 Aaron del Pozo
Aaron del Pozo Leanne Lehmann
Leanne Lehmann Kevin M. Knox
Kevin M. Knox Melissa Barker-Haliski
Melissa Barker-Haliski- Department of Pharmacy, School of Pharmacy, University of Washington, Seattle, WA, United States
Older people represent the fastest growing group with epilepsy diagnosis. For example, cerebrovascular disease may underlie roughly 30–50% of epilepsy in older adults and seizures are also an underrecognized comorbidity of Alzheimer's disease (AD). As a result, up to 10% of nursing home residents may take antiseizure medicines (ASMs). Despite the greater incidence of epilepsy in older individuals and increased risk of comorbid seizures in people with AD, aged animals with seizures are strikingly underrepresented in epilepsy drug discovery practice. Increased integration of aged animals into preclinical epilepsy drug discovery could better inform the potential tolerability and pharmacokinetic interactions in aged individuals as the global population becomes increasingly older. Quite simply, the ASMs on the market today were brought forth based on efficacy in young adult, neurologically intact rodents; preclinical information concerning the efficacy and safety of promising ASMs is not routinely evaluated in aged animals. Integrating aged animals more often into basic epilepsy research may also uncover novel treatments for hyperexcitability. For example, cannabidiol and fenfluramine demonstrated clear efficacy in syndrome-specific pediatric models that led to a paradigm shift in the perceived value of pediatric models for ASM discovery practice; aged rodents with seizures or rodents with aging-related neuropathology represent an untapped resource that could similarly change epilepsy drug discovery. This review, therefore, summarizes how aged rodent models have thus far been used for epilepsy research, what studies have been conducted to assess ASM efficacy in aged rodent seizure and epilepsy models, and lastly to identify remaining gaps to engage aging-related neurological disease models for ASM discovery, which may simultaneously reveal novel mechanisms associated with epilepsy.
Epilepsy and Seizures
Seizures and epilepsy have a high incidence worldwide. At least 10% of the population will experience seizures at some point in life (1). Many factors can contribute to a seizure: metabolic disorders (2), alcohol withdrawal (3), illicit drug use (4), acute neurological insults, or high fever (1, 5). Regardless of the inciting mechanism, seizures can be generalized or focal (6, 7). On the other hand, epilepsy is a chronic disease with a cumulative incidence of 3% worldwide. Many factors increase the risk of developing epilepsy: congenital disorders (5), infections (8), trauma (9), tumors (10), vascular accidents (11), and neurodegenerative diseases (12, 13). The incidence of focal seizures increases significantly in individuals aged 65+ (14), resulting in a higher socioeconomic impact for this group vs. younger individuals (15). Epilepsy in older adults can arise secondary to other disorders; ~10–20% of cases of epilepsy syndromes are comorbid with neurodegenerative diseases, such as Alzheimer's Disease (AD) (13, 16). Despite this, the epileptic syndromes most frequently studied in preclinical practice generally favor disease or symptoms that occur in infant or adolescent stages of neurodevelopment, (17, 18). However, despite the fact that older populations have an equal or higher prevalence of epilepsy compared with younger populations (16), comprehensive studies in older animals are rather limited (19, 20). Altogether, epilepsy in older adults is a problem that is more frequently documented in clinical settings than is investigated in pre-clinical practice.
Drug-Resistant Epilepsy: the Importance of Aging
Preclinical antiseizure drug discovery is booming (21, 22). Numerous antiseizure medicines (ASMs) improve the quality of life in people with epilepsy by reducing the number of seizures, as well as generally increasing seizure threshold (19, 23–25). ASMs can regulate the expression and activity of ion channels (especially sodium and calcium channels), inhibit hyperexcitability by increasing GABA levels and/or reducing glutamate levels or regulate the trafficking and release of other neurotransmitters (26). However, about one-third of people with epilepsy suffer from uncontrolled seizures despite pharmacotherapy and are considered “drug-resistant” (27, 28); i.e., when the control of seizures is not achieved with at least two or three appropriately chosen ASMs. One of the most severe consequences of uncontrolled chronic seizures is the increased risk for sudden unexpected death in epilepsy (SUDEP). Unfortunately, the precise mechanisms of drug-resistant epilepsy (DRE) are still unknown. Despite the approval of many new ASMs in the last 20+ years, the percentage of people with DRE has not significantly changed (29). New strategies are needed to improve twenty-first century ASM discovery and meaningfully shift this percentage.
Seizure vulnerability is not uniform with advanced age (15, 16), therefore pharmacological considerations must also be addressed for this group. Older populations respond more effectively to pharmacological treatment than younger populations (30–32). The higher efficacy in seizure control among older people with epilepsy may be due, in part, to age-related differences in etiology. For example, perampanel administration to older people with epilepsy leads to a 50% responder rate of 57% (33), whereas this responder rate is 35% in younger people. At the same time, the absence of ASM treatment may exacerbate the burden of neurodegenerative diseases that have comorbid seizures, such as AD (13, 34). However, older people with epilepsy are at considerable risk for drug-drug interactions due to changes in renal, hepatic, or metabolic function. There must be, as a result, careful consideration of the ASMs to be used for older people with epilepsy. Aged seizure and epilepsy pre-clinical models could be appropriate to elucidate the differences between the old and young brain, determine the potential for adverse effects liability, and identify the genes and pathways involved in both epilepsy and neurodegenerative disorders. Precision medicine approaches for epilepsy to-date have relied on the identification of novel therapies in syndrome-specific models of genetically acquired pediatric epilepsy, with a high degree of success translating those discoveries to clinical populations [e.g., fenfluramine (35) and cannabidiol (36, 37)]. Whether older people with epilepsy represent a similarly rich population to model diverse causes of hyperexcitability and potentially uncover novel pharmacological targets for disease represents an untapped opportunity.
Unfortunately, most studies on epilepsy have employed younger adult animals; the study of epilepsy (and other comorbid conditions) in aged animals is rather limited (32, 38). Thus, this review will update the studies on epilepsy and seizures performed in aged rodents, describe the existing gaps in the study of epilepsy, and propose new targets relevant to the aged brain, information which may help to increase the understanding of epilepsy and discover new pharmacological therapies that are useful in a diversity of epilepsy syndromes.
The Relationship Between Aging and Epilepsy
Modeling the sequelae of epilepsy in aged animals is challenging from a variety of perspectives, including species-specific physiological changes with aging, the interaction of comorbidities observed in people with epilepsy, and the feasibility and cost limitations of inducing clinically relevant neurological disease in aged animals. These factors limit current models, especially concerning the replication of age-related conditions (32, 38).
Studies of Seizures and Epilepsy in Aged Mice
Most of the studies conducted in older animals have compared seizure susceptibility between sexes (Table 1). Few have explored the relationship between aging and epilepsy. Table 1 details the studies that have been conducted since 2010 (32) and their major findings summarized.
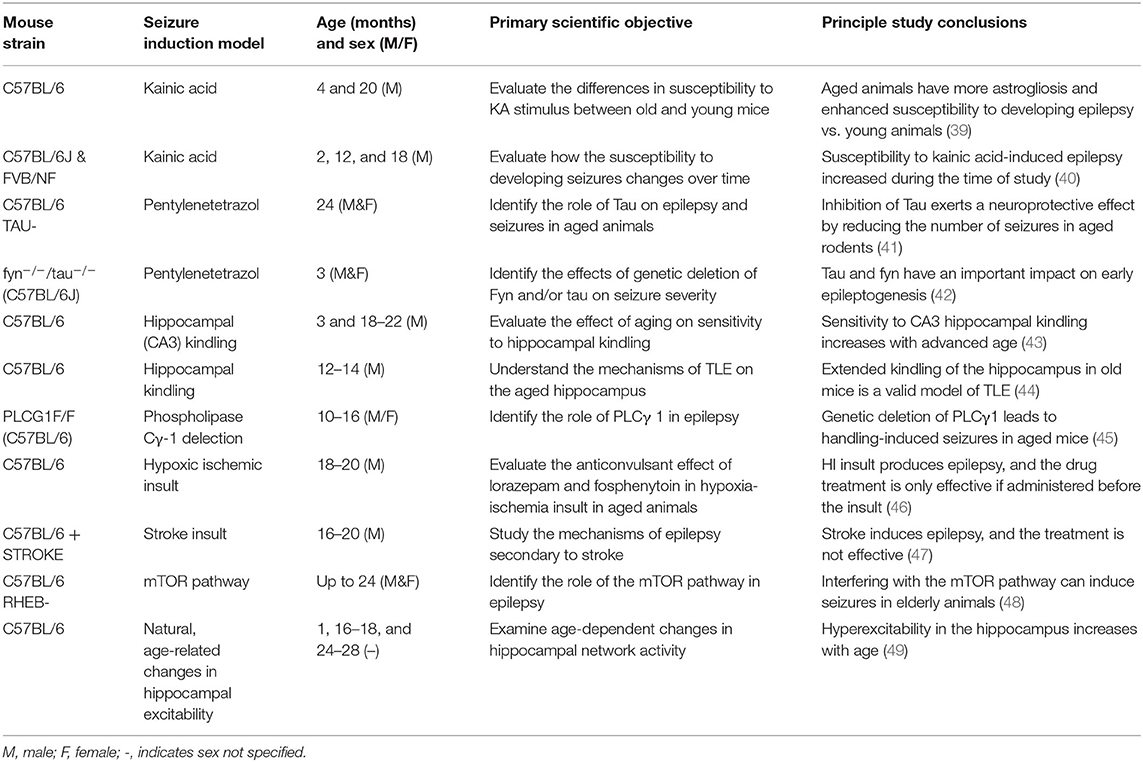
Table 1. Primary pre-clinical studies of epilepsy in aged mice.
