
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 08 March 2022
Sec. Pediatric Neurology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.826211
This article is part of the Research Topic Genetically Determined Epilepsies: Perspectives in the Era of Precision Medicine View all 19 articles
 Giulia Spoto1
Giulia Spoto1 Giulia Valentini1
Giulia Valentini1 Maria Concetta Saia1
Maria Concetta Saia1 Ambra Butera1
Ambra Butera1 Greta Amore1
Greta Amore1 Vincenzo Salpietro2,3,4*
Vincenzo Salpietro2,3,4* Antonio Gennaro Nicotera1
Antonio Gennaro Nicotera1 Gabriella Di Rosa1
Gabriella Di Rosa1The proper connection between the pre- and post-synaptic nervous cells depends on any element constituting the synapse: the pre- and post-synaptic membranes, the synaptic cleft, and the surrounding glial cells and extracellular matrix. An alteration of the mechanisms regulating the physiological synergy among these synaptic components is defined as “synaptopathy.” Mutations in the genes encoding for proteins involved in neuronal transmission are associated with several neuropsychiatric disorders, but only some of them are associated with Developmental and Epileptic Encephalopathies (DEEs). These conditions include a heterogeneous group of epilepsy syndromes associated with cognitive disturbances/intellectual disability, autistic features, and movement disorders. This review aims to elucidate the pathogenesis of these conditions, focusing on mechanisms affecting the neuronal pre-synaptic terminal and its role in the onset of DEEs, including potential therapeutic approaches.
Optimal synaptic communication is a complex and finely regulated process that is fundamental for proper nervous system physiology (1–3). The expanding knowledge in the neurobiological field has allowed to increase the accuracy of the etiopathogenesis definition of nervous system diseases: from the macroscopic involvement of anatomical structures and circuitry, the focus shifted to the microscopical elements of this system, including subcellular ones, as transporting proteins, signaling superficial molecules, receptors, and neurotransmitters. The synapse plays a central role in this exchange of information and represents the essential signal transmitting unit of the nervous system (1, 3). The neurotransmitters release requires the availability of synaptic vesicles, which undergo immediate fusion with the pre-synaptic membrane when the action potential arrives (4). The synaptic vesicles undergo repeated recycling, and this process involves the sequential participation of several proteins (see Figure 1) (2, 5).
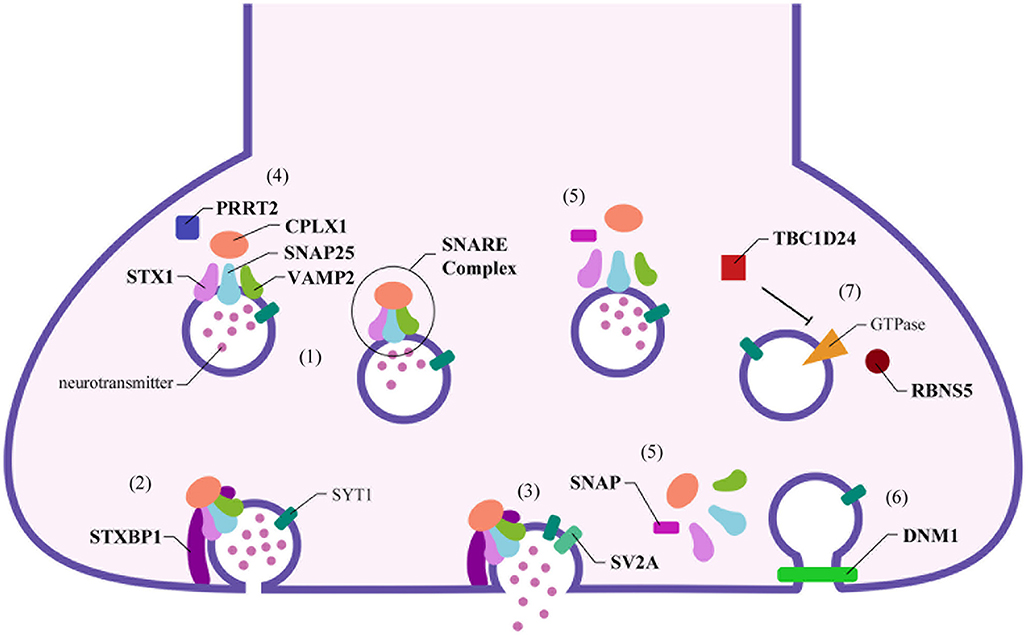
Figure 1. Schematic representation of pre-synaptic proteins localization and their role in the synaptic functioning. Numerous proteins interact together and with the cellular membrane on the pre-synaptic terminal to release the neurotransmitters in the synaptic cleft. (1) Stx1, Snap25, and Vamp2 form the core of the SNARE complex, which is stabilized by Cplx1. (2) When the synaptic vesicle reaches the cellular membrane, Stxbp1 binds Stx1, starting the priming and the rapid fusion of the vesicle. (3) Sv2a, the only specific synaptic protein, interacts