 Anniina Snellman1,2*
Anniina Snellman1,2* Laura L. Ekblad1
Laura L. Ekblad1 Mikko Koivumäki1
Mikko Koivumäki1 Noora Lindgrén1
Noora Lindgrén1 Jouni Tuisku1
Jouni Tuisku1 Merja Perälä3Lila Kallio3Riina Lehtonen1
Merja Perälä3Lila Kallio3Riina Lehtonen1 Virva Saunavaara1,4Jani Saunavaara4Vesa Oikonen1Richard Aarnio1
Virva Saunavaara1,4Jani Saunavaara4Vesa Oikonen1Richard Aarnio1 Eliisa Löyttyniemi5
Eliisa Löyttyniemi5 Riitta Parkkola6Mira Karrasch7
Riitta Parkkola6Mira Karrasch7 Henrik Zetterberg2,8,9,10,11
Henrik Zetterberg2,8,9,10,11 Kaj Blennow2,8
Kaj Blennow2,8 Juha O. Rinne1
Juha O. Rinne1- 1Turku PET Centre, University of Turku and Turku University Hospital, Turku, Finland
- 2Department of Psychiatry and Neurochemistry, Institute of Neuroscience and Physiology, The Sahlgrenska Academy at the University of Gothenburg, Mölndal, Sweden
- 3Auria Biobank, Turku University Hospital, University of Turku, Turku, Finland
- 4Department of Medical Physics, Turku University Hospital, Turku, Finland
- 5Department of Biostatistics, University of Turku, Turku, Finland
- 6Department of Radiology, Turku University Hospital, University of Turku, Turku, Finland
- 7Department of Psychology, Åbo Akademi University, Turku, Finland
- 8Clinical Neurochemistry Laboratory, Sahlgrenska University Hospital, Mölndal, Sweden
- 9Department of Neurodegenerative Disease, UCL Institute of Neurology, London, United Kingdom
- 10UK Dementia Research Institute at UCL, London, United Kingdom
- 11Hong Kong Center for Neurodegenerative Diseases, Hong Kong, China
Background: Detailed characterization of early pathophysiological changes in preclinical Alzheimer's disease (AD) is necessary to enable development of correctly targeted and timed disease-modifying treatments. ASIC-E4 study (“Beta-Amyloid, Synaptic loss, Inflammation and Cognition in healthy APOE ε4 carriers”) combines state-of-the-art neuroimaging and fluid-based biomarker measurements to study the early interplay of three key pathological features of AD, i.e., beta-amyloid (Aβ) deposition, neuroinflammation and synaptic dysfunction and loss in cognitively normal volunteers with three different levels of genetic (APOE-related) risk for late-onset AD.
Objective: Here, our objective is to describe the study design, used protocols and baseline demographics of the ASIC-E4 study.
Methods/Design: ASIC-E4 is a prospective observational multimodal imaging study performed in Turku PET Centre in collaboration with University of Gothenburg. Cognitively normal 60–75-year-old-individuals with known APOE ε4/ε4 genotype were recruited via local Auria Biobank (Turku, Finland). Recruitment of the project has been completed in July 2020 and 63 individuals were enrolled to three study groups (Group 1: APOE ε4/ε4, N = 19; Group 2: APOE ε4/ε3, N = 22; Group 3: APOE ε3/ε3, N = 22). At baseline, all participants will undergo positron emission tomography imaging with tracers targeted against Aβ deposition (11C-PIB), activated glia (11C-PK11195) and synaptic vesicle glycoprotein 2A (11C-UCB-J), two brain magnetic resonance imaging scans, and extensive cognitive testing. In addition, blood samples are collected for various laboratory measurements and blood biomarker analysis and cerebrospinal fluid samples are collected from a subset of participants based on additional voluntary informed consent. To evaluate the predictive value of the early neuroimaging findings, neuropsychological evaluation and blood biomarker measurements will be repeated after a 4-year follow-up period.
Discussion: Results of the ASIC-E4 project will bridge the gap related to limited knowledge of the synaptic and inflammatory changes and their association with each other and Aβ in “at-risk” individuals. Thorough in vivo characterization of the biomarker profiles in this population will produce valuable information for diagnostic purposes and future drug development, where the field has already started to look beyond Aβ.
Introduction
Over fifty million people worldwide live with dementia, and the number is expected to rise to 78 million by 2030 (1). Alzheimer's disease (AD) causes over two thirds of the dementia cases (2). To cope with this challenge, AD drug development has shifted its focus toward disease-modifying treatments and targeting the underlying pathophysiological events as early as possible, even before the onset of clinical symptoms (3, 4). Recent approval of Aducanumab by the U.S. Food and Drug Administration as the first disease-modifying treatment for AD (https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug) gives hope for new drugs targeting various aspects of AD pathophysiology to become available in the future. However, this development also creates an increasing need for suitable biomarkers to better characterize the different biological stages of AD and substitute clinical endpoints in future therapeutic trials with varying targets.
Biologically, AD is characterized by extracellular beta-amyloid (Aβ) plaques and intracellular tau aggregates in the brain, however, complexity behind the disease process is increasingly recognized (5). Alterations in Aβ metabolism are traditionally presented to be the earliest changes, leading to further hyperphosphorylation and aggregation of tau into intraneuronal neurofibrillary tangles, but the relationship between the two pathologies is now known to be more complex and synergistic (6). In addition, neuroinflammation, characterized by activation of the immune cells in the central nervous system (CNS), and synaptic dysfunction and loss are known to be present throughout the AD pathophysiological process (7). These alterations in the brain begin to develop decades before clinical symptoms arise, and the course of AD has been re-evaluated to be a progressive continuum from a preclinical “silent” phase to a clinically manifested dementia phase, characterized by the early presence of various biological markers of the ongoing disease process (8, 9). Thus, the earliest brain changes and their associations with each other can only be investigated in pre-symptomatic individuals, who are at increased risk of AD in the future.
The strongest genetic risk factor for sporadic AD is the ε4 allele of the apolipoprotein E gene [APOE, (10)]. APOE is polymorphic, and from the three different alleles (APOE ε2, APOE ε3 and APOE ε4), one ε4 allele increases the lifetime risk of AD 3–4 fold, and two ε4 alleles 9–15 fold (11). In the CNS, Apolipoprotein E is produced mainly by astrocytes and microglia, and it functions as a lipid transporter delivering cholesterol and other lipids to neurons, thus maintaining and restoring membranes and synaptic integrity (12). In AD pathogenesis, APOE ε4 is involved with both gain of toxic functions, such as increased Aβ aggregation, tangle formation and brain atrophy, as well as loss of neuroprotective functions, such as reduced synaptic and vascular function (13). Especially early Aβ deposition has repeatedly been shown to be present in the brain of APOE ε4 carriers (14, 15) already prior to any cognitive changes (16). Even though the mechanisms are still not understood in detail, APOE ε4 most likely contributes to increased amyloid aggregation by deficient clearance of Aβ from the brain compared to ε3 and ε2 (17). In addition to Aβ and tau pathology, neuroinflammation has been suggested to be one link between APOE mediated increased risk for AD; Since APOE is mainly expressed in astrocytes and microglia, the cells responsible for important immune functions in the CNS, APOE ε4 is likely to have also a direct effect on glial functions, independent of Aβ or tau (17). Throughout this article we describe individuals with APOE ε4ε4, APOE ε4ε3 and APOE ε3ε3 genotype as having high, intermediate and low risk for sporadic AD, respectively. However, it is important to note that this description only highlights the differences in relative APOE-related risk associated with each genotype and is not used for describing the absolute risk of future AD of individuals with these genotypes.
Development of imaging biomarkers for AD-related pathophysiological change has provided the research community with a valuable tool for investigating the regional and temporal course of different brain changes in AD in vivo. Positron emission tomography (PET) with specific radioligands targeting Aβ deposition and tau aggregates are already well-established imaging biomarkers of AD. They are used for biological definition of the disease and for studying the progression and relationship between these core AD pathophysiological changes (9). In addition to the core pathologies, PET ligands targeting TSPO expressed in activated glial cells (18–20), and recently also synaptic vesicle glycoprotein 2A (SV2A) in presynaptic vesicles (21–24), provide a way for also investigating the neuroinflammatory and synaptic components of the disease process in vivo. Despite the obvious value of various imaging biomarkers for diagnostic and research purposes, the methods are often expensive, invasive, result in a radiation dose and have limited availability. Thus, more easily accessible blood biomarkers for AD have recently been under vigorous research and shown promise as early diagnostic markers [Reviewed in (25) and (26)]. Ultrasensitive immunoassays and immunoprecipitation combined with mass spectrometry have made it possible to measure various proteins characteristic for AD pathophysiology from blood, including different soluble Aβ species (27), soluble tau and phosphorylated tau species (28–32), neurodegeneration by plasma neurofilament light chain (NfL) (33), and gliosis by plasma glial fibrillary acidic protein (GFAP) (34–37). However, it is still important to validate the performance of these blood-based biomarkers in different clinical and research populations, against gold standard PET methods and in longitudinal studies across the AD continuum.