Pentylenetetrazol (PTZ) Studies
The chemoconvulsant pentylenetetrazol (PTZ), has provided insight into some of the pathways and genes involved in epilepsy in the aged and degenerating brain (41). In the PTZ model, older mice required a lower dose of this chemoconvulsant vs. younger mice; older mice responded to GABAA agonists better than juvenile mice, mimicking patient responses (50). Further, PTZ-induced epilepsy in accelerated senescence mice (SAM) affects different cell populations, brain areas, and neurotransmitters relative to younger animals (51). During the juvenile and adult stage, the primary cell population affected by PTZ administration is astrocytes, whereas in aged animals, this cell population are neurons. Notably, the GABAergic pathway to prevent seizures in older animals was found to be particularly relevant to prevent seizures (51).
The relevance of tau in aging-related epilepsy has also been interrogated using the acute PTZ seizure model. Tau, a microtubule-associated protein, is a natively unfolded protein in human brains and has several biological roles, including microtubule assembly and stabilization (52). There are age-accelerated changes in neurofibrillary tangle (hyperphosphorylated tau) pathology in people with chronic DRE compared to age-matched individuals without epilepsy (53, 54). Aged tau-knockout (KO) mice that received PTZ administration have increased latency for acute PTZ-induced seizures and reduced seizure severity compared to controls (41). Putra and colleagues confirmed these findings with aged tau KO mice, but then went on to demonstrate that simultaneous deletion of Fyn, a member of the Src family tyrosine kinases (SFKs) that interacts with tau protein, also beneficially alters PTZ-induced seizures severity and latency (42). This beneficial effect of tau deletion seems to be due to the relationship of tau over hyperexcitability, neuroinflammation, and neuronal death (41, 42). These studies reveal that this AD-associated protein and tau-related pathophysiological mechanisms are a relevant target in epilepsy. Thus, PTZ-induced seizures evoked in old mice may affect different cell populations than in younger mice.
KA Studies
Kainic acid (KA)-induced status epilepticus (SE) in mice revealed that some of the most affected areas secondary to seizure induction are the hippocampus, cortex, amygdala, and thalamus (39, 40). Nonetheless, vulnerability to seizures is higher in aged mice, supporting the bimodal distribution of seizure susceptibility in the very young and the very old (40). Furthermore, in FVB mice, seizure stimulation results in greater cell death with advanced age vs. younger animals. Further, glia may play an important role in setting the epileptogenic threshold and determining levels of neurodegeneration (39, 40). In FVB/NJ SAM (32, 55, 56) or melatonin III KO mice (57), the electroconvulsive shock (58) or KA administration increases brain damage (specifically in the hippocampus), as well as the susceptibility to epilepsy in aged animals (39, 40, 58). While studies have more recently also assessed the contributions of tau in the intra-amygdala KA-induced SE model evoked in young-adult mice (59), similar studies to assess the additive impacts of KA-SE and AD-associated proteins (i.e., tau) have yet to be reported in aged animals. Thus, the KA SE model is useful to assess the consequences of aging on seizure susceptibility and epilepsy, as well as to understand the pathophysiological pathways and mechanisms involved.
Kindling Studies
Kindling of aged mice elicits similar biological outcomes as the chemoconvulsants mentioned above. The severity of and susceptibility to hippocampal and CA3 kindling in old C57BL/6 mice increases with advanced age (43, 44). Moreover, these animals not only present alterations on the EEG but also develop cognitive impairment (43, 44). Thus, hippocampal degeneration may explain the detrimental effect of epilepsy in older people with AD (60, 61). Post-mortem confirmed AD cases also exhibit reduced pyramidal cell counts in the hippocampus (62); whether chronic seizures in an aging model evoke similar hippocampal deficits is worthy of further scrutiny.
Other Studies
Aligning with clinical risk factors for epilepsy in old individuals (16), traumatic brain injury (TBI) and stroke are risk factors for epilepsy in aged animals (46, 47). In animal models, stroke-induced epilepsy can also result in death. Thus, animals with stroke or hypoxic-ischemic insult may not only be a valid model to study the effect of age on epilepsy in mouse models (Table 1), but they may also represent a possible SUDEP model (46, 47).
The Ras homolog enriched in brain protein (RHEB; one of the major proteins involved in the mTOR pathway; Table 1) may represent a novel driver of age- and sex-dependent susceptibility to handling-induced seizures (48). This mutation is mechanistically related to everolimus, which is approved as a disease-modifying treatment for tuberous sclerosis complex, a condition characterized by chronic seizures and epilepsy in pediatric populations. Rapalogues, such as everolimus, have been found to be disease-modifying in some (63), but not all (25, 64, 65), animal models of acquired spontaneous recurrent seizures. Notably, the mTOR pathway also plays a critical role in epilepsy and seizures susceptibility in aged rodents (48), but further investigations are certainly needed to more comprehensively define the role of mTOR signaling in spontaneous seizures in aged mice.
Phospholipase Cγ-1 protein in mice (a protein expressed in GABAergic neurons) may explain some differences in epilepsy development between young and older individuals (45). Aged mice with selective deletion of phospholipase Cγ-1 in GABAergic neurons have increased susceptibility to handling-induced spontaneous seizures (45). Further, aged mice with selective phospholipase Cγ-1 deletion in GABAergic neurons have increased spontaneous seizures, but reduced anxiety and deficits in contextual fear memory, despite the reduction in inhibitory synapses observed in the hippocampus (45). Thus, the GABAergic pathway may be particularly relevant to spontaneous seizures, but not epilepsy-related behavioral comorbidities, in aged animals.
Other studies revealed that there is a progressive age-related increase in hippocampal hyperexcitability (specifically in CA3). Variation in the levels of the proto-oncogene c-Fos, responsible, among other things, for synaptic plasticity (49) and the modification of glial phenotype and function are some of the causes that may be involved in this progressive increase in hyperexcitability. Moreover, age-related changes in gliosis and synaptic plasticity can impact the integrity of synapses and neurons (66).
In conclusion, although few additional aged mouse studies have been reported since the last comprehensive review was published (32), these studies reported have shown that the release of neurotransmitters, synaptic plasticity, and metabolic pathways enhance seizure susceptibility with advanced age (Table 1). Despite the differences in mice strain, response to epileptic stimuli, and experimental seizure induction paradigm, these studies are nonetheless influential to broaden understanding of the pathophysiological mechanisms of seizures and epilepsy in the aged brain. These works establish new models to improve the knowledge of the mechanisms of epilepsy and its relationship with old age.
Studies of Seizures and Epilepsy in Aged Rats
During the last decade, studies of epilepsy and aging effects in rats have been even less frequently conducted than in mice, likely as a result of advances in genetic models of aging-related neurological disorders favoring mice, as well as the financial constraints and resource limitations associated with aged rats. Nonetheless, the most notable studies carried out in recent years are summarized in Table 2.
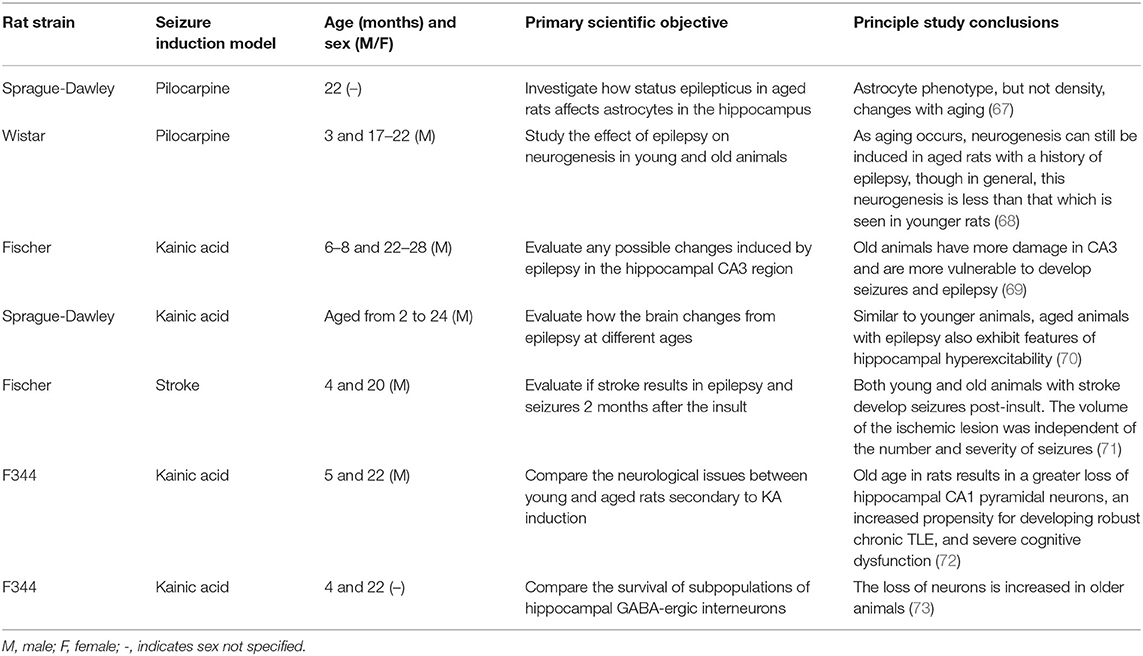
Table 2. Primary pre-clinical studies of epilepsy in aged rats.
