 Yu-lei Gao
Yu-lei Gao Yan-cun Liu
Yan-cun Liu Xiang Zhang2
Xiang Zhang2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 16 March 2022
Sec. Multiple Sclerosis and Neuroimmunology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.830784
This article is part of the Research Topic Causative Mechanism and Potential Pharmacological and Non-pharmacological Interventions in Sepsis-Associated Encephalopathy View all 7 articles
Sepsis-associated encephalopathy (SAE) is a diffuse central nervous system (CNS) dysfunction during sepsis, and is associated with increased mortality and poor outcomes in septic patients. Despite the high incidence and clinical relevance, the exact mechanisms driving SAE pathogenesis are not yet fully understood, and no specific therapeutic strategies are available. Regulatory T cells (Tregs) have a role in SAE pathogenesis, thought to be related with alleviation of sepsis-induced hyper-inflammation and immune responses, promotion of T helper (Th) 2 cells functional shift, neuroinflammation resolution, improvement of the blood-brain barrier (BBB) function, among others. Moreover, in a clinical point of view, these cells have the potential value of improving neurological and psychiatric/mental symptoms in SAE patients. This review aims to provide a general overview of SAE from its initial clinical presentation to long-term cognitive impairment and summarizes the main features of its pathogenesis. Additionally, a detailed overview on the main mechanisms by which Tregs may impact SAE pathogenesis is given. Finally, and considering that Tregs may be a novel target for immunomodulatory intervention in SAE, different therapeutic options, aiming to boost peripheral and brain infiltration of Tregs, are discussed.
Sepsis-associated encephalopathy (SAE) is a common, but poorly understood, diffuse central nervous system (CNS) dysfunction, that commonly appears in the setting of sepsis or systemic inflammatory response syndrome (SIRS) (1–3). Clinically, SAE is characterized by episodes of delirium, seizures, mild or deep unconsciousness, cognitive impairments, depression, decreased attention and motor coordination and social interaction problems (4, 5). This is the most common type of encephalopathy and one of the foremost causes of morbidity and mortality in patients in intensive-care units (ICU) worldwide. Moreover, SAE was shown to be associated with extensive in-hospital costs and prolonged hospitalization (6–8). Nevertheless, the epidemiological features and risk factors of SAE are not yet fully understood, and should be further explored in order to define suitable clinical interventions to reduce risk factors and thus, morbidity and mortality (9).
The pathogenesis of SAE involves multiple intertwined factors, namely excessive production of pro-inflammatory cytokines, chemokine and acute phase proteins, including, but not restricted to interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, Interferon (IFN)-gamma (γ), CXC-chemokine ligand 10 (CXCL10), C-reactive protein (CRP) and complement factors; endothelial cells activation/blood-brain barrier (BBB) collapse; glial cells activation; uncontrolled neuroinflammation; ischemic processes/injury; altered neurotransmission; mitochondrial dysfunction; apoptosis; and cognitive impairment (2–5, 7, 8, 10–13) (Figure 1). The role of each factor is patient-dependent and varies according to the clinical situation. Further studies need to be conducted to better understand the SAE pathophysiology and potentially develop new and more efficient therapeutic strategies.
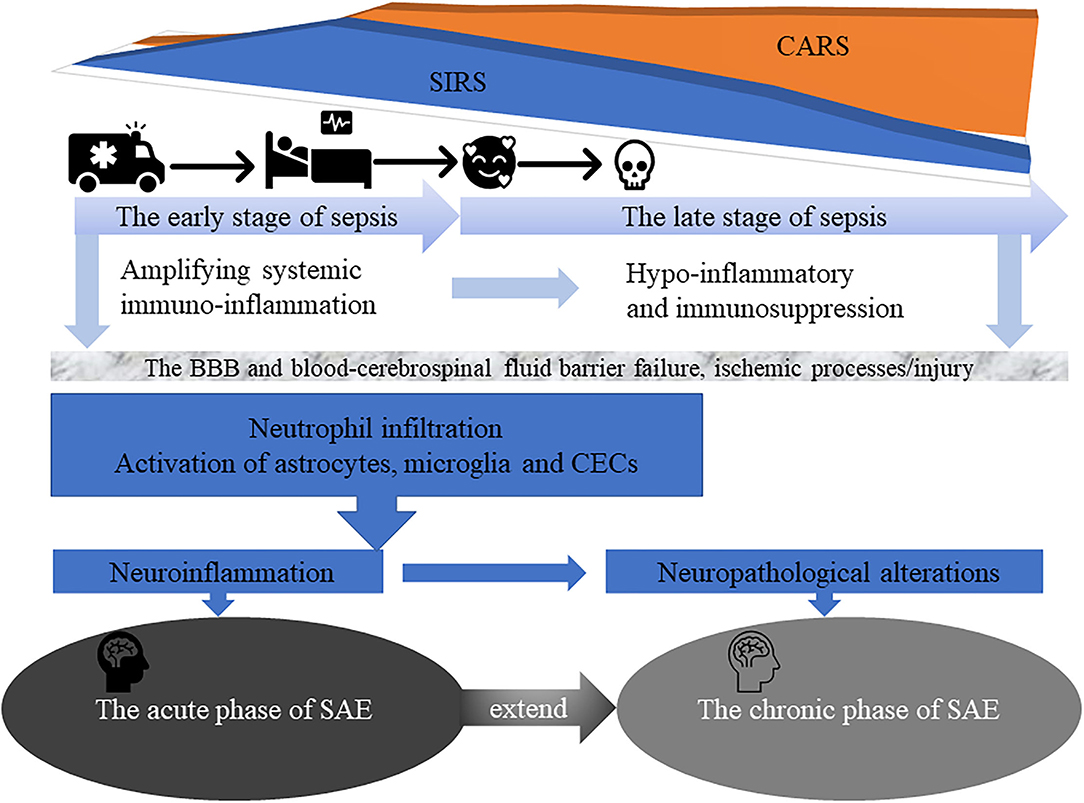
Figure 1. The early stage of sepsis is often accompanied by an acute phase of SAE characterized by delirium symptoms and acute changes in the patient's consciousness. In addition, in the late stage of sepsis, more than half of surviving patients gradually progress to the chronic phase of SAE suffering from severe and long-term cognitive deficits, and even depression, anxiety, post-traumatic stress disorder, and self-destructive tendencies, that affect their daily quality of life and place a significant burden on families and society.
From a clinical point a view, the disease course of SAE can be divided into acute and chronic phase (7, 9, 11, 14, 15). The acute phase of SAE is commonly characterized by delirium symptoms and acute changes in patient's consciousness (7, 9, 16, 17) and by the occurrence of different complex pathophysiological mechanisms, including uncontrolled neuroinflammation, as result of neutrophils, monocytes and resident glial cells activation and infiltration; long-term expression of multiple cytokines and chemokine genes, also called “cytokine storm” (18) and is thought to be the major cause of organ dysfunction and acute symptoms like fever, fatigue and anorexia; and enhanced T helper (Th)1 and Th17 cells activity (4) (Figure 1). These lead to dysfunction and massive apoptosis of brain cells, especially microglia, astrocytes, neurons and cerebral endothelial cells (CECs) (15, 19, 20). Indeed/accordingly, a study by Saito et al. (12) showed that in a cecal slurry (CS)-induced septic mouse model, neutrophils infiltration, an hallmark feature of SAE acute phase, favors CD4+ and CD8+ T cells accumulation, microglial activation and neuroinflammation (IL-1β and IL-6) in the cerebral cortex, BBB disruption, and increase anxiety-like symptoms. On the other hand, the number of astrocytes in the cerebral cortex and hippocampus was showed to gradually decrease. The complex pathophysiological mechanisms of SAE acute phase, may also have a long-term and chronic impact on the cognitive ability of sepsis survivors, and even in the development of dementia or neurodegenerative diseases (12, 21, 22). Sepsis may modulate neurodegenerative changes associated with Alzheimer's disease (AD), by favoring amyloid deposition and neuroinflammation in the brain (22, 23). Induction of polymicrobial sepsis by cecal ligation and puncture (CLP) significantly increased the formation of fibrillary amyloid plaques in the hippocampus of mouse models of AD-associated β -amyloidosis, enhanced intracranial plaque- related astrocytes activation and complement C4b expression, both of which regulate amyloid formation. Additional large-scale changes in the intestinal microbiota of mice were also associated with pro-amyloidosis and neuroinflammatory states (23).
As result of an increasing incidence and a decrease in mortality rates, sepsis survivors gradually progress to the chronic phase of SAE. This is often accompanied by severe and long-term cognitive deficits, at the level of memory, attention and verbal fluency; increased incidence of psychiatric disorders including, depression, anxiety, post-traumatic stress disorder and self-destructive tendencies; and inability to live independently. This strongly affects patient's quality of life and cause a significant burden to the families. Moreover, the need to keep a continuous monitoring on sepsis survivors, constitutes a major burden to the health care system, with high socio-economic costs (7, 9, 17, 24–26). Post mortem histology in humans who died from septic shock at the early stage of sepsis, indicate that the neuropathological alterations may be the starting point for permanent neurological damage in survivors, leading to long-term SAE - associated symptoms (4, 5, 16, 27–29). Early-onset neonatal sepsis can lead to an extremely complex set of events, like brain injury, and survivors remain vulnerable to both short- and long-term neurodevelopmental morbidity, due to long-term changes in cerebral blood flow, the release of neuroinflammatory proteins and altered metabolism (30).
Regulatory T cells (Tregs) are key players in immune regulation of both physiological and pathophysiological conditions, and are characterized by the expression of CD4, CD25 and forkhead box protein P3 (Foxp3). These cells have a negative immunomodulatory function, essential to maintain peripheral immune tolerance, prevent autoimmunity and limit chronic inflammatory diseases (31–34).
Tregs are one of the major subtypes of T cells, showing potent anti-inflammatory activity and mediating specialized functions in tissue remodeling (35). Several groups have been exploring the role of these cells on sepsis-induced immuno-inflammatory dysfunction (36–39) (Figure 2), however, their contribution in SAE pathogenesis, still remains unclear.
