stafstrom, Johns Hopkins Medicine, United StatesReviewed by:
Salvatore Mangano, Università degli Studi di Palermo, ItalyDario Pruna, G. Brotzu Hospital, Italy
Copyright © 2022 Liu, Tang, Wan, Luo, Guan, Li, Liu, Li, Liu, Xie and Yi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-Hong Yi, eXloMTY4JiN4MDAwNDA7c2luYS5jb20=; Long-Shan Xie, MTM5Mjk5NTE2ODUmI3gwMDA0MDsxMjYuY29t
†These authors have contributed equally to this work and share first authorship