The aim of the ASIC-E4 (“Beta-Amyloid, Synaptic loss, Inflammation and Cognition in healthy APOE ε4 carriers”) study is to uncover the early interplay of three key pathological features of AD, i.e., synaptic dysfunction and loss, neuroinflammation and Aβ deposition, in cognitively normal individuals with either high, intermediate, or typical APOE-related genetic risk of late-onset AD. By utilizing state-of-the art neuroimaging and fluid biomarker measurements, the ASIC-E4 study aims to clarify whether differences in regional neuroinflammation and synaptic density can be detected in vivo already in cognitively normal subjects differentiated only by their APOE genotype and how they associate with each other, Aβ deposition, cognitive performance, and fluid blood biomarker findings in different APOE genotypes. In addition, after a 4-year follow-up, our aim is to evaluate how the early neuroimaging findings associate with changes in cognitive performance and blood biomarker levels in this valuable at-risk population.
Methods and Analysis
Study Design
ASIC-E4 is a prospective multimodal imaging study aiming to investigate the interplay of Aβ deposition, neuroinflammation and synaptic changes in cognitively normal individuals at different APOE-related genetic risk of AD (Figure 1). The study has been approved by Hospital District of South-West Finland (27.4.2018), and Scientific Advisory Boards of Turku PET Centre (26.3.2018) and Auria Biobank (30.1.2018). ASIC-E4 study is conducted following the guidelines of both National Advisory Board on Research Ethics in Finland, and the European Code of Conduct for Research Integrity by All European Academics. In addition, Declaration of Helsinki, Good Clinical Practice, and EU legislation and General Data Protection Regulation are followed. All used research material and data will be collected and analyzed during the project from participants recruited for the study. This study is observational, and does not include any studied interventions, thus it has not been prospectively registered to ClinicalTrials.gov.
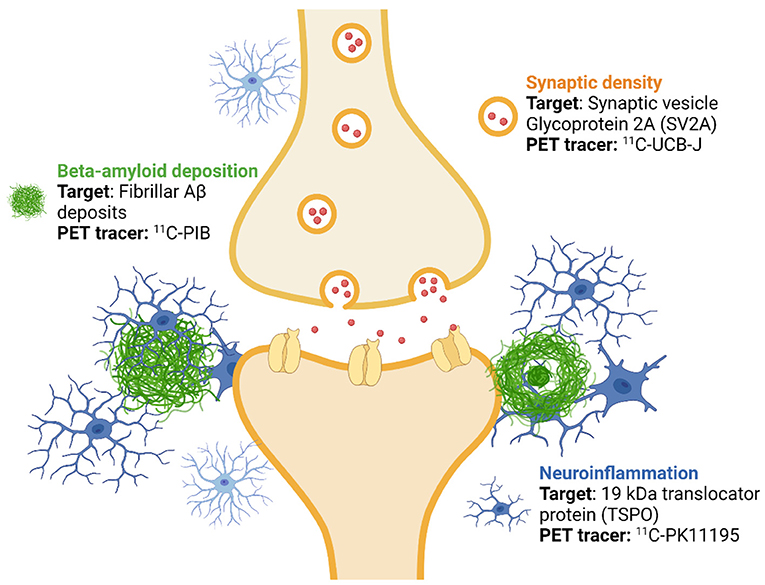
Figure 1. Imaging targets and used positron emission tomography (PET) tracers. ASIC-E4 is a prospective, observational multi-tracer imaging study, where three different positron emission tomography (PET) tracers are used to investigate beta-amyloid (Aβ) deposition, activation of glial cells and synaptic density in individuals with three levels of genetic (APOE-related) risk for sporadic Alzheimer's disease. The figure was created with BioRender.com.
Power Calculation
We performed power calculations based on previously published data with the three used PET tracers, aiming for 90 % power (1-β = 0.9, α = 0.05) to detect 10–20 % differences in regional tracer binding between the cognitively normal APOE ε4/ε4 and APOE ε3/ε3 groups. Based on the calculations, with nine participants per group, we would be able to detect a 20 % difference in mean 11C-PIB standardized uptake value ratios (SUVRs) (1.2 vs. 1.5, standard deviation [SD] 0.2) (38); with 21 participants a 20 % difference in 11C-PK11195 binding potentials (0.37 vs. 0.44, SD 0.07) (18); and with 17 participants a 10 % difference in 11C-UCB-J binding potentials (3.0 vs. 2.7, SD 1.9) (21).
Due to the used multi-tracer approach, and thus higher risk that subjects will suspend their participation before completing all three PET scans, we aimed for recruiting 20–25 participants per group. The calculations were performed for differences between the homozygous APOE ε4/ε4 and APOE ε3/ε3 carriers, however, a gene dose effect is expected to be seen in heterozygous APOE ε4/ε3 carriers, even though it might not reach statistical significance.
Objectives and Hypothesis
Our pre-defined objectives for the study are:
(i) To quantify differences in regional Aβ deposition (11C-PIB binding), neuroinflammation (11C-PK11195 binding), synaptic density (11C-UCB-J binding) and structural brain changes between APOE ε4/ε4 and APOE ε4/ε3 carriers with increased APOE-related genetic risk for sporadic AD, and homozygous APOE ε3/ε3 controls with typical APOE-related genetic risk for sporadic AD.
(ii) To study differences in blood biomarker levels between the three different APOE genotypes and cognitively normal amyloid positive and amyloid negative individuals.
(iii) To investigate regional and voxel-wise associations between brain Aβ deposition, neuroinflammation, and synaptic density with each other, with cognitive performance, with structural brain changes, and with blood biomarker levels both in the whole cohort and within the three different APOE genotypes.
(iv) To investigate the association between baseline neuroimaging findings and changes in cognitive performance and blood biomarker concentrations after 4-year follow-up.
Based on current knowledge and theoretical premise behind the research we hypothesize that:
(i) Cognitively normal APOE ε4/ε4 and APOE ε4/ε3 carriers show regionally increased 11C-PIB and 11C-PK11195 binding and decreased 11C-UCB-J binding compared to APOE ε3/ε3 carriers in brain regions typical for early tau and Aβ pathology. Risk allele dose effect is visible both in 11C-UCB-J (APOE ε4/ε4 < APOE ε4/ε3 < APOE ε3/ε3) and 11C-PIB and 11C-PK11195 uptake (APOE ε4/ε4 > APOE ε4/ε3 > APOE ε3/ε3).
(ii) Due to direct effect of toxic Aβ and tau oligomers on synapses, also 11C-PIB-negative APOE ε4 carriers with less fibrillar amyloid accumulation show decreased 11C-UCB-J binding in regions of early Aβ accumulation. Low synaptic density and high glial activation in APOE ε4 carriers are associated with lower baseline cognitive performance, and increased abnormality in blood biomarkers for AD pathology.
(iii) Increased abnormality in blood biomarker levels is present already in cognitive normal APOE ε4 carriers compared to APOE ε3/ε3 controls.
(iv) Abnormal baseline neuroimaging findings are associated with 4-year temporal changes in cognitive performance.
Selection of Subjects
Inclusion and Exclusion Criteria
Predefined inclusion criteria for the ASIC-E4 study were:
(i) APOE ε4/ε4, APOE ε4/ε3 or APOE ε3/ε3 genotype.
(ii) 60–75-years of age.
(iii) Mini-Mental State Examination (MMSE) score ≥ 25 points.
(iv) Consortium to Establish a Registry for Alzheimer's Disease (CERAD) neuropsychological battery total score > 62 points (39).
The cut point for CERAD total score is defined as two standard deviations below the mean score of cognitively normal individuals in a previously published study on an European population (40). Participants are expected to be cognitively unimpaired as described in the syndromal staging of cognitive continuum in the AD research framework, i.e., cognitive performance of all participants was within expected range of that individual based on all available information (9).
Predefined main exclusion criteria for the ASIC-E4 study were:
(i) Dementia or cognitive impairment, including reported subjective memory complaints.
(ii) Previous diagnosis of other neurological or psychiatric diseases.
(iii) Previous diagnosis of diabetes.
(iv) Previous diagnosis of chronic inflammatory condition and related continuous use of anti-inflammatory medication
(v) Contraindication for magnetic resonance imaging (MRI) or PET (e.g., claustrophobia, presence of ferromagnetic objects in the body, previous high radiation doses).