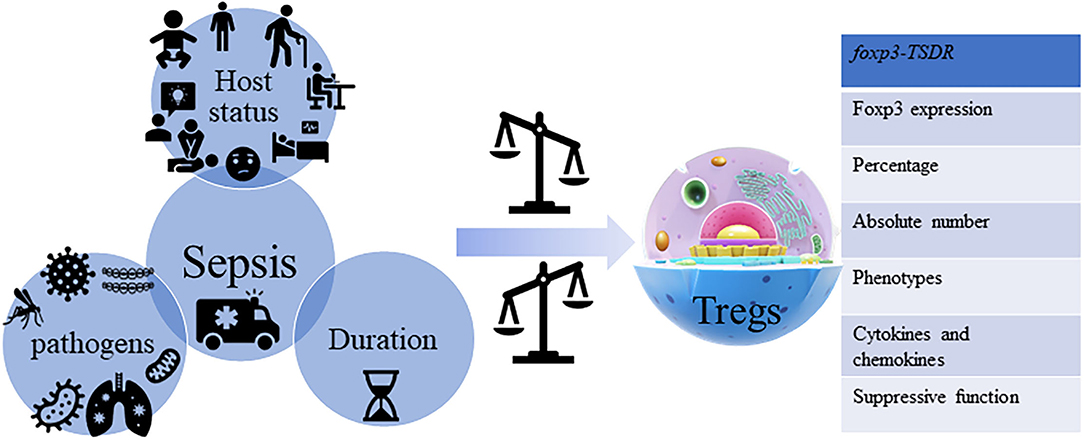
Figure 2. Sepsis and Tregs. Sepsis is a kind of multi-dimensional heterogeneous syndrome, which is not only reflected in the host's demographics, chronic illness, comorbidities, laboratory abnormalities, infections position, patterns of organ dysfunction and severity of illness, and different types of the pathogen but also reflected in the protean host immune responses, where each is not identical. Sepsis influences the heterogeneous characteristics of Tregs from the aspects of percentage (CD4+CD25+/CD4+), absolute number, phenotypes [cytotoxic T lymphocyte antigen (CTLA)-4, CD25, PD-1, CD43, B- and T-lymphocyte attenuator (BTLA), neuropilin (Nrp)-1, G protein-coupled receptor (GPR) 174, lymphocyte activation gene (LAG)-3 and membrane-associated transforming growth factor-β (TGF-βm+), etc.], cytokines and chemokines [IL-10, TGF-β, IL-3, IL-35, and chemokine (C-X-C motif) ligand (CXCL)-4, etc.] secretion, and stability [Foxp3 expression, suppressive function, and methylation status of the foxp3-Tregs-specific demethylated region (TSDR), etc.].
Tregs phenotype and function heterogeneity makes hard to define an universal comprehensive classification of the different Tregs subpopulations.
Based on their origin, Tregs can be divided into two subsets: thymus-derived Tregs (tTregs) and peripheral-derived Tregs (pTregs) (40–43). tTregs (44) are characterized by their constitutive expression of Foxp3 and stable suppressive function, whereas pTregs (45) exhibit unstable Foxp3 expression, unstable suppressive function and can differentiate into cells with an effector function. In a study conducted by Drechsler et al. it was demonstrated that pTregs, rather than tTregs, may play a role in improving septic survival (46). Increasing evidence, directly or indirectly, confirmed that pTregs and brain-specific Tregs are involved in the acute and chronic phase of SAE by mitigating sepsis-induced hyper-inflammatory and immune responses, promoting Th2 functional shift, resolving neuroinflammation and improving the barrier function of the BBB endothelial cells, and even have the potential value of improving SAE-induced cognitive, behavioral and mental abnormalities (10, 12, 21, 47).
Other subsets of Tregs were defined based on cell phenotype and characteristic markers, secreted cytokines and chemokines and specific immune regulation functions (Table 1) (43, 48). These include Resting Tregs (rTregs), Activated Tregs (aTregs), Non-suppressive Tregs (non-Tregs) and helper-like Tregs (Th-Tregs), and three recently discovered Foxp3-Tregs. Althoug there is still, to date, available studies exploring the impact of these subtypes in SAE, different studies demonstrated that distinct subsets with different functions could have a significant role on the control of the immune response and induction of peripheral tolerance (42).
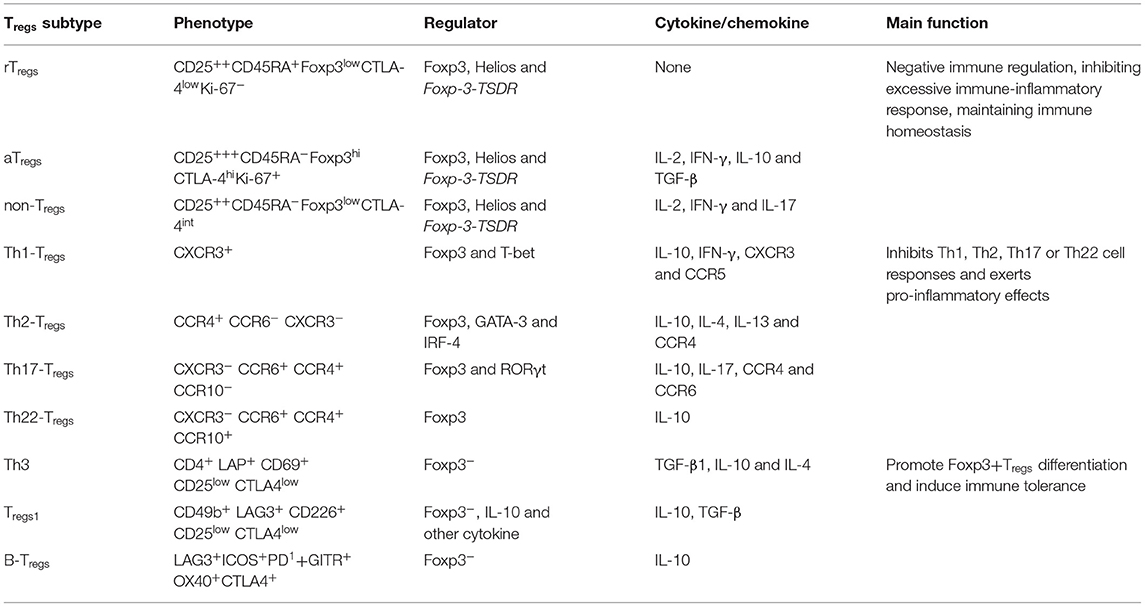
Table 1. Tregs heterogeneity.
Tregs may impact the pathophysiological mechanism of sepsis by acting on the innate and the adaptive immune system, weakening immune function, causing immunoparalysis, and eventually leading to multiple organ dysfunction syndromes and death in sepsis (37, 49). Tregs seem to be key players in the development of sepsis, as well as the hotspot strategies in immunotherapy and immune checkpoints of sepsis and sepsis-associated complications. However, the dual functions of Tregs in infections may provide beneficial or harmful effects even though the number of CD3+ CD4+ CD25hi CD127loTregs in the early stage of sepsis (within 3 days) is not associated with the outcomes (36–39, 50–59).
The brain-specific Tregs of normal rat are memory T cells that account for a larger proportion of cerebral T cells than in the periphery, and, as in the periphery, also play a key role in immune tolerance. The brain-specific Tregs have immunosuppressive effects, including control glial cells activation, with high expression of anti-inflammatory cytokines (IL-10 and IL-35) and immunosuppressive molecules (Foxp3, CTLA-4, CD39, and CD73) (60). Complex systems between the nervous system and the immune system have evolved to mitigate the effects and facilitate recovery from brain injuries caused by diseases such as traumatic brain injury (TBI), ischemic stroke, AD and other degenerative neuropathies, as well as different kind of encephalopathies, including SAE, etc. (16, 17, 61, 62). Tregs infiltrated the brain 1–5 weeks after experimental ischemic stroke in mice and had potent immunomodulatory effects on other immune cells, including monocytes. The interaction between infiltrated Tregs and microglia via Tregs-derived osteopontin in the brain is crucial in behavioral recovery, brain repair, and long-term outcomes (62).
SAE is partly mediated by the dysregulated immune response to sepsis, nevertheless, the pathophysiology underlying it, is still largely unknown. With this in mind and extrapolating the knowledge that Tregs may positively impact different processes, in this section, we give an overview of the main mechanisms by which Tregs may regulate SAE pathogenesis (Figure 3; Table 2).
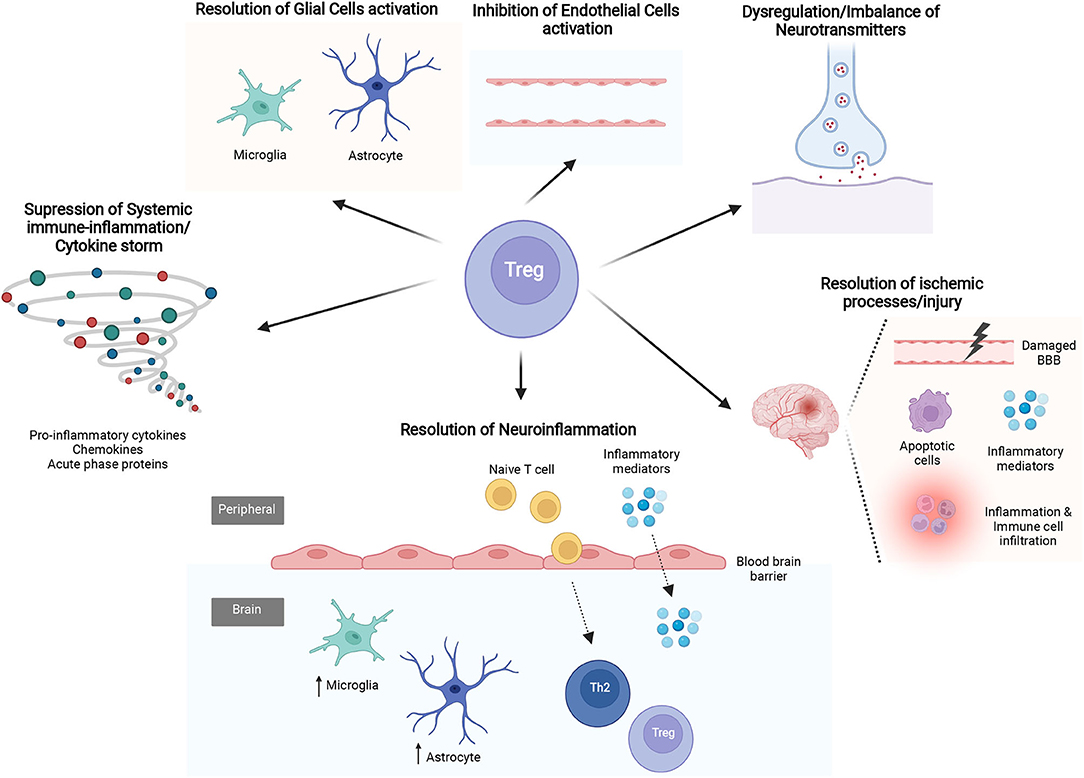
Figure 3. The mechanisms of Regulatory T cells (Tregs) in sepsis-associated encephalopathy (SAE). Several studies evidence the mechanisms by which Tregs may affect different processes know to be related with SAE pathophysiology. The latter include suppression of systemic immune-inflammation/cytokine storm, resolution of excessive neuroinflammation and ischemic processes/injury, regulate the imbalance of neurotransmitters and regulation of glial cells and cerebral endothelial cells (CECs) activity.