Studies in rats highlight the relevance of other pathological mechanisms that are different from those observed in mice. Pilocarpine-induced epilepsy in aged rats triggers an important switch in glial phenotype and response (67). Further, epilepsy-induced in old rats leads to a reduction of their neurogenic properties, resulting in a lower density of basal dendrites and dentate granule cells compared with younger animals (68). Moreover, KA-induced epilepsy in aged rats results in a greater loss of pyramidal neurons in CA3 compared with younger rats (72, 73). Altogether, seizures promote a massive glutamate release, oxidative stress, and inflammation, similar to the impacts of other aging-related disorders, such as AD or stroke (32, 69). Further, in parallel with mice, hippocampal hyperexcitability in aged rats is the most affected biological outcome (69, 70). Epilepsy in aged mice and rats clearly reproduce similar pathophysiology as in humans.
Clearly, there is untapped value in integrating aged models more frequently to better understand the molecular pathways of epilepsy and seizure. As we have been able to successfully uncover many novel therapeutic targets based on findings in pediatric genetic epilepsy models, it is entirely plausible that integrating aged rodents may similarly carry broad relevance to other epilepsy indications, including developmental epileptic encephalopathies (DEE).
Potential Neurobiological Mechanisms
Aging, in general, is associated with cellular, molecular, anatomical, and physiological changes (74). There are likely also intrinsic processes unique to the aged brain that may promote seizure susceptibility (75). For example, age-related increases in tau and β-amyloid protein accumulation may contribute to the enhanced incidence of seizures (75), as well as glial response and DNA alterations (76, 77). Below, we will highlight the major physiological mechanisms related to brain aging and explain how these factors may bidirectionally influence susceptibility to seizures and epilepsy (Figure 1). Moreover, these biomolecular changes may identify the pathways that contribute to the better drug response seen in older people with epilepsy, as well as the potential factors that may promote DRE.
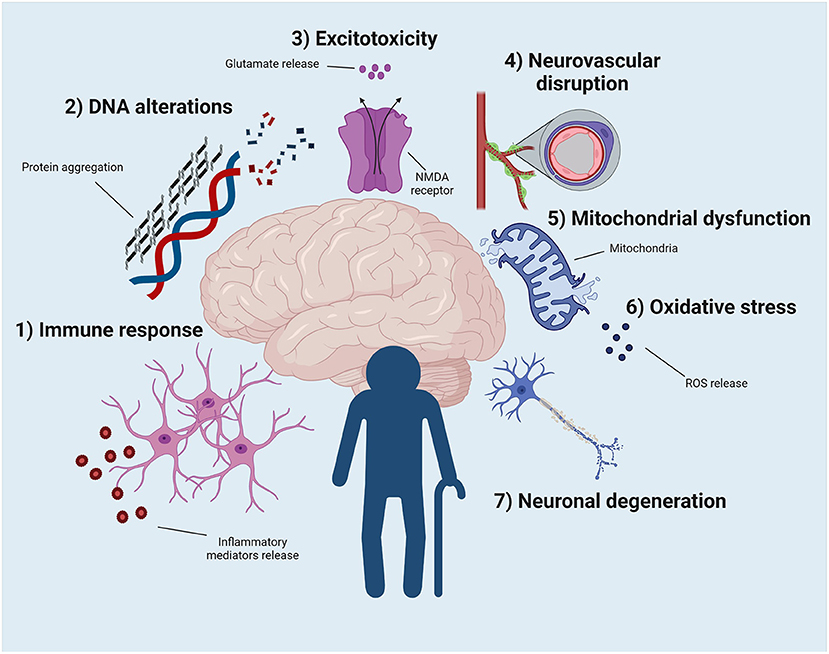
Figure 1. Potential physiological events that can contribute to the development of epilepsy and to the better pharmacological response of older people with epilepsy. (1) The exacerbated immune response, as well as the change in the phenotype and properties of microglia and astrocytes, seem to contribute to this phenomenon. (2) DNA modification due to protein aggregation contributes to enhance epilepsy vulnerability in the elderly population. (3) The uncontrolled release of glutamate ultimately leads to increased excitotoxicity, resulting in a new pathway that explains old brain function. (4) Blood-brain barrier permeability increases with aging. This event may increase the vulnerability to developing epilepsy but may also improve pharmacological sensitivity in older adults. (5) Mitochondrial dysfunction and oxidative stress (6) can be related to epilepsy, age, and drug response. (7) Neuronal death secondary to aging increases the susceptibility to epilepsy. Illustration created with BioRender.com under a paid subscription.
Immune Response
Aging shifts the responsiveness of the immune system (16, 78), i.e., immunosenescence. With aging, there is decreased immune competence to respond to a stimulus and a generally elevated inflammatory state (78, 79). Although the biological mechanisms involved are the subject of active investigation and beyond the scope of this review, aging affects the phenotype and function of the cells comprising innate and adaptive immune cells (78). In both humans and rodents of advanced age, inflammation increases in different areas of the brain (16, 78, 80–82) (Figure 1). In aged individuals, microglia and astrocytes promote inflammatory cytokine release; glial cells also have reduced phagocytic properties (16, 78, 80–82). Additionally, the release of anti-inflammatory cytokines in aged individuals is significantly lower than in younger populations. As in other neurological disorders, the immune response is not only restricted to the brain in epilepsy. Neutrophils and monocytes/macrophages in older adults show impaired phagocytosis, chemotaxis, and immune capacity in response to injury or pathogenic signal in the periphery (16, 78, 80–82). Studying the interaction between the brain and systemic inflammation in older people is thus essential to reproduce the course of different pathologies, such as epilepsy. It is worth mentioning that this effect is sex-dependent, such that estradiol helps to improve the function of immune system cells until menopause occurs, wherein the aging of the immune environment begins (78).
Microglia, particularly when activated, are the primary source of inflammatory cytokines in the brain. IL-1, IL-6, and IFN-α levels are increased in the brains of old vs. younger adult mice and rats (78, 83). Additionally, primed and activated microglia residing in the aged brain often show an exacerbated response to external stimuli, such as infection or stress (78, 83). Peripheral markers of inflammation, including serum cytokine levels, are also elevated in older population. In addition, peripheral inflammation serves as a risk factor for the development of age-related neurodegenerative disease (78, 83) and may play a primary role in the etiology or progression of age-associated pathologies. Thus, inflammation may play an important role in aging-related disorders, as it does in as epilepsy and seizures.
Protein Dysregulation
Proteins are repaired by the chaperone system and eliminated by autophagy or ubiquitin systems (78, 84). These systems become compromised with advanced age. Folding and accumulation of aberrant protein aggregates are thus common in aged individuals (78, 84) (Figure 1). Protein folding or aggregation can have a detrimental effect on the cell, resulting in loss or gain of function. Indeed, β-amyloid and tau proteins progressively aggregate with age, and are associated with the presence of cellular dysfunction, degeneration, and epilepsy (78, 84).
The aggregation of tau and β-amyloid, two proteins implicated in AD, can similarly explain the close relationship between neurodegeneration and epilepsy. Although the mechanisms by which these proteins may act on epilepsy are not fully understood, tau reduces PP2A activity, increasing GSK3β and CDK5 activity, activating PI3K/AKT pathways, and promotes NMDAR subtypes to drive glutamate release (85). These findings support the role of Aβ and tau in epilepsy and indicate that these neurodegenerative proteins may be relevant therapeutic targets in older people with epilepsy.
Oxidative Stress
Aging and oxidative stress are closely related (78) (Figure 1). Aging is the progressive loss of tissue and organ function over time (86). The free radical or the oxidative stress theory of aging, is based on the structural damage-based hypothesis that age-associated functional losses are due to the accumulation of oxidative damage to macromolecules (i.e., lipids, DNA, and proteins) by reactive oxygen species (ROS) (86). The exact mechanism of oxidative stress-induced aging is still not clear but is likely due to increased ROS levels leading to cellular senescence, a physiological mechanism that stops cellular proliferation in response to damages that occur during replication. Senescent cells promote the secretion of soluble factors (interleukins, chemokines, and growth factors), degradative enzymes and insoluble proteins/extracellular matrix components (86).
Studies of naturally aged rodents and genetic SAM models demonstrate that there is an accumulation of oxidation products in the brain over time (78). Oxidative stress is the result of the aggregation of ROS (87, 88). Several factors are involved in ROS accumulation (78). However, a drastic reduction in the levels of antioxidant enzymes, such as superoxide dismutase, and increased inflammation particularly impact epilepsy in the aged brain (87, 88).