Recruitment
Recruitment was done in collaboration with the local biobank (Auria Biobank, Turku, Finland, study number: AB17-2549). The biobank has access to blood samples from individuals who had previously signed a biobank consent, and an additional informed consent allowing the biobank to contact them if they are suitable for participating in a research study. APOE genotype had already been determined from a subset of the blood samples (APOE genotyping for methodological details), allowing the biobank to directly contact persons with either APOE ε4/ε4, APOE ε3/ε4 or APOE ε3/ε3 genotype. As only ~ 2–3% of the blood samples are expected to be homozygous APOE ε4/ε4, it would not have been feasible to recruit a group of APOE ε4/ε4 homozygotes without this valuable collaboration.
Altogether 199 invitation letters were sent based on a biobank consent to people with either APOE ε3/ε3 (76 letters), APOE ε3/ε4 (66 letters) or APOE ε4/ε4 (57 letters) genotype during years 2018–2020 (Figure 2). Letters were sent in four batches balanced by age and gender, depending on the recruitment status after previous batches. From the individuals who received the invitation to participate, 109 returned a signed letter of interest and wanted to receive more information about the study. At this point, interested individuals also gave a written informed consent that the APOE status can be given from the biobank to the study site to ensure enrolment to different groups in a balanced way. Detailed study plan and informed consent were sent to all interested candidates, and everyone was subsequently contacted via telephone. During the conversation, the full study protocol was discussed, and if no clear exclusion criteria were found based on the telephone interview, and if the candidate was still interested in participating, they were invited to a screening visit to Turku PET Center.
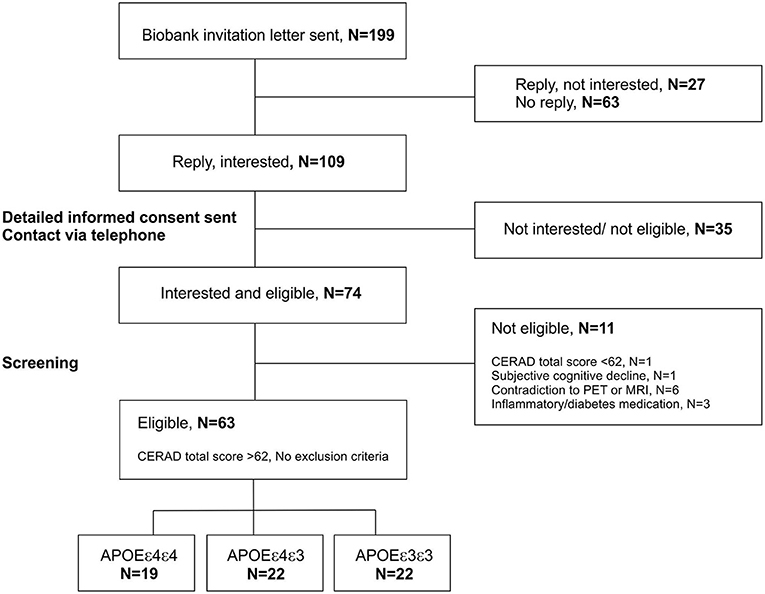
Figure 2. Recruitment scheme for the ASIC-E4 study. Recruitment is done in collaboration with the local biobank (Auria Biobank, Turku, Finland). The biobank was able to send invitation letters based on a biobank consent, directly to individuals with known APOE genotypes. The signed letters of interest including the contact information of the invited individuals, and written informed consent for transforming the APOE status from the biobank to the study site were returned to the responsible researcher by the invited individuals. Subsequently, researchers contacted interested individuals by telephone and if no obvious exclusion criteria was present, invited them to a screening visit. At screening, all interested individuals were interviewed and evaluated with Consortium to Establish a Registry for Alzheimer's Disease (CERAD) neuropsychological test battery. If calculated CERAD total score was >62 points, and no other exclusion criteria was present, individuals were enrolled to the study. APOE, apolipoprotein E gene; PET, positron emission tomography; MRI, magnetic resonance imaging.
Screening
During the screening visit, ASIC-E4 study protocol was discussed in detail and the informed consent was signed by all potential participants. Subsequently, more detailed medical history (e.g., current and past medications, previous medical diagnosis, past head traumas) and general health related personal information (education, smoking, family history for AD) were inquired. After the interview, CERAD neuropsychological battery was performed to all potential participants, to ensure that only cognitively unimpaired individuals were enrolled.
All medical information, CERAD total score and individual subtest scores collected during the screening visit were reviewed by a neurologist, who together with the person performing the screening based on this information made the final enrolment decision.
APOE Genotyping
APOE genotype data used for the ASIC-E4 study were obtained from Auria Biobank, Turku, Finland (Study number: AB17-2549). Genomic DNA was extracted from EDTA-anticoagulated whole blood samples from consented donors using the Chemagic DNA Blood Kit (CMG-1091, PerkinElmer) according to the manufacturer's instructions. APOE genotype analysis was performed at Turku University Hospital, Clinical Microbiology and Immunology Laboratory with a Taqman SNP genotyping assay (Applied Biosystems, ThermoFisher). The genotypes were determined using the TaqMan Genotyper Software (ThermoFisher).
Study Protocol and Timeline
Detailed study protocol and planned visits to study site are illustrated in (Figure 3).
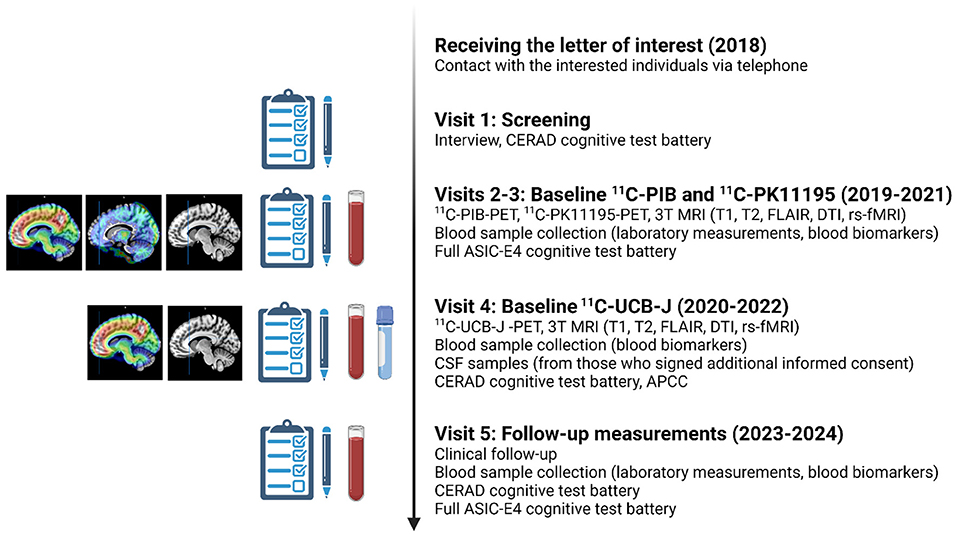
Figure 3. Planned study visits during the ASIC-E4 study. All included ASIC-E4 participants will visit Turku PET Center and Turku University Hospital 3–5 times during years 2019–2022. In addition, all will be invited to a follow-up visit approximately 4 years from screening (starting 2023). APCC, Alzheimer's Prevention Initiative Composite Cognitive Test Score; CERAD, Consortium to Establish a Registry for Alzheimer's Disease; CSF, cerebrospinal fluid; DTI, diffusion tensor imaging; rs-fMRI, resting state functional magnetic resonance imaging; FLAIR, fluid attenuated inversion recovery PET, positron emission tomography; MRI, magnetic resonance imaging. The figure was created with BioRender.com.
All enrolled participants will visit the PET Center 3–5 times for baseline PET imaging, MRI, blood sample collection and extensive neuropsychological evaluation. All measurements are aimed to be performed during a time interval as short as possible, preferably within a 3-month period. However, due to a delay in 11C-UCB-J availability, baseline 11C-UCB-J scans will be acquired approximately 12-18 months after baseline 11C-PK11195/11C-PIB/MRI for each participant. Due to this, an amendment to the study protocol was performed, and additional MRI, revised cognitive test battery and blood samples will be collected with the 11C-UCB-J scan to enable accurate analysis of the 11C-UCB-J PET data, and evaluation of association between 11C-UCB-J binding and blood biomarker levels at the same time point (see Figure 3 for details). It should be noted that the additional MRI scans and cognitive testing was not designed for follow-up purposes.
CSF samples are collected near 11C-UCB-J scans from those who give an additional informed consent for a lumbar puncture.
Four years after the initial visits (starting in January 2023), all participants will be invited for a follow-up visit. All participants will come to Turku PET Center for a clinical follow-up, interview and CERAD neuropsychological battery, similarly to the screening visit. New venous blood samples for laboratory measurements and blood biomarker analysis will be taken and stored for follow-up analysis. In addition to the CERAD test battery, all participants will go through another round of neuropsychological testing with the same ASIC-E4 test battery performed at baseline.