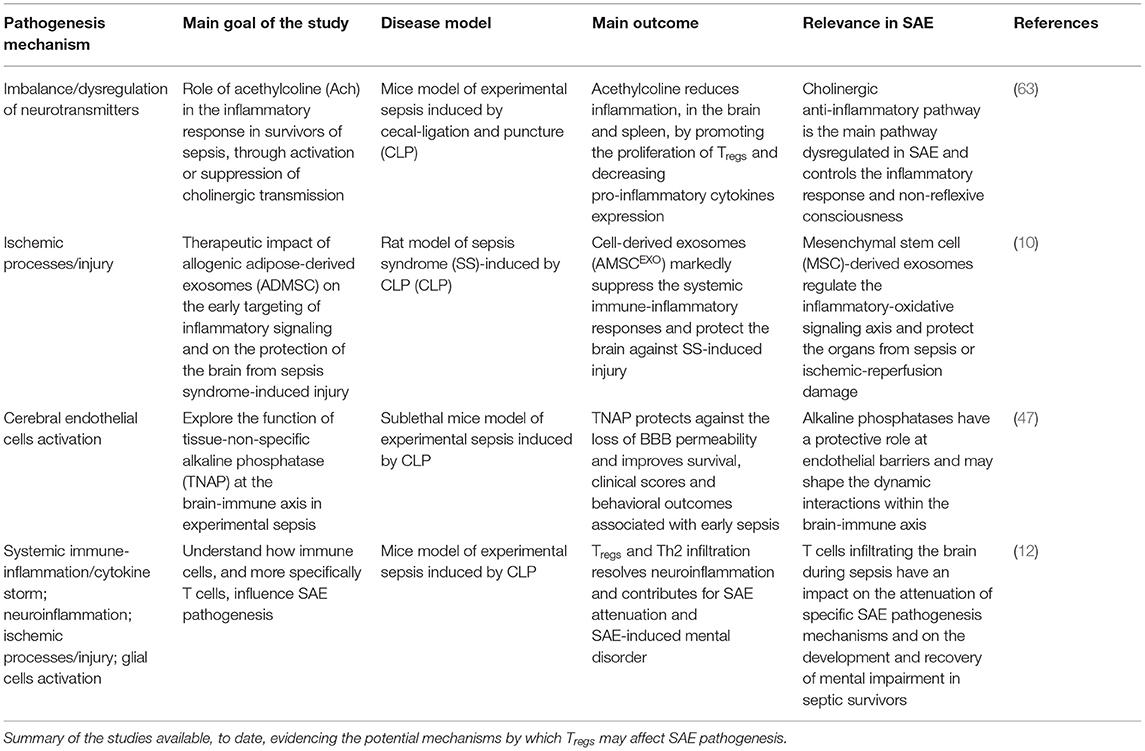
Table 2. Mechanisms of Tregs in SAE.
The systemic immune-inflammation/cytokine storm, which constitutes an exaggerated host immune response associated with excessive production of pro-inflammatory cytokines, chemokines and acute phase proteins, is a hallmark feature in the acute phase of sepsis (18, 64–71). This is one of the main causes of death and ICU acquired complications, including SIRS and compensatory anti-inflammatory response syndrome (CARS), and is defined by the host status (57, 72–76), pathogens (77, 78) and the time span of sepsis (71, 79–84). Tregs have been shown to have a role on the inhibition of sepsis-induced amplifying systemic immuno-inflammation/cytokine storm and on the protection of organs in the early stage of sepsis (53, 85, 86). More specifically, a study conducted by Tatura et al. (51), evaluated the role of Tregs in the early and late stage of sepsis, using a DEREG (Depletion of REGulatory T cells) mouse model, in which sepsis was induced by CLP and subsequent Pseudomonas aeruginosa pulmonary infection. DEREG mouse exhibit high disease scores, mortality, and IL-6 expression in the early stage of sepsis. This study thus, corroborates the hypothesis that Tregs can be explored to limit sepsis-induced amplifying systemic immuno-inflammation/cytokine storm and accelerate a positive outcome in the early stage of sepsis. The suppressive role of Tregs in the sepsis-induced immune-inflammation/cytokine storm was also demonstrated in a study using G protein-coupled receptor 174 (GPR174)-deficient mice (53). In an initial phase of sepsis, this mouse model present a higher expression of IL-10 and CTLA-4 in Tregs, reduced tissue damage and promotion of macrophage polarization to an M2 phenotype induced by sepsis via Tregs. Moreover, an enhanced supressive function of Tregs on IL-6 and TNF-a secretion was also showed. Overall, this study corrobarates the prominent role of Tregs in autoimmune tolerance and in restraining the exagerated immune activation in a sepsis context.
Neuroinflammation is a critical mechanism in the pathogenesis of SAE, characterized by microglia activation, astrogliosis and infiltration of peripheral inflammatory mediators and immune cells (5, 20). Therefore, studies exploring mechanisms that allow the amelioration of this process, may be of great interest in the context of SAE. With this in mind, and considering the evidences suggesting that Tregs may contribute to recovery from SAE and mental impairment, Saito and colleagues (12), hypothesize that T cells, and more specifically Tregs infiltrating the brain, may contribute to the attenuation of SAE and the alleviation of SAE-induced anxiety-like behavior. For that, they used a cecal slurry-induced septic mouse model. A batch of behavioral tests (open-field test, marble burial-test and forced swimming test) was conducted to assess the anxiety-like behavior/state in the septic mouse model. In addition, neuroinflammation and T cell infiltration were also examined in the cerebral cortex. They reported that the infiltration of T cells, and more specifically Tregs and Th2 cells, in the chronic phase of SAE, reduced SAE-induced mental impairment by alleviating neuroinflammation (12, 61). Interestingly, they also showed that the administration of Fingolimod (FTY720), a sphingosine 1-phosphate antagonist that can inhibit lymphocyte escape from lymph nodes, delayed resolution of neuroinflammation and remission of depression in septic mice. This finding was showed to be associated with FTY720's ability to decrease the infiltration of Tregs and Th2 cells and increase Th17 cells in the brain (12). Overall, these findings suggest that infiltrated Tregs and Th2 cells have a promising role in the resolution of neuroinflammation and associated anxious/depressive behavior, which can be of great interest for the investigation of new potential therapeutic targets.
Ischemic processes/injury have been, together with neuroinflammation, identified as playing a critical role in SAE pathogenesis. These mechanisms are directly associated with BBB disruption, brain edema, monocytes and macrophages infiltration, astrogliosis and brain cells apoptosis (11, 14, 15). The aforementioned issues raise the hypothesis that early targeting of the inflammatory signaling may have a potential therapeutic role in the protection against the acute phase of SAE. In this regard, Chang and colleagues, showed that the use of allogenic adipose-derived mesenchymal stem cell-derived exosomes (AMSCEXO) markedly protect the brain against sepsis-induced ischemic injury in rat with sepsis syndrome (10).
Reductions in T lymphocytes, especially the cerebral infiltration of Tregs, is consistent with increased infiltration of natural immune cells, including neutrophils and inflammatory macrophages, and hypoxic-ischemic injury-induced brain injury, including loss of gray and white matter structures (12, 87). C-C chemokine receptors (CCR), such as CCR2, CCR5, and CCR7, are critical molecules for Tregs-mediated BBB protection and potential targets for optimizing Tregs therapy for the treatment of neurological diseases such as ischemic stroke and AD (88–90). CCR5 is critical for Tregs docking at the injured vascular wall, where they interact with blood-borne neutrophil/macrophage. Donor Tregs were deficient in CCR5 and lost their early protective effect against cerebral ischemic processes/injury (88).
Neurotransmitters are likely to be involved in the development and maintenance of SAE. Indeed, different studies have highlighted cholinergic pathway as the main pathway being dysregulated in an SAE context. However, complementary studies showed that other pathways, including gamma-aminobutyric acid, norepinephrine, serotonin and dopamine, seem also to be compromised (91, 92).
Neurotransmitters are traditionally referred as chemical messengers that trigger or inhibit the functions of neurons. However, they can also bind neurotransmitter receptors in T cells and directly activate or suppress their function. Additionally, T cells produce endogenous neurotransmitters and can be regulated by them in an autocrine/paracrine manner (33, 93). More specifically, Tregs were shown to selectively express tyrosine hydroxylase and contain endogenous catecholamines, which function in the autocrine/paracrine inhibitory loop (94). Dopamine down-regulates Tregs activity through the extracellular signal-regulated kinase pathway (95). Acetylcholine reduces inflammation by promoting the proliferation of Tregs in the hippocampus and spleen (63). The α7 nicotinic acetylcholine receptor (α7nAChR), a ligand-gated ion channel, plays an important role in inflammatory responses and is also expressed on the surface of Tregs. Activation of α7nAChR could increase the number and activation of Tregs through the α7nAChR/p-Erk/Foxp3 signaling pathway, and play an anti- neuroinflammatory role in the process of 6-hydroxydopamine (6-OHDA)-induced injury (96). Nitric oxide (NO) production seems to be important for regulating metabolic homeostasis and immune response during sepsis. Arginine (Arg)-induced NO inhibition disrupted the beneficial effect of Th1/Th2 and Th17/Tregs distribution, suggesting that the Arg-NO pathway may partly regulate Th/Tregs homeostasis during sepsis (97).