SE changes the redox potential and decreases ATP levels, reducing the production and supply of energy to the brain (89). Moreover, epilepsy reduces antioxidant enzyme levels (NADH or FADH) and increases ROS production (89). Indeed, nitric oxide synthase expression is increased in the early phases of epileptogenesis in mice (90). Acute, seizure-induced reductions in hydrogen sulfide levels are detected in young adult mice during the development of the corneal kindled model of epilepsy, and similar studies in aged mice are needed (90). Thus, oxidative stress is highly associated with both aging and epilepsy.
Mitochondrial Dysfunction
Mitochondrial dysfunction is another pathological feature of both aging and epilepsy (78, 91) (Figure 1), which can lead to endogenous production of ROS and apoptosis (74). Mitochondria fulfill their diverse functions partly through constant remodeling via fission and fusion (78, 91). Dysregulation of mitochondrial fission and fusion contributes to numerous types of nervous system disease, including epilepsy (78, 91) Scavenging of ROS may be a valid anticonvulsant strategy (92), in general. Mitochondrial quality control is compromised with aging, and there is evidence of reduced mitochondrial membrane potential, probably due to alteration of the DNA encoding the essential subunits of the electron transport chain. All these detrimental factors increase ROS accumulation (78, 91). Mitochondrial mRNA and protein expression are dynamically altered in response to epileptogenesis in the young-adult brain (90), but whether this is similarly impacted by age remains to be determined. Unfortunately, few studies have sufficiently assessed how aging and epilepsy additively or synergistically influence mitochondrial membrane integrity dynamics or quality control.
Excitotoxicity
Excitotoxicity describes the toxic actions of excitatory neurotransmitters, mainly glutamate, where exacerbated or prolonged activation of glutamate receptors initiates a cascade of neurotoxicity that ultimately leads to loss of neuronal function and cell death (88, 93) (Figure 1). Both inflammation and oxidative stress can induce massive glutamate release. During aging, the brain is in an environment in which both inflammation and oxidative stress play a key role, producing, among other things, excitotoxic damage (88). Neurodegenerative changes associated with human epilepsy, may in part, result from persistent glutamatergic activity (94). The repetitive depolarization of glutamate terminals causes release of excess glutamate resulting in excitotoxic degeneration of the postsynaptic neuron (94). Astrocytes, known to be responsible for the maintenance of synaptic glutamate levels, display phenotype and function changes with advanced age (95). As a result, excitotoxicity and late-onset epilepsy may be closely interrelated.
Neurovascular Dysfunction
The blood-brain barrier (BBB) consists of endothelial cells, tight junctions, astrocytes, pericytes, and the extracellular matrix (78, 96–98). Together, these elements maintain cerebral permeability to blood components and protect the brain from infiltration of neurotoxic compounds (78, 96–98). Physiological aging is associated with deterioration of the cerebrovascular system and increased BBB permeability, making the brain more vulnerable to neurological insults (78). Similarly, aging-related oxidative stress, inflammation, and excitotoxicity contribute to increased BBB permeability (97) (Figure 1).
Although BBB disruption is associated with other neurological disorders, increased permeability may be relevant in understanding differences in drug response between older and younger people with epilepsy. The increased permeability may contribute to better drug uptake, resulting in differential response (99). This effect has already been observed in the treatment of brain tumors. Treatment with BBB disruptors in people with brain tumors promotes a better response to traditional drugs, improving disease prognosis (99). For this reason, the CNS bioavailability in people with epilepsy may be insufficient and may be one mechanism underlying DRE (28). Age-related differences in BBB permeability could contribute to differences in drug response in old vs. young people with epilepsy, but rigorous preclinical studies to assess this are needed.
DNA Alterations and Cell Death
Aging is accompanied by progressive atrophy of tissues and aggregation of stochastic damage to DNA, RNA, protein, and lipid macromolecules (78, 100). Loss of genomic maintenance may contribute causally to aging (Figure 1). Several premature aging syndromes have underlying genetic defects in DNA repair (78, 100). Accumulation of DNA damage may be particularly prevalent in the CNS due to low DNA repair capacity in post-mitotic brain tissue (78, 100). In general, the cumulative effects of detrimental changes that occur in aging, primarily after the reproductive phase, are thought to contribute to species-specific aging rates. In addition, there is also abundant evidence of a causal link between mitochondrial DNA damage and aging phenotypes (78, 100).
In epilepsy, studies have not yet conclusively revealed the effect of an acute seizure on neuronal death and the role of seizure-induced neuronal death in acquired epileptogenesis. Isolated seizures probably do not kill neurons. However, severe, and repetitive seizures do [i.e., SE (78, 100)]. Acquired epileptogenesis does not necessarily require neuronal death (78, 100). Models with chronic seizures (e.g., kindling) are not always defined by neurodegeneration (101) in the early phases of disease, but are nonetheless valid epilepsy models that are incredibly useful to evaluate disease mechanisms and seizure susceptibility (20, 102–104), comorbidities (105), and identify therapeutic agents (106). Indeed, the absence of neuronal death is difficult, if not impossible, to prove, so studies are increasingly challenging the concept that loss of synaptic input from dying neurons is critical for inducing axonal sprouting and synaptic circuit reorganization (78, 100). An alternative hypothesis may be more relevant; biochemical pathways causing programmed neurodegeneration, rather than neuronal death, are responsible for, or contribute to, epileptogenesis. The reprogramming of neuronal death pathways derives from necroptosis or pyroptosis (a form of cell death triggered by inflammatory signals and associated with inflammation) (78, 100). This hypothesis explains why neuronal death appears to be closely related to epileptogenesis, but not always (107). Age-related neurodegeneration likely increases seizure susceptibility, but more studies will better define how these two pathological processes functionally converge.
Neurotrophic and Neurotransmitter Contributions to Aging
Normal aging evokes molecular and cellular changes in the brain that may also delay recovery after injury (74). Aging decreases monoamine densities due to alterations in neuronal number, synapses, and protein levels. Indeed, the aging brain has altered the expression of neurotrophic factors and neurotransmitters: i.e., brain-derived neurotrophic factor, norepinephrine, and serotonin (74, 108). Lastly, elevated monoamine oxidase is associated with the release of free radicals that can also further promote neuronal death via apoptosis (74).
Preclinical and clinical evidence increasingly supports that serotonin (5-HT) has an important role in epilepsy and SUDEP (109, 110). Clinical studies have focused on the effect of 5-HT agonism on seizure semiology, frequency, and autonomic function, whereas studies in animal models have focused on the effect of restoring 5-HT signaling on seizure susceptibility and/or seizure-associated mortality (111). Increased 5-HT levels may generally protect against seizures and SUDEP. Conversely, uncontrolled seizures and epilepsy may reduce 5-HT tone and elevate risk of SUDEP (111–113). The clinical efficacy of 5-HT reuptake inhibitors, such as fluoxetine or citalopram, to control seizures or prevent SUDEP is still controversial (104, 114).
Epilepsy in older people is particularly concerning when it arises due to comorbid conditions because of an elevated mortality risk. Mortality specifically from AD in people with epilepsy has increased over 216% in the last 20 years, whereas people with epilepsy alone have seen a significant (27%) decrease in mortality (115). Whether epilepsy and AD are similarly impacted by changes in 5-HT-related signaling, which is known to be associated with SUDEP risk, remains to be determined.
Aging causes substantial biological changes that play an important role in other related neurological disorders, such as epilepsy or AD. These physiological events may be relevant to understand differential sensitivity to ASMs of older adults with epilepsy compared to younger individuals, as well as elucidate the reason for the increased incidence of epilepsy in this demographic.
Relationship Between Epilepsy and Aging-Related Neurological Disorders
Older adults represent the fastest-growing demographic with epilepsy (16, 34). While seizures in older people with epilepsy are generally not resistant to available ASMs, many pathological processes can contribute to hyperexcitability. In this regard, aged rodent models are an important resource to better understand mechanisms of epilepsy and identify novel and broadly relevant therapeutic treatments.
New strategies for innovative epilepsy drug discovery and/or prevention are needed. A disruptive strategy to ASM discovery came from the US FDA-approval of cannabidiol in 2018. While early limited clinical studies had suggested potential anticonvulsant efficacy (116), there were few well-controlled studies in human subjects or animal models. The seminal demonstration of efficacy of this agent came from a pediatric epileptic syndrome-specific model, the Scn1a+/− mouse model of Dravet syndrome (36). This supported cannabidiol's further characterization in established seizure and epilepsy models in wild-type, adult rodents (37) that had otherwise advanced the clinical study of all FDA-approved ASMs on the market until that point (19). Similarly, fenfluramine was initially identified in a zebrafish model of Dravet syndrome (35) and subsequently approved by the US FDA in 2020. As a result, syndrome-specific pediatric models will assume increased prominence in twenty-first century ASM discovery. These pediatric models present an unparalleled opportunity to identify novel molecular targets and contributors to epilepsy that may be broadly relevant to epileptogenesis and ictogenesis (117). However, we herein argue that this approach could be similarly applied to the opposite end of life. Translational science must better align the use of preclinical models with the heterogeneity of clinical epilepsy (118). In the twentieth century, numerous models of acute seizure in seizure naïve and wild-type rodents were indispensable to the identification of symptomatic treatments. In the twenty-first century, impactful therapies for epilepsy will be discovered in disease models that represent all facets by which epilepsy can arise.