Imaging
MRI
Non-contrast brain MRI is performed with a 3 Tesla MRI scanner for all study participants. Due to an unexpected change in the MR scanner availability at Turku PET Center during the data collecting period, imaging will be performed with two different devices. First batch of baseline scans are done using Philips Ingenuity 3.0 T TF PET-MR (Philips Healthcare, Amsterdam, the Netherlands), and the rest of the baseline scans will be performed with Philips Ingenia 3.0 T systems (Philips Healthcare, Amsterdam, the Netherlands). A 32-channel head coil will be used with Ingenuity PET-MR system and a 20-channel dS head coil with Ingenia system.
All participants will undergo MRI at two different time points; First MRI scan is done at proximity (± 3 months) to the baseline 11C-PK11195 and 11C-PIB PET scans (with either Philips Ingenuity and or Philips Ingenia). Due to the unexpected delay in 11C-UCB-J availability, another MRI with the exact same imaging protocol will be done at the time of 11C-UCB-J scan (all performed with Philips Ingenia) to ensure timely reference image for PET image analysis and volumetric data to be combined with 11C-UCB-J data.
MRI sequences included in the ASIC-E4 imaging protocol include 3D T1-weighted sequence, T2-weighted sequence, T2-weighted fluid-attenuated inversion recovery (FLAIR) sequence, resting state functional MRI (rs-fMRI) sequence, and diffusion tensor imaging (DTI) sequence. In addition, B0 Field Mapping and DTI TOPUP sequences will be acquired and their data will be utilized in analysis to minimize image distortions in rs-fMRI and DTI data. Used parameters and details of each sequence for both of the used MR scanners are listed in Tables 1, 2.
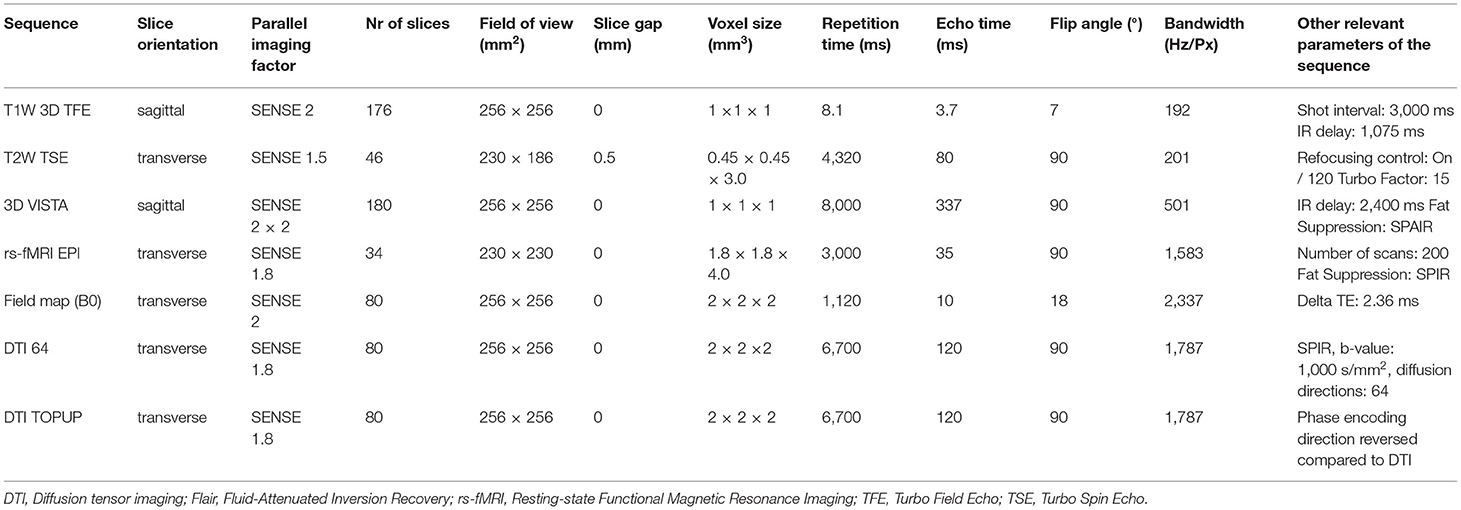
Table 1. Used MRI sequences using Philips Ingenuity 3.0 T TF PET-MR (Philips Healthcare, Amsterdam, the Netherlands).
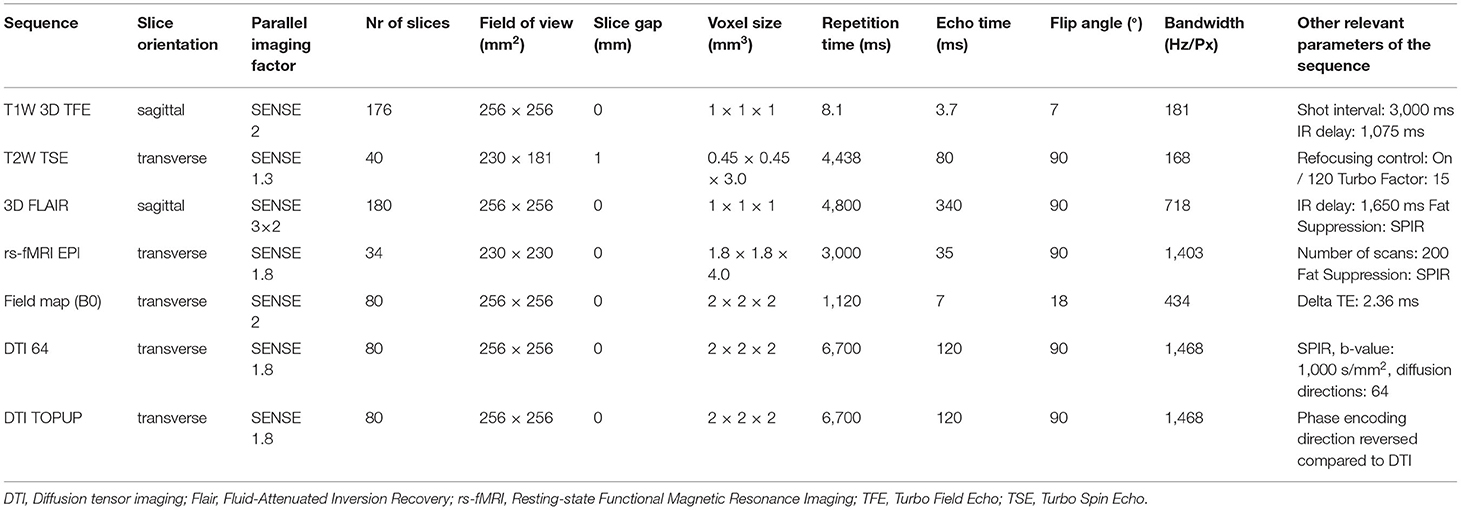
Table 2. Used MRI sequences using Philips Ingenia 3.0 T systems (Philips Healthcare, Amsterdam, the Netherlands).
All MRI images are read by a neuroradiologist, for identification of any possible other pathologies in the brain.
PET Imaging
All PET scans will be performed with brain dedicated ECAT high-resolution research tomograph (HRRT, Siemens Medical Solutions, Knoxville, TN), with a spatial resolution of 2.5 mm. First, antecubital veins are cannulated to enable intravenous injection of the used radioligands. An individual thermoplastic mask is prepared and used to reduce head motion during the scans. Each participant is imaged with three different PET tracers specified below.
11C-PIB-PET
The first PET scan is performed with 11C-PIB, a gold standard radioligand developed for imaging fibrillar Aβ aggregates (41). 11C-PIB is produced in-house with a previously published protocol (42). After intravenous administration of the tracer (dose aimed at 500 MBq, minimum 250 MBq) participants are advised to wait 30 min sitting or lying down, before they are moved into the scanner. Collection of the emission data is initiated at 40 min post injection and the scan duration will be 50 min. PET scan is followed by a 6 min transmission scan using a 137Cs point source for attenuation correction. List-mode data is histogrammed into 6 × 5 + 2 × 10 min time frames, and reconstructed with 3D ordinary Poisson ordered subset expectation maximization algorithm (OP-OSEM3D) with 16 subsets and 8 iterations and a voxel size of 1.22 × 1.22 × 1.22 mm.
11C-PK11195-PET
The second PET scan performed to evaluate the early differences in regional neuroinflammation is performed using 11C-(R)-PK11195, a radioligand targeting the 18 kDa translocator protein (TSPO) known to be expressed in the mitochondrial membrane of activated glial cells (43). 11C-PK11195 is produced in-house with previously published method (44). A transmission scan is acquired first, and 60 min dynamic emission data collection will be started simultaneously with intravenous injection of the tracer (dose aimed at 500 MBq, minimum 250 MBq). List mode data is histogrammed into 17 timeframes (2 × 15; 3 × 30; 3 × 60; 7 × 300; 2 × 600 s) and reconstructed using the same method as with 11C-PIB imaging. No blood samples are collected during the scans.