The activation of microglia, the brain's macrophages, is involved in the progression of SAE by disrupting the BBB function, increasing acute hippocampal neuroinflammation, and enhancing the release of reactive oxygen species (ROS) and consequently, mitochondrial injury (19, 47, 98, 99). Interestingly, while on the seek to better understand SAE pathogenesis and found novel molecular treatments and therapeutic strategies, a recent study explored the protective role of stanniocalcin-1 (STC-1), a glycoprotein-secreted hormone, present in brain, as well as other tissues, in a rat model of sepsis (99). STC-1 decreased microglia-induced acute hippocampal inflammation and oxidative stress and increased the activity of mitochondrial respiratory chain and creatine kinase, after 24 h, thus conferring protection against SAE. Overall, this work demonstrates that strategies promoting neuroprotection by inhibiting the inflammatory response in microglia and protecting against sepsis-associated encephalopathy, have great potential on the treatment of SAE.
The use of cell therapies to reduce microglia-mediated inflammatory response have been explored and can be of great interest as a therapeutic strategy for SAE. More specifically, the use of human mesenchymal stem cells (MSCs) in combination with human cord blood Tregs, favored a reduction of microglial proliferation and activation after TBI and altered the systemic immuno-inflammation (100). The brain-specific Tregs inhibit microglia/macrophage-mediated inflammation via IL-10 and TGF-β, while the homeostatic astrocytes maintain the heterogeneous characteristics of brain-specific Tregs, via IL-2/STAT5, IL-33, CCL1-CCR8, CCL20-CCR6 signaling pathway, as helper cells (60, 101).
Astrocyte activation was detected in brain tissues 4 h after sepsis, peaking at 24 h, and thus, favoring abnormal responses, including the decrease of mitochondria biogenesis and the secretion of inflammatory cytokines through nuclear factor (NF)-κB and other signaling pathways in the astrocytes of the cerebral cortex. Consequently, inflammatory brain injury, refractory neuroinflammation and cognitive impairment are observed (98, 102–104). In addition, extensive structural changes of astrocytes, such as structural remodeling and loss of endfeet, are responsible for BBB collapse (105). Functional Tregs play a critical role in inhibiting astrocyte activation through the production of amphiregulin (AREG), which binds epidermal growth factor receptor (EGFR) on astrocytes and inhibits IL-6 production from astrocytes STAT3 pathway (106). In FTY720-treated septic mice, the number of astrocytes in the cerebral cortex is still reduced at day 30, suggesting a role of infiltrated Tregs on the number and activity of astrocyte in sepsis (12).
Abnormal function and cell death of CECs, as result of neuroinflammation, showed great potential to accelerate SAE in the early stage of sepsis. This happens as result of the formation of a direct link between neurovascular inflammation and brain injury through the P2RX7 pathway, increased vascular inflammation and infiltration of inflammatory cells (CD11b/CD18-expressing leukocytes), abnormal migration of microglia and excess of ROS and NO (47, 98, 103, 107). Gene expression profiles of cerebral vessels isolated from the brains of peripheral LPS-treated mice revealed that, cerebral vessels respond to acute systemic inflammation within minutes by up-regulating the expression of immediate early response genes, followed by the activation of NF-κB pathway. Activation of CECs is the earliest event at the onset of SAE and is the most likely the primordial source of neuroinflammation favoring glial activation. Subsequently, apoptotic signals are activated in CECs, which are thought to result in the BBB disruption and allow leakage of peripheral cytokines into the CNS, exacerbating gliosis and leading to a malignant neuroinflammatory cascade (20). In the setting of cholestatic liver injury, Tregs have the ability to modulate the development of sickness behavior (social investigative behavior and immobility), primarily by inhibiting circulating monocytes and hepatic IL-6 production, and subsequently by circulating IL-6/STAT3 signaling that acts at the level of hippocampal CECs (108). The expression of CCL5, a CCR5 ligand, is significantly increased on the injured CECs after cerebral ischemia and is accompanied by upregulation of CCR5 expression on circulating Tregs. In the co-culture of Tregs-CECs, when Tregs are exposed to ischemia-injured CECs, CCR5 expression is induced. In addition, CCR5 induction on Tregs enhanced the expression of inhibitory molecular PD-1L, thereby inhibiting neutrophil-derived matrix metallopeptidase 9 (88).
The prominent role of Tregs in the regulation of SAE pathogenesis, makes these cells highly attractive for the development of novel therapeutic strategies aiming to modulate the immune response, improve patient's clinical outcome and reduce the associated mortality. However, there is still a lack of knowledge that limits the application of these cells in a SAE therapeutic context. With this in mind, and considering the reduced number of studies available to date exploring Tregs therapeutic potential in SAE, in this section we give an overview on potential ways in which Tregs can be therapeutically used.
For decades, dementia has been characterized by a buildup of waste in the brain and mild inflammation, and was shown to be influenced by the immune system (89). In a rat model of sepsis, exposure to LPS for 7 days leads to deposition of amyloid-β plaques and phosphorylated tauopathy in the hippocampus (23, 109). In acute pathologies, such as TBI and ischemic stroke, CD4+ T cells and Foxp3+ Tregs are recruited into the injured tissue to promote its repair (61, 62, 88, 110). Blocking the programmed cell death Ligand 1 (PD-L1) pathway for 12 days in a DM-hTAU transgenic mice model (a mouse model of tauopathy), favored an increased accumulation of Foxp3+Tregs in the brain via the CCR 2/CCL 2 axis. Simultaneously, an improvement of the cognitive behavior, disease pathology (mainly phosphorylated tauopathy deposition in the hippocampus), neuronal survival and hippocampal inflammation, was also observed (89). Nevertheless, the increased accumulation of Foxp3+Tregs in the brain can also have detrimental effects. With this in mind, it was shown that anti-CD25 antibodies can be used to mitigate the accumulation of Foxp3+Tregs, which in turn can alleviate aging and neurodegenerative diseases caused by CCR7-dependent egress of immune cells (90). Increasing the number of Tregs, by delivering the IL-2 antibody complex after ischemic stroke, improves white matter integrity and rescue neurological functions over the long term. These findings suggest that Tregs are the neurorestorative target for stroke recovery (62). Gold nanoparticles, such as 20 nm citrate-covered gold nanoparticles (cit-AuNP), have been demonstrated to have important anti-inflammatory properties in the brain of sepsis and are promising as adjunctive agents in sepsis treatment with antibiotics to avoid SAE (111). Hyperforin-loaded gold nanoparticles, gold nanorods/compounds and anti-inflammatory nanoparticles have the ability to improve symptoms by inhibiting the differentiation of Th1 and Th17, while promoting the accumulation of Tregs and Th2 in the treatment of EAE, cancer, and muscular dystrophy (112–114). Therefore, gold nanoparticles can have therapeutic interest to promote Tregs accumulation in the context of SAE.
The primary role of Tregs in lymphoid tissues is defense, while the primary role of tissue-specific Tregs located in non-lymphoid parenchymal tissues (e.g., skin, muscle, gastrointestinal tract, lung, adipose tissue, central nervous system, etc.) is to maintain homeostasis (12, 35, 115). Many of these tissue-specific Tregs functions, go beyond our initial understanding of Tregs as immuno-inflammation-specific inhibitors (84, 86), whereas a large number of previous intervention and observational studies on sepsis have focused on the functions and characteristics of Tregs in the peripheral circulation and spleen (51–53, 75, 77). The timing and location of tissue-specific Tregs-mediated immune homeostasis and regulation, remain undefined. In the EAE model, at the peak of EAE-related symptoms, infiltrated brain-specific Tregs in the brain tissue, such as Blimp1-expressing follicular Tregs and TNF receptor (TNFR2) 2 -expressing Tregs, are critical in suppressing EAE, maintaining the continuous expression of CTLA-4 and Blimp-1, and allowing active suppression of pathogenic T cells in the brain, but have no effect on T cell in peripheral lymphoid tissue (116, 117). Selective depletion of the brain-specific Tregs decreases oligodendrogenesis, white matter repair and functional recovery, after experimental ischemic stroke. The beneficial effects of Tregs on white matter regeneration were mitigated by microglia depletion. Tregs-derived osteopontin acts on microglia via integrin receptors to enhance microglial reparative activity (62).
CNS and endocrine system dysfunction, as well as peripheral immuno-inflammatory system collapse, are mutually causal in sepsis. In addition, the CNS through the hypothalamic-pituitary-adrenal (HPA) axis, gut-brain axis, sympathetic and parasympathetic nervous system, function as a transportation hub (8, 13). The immuno-inflammatory signals affect different regions of the brain, mainly through humoral and neural pathways, which mostly involve damage of the BBB and activation of vagal afferent fibers, respectively (4, 5, 8, 59). At the same time, CNS dysfunction may be an important cause of neuroendocrine-immune network breakdown, as well as a potential therapeutic target (such as cholinergic anti-inflammatory pathway, humoral pathway mediated by vasopressin, and reconstruction of the HPA axis) for sepsis-induced immunosuppression or endocrine dysfunction (19, 63, 118). The dysregulation of cholinergic and inflammatory systems are the main pathophysiologic mechanisms regulating other brain injuries (such as hepatic encephalopathy and ischemic encephalopathy). The cholinergic anti-inflammatory pathways, control inflammatory responses and non-reflexive consciousness through two-way communication between the brain and the immune system. In this regard the activity of brain acetylcholinesterase is especially important, which is of great benefit to brain injury and is a promising neuroprotective therapy (8, 19, 63, 119). The cholinergic transmission, stimulated by administration of Donepezil, reduces the inflammatory response, 15 days after CLP, by promoting the proliferation of Tregs in the hippocampus and spleen, whereas the homozygous mutant vesicular acetylcholine transporter-knockdown (VAChT-KD) reduced the number of Tregs in the hippocampus and increased inflammation (63).