with Syt1 and induces neurotransmitter release. (4) On the contrary, Prrt2 inhibits this process by reducing the formation of the SNARE complex. (5) The disassembly of the SNARE complex is mediated by Snap proteins, (6) allowing the subsequent clathrin-mediated endocytosis, with the participation of the Dnm1. (7) This process is coordinated by several GTPases: Tbc1d24 regulates Rab35, while Rbns5 interacts with Rab4 and Rab5 to lead the empty synaptic vesicle toward the endosome for recycling.
A synaptopathy is defined as an alteration in the functionality of any element constituting the synapse: the pre- and post-synaptic terminals, the synaptic cleft, and all the surrounding components, such as glial cells and extracellular matrix (1, 3, 6). Although the first reference to the term “synaptopathy” was made in 2003 by Li et al. regarding the Huntington Disease (7), in the last few decades, pathogenic variants in genes encoding synaptic proteins have been demonstrated to determine altered protein levels/function in several neuropsychiatric diseases, such as epilepsy, intellectual disability (ID), and autism spectrum disorder (1, 8).
The term “epileptic encephalopathy” describes a catastrophic form of epilepsy, with a frequent onset in infancy or early childhood, in which the epileptic activity itself significantly contributes to severe developmental delay (DD) and behavioral impairments (9, 10). In those patients presenting with pre-existing DD, the effect of the epileptic activity causes a worsening of the developmental consequences arising directly from the genetic mutation, configuring a clinical phenotype called “developmental and epileptic encephalopathy” (DEE) (10).
Since 2001, when a genetic cause for an epileptic encephalopathy was first reported (11), numerous genes have been associated with DEEs, and several synaptopathies have been described (9). Given that numerous proteins participate in the mechanisms underlying the correct functioning of the synapse, these disorders are generally studied centering the attention on the single affected gene (2). We focused our research on those genes involved in the pathogenesis of DEEs affecting the pre-synaptic compartment, consisting of the axon terminal and the proteins implicated in releasing neurotransmitters. Since these genes are often identified in the context of large cohorts of patients tested with genetic panels, an accurate description is not always available, making it challenging to characterize clinical phenotypes and to identify the DEEs. Therefore, we considered all patients presenting with epilepsy and intellectual disability (ID).
Syntaxin1 (Stx1) is a protein widely expressed in the nervous system (12, 13) and, together with Snap25 (encoded by SNAP25) and synaptobrevin-2 (encoded by VAMP2), form a stable complex called soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE) complex, made by a four-helix bundle implicated in Ca2+-dependent exocytosis of the synaptic vesicles and neurotransmitter release (14, 15). Vamp2 protein represents the vesicle membrane portion of SNARE neuronal complex (v-SNARE), while the plasma membrane of SNARE (t-SNARE) is constituted by Stx1a and Snap25 (16).