11C-UCB-J-PET
The third PET scan utilizes 11C-UCB-J, a tracer targeting synaptic vesicle protein SV2A. First scans (N < 10) will be performed with arterial blood sampling, allowing for full kinetic modeling of the data and subsequent comparison of the data to simplified methods used for the remaining scans. Arterial blood samples are collected manually every 15 s for the first 3 min, and thereafter at 4, 6, 8, 10, 15, 20, 25, 30, 45, 60, and 90 min post injection. Additional samples for analyzing radioactive metabolites and unchanged tracer fraction are collected at 3, 8, 15, 30, 45, 60, and 90 min post injection. After evaluating the first scans, protocol will be simplified, and arterial blood samples will no longer be collected. After intravenous tracer injection (dose aimed at 500 MBq, minimum 250 MBq), a 90 min dynamic emission scan is performed, followed by a 5-min transmission scan. List mode data is histogrammed into 29 timeframes (6 x 30, 7 x 60, 16 x 300 s) and reconstructed using OP-OSEM3D algorithm with 10 subsets and eight iterations and with point spread function modeling to improve further the spatial resolution.
Biofluid Collection
Blood Sample Collection
Venous blood samples are collected in the morning after a 10–12 h fasting period from all participants according to in-house standard operating procedures. Various laboratory values (e.g., plasma cholesterol, plasma triglycerides, plasma insulin and serum high sensitivity c-reactive protein levels) are measured in a local testing laboratory [TYKSLAB, accredited according to SFS-EN ISO 15189:2013 standard by Finnish Accreditation Service (FINAS)] according to its protocols and analysis measures. All performed laboratory measurements, used methods and instruments are listed in Table 3.

Table 3. Laboratory measurements performed at ASIC-E4 baseline and 4-year follow-up.
Additional EDTA-plasma (Vacuette EDTA-K2 tube no. 454411) and serum (Vacuette gel tube no. 454420) samples will be collected during the same session and used later for various blood biomarker measurements. Samples are gently inverted 5–10 times, centrifuged (2,200 × g, 10 min), aliquoted and stored in −80°C as 500 μl aliquots. Frozen samples will be shipped on dry ice to Clinical Neurochemistry Laboratory, Gothenburg, Sweden and stored in −80°C prior to analysis.
CSF Sample Collection
CSF samples will be obtained only for a subset of participants who give an additional informed consent for lumbar puncture. Refusal from the CSF sample is not an exclusion criterion for participating in the rest of the study. The CSF samples will be collected in the morning (A.M.). Fasting is not needed. The lumbar puncture will be performed under sterile conditions with the study participant either lying down or sitting, with a 20 Gauge needle. Approximately 10 ml of CSF will be tapped into a Sarstedt polypropylene tube, whereafter it will be centrifuged at 2,200 × g for 10 min, with the temperature set at +20°C. Then, 0.5–1 ml aliquots will be pipetted into cryotubes and stored at −80°C. CSF samples will be collected only once, at a time close after the participants 11C-UCB-J PET scan (see Figure 3 for details).
Neuropsychological Testing
As previously described, the CERAD neuropsychological battery was completed by each participant during the screening visit, and CERAD total score > 62 was set as an inclusion criterion to the study.
In addition, an extensive neuropsychological test battery will be performed for each participant at baseline and at follow-up according to the ASIC-E4 study plan. All neuropsychological tests included in the ASIC-E4 test battery are listed in detail in Supplementary Table 1. The used tests include, e.g., the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) battery and a subset of Ravens matrices that together with CERAD orientation to time and place are used to calculate the Alzheimer's Prevention Initiative Preclinical Cognitive Composite score (APCC). The APCC is a cognitive composite score that has been shown to be sensitive to decline in preclinical AD and was used as a primary outcome measure in the API Generation Program clinical trials (45, 46).
Data Analysis
Image Analysis
Pre-processing and kinetic modeling of the images will be done using Magia, an automated neuroimage analysis pipeline developed at the Human Emotion systems Laboratory, Turku PET Center (47) and in-house analysis software. Magia runs on MATLAB (The MathWorks, Inc., Natick, MA, USA), and combines methods from SPM12 (www.fil.ion.ucl.ac.uk/spm/) and FreeSurfer (https://surfer.nmr.mgh.harvard.edu/) with in-house software developed for kinetic modeling (47). Within the Magia pipeline, PET images are first motion-corrected, and co-registrated with the closest-in-time T1 MR images of the same subjects using SPM. MRI is then processed with Freesurfer for anatomical parcelation and defining both regions of interest (ROI) and the used reference region by using the Desikan-Killiany atlas (47).
11C-PIB uptake is quantified as standardized uptake value ratios (SUVRs) calculated 60–90 min post-injection, using cerebellar cortex as a reference region. When correlating 11C-PIB PET findings with other PET biomarkers, cognitive scores and fluid biomarker findings, PiB uptake will be treated as a continuous variable. However, amyloid positivity will also be defined by global 11C-PIB SUVR >1.5, similar to previous studies on cognitively healthy elderly populations (48, 49) and by visual read by at least two individual experts, resulting in positivity if one of the predefined cortical regions are classified as amyloid positive visually. 11C-PK11195 uptake is quantified as a distribution volume ratio (DVR) using a reference tissue input Logan's method within 20–60 min, where supervised clustering algorithm (SCA4) is used to calculate the clustered reference region (50). 11C-UCB-J uptake is quantified as distribution volume (VT) using a two-tissue compartment model with arterial input data. Additionally, reference tissue input modeling is done with simplified reference tissue model (SRTM) where non-displaceable binding potential (BPND) is estimated with respect to centrum semiovale.
For regional analysis, ROIs presenting regions typical for Aβ deposition (pre-frontal cortex, parietal cortex, anterior cingulum, posterior cingulum, precuneus and lateral temporal cortex) as well as volume weighted amyloid composite VOI containing all the aforementioned regions as well as cerebellar cortex for reference region are created using Freesurfer and used for primary analysis. Cerebellar reference region mask created by Magia goes through two-step correction; anatomical correction removing voxels most prone to partial volume effects, and tail-exclusion removing voxels whose intensity is on the end of the radioactivity distribution (47). For 11C-UCB-J, a centrum semiovale ROI is drawn manually for each subject. In addition, also medial temporal cortex, and volume weighted composite VOIs for Braak I-II (entorhinal), Braak III-IV (limbic) and Braak V-VI (neocortical) will be used to investigate differences in glial activation and synaptic density in regions known to be associated with early tau deposition (51). Freesurfer regions included in each a priori amyloid and Braak VOIs are listed in Supplementary Tables 2, 3, respectively.
Cortical thickness and hippocampal volumes will be obtained from Freesurfer and adjusted for total intracranial volume (TIV) and expressed as %TIV. In addition, T1 and FLAIR sequences will be analyzed using an automatic image analysis tool (eNeuro, Combinostics Oy, Tampere, Finland), to obtain quantified total and regional white matter hyperintensities, atrophy measures, computational Fazekas scores and volumetric parameters. Lesion load, Fazekas scores and the used MRI scanner can further be used as covariates in the baseline linear models to account for vascular component and difference between the used MRI scanners.
Biofluid Analysis
Blood-based biomarkers for AD pathophysiology and neurodegeneration (e.g., p-tau181, p-tau231, GFAP, NfL, and soluble Aβ) will be analyzed from the blood samples using ultrasensitive Single molecule array (Simoa) technology and in-house immunoprecipitation-mass spectrometry methods both at baseline and after follow-up. Subsequently, association of these biomarkers with each other and early phase neuroimaging findings will be examined in the whole cohort and within the different APOE genotypes.
CSF samples will be obtained only for a subset of participants who will give an additional informed consent for lumbar punction. Due to this, number of CSF samples in the study are expected to be lower. However, we aim to measure concentrations of core CSF biomarkers for AD (total-tau, phosphorylated-tau, Aβ1−42/1−40) and levels of novel synaptic biomarkers from these samples.
Statistical Plan
For descriptive statistics reported here, normally distributed data is presented as mean (SD) and non-normal data as median (interquartile range, IQR). Differences in continuous variables between the three groups were tested either with 1-way ANOVA or non-parametric Kruskal-Wallis test. Differences in categorical variables were tested using χ2 test. All analysis were performed using JMP Pro 16.0.0 (SAS Institute Inc.).