The gut-brain axis is a bidirectional signaling network of neurons, hormones, immune cells, and microbial molecules that communicate in different ways, namely through the vagus nerve and the enteric nervous system, which can communicate to the brain using gut metabolites (120). The enteric nervous system (ENS) is the largest nerve organ outside the brain and operates autonomously, responding and adapting to local challenges. The immune system and ENS monitor the boundaries between commensal and pathogenic microorganisms in the colon, and FoxP3+ Tregs functionally interact with ENS (121, 122). In the colonic lamina propria, microbial-responsive RORγ+ and Helios+ subsets of Tregs, closely respond to nitrogen energy and peptide nerve fibers. Intestinal neurons inhibit pTregs differentiation and regulate RORγ+ Tregs ratio by secreting IL-6. Tregs and ENS constitute a regulatory circuit in which microbial signals regulate neuronal density and activation, thereby regulating Tregs generation and immune tolerance in the gut (120). The immune signals following sepsis or altered gut composition can cause the brain to respond to a perceived threat of infection and trigger an inflammatory response (123). Patients with sepsis, experience ectopic intestinal flora caused by acute stress, while survivors also experience chronic stress-induced depression and anxiety initiation mediated by gut-brain-axis immunity. The gut-brain-microbe axis is a promising therapeutic target for stress-induced behavioral injury, as it modulates both the peripheral and cerebral immune landscapes (121–123). The combination of probiotics and prebiotics promotes behavioral resilience to chronic stress by normalizing the gut microbiome and promoting Tregs expansion, an impact reflecting behavioral responses better than neuroinflammation in limbic regions of brain. The ratio of ileal Tregs/Th17 is associated with the production of hippocampal chemokines and IL-1β in the prefrontal cortex (124). Traditional Chinese Medicine, such as resveratrol, may inhibit neuroinflammation by regulating microbiota-gut-brain axis mediated Th17/Tregs and Th1/Th2 polarity shift (125).
Commensal gut bacteria impact the host immune homeostasis and can affect disease processes in the brain, through the microbiota-gut-brain axis (120). IL-17+ γδ T cells can be transported from the gut to the brain, especially to the leptomeninges, and enhance ischemic injury through the secretion of IL-17. This results in increased levels of chemokines in the brain parenchyma and subsequent infiltration of cytotoxic immune cells, including neutrophils. Amoxicillin/clavulanate-induced alterations in the intestinal flora or fecal transplants, favors a reduction of the ischemic processes/injury. This happens as result of an increase of intestinal Tregs and a reduction of IL-17+ γδ T cells in the small intestine. Indeed, Tregs induction could be the main mechanism leading to IL-17+ γδ T cells inhibition (126). The farnesoid X receptor (FXR) signaling system closely links gastrointestinal and non-gastrointestinal tissues, especially the gut-brain-immune axis. Regulation of FXR promotes the production of Tregs and protects the brain against ischemic processes/injury (122).
Overall, the studies described, showed us the importance of further studies to better understand the effects of the intestinal microbiome on the induction and function of Tregs, and more specifically, study the crosstalk between the enteric nervous system and Tregs in the context of SAE.
Adoptive cell transfer (ACT) therapies with Tregs have been used in the clinic to manage different immune-related diseases, especially autoimmune diseases and tumors, and animal studies have shown great promise against neurological disorders such as AD, Parkinson's disease, and ischemic stroke (127, 128). In adult offspring with maternal-immune activation (MIA), ACT therapies using activated Tregs (CD4+CD25+Foxp3+) largely restores MIA-induced pro-inflammatory profile and reverses behavioral abnormalities. Moreover, through the use of pathogen-activated maternal Tregs (pathogen-specific maternal Tregs), ACT therapies favor an increase of anti-inflammatory IL-10 level by ~13-fold in the hippocampus (128). Tregs depletion exacerbates inflammation and increases cerebral infarction after ischemic stroke, while the increase in Tregs levels after stroke can prevent further exacerbation of stroke (62, 88). In the acute phase of ischemic stroke, Tregs-based ACT therapies decreases the infiltration of peripheral leukocytes into the infarct area, inhibits cerebral inflammation, and improve the integrity of the damaged BBB (88). In a model of neuromyelitis optica spectrum disorder (NMOSD), Tregs-based ACT therapy, through intraperitoneal injection, immediately after induction of NMOSD can have a protective effect on the brain injury by reducing the systemic inflammatory response in the acute phase of the disorder. At the same time, it can reduce the infiltration of macrophages, neutrophils, and T cells, and decrease the level of chemokines and pro-inflammatory cytokines in brain tissue (129).
Several studies have shown that stem cell-derived small extracellular vesicles have angiogenesis, immunomodulatory, anti-inflammatory, neurological and paracrine effects. These may have potential therapeutic value for SAE, as they may act by regulating sepsis-induced systemic immuno-inflammatory response, dysfunction of the neuroendocrine network, diffuse neuroinflammation, ischemia and imbalance of neurotransmitters (10, 81, 98, 110). The therapeutic effect of human adult stem cells in protecting against the early stage of severe sepsis is partly related to the induction of IL-10-secreting Tregs, thereby reducing inflammatory cells infiltration in different target organs and down-regulating the production of various inflammatory mediators (85). In this regard, Chang et al. showed that use of AMSCEXOcan effectively inhibit systemic inflammation induced by sepsis in rats, by increasing the number of circulating and splenic Tregs, while decreases the number of CD3+/CD4+ and CD3+/CD8+ cells, thus having a significant protective effect on brain injury induced by sepsis (10). Embryonic stem cell-derived small extracellular vesicles (ESC-sEVs) protect ischemic stroke and modulate post-stroke immune responses by regulating Tregs through the TGF-β/Smads signaling pathway, and the depletion of Tregs almost completely abrogates the protective effects of ESC-sEVs (110).
SAE is a multifaceted disorder with significant impact on patient's morbidity and mortality, and whose pathogenesis is complex and multifactorial and not yet completely understood. Tregs have been shown to play a critical role in SAE and thus are gaining increased attention as subject of research in its pathogenesis and treatment. However, the heterogeneity and plasticity of these cells, together with the complexity of the immune system, constitute a significant challenge when envisaging their therapeutic use. With this in mind, an increased understating of the alterations in human Tregs function in patients during SAE, together with animal studies addressing mechanisms underlying this phenomenon, will be greatly helpful to clarify the potential role of Tregs in patient's outcome after SAE.
Y-lG, Y-cL, and Y-fC generated the concept for this review and performed the literature search. Y-lG, Y-cL, XZ, and S-tS revised the draft. S-tS and Y-fC guided the study. All authors have read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (Nos. 81871593 and 81701931), and the National Natural Science Foundation of Tianjin (No. 18JCQNJC10500).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Prof. Shu-Zhang Cui of Tianjin Medical University General Hospital for the help with the experimental design.
1. Mazeraud A, Righy C, Bouchereau E, Benghanem S, Bozza F A., et al. Septic-Associated encephalopathy: a comprehensive review. Neurotherapeutics. (2020) 17:392–403. doi: 10.1007/s13311-020-00862-1
2. Chaudhry N, Duggal AK. Sepsis associated encephalopathy. Adv Med. (2014) 2014:762320. doi: 10.1155/2014/762320
3. Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol. (2012) 8:557–66. doi: 10.1038/nrneurol.2012.183
4. Gu M, Mei XL, Zhao YN. Sepsis and cerebral dysfunction: BBB damage, neuroinflammation, oxidative stress, apoptosis and autophagy as key mediators and the potential therapeutic approaches. Neurotox Res. (2021) 39:489–503. doi: 10.1007/s12640-020-00270-5
5. Tauber SC, Djukic M, Gossner J, Eiffert H, Bruck W, Nau R. Sepsis-associated encephalopathy and septic encephalitis: an update. Expert Rev Anti Infect Ther. (2021) 19:215–31. doi: 10.1080/14787210.2020.1812384
6. Siddiqi N, House AO, Holmes JD. Occurrence and outcome of delirium in medical in-patients: a systematic literature review. Age Ageing. (2006) 35:350–64. doi: 10.1093/ageing/afl005
7. Feng Q, Ai YH, Gong H, Wu L, Ai M, et al. Characterization of sepsis and sepsis-associated encephalopathy. J Intensive Care Med. (2019) 34:938–45. doi: 10.1177/0885066617719750
8. Ren C, Yao R Q., Zhang H, Feng Y W., et al. Sepsis-associated encephalopathy: a vicious cycle of immunosuppression. J Neuroinflammation. (2020) 17:14. doi: 10.1186/s12974-020-1701-3
9. Zhang LN, Wang XT, Ai YH, Guo Q, et al. Epidemiological features and risk factors of sepsis-associated encephalopathy in intensive care unit patients: 2008-2011. Chin Med J. (2012) 125:828–31.
10. Chang CL, Chen H H., Chen K H., Chiang J, et al. Adipose-derived mesenchymal stem cell-derived exosomes markedly protected the brain against sepsis syndrome induced injury in rat. Am J Transl Res. (2019) 11:3955–71.