To address this untapped opportunity, additional emphasis on aging-related models of epilepsy affords a novel precision medicine strategy for early drug discovery and/or to identify new molecular contributors to epilepsy, in general. Many of the animal models used to understand the effect of epilepsy in the context of aging involve pathways that are also relevant to the pathophysiology of cerebrovascular disease (i.e., stroke) or AD (13, 32). A high percentage of patients usually develop epilepsy and seizures because of either condition (16). Age exacerbates the associated pathological factors. We propose a strategy of increased prioritization of aged animals in epilepsy research and drug discovery, with an explicit focus on the two primary drivers of epilepsy in older adults: cerebrovascular disease and AD.
Stroke
Stroke and cerebrovascular disease are highly associated with epilepsy (16, 71, 119). Post-stroke seizure refers to seizures or epilepsy that develops after a stroke without a previous history of epilepsy, which is due to reversible or irreversible damage caused by the ischemic or hemorrhagic infarction and not to other structural brain abnormalities or metabolic disorders (119). Stroke is the most identified cause of epilepsy in adults older than 35 years. In older adults, stroke accounts for more than half of the newly diagnosed cases of epilepsy in which a cause is determined, ahead of degenerative disorders, brain tumors, and head trauma (120). From stroke registry data, about 5–20% of all individuals who have a stroke will have subsequent seizures, but epilepsy (recurrent seizures) will develop in only a small subset of this group (120). Each year, more than 730,000 people in the United States have a stroke; a yearly conservative incidence of post-stroke seizures is about 36,500 new cases per year (120).
Alzheimer's Disease
Epilepsy is an under-recognized comorbidity of AD (13). Roughly 44 million people worldwide live with AD. Of those, around 5% have early-onset AD (EOAD), which arises due to a mutation in one of three deterministic risk genes: amyloid precursor protein (APP), Presenilin-1 (PSEN1), or Presenilin-2 (PSEN2). The precise mechanisms leading to the development of seizures in AD are still under investigation and have been extensively reviewed elsewhere (13, 121). Nonetheless, the relative risk of unprovoked seizures is markedly increased in people with EOAD-associated risk factors, reaching up to 87 times the risk of seizures in individuals with AD onset between the ages of 50 and 59 years vs. the general population. For comparison, the relative incidence of Dravet syndrome is 1:15,700 individuals in the U.S, whereas early-onset AD incidence is estimated to be 13.2 per 100,000/year in individuals aged 30–64 (1:7,575 individuals) (122), highlighting a significant disparity in the allocation of resources to investigate a major contributor to epilepsy, as well as an untapped opportunity to identify new molecular targets that could be broadly relevant to a variety of epilepsy syndromes.
Recommendations for Incorporating Aged Animal Models of Epilepsy
Unfortunately, the number of preclinical studies of the effect of aging on epilepsy is limited. There are several plausible limitations. One reason is the resource limitations associated with mimicking the physiological conditions of aging itself. Second, aging naturally deteriorates biological resilience and processes. The coexistence of chronic comorbidities, such as high cholesterol or hypertension, results in a “new physiological state” that is difficult to mimic in rodents. Further, such reduced systems as rodent models may infrequently consider additive impacts such as environmental conditions to reduce variables in a reasonably powered study. Third, older adults (over 65 years of age) are a more frequently polymedicated population (123, 124). Identifying the potential therapeutic effect of an investigational agent in an aging-related disorder model with the added consideration of polypharmacy is an experimental constraint. The cost of keeping older animals is more expensive compared with younger animals. Finally, studying older animals simply takes a lot of time. Investigators must anticipate experiments more in advance than if they were performed in young-adult animal models. These factors altogether limit the feasibility of old preclinical models, reducing capacity to identify new potential drugs or targets.
Ultimately, twenty-first century epilepsy drug discovery could strategically rely on developing new animal models based on the mutations or pathophysiological events produced in both stroke and AD to better reproduce late-onset epilepsy in an efficient manner. These models would fill the above-detailed preclinical void. Interdisciplinary integration of clinical observations into preclinical research would dynamically improve translational research (118). Rather than a reliance on a bench-to-patient approach that has defined twentieth century ASM discovery, twenty-first century efforts must rely on clinical findings to inform translational scientific investigation for epilepsy. Advanced age negatively influences epilepsy susceptibility. Thus, the study of epilepsy in aged animal models remains an area of considerable opportunity for future therapeutic innovation.
We propose that some of the characteristics that animal models should have to study the role of epilepsy in advanced age and vice versa are:
• Use a range of ages of “old” animals to understand heterogeneity in the geriatric population. This includes rodents older than 16 months.
• Studies should be performed with both sexes to dissect the age-related effects of gonadal hormones or other sex-specific factors on functional outcomes and susceptibility to epilepsy.
• Use epilepsy models that phenotypically reproduce the symptomatology observed in older people with epilepsy, including those age-associated comorbidities such as stroke and AD-associated genotypes.
• Increase emphasis on establishing a pharmacokinetic/pharmacodynamic relationship in aged animals to inform on the potential for adverse effects liability in clinical populations. This is particularly critical to older population, who have decreased renal and hepatic function and may be subject to polypharmacy.
• Study the response to repeated seizures both acutely and chronically.
• Include biomarkers to validate of age-related changes that are translationally feasible.
Conclusions
Older adults are more vulnerable to seizures and epilepsy, but less resistant to ASMs than young people with epilepsy. Preclinical studies in aged animals and the effect of advanced age on epilepsy and seizures are unfortunately limited. Little work has been conducted to define ASM efficacy in preclinical models of late-onset epilepsy. This review updates the comprehensive review by Kelly (32) and extends upon that report to highlight new avenues for therapeutic innovation. We suggest incorporation of aged animals and new pathways and pathophysiological mechanisms involved in epilepsy and seizures in the aged and degenerating brain, which may vary with the age of the individuals. If nothing else, the intensity and effort of the epilepsy research community should match the clinical need in this patient group to a similar degree as has, in recent years, been dedicated to pediatric patients, whose seizure incidence is similar (13). Likewise, this review highlights the importance of acting on the biomolecular mechanisms that naturally vary with advanced age because they play a fundamental role in age-related neurological disorders. Finally, we conclude that, as in humans, animals with stroke or AD variants could be more frequently integrated into the study of mechanisms of late-onset epilepsy, which may extend more broadly to other syndromes and advance new therapeutic targets to people with epilepsy.
Author Contributions
AP, LL, KK, and MB-H contributed to the literature review and writing of this manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by ITHS KL2 (KL2TR002317) and R01AG067788 to MB-H.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Pearl PL, Bennett HD, Khademian Z. Seizures and metabolic disease. Curr Neurol Neurosci Rep. (2005) 5:127–33. doi: 10.1007/s11910-005-0010-7