For subsequent data analysis in the project, analysis will include data from all study participants. If a participant will suspend the study, all data collected so far will be included. Normality of continuous variables will be inspected visually and from residuals. If required, appropriate transformations will be applied to gain normality and allow the use of parametric methods. Non-parametric methods will be applied for data that still does not follow normality assumption after transformation. If two groups are compared, student's t-test, Mann Whitney U-test or linear models taking account different co-variants (age, genotype, gender etc.) can be used. For three groups, 1-way ANOVA, Kruskal-Wallis test by ranks or linear models taking account different co-variants are used. For comparison of categorical variables, χ2 test can be used. Association between variables that are measured at one time point can be analyzed using correlations both within the whole cohort and within the three APOE genotypes. Pearson's correlation is applied for normally distributed data and Spearman's rank correlation for data that does not fulfill normality assumption. Association between numerical variables that will be measured more than once can be analyzed using hierarchical linear mixed models, taking into account both correlation between time points, and co-variants that affect the change.
Voxel-wise analysis for collected imaging data will be performed using SPM12 (or later) and normalized parametric images in Montreal Neurological Institute space. Results will be adjusted for multiple comparisons using false discovery rate at P < 0.05.
All results are considered statistically significant if P < 0.05 (two-sided).
Results
Recruitment started in October 2018 and was finished in July 2020. From the 74 screened individuals, 11(14.9 %) were not eligible to the study; six individuals had contraindication to PET or MRI scan, one had CERAD total score lower than the inclusion limit of 62 points, one experienced subjective cognitive decline and three were on chronic inflammatory medication.
Sixty-three screened individuals filled all inclusion criteria and were enrolled to the following three study groups: Group 1: high risk, APOE ε4/ε4, N = 19; Group 2: intermediate risk, APOE ε4/ε3, N = 22; Group 3: typical risk, APOE ε3/ε3, N = 22.
Sociodemographic Characteristics
Demographic data from the participants are presented in Table 4. A majority (61.9 %) of all participants were females and the mean age in our cohort was 67.5 (SD 4.6, range 60–75) years. The cohort includes individuals from all education levels; 30.2 % had completed primary school, 19.1 % middle or comprehensive school, 33.3% high school and 17.5 % college or university as their highest degree. The three study groups were well-matched for age (P = 0.91, 1-way ANOVA), sex (P = 0.94, χ2 test), education level (P = 0.30, χ2 test) and body mass index (P = 0.61, Kruskal-Wallis test). Approximately half (49.2 %) of all participants had a family member with AD or other memory disorder diagnosis, and the frequency was similar in all groups (P = 0.62, χ2 test). All participants were non-smokers at the time of recruitment.
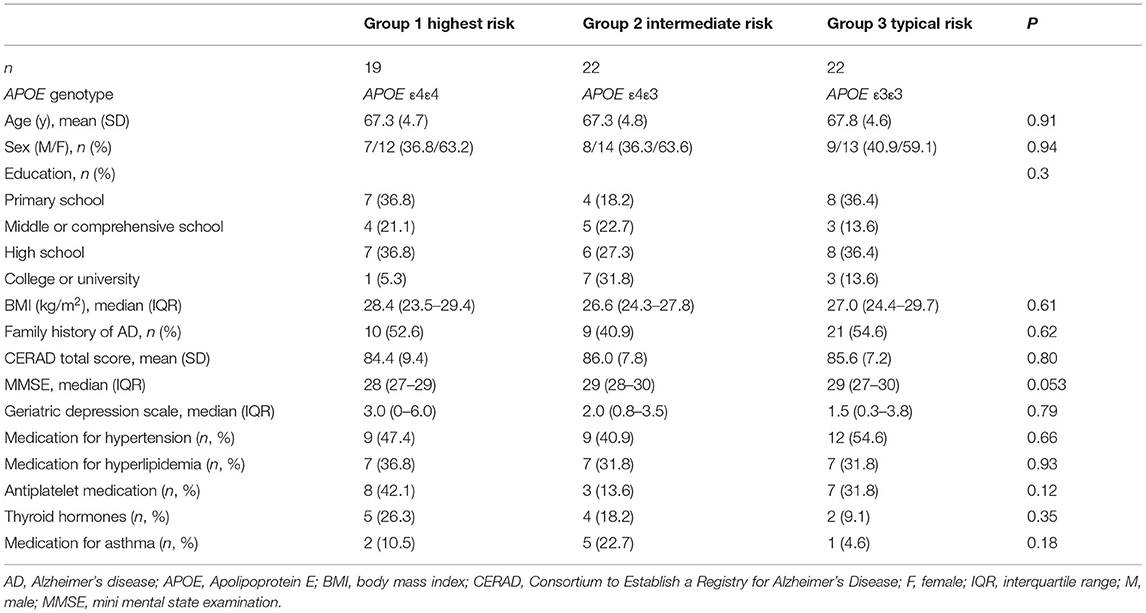
Table 4. Baseline demographics of the ASIC-E4 study.
Clinical and Cognitive Characteristics
Clinical and cognitive characteristics of the participants are presented in Table 4. No significant differences between groups were present in mean CERAD total score (P = 0.80, 1-way ANOVA) or geriatric depression scale score (P = 0.79, Kruskal-Wallis test). The difference in MMSE score at baseline was near the level of statistical significance (P = 0.053, Kruskal-Wallis test), and in examining pair-wise comparisons, APOE ε4/ε3 heterozygotes showed higher median MMSE at baseline (29) as compared to APOE ε4/ε4 homozygotes (28, P = 0.042).
Most used medications in our cohort were for treating cardiovascular diseases; These included anti-hypertensive medication used by 47.6% (30/63), hypolipidemic drugs used by 33.3 % (21/63) and antiplatelet drugs used by 28.6 % (18/63) of all included participants. In addition, 17.5 % (11/63) used thyroid hormones and 12.7 % (8/63) had medication for asthma. Other medications used by more than one individual included low doses of anxiolytics [7.9 % (5/63)], migraine prevention [3.2 % (2/63)], treatment for insomnia [3.2% (2/63)], B12 vitamin [3.2% (3/63)] and analgesics [3.2 % (2/63)]. There were no differences between groups in any of the most frequently used medications (Table 4).
Discussion
The ASIC-E4 study aims to investigate the early interplay of Aβ deposition, neuroinflammation and synaptic loss in cognitively unimpaired elderly with varying levels of genetic risk for AD, based on their APOE ε4 gene dose. We will utilize the information available at the local biobank, and the relatively high prevalence of APOE ε4/ε4 homozygotes in the Finnish population to recruit three groups (matched at group-level for age and sex) with different APOE genotype, and thus different genetic risk for future sporadic AD. Results obtained from the study will produce novel information about the gene-dose effect and possibility for early detection of AD pathological changes using various molecular imaging biomarkers, and fluid-based biomarkers more easily accessible for pharma industry and general clinical practice. The at-risk population investigated in the ASIC-E4 study carry the APOE ε4 risk allele for sporadic old age AD rather than the rare familial mutations causing the early-onset form of AD. Thus, the results are better generalizable at population level, especially in countries like Finland, with relatively high frequency of APOE ε4 carriers (approximately 32 % carry at least one ε4 allele based on the nationwide, population-based Health 2000 Health Examination Survey).
The project has many strengths that are worth mentioning. First, because of the valuable biobank collaboration, we were able to recruit also rare homozygotic carriers of the APOE ε4. Without this collaboration, by only relying on genotyping individual samples aiming to find the same number of such rare individuals would not be feasible. Due to the known risk effect of the APOE ε4 allele, most studies include APOE ε4 carrier status as a covariate in their analysis, or group participants as APOE ε4 carriers and non-carriers. However, fewer studies have investigated the gene dose effect to imaging biomarkers and compared homozygotic and heterozygotic APOE ε4 carriers. Aβ deposition is known to increase in a gene dose related fashion (14, 16) but less is known about its association with neuroinflammation and synaptic function in vivo. That said, another obvious strength of this study is the multimodal and multitracer approach that allows us to combine information from various pathophysiological routes that have been connected with AD in individuals with various levels of genetic risk for AD. Even though at this point no imaging follow-up has been planned, repeated neuropsychological testing and blood sampling allow us to investigate also effect of baseline imaging findings to future cognitive decline and blood biomarker changes.
The ASIC-E4 project does not go without limitations; first, due to delays in PET tracer production we will be unable to schedule all three PET scans within the originally planned 3-month period, but rather the 11C-UCB-J PET scans will be performed after 11C-PIB and 11C-PK11195. However, due to this, an amendment to the study protocol has been made; Participants gave an additional informed consent and repeated MRI scan, CERAD test battery, neuropsychological tests included in the APCC battery and gave additional blood samples. In this way, we are able to get more timely structural reference image and cognitive performance measures for the 11C-UCB-J PET analysis, and correct for the the possible change between baseline 11C-PIB/11C-PK11195 and 11C-UCB-J scans in further analysis. Secondly, even though CSF samples are included in the study protocol, they are not mandatory, and such samples are expected to be obtained from only a small subgroup of the participants. However, even though the CSF sample number will be lower and group wise comparisons most likely are not possible, they will still provide valuable way of evaluating the associations between, e.g., synaptic biomarkers in CSF with the SV2A PET imaging findings in the whole cohort. Thirdly, power analysis for the study were performed based on previously published data with the used PET tracers, thus the study might not be powered for blood biomarker comparisons between groups. Associations between the biomarker concentrations, cognitive performance and various imaging parameters can be evaluated within the whole cohort, and due to planned follow-up sample collection, longitudinal biomarker data allows us to evaluate differences in temporal change rather than solely cross-sectional differences between the groups. In addition, because the study does not include tau PET, we cannot include information on participants level of tangle pathology for our analysis.