11. Bluemel P, Wickel J, Grunewald B, Ceanga M, Keiner S, Witte O, et al. Sepsis promotes gliogenesis and depletes the pool of radial glia like stem cells in the hippocampus. Exp Neurol. (2021) 338:113591. doi: 10.1016/j.expneurol.2020.113591
12. Saito M, Fujinami Y, Ono Y, Ohyama S, Fujioka K, Yamashita K, et al. Infiltrated regulatory T cells and Th2 cells in the brain contribute to attenuation of sepsis-associated encephalopathy and alleviation of mental impairments in mice with polymicrobial sepsis. Brain Behav Immun. (2021) 92:25–38. doi: 10.1016/j.bbi.2020.11.010
13. Zhao J, Liu Y, Hu JN, Peng M, Dong N, Zhu XM, et al. Autocrine regulation of interleukin-3 in the activity of regulatory T cells and its effectiveness in the pathophysiology of sepsis. J Infect Dis. (2021) 223:893–904. doi: 10.1093/infdis/jiaa441
14. Chung HY, Wickel J, Brunkhorst FM, Geis C. Sepsis-associated encephalopathy: from delirium to Dementia? J Clin Med. (2020) 9:703. doi: 10.3390/jcm9030703
15. Hoshino K, Uchinami Y, Uchida Y, Saito H, Morimoto Y. Interleukin-1beta modulates synaptic transmission and synaptic plasticity during the acute phase of sepsis in the senescence-accelerated mouse hippocampus. Front Aging Neurosci. (2021) 13:637703. doi: 10.3389/fnagi.2021.637703
16. Eidelman LA, Putterman D, Putterman C, Sprung C L. The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA. (1996) 275:470–3. doi: 10.1001/jama.1996.03530300054040
17. Iacobone E, Bailly-Salin J, Polito A, Friedman D, Stevens R, Sharsher D. Sepsis-associated encephalopathy and its differential diagnosis. Crit Care Med. (2009) 37(Suppl. 10):S331–6. doi: 10.1097/CCM.0b013e3181b6ed58
18. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
19. Zaghloul N, Addorisio ME, Silverman HA, Patel HL, Valdes-Ferrer SI, Ayasolla KR, et al. Forebrain cholinergic dysfunction and systemic and brain inflammation in murine sepsis survivors. Front Immunol. (2017) 8:1673. doi: 10.3389/fimmu.2017.01673
20. Kodali MC, Chen H, Liao FF. Temporal unsnarling of brain's acute neuroinflammatory transcriptional profiles reveals panendothelitis as the earliest event preceding microgliosis. Mol Psychiatry. (2021) 26:3905–19. doi: 10.1038/s41380-020-00955-5
21. Zhang R, Miao J, Zhu P. Regulatory T cell heterogeneity and therapy in autoimmune diseases. Autoimmun Rev. (2021) 20:102715. doi: 10.1016/j.autrev.2020.102715
22. Widmann CN, Heneka MT. Long-term cerebral consequences of sepsis. Lancet Neurol. (2014) 13:630–6. doi: 10.1016/S1474-4422(14)70017-1
23. Basak JM, Ferreiro A, Cohen L S., Sheehan P W., et al. Bacterial sepsis increases hippocampal fibrillar amyloid plaque load and neuroinflammation in a mouse model of Alzheimer's disease. Neurobiol Dis. (2021) 152:105292. doi: 10.1016/j.nbd.2021.105292
24. Wintermann GB, Brunkhorst FM, Petrowski K, Strauss B, Oehmichen F, Pohl M, et al. Stress disorders following prolonged critical illness in survivors of severe sepsis. Crit Care Med. (2015) 43:1213–22. doi: 10.1097/CCM.0000000000000936
25. Weng L, Zeng XY, Yin P, Wang LJ, Wang CY, Jiang W, et al. Sepsis-related mortality in China: a descriptive analysis. Intensive Care Med. (2018) 44:1071–80. doi: 10.1007/s00134-018-5203-z
26. Xie J, Wang H, Kang Y, Zhou L, Liu Z, Qin B, et al. The epidemiology of sepsis in Chinese ICUs: a national cross-sectional survey. Crit Care Med. (2020) 48:e209–18. doi: 10.1097/CCM.0000000000004155
27. Sharshar T, Annane D, de la Grandmaison GL, Brouland JP, Hoskinson NS, Francoise G, et al. The neuropathology of septic shock. Brain Pathol. (2004) 14:21–33. doi: 10.1111/j.1750-3639.2004.tb00494.x
28. Ehler J, Barrett LK, Taylor V, Groves M, Scaravilli F, Wittstock M, et al. Translational evidence for two distinct patterns of neuroaxonal injury in sepsis: a longitudinal, prospective translational study. Crit Care. (2017) 21:262. doi: 10.1186/s13054-017-1850-7
29. Darden DB, Fenner BP, Foster T, Larson S, Efron P. Chronic sepsis brain and regulatory T cells - a promising therapeutic target. Brain Behav Immun. (2021) 93:10–1. doi: 10.1016/j.bbi.2021.01.017
30. El Shimy MS, El-Raggal NM, El-Farrash RA, Shaaban H, Mohamed E, et al. Cerebral blood flow and serum neuron-specific enolase in early-onset neonatal sepsis. Pediatr Res. (2018) 84:261–6. doi: 10.1038/s41390-018-0062-4
31. Cortez JT, Montauti E, Shifrut E, Gatchalian J, Zhang Y, Shaked O, et al. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature. (2020) 582:416–20. doi: 10.1038/s41586-020-2246-4
32. von Knethen A, Heinicke U, Weigert A, Zacharowski K, Brune B. Histone deacetylation inhibitors as modulators of regulatory T Cells. Int J Mol Sci. (2020) 21:2356. doi: 10.3390/ijms21072356
33. Gao Y, Wang C, Wang Z, Li W, Liu Y, Shou S, et al. Semaphorin 3A contributes to sepsisinduced immunosuppression by impairing CD4(+) T cell anergy. Mol Med Rep. (2021) 23:302. doi: 10.3892/mmr.2021.11941
34. Ramsdell F, Rudensky AY. Foxp3: a genetic foundation for regulatory T cell differentiation and function. Nat Immunol. (2020) 21:708–9. doi: 10.1038/s41590-020-0694-5
35. Panduro M, Benoist C, Mathis D. Tissue tregs. Annu Rev Immunol. (2016) 34:609–33. doi: 10.1146/annurev-immunol-032712-095948
36. McBride MA, Patil TK, Bohannon JK, Hernandez A, Sherwood ER, Patil NK. Immune checkpoints: novel therapeutic targets to attenuate sepsis-induced immunosuppression. Front Immunol. (2020) 11:624272. doi: 10.3389/fimmu.2020.624272
37. Yao RQ, Ren C, Wang JN, Wu GS, Zhu XM, Xia ZF, et al. Publication trends of research on sepsis and host immune response during 1999-2019: a 20-year bibliometric analysis. Int J Biol Sci. (2020) 16:27–37. doi: 10.7150/ijbs.37496
38. de Lima MHF, Hiroki CH, de Fatima Borges V, Cebinelli GCM, Santos J, Rosa MH, et al. Sepsis-induced immunosuppression is marked by an expansion of a highly suppressive repertoire of FOXP3 +T regulatory cells-expressing TIGIT. J Infect Dis. (2021) 225:531–41. doi: 10.1093/infdis/jiab405
39. Guinault D, Nicolau-Travers ML, Silva S, Cointault O, Daniau B, Del Bello A, et al. Expression of exhaustion markers on CD8+ T-Cell patterns predict outcomes in septic patients admitted to the ICU. Crit Care Med. (2021) 49:1513–23. doi: 10.1097/CCM.0000000000005047
40. Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. (2004) 21:267–77. doi: 10.1016/j.immuni.2004.07.009
41. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. (2010) 10:490–500. doi: 10.1038/nri2785
42. Giganti G, Atif M, Mohseni Y, Mastronicola D, Grageda N, Povoleri G, et al. Treg cell therapy: how cell heterogeneity can make the difference. Eur J Immunol. (2021) 51:39–55. doi: 10.1002/eji.201948131
43. Zhang X, Zhang X, Qiu C, Shen H, Zhang H, He Z, et al. The imbalance of Th17/Treg via STAT3 activation modulates cognitive impairment in P. gingivalis LPS-induced periodontitis mice. J Leukoc Biol. (2021) 110:511–24. doi: 10.1002/JLB.3MA0521-742RRR
44. Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol. (2009) 9:83–9. doi: 10.1038/nri2474
45. Hoechst B, Gamrekelashvili J, Manns MP, Greten TF, Korangy F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood. (2011) 117:6532–41. doi: 10.1182/blood-2010-11-317321
46. Drechsler S, Zipperle J, Rademann P, Jafarmadar M, Klotz A, Bahrami S, et al. Splenectomy modulates early immuno-inflammatory responses to trauma-hemorrhage and protects mice against secondary sepsis. Sci Rep. (2018) 8:14890. doi: 10.1038/s41598-018-33232-1
47. Brichacek AL, Benkovic SA, Chakraborty S, Nwafor DC, Wang W, Jun S, et al. Systemic inhibition of tissue-nonspecific alkaline phosphatase alters the brain-immune axis in experimental sepsis. Sci Rep. (2019) 9:18788. doi: 10.1038/s41598-019-55154-2
48. Santamaria JC, Borelli A, Irla M. Regulatory T cell heterogeneity in the thymus: impact on their functional activities. Front Immunol. (2021) 12:643153. doi: 10.3389/fimmu.2021.643153
49. Monneret G, Debard AL, Venet F, Bohe J, Hequet O, Bienvenu J, Lepape A. Marked elevation of human circulating CD4+CD25+ regulatory T cells in sepsis-induced immunoparalysis. Crit Care Med. (2003) 31:2068–71. doi: 10.1097/01.CCM.0000069345.78884.0F
50. Chang KC, Burnham CA, Compton SM, Rasche D, Mazuski RJ, McDonough JS, et al. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care. (2013) 17:R85. doi: 10.1186/cc12711
51. Tatura R, Zeschnigk M, Hansen W, Steinmann J, Vidigal PG, Hutzler M, et al. Relevance of Foxp3(+) regulatory T cells for early and late phases of murine sepsis. Immunology. (2015) 146:144–56. doi: 10.1111/imm.12490
52. Liu Q, Lu Y, An L, Li CS. B- and T-lymphocyte attenuator expression on regulatory T-cells in patients with severe sepsis. Chin Med J. (2018) 131:2637–9. doi: 10.4103/0366-6999.244104
53. Qiu D, Chu X, Hua L, Yang Y, Li K, Han Y, et al. Gpr174-deficient regulatory T cells decrease cytokine storm in septic mice. Cell Death Dis. (2019) 10:233. doi: 10.1038/s41419-019-1462-z
54. Yu Q, Li Y, Wang H, Xiong H. TSLP induces a proinflammatory phenotype in circulating innate cells and predicts prognosis in sepsis patients. FEBS Open Bio. (2019) 9:2137–48. doi: 10.1002/2211-5463.12746
55. Gao Y, Li L, Liu Y, Li W, Wang Z, Shou S, et al. [Effect of semaphorin-3A on the cellular stability of CD4(+)CD25(+) regulatory T cells induced by lipopolysaccharide]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. (2020) 32:1454–60. doi: 10.3760/cma.j.cn121430-20200706-00501
56. Lou JS, Wang JF, Fei MM, Zhang Y, Wang J, Guo Y, et al. Targeting lymphocyte activation gene 3 to reverse T-lymphocyte dysfunction and improve survival in murine polymicrobial sepsis. J Infect Dis. (2020) 222:1051–61. doi: 10.1093/infdis/jiaa191
57. Xu T, Zhao J, Wang X, Meng Y, Zhao Z, Bao R, et al. CXCL4 promoted the production of CD4(+)CD25(+)FOXP3(+)treg cells in mouse sepsis model through regulating STAT5/FOXP3 pathway. Autoimmunity. (2020) 53:289–96. doi: 10.1080/08916934.2020.1777283
58. Gao YL, Wang CX, Wang ZY, Li W, Liu YC, Shou ST, et al. Targeting neuropilin-1 suppresses the stability of CD4(+) CD25(+) regulatory T cells via the NF-kappaB signaling pathway in sepsis. Infect Immun. (2021) 89:e00399–20. doi: 10.1128/IAI.00399-20
59. Zhao L, Gao Y, Guo S, Lu X, Yu S, Ge Z, et al. Sepsis-Associated encephalopathy: insight into injury and pathogenesis. CNS Neurol Disord Drug Targets. (2021) 20:112–24. doi: 10.2174/1871527319999201117122158