3. Jesse S, Bråthen G, Ferrara M, Keindl M, Ben-Menachem E, Tanasescu R, et al. Alcohol withdrawal syndrome: mechanisms, manifestations, and management. Acta Neurol Scand. (2017) 135:4–16. doi: 10.1111/ane.12671
4. Alldredge BK, Lowenstein DH, Simon RP. Seizures associated with recreational drug abuse. Neurology. (1989) 39:1037–9. doi: 10.1212/WNL.39.8.1037
5. Sirven JI. Epilepsy: a spectrum disorder. Cold Spring Harb Perspect Med. (2015) 5:a022848. doi: 10.1101/cshperspect.a022848
6. Edwards JC. Seizure types, epilepsy syndromes, etiology, and diagnosis. CNS Spectr. (2001) 6:750–5. doi: 10.1017/S1092852900001498
8. Vezzani A, Friedman A. Brain inflammation as a biomarker in epilepsy. Biomark Med. (2011) 5:607–14. doi: 10.2217/bmm.11.61
9. Lucke-Wold BP, Nguyen L, Turner RC, Logsdon AF, Chen YW, Smith KE, et al. Traumatic brain injury and epilepsy: underlying mechanisms leading to seizure. Seizure. (2015) 33:13–23. doi: 10.1016/j.seizure.2015.10.002
10. Erturk Cetin O, Isler C, Uzan M, Ozkara C. Epilepsy-related brain tumors. Seizure. (2017) 44:93–7. doi: 10.1016/j.seizure.2016.12.012
11. Camilo O, Goldstein LB. Seizures and epilepsy after ischemic stroke. Stroke. (2004) 35:1769–75. doi: 10.1161/01.STR.0000130989.17100.96
12. Annegers JF, Hauser WA, Lee JR, Rocca WA. Incidence of acute symptomatic seizures in Rochester, Minnesota, 1935-1984. Epilepsia. (1995) 36:327–33. doi: 10.1111/j.1528-1157.1995.tb01005.x
13. Lehmann L, Lo A, Knox KM, Barker-Haliski M. Alzheimer's Disease and epilepsy: a perspective on the opportunities for overlapping therapeutic innovation. Neurochem Res. (2021) 46:1895–912. doi: 10.1007/s11064-021-03332-y
14. Acharya JN, Acharya VJ. Epilepsy in the elderly: special considerations and challenges. Ann Indian Acad Neurol. (2014) 17:S18–26. doi: 10.4103/0972-2327.128645
15. Jennum P, Gyllenborg J, Kjellberg J. The social and economic consequences of epilepsy: a controlled national study. Epilepsia. (2011) 52:949–56. doi: 10.1111/j.1528-1167.2010.02946.x
16. Sen A, Jette N, Husain M, Sander JW. Epilepsy in older people. Lancet. (2020) 395:735–48. doi: 10.1016/S0140-6736(19)33064-8
17. Park JT, Shahid AM, Jammoul A. Common pediatric epilepsy syndromes. Pediatr Ann. (2015) 44:e30–5. doi: 10.3928/00904481-20150203-09
18. Wirrell E. Infantile, childhood, adolescent epilepsies. Continuum. (2016) 22:60–93. doi: 10.1212/CON.0000000000000269
19. Barker-Haliski M, White HS. Validated animal models for antiseizure drug (ASD) discovery: advantages and potential pitfalls in ASD screening. Neuropharmacology. (2019) 167:107750. doi: 10.1016/j.neuropharm.2019.107750
20. Beckman M, Knox K, Koneval Z, Smith C, Jayadev S, Barker-Haliski M. Loss of presenilin 2 age-dependently alters susceptibility to acute seizures and kindling acquisition. Neurobiol Dis. (2020) 136:104719. doi: 10.1016/j.nbd.2019.104719
21. Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Perucca P, et al. Progress report on new antiepileptic drugs: a summary of the fifteenth eilat conference on new antiepileptic drugs and devices (EILAT XV). I. Drugs in preclinical and early clinical development. Epilepsia. (2020) 61:2340–64. doi: 10.1111/epi.16725
22. Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Perucca P, et al. Progress report on new antiepileptic drugs: a summary of the fifteenth eilat conference on new antiepileptic drugs and devices (EILAT XV). II. Drugs in more advanced clinical development. Epilepsia. (2020) 61:2365–85. doi: 10.1111/epi.16726
23. Klein P, Dingledine R, Aronica E, Bernard C, Blumcke I, Boison D, et al. Commonalities in epileptogenic processes from different acute brain insults: do they translate? Epilepsia. (2018) 59:37–66. doi: 10.1111/epi.13965
24. Barker-Haliski M. How do we choose the appropriate animal model for antiseizure therapy development? Expert Opin Drug Discov. (2019) 14:499–509. doi: 10.1080/17460441.2019.1636782
25. Barker-Haliski M, Knox K, Zierath DK, Koneval Z, Metcalf CS, Wilcox KS, et al. Development of an antiepileptogenesis drug screening platform: effects of everolimus and phenobarbital. Epilepsia. (2021) 62:1677–88. doi: 10.1111/epi.16955
26. Sills GJ, Rogawski MA. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology. (2020) 168:107966. doi: 10.1016/j.neuropharm.2020.107966
27. Kwan P, Brodie MJ. Epilepsy after the first drug fails: substitution or add-on? Seizure. (2000) 9:464–8. doi: 10.1053/seiz.2000.0442
28. Tang F, Hartz AMS, Bauer B. Drug-Resistant epilepsy: multiple hypotheses, few answers. Front Neurol. (2017) 8:301. doi: 10.3389/fneur.2017.00301
29. Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia. (2010) 51:1069–77. doi: 10.1111/j.1528-1167.2009.02397.x
30. Thomas RJ. Seizures and epilepsy in the elderly. Arch Intern Med. (1997) 157:605–17. doi: 10.1001/archinte.1997.00440270035003
31. Arif H, Buchsbaum R, Pierro J, Whalen M, Sims J, Resor R Jr, et al. Comparative effectiveness of 10 antiepileptic drugs in older adults with epilepsy. Arch Neurol. (2010) 67:408–15. doi: 10.1001/archneurol.2010.49
32. Kelly KM. Aging models of acute seizures and epilepsy. Epilepsy Curr. (2010) 10:15–20. doi: 10.1111/j.1535-7511.2009.01341.x
33. Lattanzi S, Cagnetti C, Foschi N, Ciuffini R, Osanni E, Chiesa V, et al. Adjunctive perampanel in older patients with epilepsy: a multicenter study of clinical practice. Drugs Aging. (2021) 38:603–10. doi: 10.1007/s40266-021-00865-3
34. Mcareavey MJ, Ballinger BR, Fenton GW. Epileptic seizures in elderly patients with dementia. Epilepsia. (1992) 33:657–60. doi: 10.1111/j.1528-1157.1992.tb02343.x
35. Zhang Y, Kecskes A, Copmans D, Langlois M, Crawford AD, Ceulemans B, et al. Pharmacological characterization of an antisense knockdown zebrafish model of Dravet syndrome: inhibition of epileptic seizures by the serotonin agonist fenfluramine. PLoS ONE. (2015) 10:e0125898. doi: 10.1371/journal.pone.0125898
36. Kaplan JS, Stella N, Catterall WA, Westenbroek RE. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA. (2017) 114:11229–34. doi: 10.1073/pnas.1711351114
37. Patra PH, Barker-Haliski M, White HS, Whalley BJ, Glyn S, Sandhu H, et al. Cannabidiol reduces seizures and associated behavioral comorbidities in a range of animal seizure and epilepsy models. Epilepsia. (2019) 60:303–14. doi: 10.1111/epi.14629
38. Murphree LJ, Rundhaugen LM, Kelly KM. Animal models of geriatric epilepsy. Int Rev Neurobiol. (2007) 81:29–40. doi: 10.1016/S0074-7742(06)81003-2
39. Benkovic SA, O'callaghan JP, Miller DB. Regional neuropathology following kainic acid intoxication in adult and aged C57BL/6J mice. Brain Res. (2006) 1070:215–31. doi: 10.1016/j.brainres.2005.11.065
40. Mccord MC, Lorenzana A, Bloom CS, Chancer ZO, Schauwecker PE. Everity and cell death. Neuroscience. (2008) 154:1143–53. doi: 10.1016/j.neuroscience.2008.03.082
41. Li Z, Hall AM, Kelinske M, Roberson ED. Seizure resistance without parkinsonism in aged mice after tau reduction. Neurobiol Aging. (2014) 35:2617–24. doi: 10.1016/j.neurobiolaging.2014.05.001
42. Putra M, Puttachary S, Liu G, Lee G, Thippeswamy T. Fyn-tau ablation modifies PTZ-induced seizures and post-seizure hallmarks of early epileptogenesis. Front Cell Neurosci. (2020) 14:592374. doi: 10.3389/fncel.2020.592374
43. Stover KR, Lim S, Zhou TL, Stafford PM, Chow J, Li H, et al. Susceptibility to hippocampal kindling seizures is increased in aging C57 black mice. IBRO Rep. (2017) 3:33–44. doi: 10.1016/j.ibror.2017.08.001
44. Liu AH, Chu M, Wang YP. Up-Regulation of Trem2 inhibits hippocampal neuronal apoptosis and alleviates oxidative stress in epilepsy via the PI3K/Akt pathway in mice. Neurosci Bull. (2019) 35:471–85. doi: 10.1007/s12264-018-0324-5
45. Kim HY, Yang YR, Hwang H, Lee HE, Jang HJ, Kim J, et al. Deletion of PLCgamma1 in GABAergic neurons increases seizure susceptibility in aged mice. Sci Rep. (2019) 9:17761. doi: 10.1038/s41598-019-54477-4
46. Wang J, Wu C, Peng J, Patel N, Huang Y, Gao X, et al. Early-Onset convulsive seizures induced by brain hypoxia-ischemia in aging mice: effects of anticonvulsive treatments. PLoS ONE. (2015) 10:e0144113. doi: 10.1371/journal.pone.0144113
47. Wu C, Wang J, Peng J, Patel N, Huang Y, Gao X, et al. Modeling early-onset post-ischemic seizures in aging mice. Exp Neurol. (2015) 271:1–12. doi: 10.1016/j.expneurol.2015.04.018