With three PET scans, two MRI scans and multiple sampling planned for each subject, there are also risks in the project that should be evaluated and prepared for. The dropout of people during the long study protocol, especially as the global COVID-19 pandemic started during the data collection is one obvious risk for the study. To minimize this risk, changes in schedules and updates concerning the study during the pandemic have been actively communicated with the participants. So far, we have been able to continue study procedures even during the COVID-19 pandemic e.g., by providing individual transfer to the study site and private access to study facility with contacts restricted to essential study personnel following the guidelines of local authorities. In addition, to reduce the risk of dropouts, the follow-up time for cognitive testing and blood sampling was chosen to be relatively short (4 years). However, our study protocol and informed consent allows us also to contact all the study participant for possible extension of this study in the future.
To conclude, results from the ASIC-E4 study will increase the understanding of the molecular interplay between different early AD pathologies in individuals at different risk of developing AD. The used novel imaging and fluid biomarker methods will be investigated in an “at-risk” population and against cognitive changes and the results could promote the inclusion of these biomarkers into future trials investigating the effects of novel disease-modifying drugs targeted toward synaptic restoration and neuroinflammation in preclinical AD. If the hypothesized early brain changes are detected in APOE ε4 carriers, it could be feasible to target preventative actions toward this group in the future.
Study Status
Study is ongoing. Recruitment of study subjects has been finished. Baseline data collection is ongoing and estimated to be completed in June 2022.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Hospital District of Southwest Finland (original 17.1.2018; amendment 24.9.2018). Written informed consent was obtained from each participant both before transferring the APOE status from the biobank to the researcher, and before enrolment to this study.
Author Contributions
JR and AS designed the study concept. AS drafted the manuscript with assistance from RL. JR and LE made major revisions to the manuscript. MP and LK contributed to the design and execution of recruitment. AS, JR, LE, NL, and MKo contributed to the design of screening, PET imaging, and sample collection. AS, JT, VO, and RA contributed to PET imaging, modelling, and analysis protocols. VS, JS, and RP contributed to the design of MRI data collection. EL contributed to the design of statistical analysis plan. MKa contributed to the design of the used neuropsychological test battery. HZ and KB contributed to the planning of sample collection and fluid biomarker analysis. All authors contributed to the conception and design of different parts of the study protocol, made revisions to the manuscript, and approved the submitted version.
Funding
AS has receive funding for this study from Finnish Governmental Research Funding (ERVA) for Turku University Hospital, Emil Aaltonen Foundation, Orion Research Foundation sr, Paulo Foundation and Academy of Finland (#341059). LE has received funding from the Emil Aaltonen Foundation. HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), the AD Strategic Fund and the Alzheimer's Association (#ADSF-21-831376-C, #ADSF-21-831381-C, and #ADSF-21-831377-C), the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019-0228), the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 860197 (MIRIADE), and the UK Dementia Research Institute at UCL. KB is supported by the Swedish Research Council (#2017-00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986), the European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236), the National Institute of Health (NIH), USA, (grant #1R01AG068398-01), and the Alzheimer's Association 2021 Zenith Award (ZEN-21-848495). JR has received funding from the Academy of Finland (#310962), Sigrid Juselius Foundation and Finnish Governmental Research Funding (VTR) for Turku University Hospital. Funds for open access publication fees were received from Turku University Hospital Research Services.
Conflict of Interest
HZ has served at scientific advisory boards and/or as a consultant for Abbvie, Alector, Annexon, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Pinteon Therapeutics, Red Abbey Labs, Passage Bio, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure and Biogen, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. KB has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, Biogen, JOMDD/Shimadzu. Julius Clinical, Lilly, MagQu, Novartis, Pharmatrophix, Prothena, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank all the participants of this study for their valuable contribution and dedication, without it, the study could not be performed. In addition, we thank the staff in Turku PET Center and Turku university Hospital for all their help during the data collection phase. We would also like to acknowledge hospital physicist in Turku PET Center Tuula Tolvanen for radiation dose estimations for the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.826423/full#supplementary-material
References
1. Gauthier S, Rosa-Neto P, Morais JA, Webster C. World Alzheimer Report 2021 - Journey Through the Diagnosis of Dementia. (2021). Available online at: https://www.alzint.org/resource/world-alzheimer-report-2021/ (accessed November 2, 2021).
2. Alzheimer's Association Report. 2020 Alzheimer's disease facts and figures. Alzheimers Dement. (2020) 16:391–460. doi: 10.1002/alz.12068
3. Cummings J, Lee G, Zhong K, Fonseca J, Taghva K. Alzheimer's disease drug development pipeline: 2021. Alzheimers Dement. (2021) 7:e12179. doi: 10.1002/trc2.12179
4. Crous-Bou M, Minguillon C, Gramunt N, Molinuevo JL. Alzheimer's disease prevention: from risk factors to early intervention. Alzheimers Res Ther. (2017) 9:71. doi: 10.1186/s13195-017-0297-z
5. Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, et al. Alzheimer's disease. Lancet. (2016) 388:505–17. doi: 10.1016/S0140-6736(15)01124-1
6. Busche MA, Hyman BT. Synergy between amyloid-beta and tau in Alzheimer's disease. Nat Neurosci. (2020) 23:1183–93. doi: 10.1038/s41593-020-0687-6
7. Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, et al. Early abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. (2004) 62:925–31. doi: 10.1212/01.WNL.0000115115.98960.37
8. Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. (2011) 7:280–92. doi: 10.1016/j.jalz.2011.03.003
9. Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. (2018) 14:535–62. doi: 10.1016/j.jalz.2018.02.018
10. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. (1993) 261:921–3. doi: 10.1126/science.8346443
11. Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry. (2011) 16:903–7. doi: 10.1038/mp.2011.52
12. Belloy ME, Napolioni V, Greicius MD. A quarter century of APOE and Alzheimer's disease: progress to date and the path forward. Neuron. (2019) 101:820–38. doi: 10.1016/j.neuron.2019.01.056
13. Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. (2013) 9:106–18. doi: 10.1038/nrneurol.2012.263
14. Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A. (2009) 106:6820–5. doi: 10.1073/pnas.0900345106
15. Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. (2011) 69:181–92. doi: 10.1002/ana.22248
16. Mecca AP, Barcelos NM, Wang S, Bruck A, Nabulsi N, Planeta-Wilson B, et al. Cortical beta-amyloid burden, gray matter, and memory in adults at varying APOE epsilon4 risk for Alzheimer's disease. Neurobiol Aging. (2018) 61:207–14. doi: 10.1016/j.neurobiolaging.2017.09.027
17. Fernandez CG, Hamby ME, McReynolds ML, Ray WJ. The Role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer's disease. Front Aging Neurosci. (2019) 11:14. doi: 10.3389/fnagi.2019.00014
18. Fan Z, Brooks DJ, Okello A, Edison P. An early and late peak in microglial activation in Alzheimer's disease trajectory. Brain. (2017) 140:792–803. doi: 10.1093/brain/aww349
19. Hamelin L, Lagarde J, Dorothee G, Leroy C, Labit M, Comley RA, et al. Early and protective microglial activation in Alzheimer's disease: a prospective study using 18F-DPA-714 PET imaging. Brain. (2016) 139:1252–64. doi: 10.1093/brain/aww017