60. Xie L, Choudhury GR, Winters A, Yang SH, Jin K. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur J Immunol. (2015) 45:180–91. doi: 10.1002/eji.201444823
61. Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature. (2019) 565:246–50. doi: 10.1038/s41586-018-0824-5
62. Shi L, Sun Z, Su W, Xu F, Xie D, Zhang Q, et al. Treg cell-derived osteopontin promotes microglia-mediated white matter repair after ischemic stroke. Immunity. (2021) 54:1527–42 e1528. doi: 10.1016/j.immuni.2021.04.022
63. Jeremias IC, Victorino VJ, Barbeiro HV, Kubo SA, Prodo CM, Lima TM, et al. The role of acetylcholine in the inflammatory response in animals surviving sepsis induced by cecal ligation and puncture. Mol Neurobiol. (2016) 53:6635–43. doi: 10.1007/s12035-015-9538-y
64. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. (2013) 13:862–74. doi: 10.1038/nri3552
65. Kellum JA, Pike F, Yealy DM, Huang DT, Shapiro NI, Angus DC, et al. Relationship between alternative resuscitation strategies, host response and injury biomarkers, and outcome in septic shock: analysis of the protocol-based care for early septic shock study. Crit Care Med. (2017) 45:438–45. doi: 10.1097/CCM.0000000000002206
66. Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour C, Iwashyna TJ, et al. Incidence and trends of sepsis in US hospitals using clinical vs claims data, 2009-2014. JAMA. (2017) 318:1241–9. doi: 10.1001/jama.2017.13836
67. Seymour CW, Gesten F, Prescott HC, Friedrich ME, Iwashyna TJ, Phillipps GS, et al. Time to treatment and mortality during mandated emergency care for sepsis. N Engl J Med. (2017) 376:2235–44. doi: 10.1056/NEJMoa1703058
68. Lu Y, An L, Liu Q, Li C. Expression and clinical correlations of costimulatory molecules on peripheral T lymphocyte subsets of early-stage severe sepsis: a prospective observational study. Shock. (2018) 49:631–40. doi: 10.1097/SHK.0000000000001017
69. You B, Zhang YL, Luo GX, Dang YM, Jiang B, Huang GT, et al. Early application of continuous high-volume haemofiltration can reduce sepsis and improve the prognosis of patients with severe burns. Crit Care. (2018) 22:173. doi: 10.1186/s13054-018-2095-9
70. Carvelli J, Piperoglou C, Bourenne J, Farnarier C, Banzet N, Demerle C, et al. Imbalance of circulating innate lymphoid cell subpopulations in patients with septic shock. Front Immunol. (2019) 10:2179. doi: 10.3389/fimmu.2019.02179
71. Karakike E, Kyriazopoulou E, Tsangaris I, Routsi C, Vincent JL, Giamarellos-Bourboulis EJ. The early change of SOFA score as a prognostic marker of 28-day sepsis mortality: analysis through a derivation and a validation cohort. Crit Care. (2019) 23:387. doi: 10.1186/s13054-019-2665-5
72. Ruiz LA, Espana PP, Gomez A, Bilbao A, Jaca C, Aramburu A, et al. Age-related differences in management and outcomes in hospitalized healthy and well-functioning bacteremic pneumococcal pneumonia patients: a cohort study. BMC Geriatr. (2017) 17:130. doi: 10.1186/s12877-017-0518-0
73. Ahmadi N, Ahmadi A, Kheirali E, Hossein Yadegari M, Bayat M, Shajiei A, et al. Systemic infection with Candida albicans in breast tumor bearing mice: cytokines dysregulation and induction of regulatory T cells. J Mycol Med. (2019) 29:49–55. doi: 10.1016/j.mycmed.2018.10.006
74. Yende S, Kellum JA, Talisa VB, Peck Palmer O, Chang CH, Filbin MR, et al. Long-term host immune response trajectories among hospitalized patients with sepsis. JAMA Netw Open. (2019) 2:e198686. doi: 10.1001/jamanetworkopen.2019.8686
75. Saito M, Inoue S, Yamashita K, Kakeji Y, Fukumoto T, Kotani J. IL-15 improves aging-induced persistent T cell exhaustion in mouse models of repeated sepsis. Shock. (2020) 53:228–35. doi: 10.1097/SHK.0000000000001352
76. Franekova J, Protus M, Kieslichova E, Brezina A, Komrskova J, Vymetalik J, et al. Changes in sepsis biomarkers after immunosuppressant administration in transplant patients. Mediators Inflamm. (2021) 2021:8831659. doi: 10.1155/2021/8831659
77. Li C, Spallanzani RG, Mathis D. Visceral adipose tissue tregs and the cells that nurture them. Immunol Rev. (2020) 295:114–25. doi: 10.1111/imr.12850
78. de Roquetaillade C, Mansouri S, Brumpt C, Neuwirth M, Voicu S, Le Dorze M, et al. Comparison of circulating immune cells profiles and kinetics between coronavirus disease 2019 and bacterial sepsis. Crit Care Med. (2021) 49:1717–25. doi: 10.1097/CCM.0000000000005088
79. Landelle C, Lepape A, Voirin N, Tognet E, Venet F, Bohe J, et al. Low monocyte human leukocyte antigen-DR is independently associated with nosocomial infections after septic shock. Intensive Care Med. (2010) 36:1859–66. doi: 10.1007/s00134-010-1962-x
80. Antonakos N, Tsaganos T, Oberle V, Tsangaris I, Lada M, Pistiki A, et al. Decreased cytokine production by mononuclear cells after severe gram-negative infections: early clinical signs and association with final outcome. Crit Care. (2017) 21:48. doi: 10.1186/s13054-017-1625-1
81. Bomans K, Schenz J, Sztwiertnia I, Schaack D, Weigand M A., et al. Sepsis induces a long-lasting state of trained immunity in bone marrow monocytes. Front Immunol. (2018) 9:2685. doi: 10.3389/fimmu.2018.02685
82. Schmoeckel K, Mrochen DM, Huhn J, Potschke C, Broker B. Polymicrobial sepsis and non-specific immunization induce adaptive immunosuppression to a similar degree. PLoS ONE. (2018) 13:e0192197. doi: 10.1371/journal.pone.0192197
83. Hotchkiss RS, Colston E, Yende S, Crouser ED, Martin G, Albertson T, et al. Immune checkpoint inhibition in sepsis: a Phase 1b randomized study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of nivolumab. Intensive Care Med. (2019) 45:1360–71. doi: 10.1007/s00134-019-05704-z
84. Tran DT, Jeong YY, Kim JM, Bae HB, Son SK, Kwak SH. The anti-inflammatory role of bilirubin on two-hit sepsis animal model. Int J Mol Sci. (2020) 21:8650. doi: 10.3390/ijms21228650
85. Gonzalez-Rey E, Anderson P, Gonzalez MA, Rico L, Buscher D, Delgado M. Human adult stem cells derived from adipose tissue protect against experimental colitis and sepsis. Gut. (2009) 58:929–39. doi: 10.1136/gut.2008.168534
86. Li H, Qiu D, Yang H, Yuan Y, Wu L, Chu L, et al. Therapeutic efficacy of excretory-secretory products of trichinella spiralis adult worms on sepsis-induced acute lung injury in a mouse model. Front Cell Infect Microbiol. (2021) 11:653843. doi: 10.3389/fcimb.2021.653843
87. Herz J, Koster C, Crasmoller M, Abberger H, Hansen W, Felderhoff-Muser U, et al. Peripheral T cell depletion by FTY720 exacerbates hypoxic-ischemic brain injury in neonatal mice. Front Immunol. (2018) 9:1696. doi: 10.3389/fimmu.2018.01696
88. Li P, Wang L, Zhou Y, Gan Y, Zhu W, Xia Y, et al. C-C chemokine receptor type 5. (CCR5)-mediated docking of transferred tregs protects against early blood-brain barrier disruption after stroke. J Am Heart Assoc. (2017) 6:e006387. doi: 10.1161/JAHA.117.006387
89. Ben-Yehuda H, Arad M, Peralta Ramos JM, Sharon E, Castellani G, Ferrera S, et al. Key role of the CCR2-CCL2 axis in disease modification in a mouse model of tauopathy. Mol Neurodegener. (2021) 16:39. doi: 10.1186/s13024-021-00458-z
90. Da Mesquita S, Herz J, Wall M, Dykstra T, de Lima KA, Norris GT, et al. Aging-associated deficit in CCR7 is linked to worsened glymphatic function, cognition, neuroinflammation, beta-amyloid pathology. Sci Adv. (2021) 7:eabe4601. doi: 10.1126/sciadv.abe4601
91. Semmler A, Frisch C, Debeir T, Ramanathan M, Okulla T, Klockgether T, et al. Long-term cognitive impairment, neuronal loss and reduced cortical cholinergic innervation after recovery from sepsis in a rodent model. Exp Neurol. (2007) 204:733–40. doi: 10.1016/j.expneurol.2007.01.003
92. van Gool WA, van de Beek D, Eikelenboom P. Systemic infection and delirium: when cytokines and acetylcholine collide. Lancet. (2010) 375:773–5. doi: 10.1016/S0140-6736(09)61158-2
93. Levite M. Neurotransmitters activate T-cells and elicit crucial functions via neurotransmitter receptors. Curr Opin Pharmacol. (2008) 8:460–71. doi: 10.1016/j.coph.2008.05.001
94. Cosentino M, Fietta AM, Ferrari M, Rasini E, Bombelli R, Carcano E, et al. Human CD4+CD25+ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine/paracrine inhibitory functional loop. Blood. (2007) 109:632–42. doi: 10.1182/blood-2006-01-028423
95. Kipnis J, Cardon M, Avidan H, Lewitus GM, Mordechay S, Rolls A, et al. Dopamine, through the extracellular signal-regulated kinase pathway, downregulates CD4+CD25+ regulatory T-cell activity: implications for neurodegeneration. J Neurosci. (2004) 24:6133–43. doi: 10.1523/JNEUROSCI.0600-04.2004
96. Jiang Y, Ma H, Wang X, Wang Z, Yang Y, Li L, et al. Protective effect of the alpha7 nicotinic receptor agonist PNU-282987 on dopaminergic neurons against 6-hydroxydopamine, regulating anti-neuroinflammatory and the immune balance pathways in rat. Front Aging Neurosci. (2020) 12:606927. doi: 10.3389/fnagi.2020.606927
97. Yeh CL, Tanuseputero SA, Wu JM, Tseng Y, Yang PJ, Lee PC, et al. Intravenous arginine administration benefits CD4(+) T-cell homeostasis and attenuates liver inflammation in mice with polymicrobial sepsis. Nutrients. (2020) 12:1047. doi: 10.3390/nu12041047
98. Hasegawa-Ishii S, Inaba M, Umegaki H, Unno K, Wakabayashi K, Shimada A. Endotoxemia-induced cytokine-mediated responses of hippocampal astrocytes transmitted by cells of the brain-immune interface. Sci Rep. (2016) 6:25457. doi: 10.1038/srep25457
99. Bonfante S, Joaquim L, Fileti ME, Giustina AD, de Souza Goldim MP, Danielki LG, et al. Stanniocalcin 1 inhibits the inflammatory response in microglia and protects against sepsis-associated encephalopathy. Neurotox Res. (2021) 39:119–32. doi: 10.1007/s12640-020-00293-y
100. Caplan HW, Prabhakara KS, Toledano Furman NE, Zorofchian S, Kumar A, Martin C, et al. Combination therapy with treg and mesenchymal stromal cells enhances potency and attenuation of inflammation after traumatic brain injury compared to monotherapy. Stem Cells. (2021) 39:358–70. doi: 10.1002/stem.3320
101. Corsi-Zuelli F, Deakin B. Impaired regulatory T cell control of astroglial overdrive and microglial pruning in schizophrenia. Neurosci Biobehav Rev. (2021) 125:637–53. doi: 10.1016/j.neubiorev.2021.03.004
102. Zhao YZ, Gao ZY, Ma LQ, Zhuang Y, Guan FL. Research on biogenesis of mitochondria in astrocytes in sepsis-associated encephalopathy models. Eur Rev Med Pharmacol Sci. (2017) 21:3924–34.