48. Tian Q, Gromov P, Clement JH, Wang Y, Riemann M, Weih F, et al. RHEB1 insufficiency in aged male mice is associated with stress-induced seizures. Geroscience. (2017) 39:557–70. doi: 10.1007/s11357-017-9997-3
49. El-Hayek YH, Wu C, Ye H, Wang J, Carlen PL, Zhang L. Hippocampal excitability is increased in aged mice. Exp Neurol. (2013) 247:710–9. doi: 10.1016/j.expneurol.2013.03.012
50. Kitani K, Sato Y, Kanai S, Nokubo M, Ohta M, Masuda Y. Age related increased threshold for electroshock seizure in BDF1 mice. Life Sci. (1985) 36:657–62. doi: 10.1016/0024-3205(85)90170-5
51. Kondziella D, Hammer J, Sletvold O, Sonnewald U. The pentylenetetrazole-kindling model of epilepsy in SAMP8 mice: glial-neuronal metabolic interactions. Neurochem Int. (2003) 43:629–37. doi: 10.1016/S0197-0186(03)00093-7
52. Sinsky J, Pichlerova K, Hanes J. Tau protein interaction partners and their roles in alzheimer's disease and other tauopathies. Int J Mol Sci. (2021) 22:9207. doi: 10.3390/ijms22179207
53. Thom M, Liu JY, Thompson P, Phadke R, Narkiewicz M, Martinian L, et al. Neurofibrillary tangle pathology and braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: a post-mortem study. Brain. (2011) 134:2969–81. doi: 10.1093/brain/awr209
54. Tai XY, Koepp M, Duncan JS, Fox N, Thompson P, Baxendale S, et al. Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain. (2016) 139:2441–55. doi: 10.1093/brain/aww187
55. Kim HC, Bing G, Jhoo WK, Kim WK, Shin EJ, Park ES, et al. Oxidative damage causes formation of lipofuscin-like substances in the hippocampus of the senescence-accelerated mouse after kainate treatment. Behav Brain Res. (2002) 131:211–20. doi: 10.1016/S0166-4328(01)00382-5
56. Shin EJ, Jeong JH, Bing G, Park ES, Chae JS, Yen TP, et al. Kainate-induced mitochondrial oxidative stress contributes to hippocampal degeneration in senescence-accelerated mice. Cell Signal. (2008) 20:645–58. doi: 10.1016/j.cellsig.2007.11.014
57. Erickson JC, Hollopeter G, Thomas SA, Froelick GJ, Palmiter RD. Disruption of the metallothionein-III gene in mice: analysis of brain zinc, behavior, and neuron vulnerability to metals, aging, and seizures. J Neurosci. (1997) 17:1271–81. doi: 10.1523/JNEUROSCI.17-04-01271.1997
58. D'costa A, Breese CR, Boyd RL, Booze RM, Sonntag WE. Attenuation of Fos-like immunoreactivity induced by a single electroconvulsive shock in brains of aging mice. Brain Res. (1991) 567:204–11. doi: 10.1016/0006-8993(91)90797-Y
59. Alves M, Kenny A, De Leo G, Beamer EH, Engel T. Tau phosphorylation in a mouse model of temporal lobe epilepsy. Front Aging Neurosci. (2019) 11:308. doi: 10.3389/fnagi.2019.00308
60. Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. (2013) 70:1158–66. doi: 10.1001/jamaneurol.2013.136
61. Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL. Epileptic activity in Alzheimer's disease: causes and clinical relevance. Lancet Neurol. (2017) 16:311–22. doi: 10.1016/S1474-4422(17)30044-3
62. Forstl H. Neurologic disease described in the journal of empirical psychology (gnothi sauton oder magazin zur erfahrungsseelenkunde), 1783-1793. Arch Neurol. (1992) 49:187–8. doi: 10.1001/archneur.1992.00530260089026
63. Guo D, Zeng L, Brody DL, Wong M. Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PLoS ONE. (2013) 8:e64078. doi: 10.1371/journal.pone.0064078
64. Shima A, Nitta N, Suzuki F, Laharie AM, Nozaki K, Depaulis A. Activation of mTOR signaling pathway is secondary to neuronal excitability in a mouse model of mesio-temporal lobe epilepsy. Eur J Neurosci. (2015) 41:976–88. doi: 10.1111/ejn.12835
65. Theilmann W, Gericke B, Schidlitzki A, Muneeb Anjum SM, Borsdorf S, Harries T, et al. Novel brain permeant mTORC1/2 inhibitors are as efficacious as rapamycin or everolimus in mouse models of acquired partial epilepsy and tuberous sclerosis complex. Neuropharmacology. (2020) 180:108297. doi: 10.1016/j.neuropharm.2020.108297
66. Weber M, Wu T, Hanson JE, Alam NM, Solanoy H, Ngu H, et al. Cognitive deficits, changes in synaptic function, and brain pathology in a mouse model of normal aging(1,2,3). eNeuro. (2015) 2. doi: 10.1523/ENEURO.0047-15.2015
67. Arisi GM, Ruch M, Foresti ML, Mukherjee S, Ribak CE, Shapiro LA. Astrocyte alterations in the hippocampus following pilocarpine-induced seizures in aged rats. Aging Dis. (2011) 2:294–300.
68. Avanzi RD, Cavarsan CF, Santos JG Jr, Hamani C, Mello LE, Covolan L. Basal dendrites are present in newly born dentate granule cells of young but not aged pilocarpine-treated chronic epileptic rats. Neuroscience. (2010) 170:687–91. doi: 10.1016/j.neuroscience.2010.08.004
69. Kanak DJ, Jones RT, Tokhi A, Willingham AL, Zaveri HP, Rose GM, et al. Electrical and pharmacological stimuli reveal a greater susceptibility for CA3 network excitability in hippocampal slices from aged vs. adult fischer 344 rats. Aging Dis. (2011) 2:318–31. Available online at: http://www.aginganddisease.org/EN/Y2011/V2/I4/318
70. Zhang K, Tolstykh GP, Sanchez RM, Cavazos JE. Chronic cellular hyperexcitability in elderly epileptic rats with spontaneous seizures induced by kainic acid status epilepticus while young adults. Aging Dis. (2011) 2:332–8.
71. Kelly KM, Jukkola PI, Yin G, Miller ER, Kharlamov EA, Shiau DS, et al. Poststroke epilepsy following transient unilateral middle cerebral and common carotid artery occlusion in young adult and aged F344 rats. Epilepsy Res. (2018) 141:38–47. doi: 10.1016/j.eplepsyres.2018.02.002
72. Hattiangady B, Kuruba R, Shetty AK. Acute seizures in old age leads to a greater loss of CA1 pyramidal neurons, an increased propensity for developing chronic tle and a severe cognitive dysfunction. Aging Dis. (2011) 2:1–17. Available online at: http://www.aginganddisease.org/EN/article/metrics.do?articleId=147264
73. Kuruba R, Hattiangady B, Parihar VK, Shuai B, Shetty AK. Differential susceptibility of interneurons expressing neuropeptide Y or parvalbumin in the aged hippocampus to acute seizure activity. PLoS ONE. (2011) 6:e24493. doi: 10.1371/journal.pone.0024493
74. Iboaya A, Harris JL, Arickx AN, Nudo RJ. Models of traumatic brain injury in aged animals: a clinical perspective. Neurorehabil Neural Repair. (2019) 33:975–88. doi: 10.1177/1545968319883879
75. Baram TZ. The brain, seizures and epilepsy throughout life: understanding a moving target. Epilepsy Curr. (2012) 12:7–12. doi: 10.5698/1535-7511-12.4s.7
76. Elobeid A, Libard S, Leino M, Popova SN, Alafuzoff I. Altered proteins in the aging brain. J Neuropathol Exp Neurol. (2016) 75:316–25. doi: 10.1093/jnen/nlw002
77. Munger EL, Edler MK, Hopkins WD, Ely JJ, Erwin JM, Perl DP, et al. Astrocytic changes with aging and Alzheimer's disease-type pathology in chimpanzees. J Comp Neurol. (2019) 527:1179–95. doi: 10.1002/cne.24610
78. Simen AA, Bordner KA, Martin MP, Moy LA, Barry LC. Cognitive dysfunction with aging and the role of inflammation. Ther Adv Chronic Dis. (2011) 2:175–95. doi: 10.1177/2040622311399145
79. Kaltschmidt B, Kaltschmidt C. NF-KappaB in long-term memory and structural plasticity in the adult mammalian brain. Front Mol Neurosci. (2015) 8:69. doi: 10.3389/fnmol.2015.00069
80. Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. (2013) 13:875–87. doi: 10.1038/nri3547
81. Niraula A, Sheridan JF, Godbout JP. Microglia priming with aging and stress. Neuropsychopharmacology. (2017) 42:318–33. doi: 10.1038/npp.2016.185
82. Raj D, Yin Z, Breur M, Doorduin J, Holtman IR, Olah M, et al. Increased white matter inflammation in aging- and alzheimer's disease brain. Front Mol Neurosci. (2017) 10:206. doi: 10.3389/fnmol.2017.00206
83. Campuzano O, Castillo-Ruiz MM, Acarin L, Castellano B, Gonzalez B. Increased levels of proinflammatory cytokines in the aged rat brain attenuate injury-induced cytokine response after excitotoxic damage. J Neurosci Res. (2009) 87:2484–97. doi: 10.1002/jnr.22074
84. Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK. Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci. (2019) 12:25. doi: 10.3389/fnmol.2019.00025
85. Paudel YN, Angelopoulou E, Piperi C, Othman I, Shaikh MF. Revisiting the impact of neurodegenerative proteins in epilepsy: focus on alpha-synuclein, beta-amyloid, and tau. Biology. (2020) 9:122. doi: 10.3390/biology9060122
86. Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, et al. Oxidative stress, aging, and diseases. Clin Interv Aging. (2018) 13:757–72. doi: 10.2147/CIA.S158513