20. Pascoal TA, Benedet AL, Ashton NJ, Kang MS, Therriault J, Chamoun M, et al. Microglial activation and tau propagate jointly across braak stages. Nat Med. (2021) 27:1592–9. doi: 10.1038/s41591-021-01456-w
21. Finnema SJ, Nabulsi NB, Eid T, Detyniecki K, Lin SF, Chen MK, et al. Imaging synaptic density in the living human brain. Sci Transl Med. (2016) 8:348ra96. doi: 10.1126/scitranslmed.aaf6667
22. Finnema SJ, Nabulsi NB, Mercier J, Lin SF, Chen MK, Matuskey D, et al. Kinetic evaluation and test-retest reproducibility of [C]UCB-J, a novel radioligand for positron emission tomography imaging of synaptic vesicle glycoprotein 2A in humans. J Cereb Blood Flow Metab. (2018) 38:2041–52. doi: 10.1177/0271678X17724947
23. Mecca AP, Chen MK, O'Dell RS, Naganawa M, Toyonaga T, Godek TA, et al. In vivo measurement of widespread synaptic loss in Alzheimer's disease with SV2A PET. Alzheimers Dement. (2020) 16:974–82. doi: 10.1002/alz.12097
24. Chen MK, Mecca AP, Naganawa M, Gallezot JD, Toyonaga T, Mondal J, et al. Comparison of [C]UCB-J and [F]FDG PET in Alzheimer's disease: a tracer kinetic modeling study. J Cereb Blood Flow Metab. (2021) 41:2395–409. doi: 10.1177/0271678X211004312
25. Ashton NJ, Leuzy A, Karikari TK, Mattsson-Carlgren N, Dodich A, Boccardi M, et al. The validation status of blood biomarkers of amyloid and phospho-tau assessed with the 5-phase development framework for AD biomarkers. Eur J Nucl Med Mol Imaging. (2021) 48:2140–56. doi: 10.1007/s00259-021-05253-y
26. Alawode DOT, Heslegrave AJ, Ashton NJ, Karikari TK, Simren J, Montoliu-Gaya L, et al. Transitioning from cerebrospinal fluid to blood tests to facilitate diagnosis and disease monitoring in Alzheimer's disease. J Intern Med. (2021) 290:583–601. doi: 10.1111/joim.13332
27. Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Dore V, et al. High performance plasma amyloid-beta biomarkers for Alzheimer's disease. Nature. (2018) 554:249–54. doi: 10.1038/nature25456
28. Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. (2020) 19:422–33. doi: 10.1016/S1474-4422(20)30071-5
29. Karikari TK, Benedet AL, Ashton NJ, Lantero Rodriguez J, Snellman A, Suarez-Calvet M, et al. Diagnostic performance and prediction of clinical progression of plasma phospho-tau181 in the Alzheimer's disease neuroimaging initiative. Mol Psychiatry. (2021) 26:429–42. doi: 10.1038/s41380-020-00923-z
30. Thijssen EH, La Joie R, Wolf A, Strom A, Wang P, Iaccarino L, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration. Nat Med. (2020) 26:387–97. doi: 10.1038/s41591-020-0762-2
31. Palmqvist S, Janelidze S, Quiroz YT, Zetterberg H, Lopera F, Stomrud E, et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA. (2020) 324:772–81. doi: 10.1001/jama.2020.12134
32. Ashton NJ, Pascoal TA, Karikari TK, Benedet AL, Lantero-Rodriguez J, Brinkmalm G, et al. Plasma p-tau231: a new biomarker for incipient Alzheimer's disease pathology. Acta Neuropathol. (2021) 141:709–24. doi: 10.1007/s00401-021-02275-6
33. Ashton NJ, Janelidze S, Al Khleifat A, Leuzy A, van der Ende EL, Karikari TK, et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat Commun. (2021) 12:3400. doi: 10.1038/s41467-021-23620-z
34. Oeckl P, Halbgebauer S, Anderl-Straub S, Steinacker P, Huss AM, Neugebauer H, et al. Glial fibrillary acidic protein in serum is increased in Alzheimer's disease and correlates with cognitive impairment. J Alzheimers Dis. (2019) 67:481–8. doi: 10.3233/JAD-180325
35. Cicognola C, Janelidze S, Hertze J, Zetterberg H, Blennow K, Mattsson-Carlgren N, et al. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimers Res Ther. (2021) 13:68. doi: 10.1186/s13195-021-00804-9
36. Chatterjee P, Pedrini S, Stoops E, Goozee K, Villemagne VL, Asih PR, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer's disease. Transl Psychiatry. (2021) 11:27. doi: 10.1038/s41398-020-01137-1
37. Benedet AL, Mila-Aloma M, Vrillon A, Ashton NJ, Pascoal TA, Lussier F, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. (2021) 78:1471–83. doi: 10.1001/jamaneurol.2021.3671
38. Aalto S, Scheinin NM, Kemppainen NM, Nagren K, Kailajarvi M, Leinonen M, et al. Reproducibility of automated simplified voxel-based analysis of PET amyloid ligand [11C]PIB uptake using 30-min scanning data. Eur J Nucl Med Mol Imaging. (2009) 36:1651–60. doi: 10.1007/s00259-009-1174-1
39. Chandler MJ, Lacritz LH, Hynan LS, Barnard HD, Allen G, Deschner M, et al. A total score for the CERAD neuropsychological battery. Neurology. (2005) 65:102–6. doi: 10.1212/01.wnl.0000167607.63000.38
40. Paajanen T, Hanninen T, Tunnard C, Mecocci P, Sobow T, Tsolaki M, et al. CERAD neuropsychological battery total score in multinational mild cognitive impairment and control populations: the AddNeuroMed study. J Alzheimers Dis. (2010) 22:1089–97. doi: 10.3233/JAD-2010-100459
41. Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. (2004) 55:306–19. doi: 10.1002/ana.20009
42. Snellman A, Rokka J, Lopez-Picon FR, Helin S, Re F, Loyttyniemi E, et al. Applicability of [C]PIB micro-PET imaging for in vivo follow-up of anti-amyloid treatment effects in APP23 mouse model. Neurobiol Aging. (2017) 57:84–94. doi: 10.1016/j.neurobiolaging.2017.05.008
43. Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. (2010) 9:971–88. doi: 10.1038/nrd3295
44. Rissanen E, Tuisku J, Vahlberg T, Sucksdorff M, Paavilainen T, Parkkola R, et al. Microglial activation, white matter tract damage, and disability in MS. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e443. doi: 10.1212/NXI.0000000000000443
45. Reiman EM, Langbaum JB, Tariot PN, Lopera F, Bateman RJ, Morris JC, et al. CAP–advancing the evaluation of preclinical Alzheimer disease treatments. Nat Rev Neurol. (2016) 12:56–61. doi: 10.1038/nrneurol.2015.177
46. Langbaum JB, Ellison NN, Caputo A, Thomas RG, Langlois C, Riviere ME, et al. The Alzheimer's prevention initiative composite cognitive test: a practical measure for tracking cognitive decline in preclinical Alzheimer's disease. Alzheimers Res Ther. (2020) 12:66. doi: 10.1186/s13195-020-00633-2
47. Karjalainen T, Tuisku J, Santavirta S, Kantonen T, Bucci M, Tuominen L, et al. Magia: robust automated image processing and kinetic modeling toolbox for PET neuroinformatics. Front Neuroinform. (2020) 14:3. doi: 10.3389/fninf.2020.00003
48. Toppala S, Ekblad LL, Tuisku J, Helin S, Johansson JJ, Laine H, et al. Association of early beta-amyloid accumulation and neuroinflammation measured with [C]PBR28 in elderly individuals without dementia. Neurology. (2021) 96:e1608–19. doi: 10.1212/WNL.0000000000011612
49. Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. (2010) 31:1275–83. doi: 10.1016/j.neurobiolaging.2010.04.007
50. Yaqub M, van Berckel BN, Schuitemaker A, Hinz R, Turkheimer FE, Tomasi G, et al. Optimization of supervised cluster analysis for extracting reference tissue input curves in (R)-[C]PK11195 brain PET studies. J Cereb Blood Flow Metab. (2012) 32:1600–8. doi: 10.1038/jcbfm.2012.59
Keywords: Alzheimer's disease, APOE, preclinical, biomarker, beta-amyloid, TSPO (18 kDa translocator protein), SV2A ligand, neuroinflammation
Citation: Snellman A, Ekblad LL, Koivumäki M, Lindgrén N, Tuisku J, Perälä M, Kallio L, Lehtonen R, Saunavaara V, Saunavaara J, Oikonen V, Aarnio R, Löyttyniemi E, Parkkola R, Karrasch M, Zetterberg H, Blennow K and Rinne JO (2022) ASIC-E4: Interplay of Beta-Amyloid, Synaptic Density and Neuroinflammation in Cognitively Normal Volunteers With Three Levels of Genetic Risk for Late-Onset Alzheimer's Disease – Study Protocol and Baseline Characteristics. Front. Neurol. 13:826423. doi: 10.3389/fneur.2022.826423
Received: 30 November 2021; Accepted: 06 January 2022;
Published: 09 February 2022.
Edited by:
Fabiana Novellino, National Research Council (CNR), ItalyReviewed by:
Danni Li, University of Minnesota Twin Cities, United StatesDavid Brooks, Imperial College London, United Kingdom
Copyright © 2022 Snellman, Ekblad, Koivumäki, Lindgrén, Tuisku, Perälä, Kallio, Lehtonen, Saunavaara, Saunavaara, Oikonen, Aarnio, Löyttyniemi, Parkkola, Karrasch, Zetterberg, Blennow and Rinne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anniina Snellman, YW5uaWluYS5zbmVsbG1hbkB1dHUuZmk=