103. Bellaver B, Dos Santos JP, Leffa DT, Bobermin LD, Roppa PHA, da Silva Tores IL, et al. Systemic inflammation as a driver of brain injury: the astrocyte as an emerging player. Mol Neurobiol. (2018) 55:2685–95. doi: 10.1007/s12035-017-0526-2
104. Shulyatnikova T, Verkhratsky A. Astroglia in sepsis associated encephalopathy. Neurochem Res. (2020) 45:83–99. doi: 10.1007/s11064-019-02743-2
105. Cardoso FL, Herz J, Fernandes A, Rocha J, Sepodes B, Brito M, et al. Systemic inflammation in early neonatal mice induces transient and lasting neurodegenerative effects. J Neuroinflammation. (2015) 12:82. doi: 10.1186/s12974-015-0299-3
106. Ma X, Qin C, Chen M, Yu HH, Chu YH, Chen TJ, et al. Regulatory T cells protect against brain damage by alleviating inflammatory response in neuromyelitis optica spectrum disorder. J Neuroinflammation. (2021) 18:201. doi: 10.1186/s12974-021-02266-0
107. Wang H, Hong LJ, Huang JY, Jiang Q, Tao RR, Tan C, et al. P2RX7 sensitizes Mac-1/ICAM-1-dependent leukocyte-endothelial adhesion and promotes neurovascular injury during septic encephalopathy. Cell Res. (2015) 25:674–90. doi: 10.1038/cr.2015.61
108. Nguyen K, D'Mello C, Le T, Urbanski S, Swain MG. Regulatory T cells suppress sickness behaviour development without altering liver injury in cholestatic mice. J Hepatol. (2012) 56:626–31. doi: 10.1016/j.jhep.2011.09.014
109. Kirk RA, Kesner RP, Wang LM, Wu Q, Towner RA, Hoffman JM, et al. Lipopolysaccharide exposure in a rat sepsis model results in hippocampal amyloid-beta plaque and phosphorylated tau deposition and corresponding behavioral deficits. Geroscience. (2019) 41:467–81. doi: 10.1007/s11357-019-00089-9
110. Xia Y, Hu G, Chen Y, Yuan J, Zhang J, Wang S, et al. Embryonic stem cell derived small extracellular vesicles modulate regulatory T cells to protect against ischemic stroke. ACS Nano. (2021) 15:7370–85. doi: 10.1021/acsnano.1c00672
111. Di Bella D, Ferreira JPS, Silva RNO, Echem C, Milan A, Akamine EH, et al. Gold nanoparticles reduce inflammation in cerebral microvessels of mice with sepsis. J Nanobiotechnology. (2021) 19:52. doi: 10.1186/s12951-021-00796-6
112. Nosratabadi R, Rastin M, Sankian M, Haghmorad D, Mahmoudi M. Hyperforin-loaded gold nanoparticle alleviates experimental autoimmune encephalomyelitis by suppressing Th1 and Th17 cells and upregulating regulatory T cells. Nanomedicine. (2016) 12:1961–71. doi: 10.1016/j.nano.2016.04.001
113. Safina I, Al Sudani ZAN, Hashoosh A, Darrigues E, Watanabe F, Biris AS, et al. Gold nanorods enhance different immune cells and allow for efficient targeting of CD4+ Foxp3+ tregulatory cells. PLoS ONE. (2021) 16:e0241882. doi: 10.1371/journal.pone.0241882
114. Raimondo TM, Mooney DJ. Anti-inflammatory nanoparticles significantly improve muscle function in a murine model of advanced muscular dystrophy. Sci Adv. (2021) 7:eabh3693. doi: 10.1126/sciadv.abh3693
115. Li LL, Dai B, Sun YH, Zhang TT. The activation of IL-17 signaling pathway promotes pyroptosis in pneumonia-induced sepsis. Ann Transl Med. (2020) 8:674. doi: 10.21037/atm-19-1739
116. Luo L, Hu X, Dixon ML, Pope BJ, Leavenworth JD, Raman C, et al. Dysregulated follicular regulatory T cells and antibody responses exacerbate experimental autoimmune encephalomyelitis. J Neuroinflammation. (2021) 18:27. doi: 10.1186/s12974-021-02076-4
117. Ronin E, Pouchy C, Khosravi M, Hilaire M, Gregoire S, Casrouge A, et al. Tissue-restricted control of established central nervous system autoimmunity by TNF receptor 2-expressing Treg cells. Proc Natl Acad Sci USA. (2021) 118:e2014043118. doi: 10.1073/pnas.2014043118
118. Passaglia P, de Lima Faim F, Batalhao ME, Stabile A, Brndhack LM, Antunes-Rodrigues J, et al. Central administration of angiotensin-(1-7) improves vasopressin impairment and hypotensive response in experimental endotoxemia. Cells. (2021) 10:105. doi: 10.3390/cells10010105
119. Ye J, Lin H, Mu J, Cui X, Ying H, Lin M, et al. Effect of basic fibroblast growth factor on hippocampal cholinergic neurons in a rodent model of ischaemic encephalopathy. Basic Clin Pharmacol Toxicol. (2010) 107:931–9. doi: 10.1111/j.1742-7843.2010.00603.x
120. Yan N, Yan T, Xia Y, Hao H, Wang G, Gonzalez F, et al. The pathophysiological function of non-gastrointestinal farnesoid X receptor. Pharmacol Ther. (2021) 226:107867. doi: 10.1016/j.pharmthera.2021.107867
121. Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic FJ, van der Zwan J, et al. Molecular architecture of the mouse nervous system. Cell. (2018) 174:999–1014 e1022. doi: 10.1016/j.cell.2018.06.021
122. Yan Y, Ramanan D, Rozenberg M, McGovern K, Rastelli D, Vijaykumar B, et al. Interleukin-6 produced by enteric neurons regulates the number and phenotype of microbe-responsive regulatory T cells in the gut. Immunity. (2021) 54:4991–513 e495. doi: 10.1016/j.immuni.2021.02.002
123. Moughnyeh MM, Brawner KM, Kennedy BA, Yeramilli V, Udayakumar N, Graham JA, et al. Stress and the gut-brain axis: implications for cancer, inflammation and sepsis. J Surg Res. (2021) 266:336–44. doi: 10.1016/j.jss.2021.02.055
124. Westfall S, Caracci F, Estill M, Frolinger T, Shen L, Pasinetti G, et al. Chronic stress-induced depression and anxiety priming modulated by gut-brain-axis immunity. Front Immunol. (2021) 12:670500. doi: 10.3389/fimmu.2021.670500
125. Dou Z, Rong X, Zhao E, Zhang L, Lv Y. Neuroprotection of resveratrol against focal cerebral ischemia/reperfusion injury in mice through a mechanism targeting gut-brain axis. Cell Mol Neurobiol. (2019) 39:883–98. doi: 10.1007/s10571-019-00687-3
126. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med. (2016) 22:516–23. doi: 10.1038/nm.4068
127. Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol. (2020) 20:158–72. doi: 10.1038/s41577-019-0232-6
128. Xu Z, Zhang X, Chang H, Kong Y, Ni Y, Liu R, et al. Rescue of maternal immune activation-induced behavioral abnormalities in adult mouse offspring by pathogen-activated maternal treg cells. Nat Neurosci. (2021) 24:818–30. doi: 10.1038/s41593-021-00837-1
Keywords: sepsis, sepsis-associated encephalopathy, Regulatory T cells, neuroinflammation, ischemic injury
Citation: Gao Y-l, Liu Y-c, Zhang X, Shou S-t and Chai Y-f (2022) Insight Into Regulatory T Cells in Sepsis-Associated Encephalopathy. Front. Neurol. 13:830784. doi: 10.3389/fneur.2022.830784
Received: 13 January 2022; Accepted: 18 February 2022;
Published: 16 March 2022.
Edited by:
Sundararajan Jayaraman, University of Illinois, United StatesReviewed by:
Ziyu Zhu, Shanghai Jiao Tong University School of Medicine, ChinaCopyright © 2022 Gao, Liu, Zhang, Shou and Chai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan-fen Chai, Y2hhaXlhbmZlbjIwMTJAMTI2LmNvbQ==; Yu-lei Gao, Z2FveXVsZWk4MjhAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.