87. Anton M, Rodriguez-Gonzalez A, Ballesta A, Gonzalez N, Del Pozo A, De Fonseca FR, et al. Alcohol binge disrupts the rat intestinal barrier: the partial protective role of oleoylethanolamide. Br J Pharmacol. (2018) 175:4464–79. doi: 10.1111/bph.14501
88. Martinez-Orgado J, Villa M, Del Pozo A. Cannabidiol for the treatment of neonatal hypoxic-ischemic brain injury. Front Pharmacol. (2020) 11:584533. doi: 10.3389/fphar.2020.584533
89. Aguiar CC, Almeida AB, Araujo PV, De Abreu RN, Chaves EM, Do Vale OC, et al. Oxidative stress and epilepsy: literature review. Oxid Med Cell Longev. (2012) 2012:795259. doi: 10.1155/2012/795259
90. Cho C, Zeigler M, Mizuno S, Morrison RS, Totah RA, Barker-Haliski M. Reductions in hydrogen sulfide and changes in mitochondrial quality control proteins are evident in the early phases of the corneally kindled mouse model of epilepsy. Int J Mol Sci. (2022) 23:1434. doi: 10.3390/ijms23031434
91. Srivastava S. The mitochondrial basis of aging and age-related disorders. Genes. (2017) 8:398. doi: 10.3390/genes8120398
92. Fulton RE, Pearson-Smith JN, Huynh CQ, Fabisiak T, Liang LP, Aivazidis S, et al. Neuron-specific mitochondrial oxidative stress results in epilepsy, glucose dysregulation and a striking astrocyte response. Neurobiol Dis. (2021) 158:105470. doi: 10.1016/j.nbd.2021.105470
93. Ceprian M, Vargas C, Garcia-Toscano L, Penna F, Jimenez-Sanchez L, Achicallende S, et al. Cannabidiol administration prevents hypoxia-ischemia-induced hypomyelination in newborn rats. Front Pharmacol. (2019) 10:1131. doi: 10.3389/fphar.2019.01131
94. Barker-Haliski M, White HS. Glutamatergic mechanisms associated with seizures and epilepsy. Cold Spring Harb Perspect Med. (2015) 5:a022863. doi: 10.1101/cshperspect.a022863
95. Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci USA. (2018) 115:E1896–905. doi: 10.1073/pnas.1800165115
96. Keep RF, Xiang J, Ennis SR, Andjelkovic A, Hua Y, Xi G, et al. Blood-brain barrier function in intracerebral hemorrhage. Acta Neurochir Suppl. (2008) 105:73–7. doi: 10.1007/978-3-211-09469-3_15
97. Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. (2008) 32:200–19. doi: 10.1016/j.nbd.2008.08.005
98. Montagne A, Toga AW, Zlokovic BV. Blood-Brain barrier permeability and gadolinium: benefits and potential pitfalls in research. JAMA Neurol. (2016) 73:13–4. doi: 10.1001/jamaneurol.2015.2960
99. Blethen KE, Arsiwala TA, Fladeland RA, Sprowls SA, Panchal DM, Adkins CE, et al. Modulation of the blood-tumor barrier to enhance drug delivery and efficacy for brain metastases. Neurooncol Adv. (2021) 3:v133–43. doi: 10.1093/noajnl/vdab123
100. Dingledine R, Varvel NH, Dudek FE. When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv Exp Med Biol. (2014) 813:109–22. doi: 10.1007/978-94-017-8914-1_9
101. Loewen JL, Barker-Haliski ML, Dahle EJ, White HS, Wilcox KS. Neuronal Injury, gliosis, and glial proliferation in two models of temporal lobe epilepsy. J Neuropathol Exp Neurol. (2016) 75:366–78. doi: 10.1093/jnen/nlw008
102. Albertini G, Walrave L, Demuyser T, Massie A, De Bundel D, Smolders I. 6 Hz corneal kindling in mice triggers neurobehavioral comorbidities accompanied by relevant changes in c-Fos immunoreactivity throughout the brain. Epilepsia. (2017) 55:475–82. doi: 10.1111/epi.13943
103. Remigio GJ, Loewen JL, Heuston S, Helgeson C, White HS, Wilcox KS, et al. Corneal kindled C57BL/6 mice exhibit saturated dentate gyrus long-term potentiation and associated memory deficits in the absence of overt neuron loss. Neurobiol Dis. (2017) 105:221–34. doi: 10.1016/j.nbd.2017.06.006
104. Koneval Z, Knox KM, White HS, Barker-Haliski M. Lamotrigine-resistant corneal-kindled mice: a model of pharmacoresistant partial epilepsy for moderate-throughput drug discovery. Epilepsia. (2018) 59:1245–56. doi: 10.1111/epi.14190
105. Barker-Haliski M, White HS. Antiepileptic drug development and experimental models. In: Wyllie E, Gidal BE, Goodkin HP, editors. Wyllie's Treatment of Epilepsy. 6th edition. Philadelphia, PA: Lippencott, Williams & Wilkins (2015).
106. Matagne A, Klitgaard H. Validation of corneally kindled mice: a sensitive screening model for partial epilepsy in man. Epilepsy Res. (1998) 31:59–71. doi: 10.1016/S0920-1211(98)00016-3
107. Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. (1996) 16:1337–45. doi: 10.1523/JNEUROSCI.16-04-01337.1996
108. Szapacs ME, Numis AL, Andrews AM. Late onset loss of hippocampal 5-HT and NE is accompanied by increases in BDNF protein expression in mice co-expressing mutant APP and PS1. Neurobiol Dis. (2004) 16:572–80. doi: 10.1016/j.nbd.2004.04.010
109. Tupal S, Faingold CL. Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia. (2006) 47:21–6. doi: 10.1111/j.1528-1167.2006.00365.x
110. Mdawar B, Ghossoub E, Khoury R. Selective serotonin reuptake inhibitors and Alzheimer's disease. Neural Regen Res. (2020) 15:41–6. doi: 10.4103/1673-5374.264445
111. Petrucci AN, Joyal KG, Purnell BS, Buchanan GF. Serotonin and sudden unexpected death in epilepsy. Exp Neurol. (2020) 325:113145. doi: 10.1016/j.expneurol.2019.113145
112. Bagdy G, Kecskemeti V, Riba P, Jakus R. Serotonin and epilepsy. J Neurochem. (2007) 100:857–73. doi: 10.1111/j.1471-4159.2006.04277.x
113. Richerson GB, Buchanan GF. The serotonin axis: Shared mechanisms in seizures, depression, and SUDEP. Epilepsia. (2011) 52 (Suppl. 1): 28–38. doi: 10.1111/j.1528-1167.2010.02908.x
114. Buchanan GF, Murray NM, Hajek MA, Richerson GB. Serotonin neurones have anti-convulsant effects and reduce seizure-induced mortality. J Physiol. (2014) 592:4395–410. doi: 10.1113/jphysiol.2014.277574
115. Degiorgio CM, Curtis A, Carapetian A, Hovsepian D, Krishnadasan A, Markovic D. Why are epilepsy mortality rates rising in the United States? A population-based multiple cause-of-death study. BMJ Open. (2020) 10:e035767. doi: 10.1136/bmjopen-2019-035767
116. Cunha JM, Carlini EA, Pereira AE, Ramos OL, Pimentel C, Gagliardi R, et al. Chronic administration of cannabidiol to healthy volunteers and epileptic patients. Pharmacology. (1980) 21:175–85. doi: 10.1159/000137430
117. Jones A, Barker-Haliski M, Ilie AS, Herd MB, Baxendale S, Holdsworth CJ, et al. A multiorganism pipeline for antiseizure drug discovery: identification of chlorothymol as a novel gamma-aminobutyric acidergic anticonvulsant. Epilepsia. (2020) 61:2106–18. doi: 10.1111/epi.16644
118. Barker-Haliski M, Friedman D, White HS, French JA. How clinical development can, and should, inform translational science. Neuron. (2014) 84:582–93. doi: 10.1016/j.neuron.2014.10.029
119. Yang H, Rajah G, Guo A, Wang Y, Wang Q. Pathogenesis of epileptic seizures and epilepsy after stroke. Neurol Res. (2018) 40:426–32. doi: 10.1080/01616412.2018.1455014
120. Silverman IE, Restrepo L, Mathews GC. Poststroke seizures. Arch Neurol. (2002) 59:195–201. doi: 10.1001/archneur.59.2.195
121. Horvath A, Szucs A, Barcs G, Noebels JL, Kamondi A. Epileptic seizures in alzheimer disease: a review. Alzheimer Dis Assoc Disord. (2016) 30:186–92. doi: 10.1097/WAD.0000000000000134
122. Chiari A, Vinceti G, Adani G, Tondelli M, Galli C, Fiondella L, et al. Epidemiology of early onset dementia and its clinical presentations in the province of Modena, Italy. Alzheimers Dement. (2021) 17:81–8. doi: 10.1002/alz.12177
123. Lackner TE, Cloyd JC, Thomas LW, Leppik IE. Antiepileptic drug use in nursing home residents: effect of age, gender, and comedication on patterns of use. Epilepsia. (1998) 39:1083–7. doi: 10.1111/j.1528-1157.1998.tb01294.x
Keywords: late-onset epilepsy, animal models, neuroinflammation, Alzheimer's disease, stroke
Citation: del Pozo A, Lehmann L, Knox KM and Barker-Haliski M (2022) Can Old Animals Reveal New Targets? The Aging and Degenerating Brain as a New Precision Medicine Opportunity for Epilepsy. Front. Neurol. 13:833624. doi: 10.3389/fneur.2022.833624
Received: 11 December 2021; Accepted: 07 April 2022;
Published: 28 April 2022.
Edited by:
Arjune Sen, University of Oxford, United KingdomReviewed by:
Meng-Han Tsai, Kaohsiung Chang Gung Memorial Hospital, TaiwanSimona Lattanzi, Marche Polytechnic University, Italy
Copyright © 2022 del Pozo, Lehmann, Knox and Barker-Haliski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melissa Barker-Haliski, bWhhbGlza2lAdXcuZWR1