Stx1 is an integral membrane protein composed of three functional domains: (1) the N-terminal peptide, (2) an α-helical domain which is called Habc domain, and (3) the SNARE and transmembrane motif in the C-terminal region (13, 17–20). There are two different protein configurations, namely the “closed” and the “open” Stx1. The first one is characterized by a link between the Habc domain and the N-peptide. Switching from the closed to the open conformation is crucial in regulating the exocytosis mediated by the SNARE complex, and Stx1 plays a crucial role in the initiation of synaptic exocytosis.
The first step that allows the SNARE complex assembly is represented by the binding between Munc18 (encoded by STXBP1) and Stx1 in its closed form.
This is the starting point of a process that will end with releasing the neurotransmitter in the synaptic cleft (21–23).
There are two isoforms of Stx1 called 1a and 1b. Even if they share 84% of their amino acid sequence and the basic function as neuronal t-SNAREs, Stx1b is the principal mediator for spontaneous and evoked fast synaptic vesicle exocytosis (24, 25).
The gene encoding for Stx1a is located on chromosome 7 and is one of the genes involved in Williams-Beuren Syndrome (WBS), caused by deletion 7q11.23 (26). In this regard, Gao et al. (27) showed that the level of expression of Stx1a is correlated with the degree of intelligence in patients with WBS. It has also been found that some variants of STX1A are associated with an increased chance of developing migraines (28). Moreover, data from studies conducted on many families investigated with Whole Exome Sequencing (WES) suggest that STX1A may be a possible candidate gene in the development of neurodevelopmental disorders (29).
On the contrary, less is known about the involvement of pathogenic variants of STX1A in the development of epilepsy. However, the influence that Stx1a exerts on glutamate uptake and glutamatergic transmission could partially explain the role of STX1A in epileptogenesis. Data from the literature suggest that Stx1a acts by enhancing the internalization of excitatory amino acid transporter 1 (EAAC1) responsible for glutamate re-uptake: increased internalization and reduced expression on the cell surface cause an overall reduction in glutamate uptake (30). Additionally, a Stx1a role in regulating voltage-gated K+ channel determined by physical interaction of this protein with the ion channels has been demonstrated in animal models (31). Moreover, single nucleotide polymorphisms (SNP) of STX1A and VAMP2 have been described in association with cryptogenic epilepsy (32).
Much more has been described regarding the 1b isoform. STX1B gene is located on chromosome 16 (16p11.2 region) (33) and studies conducted on mouse models have shown an early death of the animals and an altered function of the neuromuscular junction in KO mice for STX1B (34). These findings underline the critical role played by Stx1b in the proper signaling of the nervous system. Therefore, dysfunctions of this protein, whether due to mutations or deletions, are associated with the development of various disorders of the nervous system, including ID, speech disorders, and various forms of epilepsy (35).
To the best of our knowledge, STX1A-related DEEs are not reported in the literature. For this reason, we focused on the 1b isoform and identified 20 patients (35–39). Clinical information was collected about the age onset of symptoms, the type of seizures, the presence of febrile seizures (FS), electroencephalographic patterns, and the development of ID and/or movement disorders. Not all clinical information could be traced for all patients.
The age of onset of epilepsy ranged from a few days after birth to a maximum of 4 years (35–39). Most of them present multiple forms of epilepsy: the most represented type of seizures are myoclonic ones (11/20), generalized tonic-clonic ones (10/20), and absences (9/20). Other types of seizures reported are atonic (7/20), tonic (6/20), focal (3/20), spasms (1/20), myoclonic-astatic epilepsy (1/20). FS were also frequent, reported in seven (7/20) patients. The increased susceptibility to develop FS has been reported in previous studies (35, 40, 41), and more recently by Mishima et al. (42). In this cohort of patients, we report a recurrence of ataxia, present in 11 of 20 cases (35, 37, 39).
In addition, seizure control was reached in five patients, but given the large number of drugs administered, often in co-administration, it is difficult to define the most appropriate pharmacological treatment. Clinical data are summarized in Table 1.
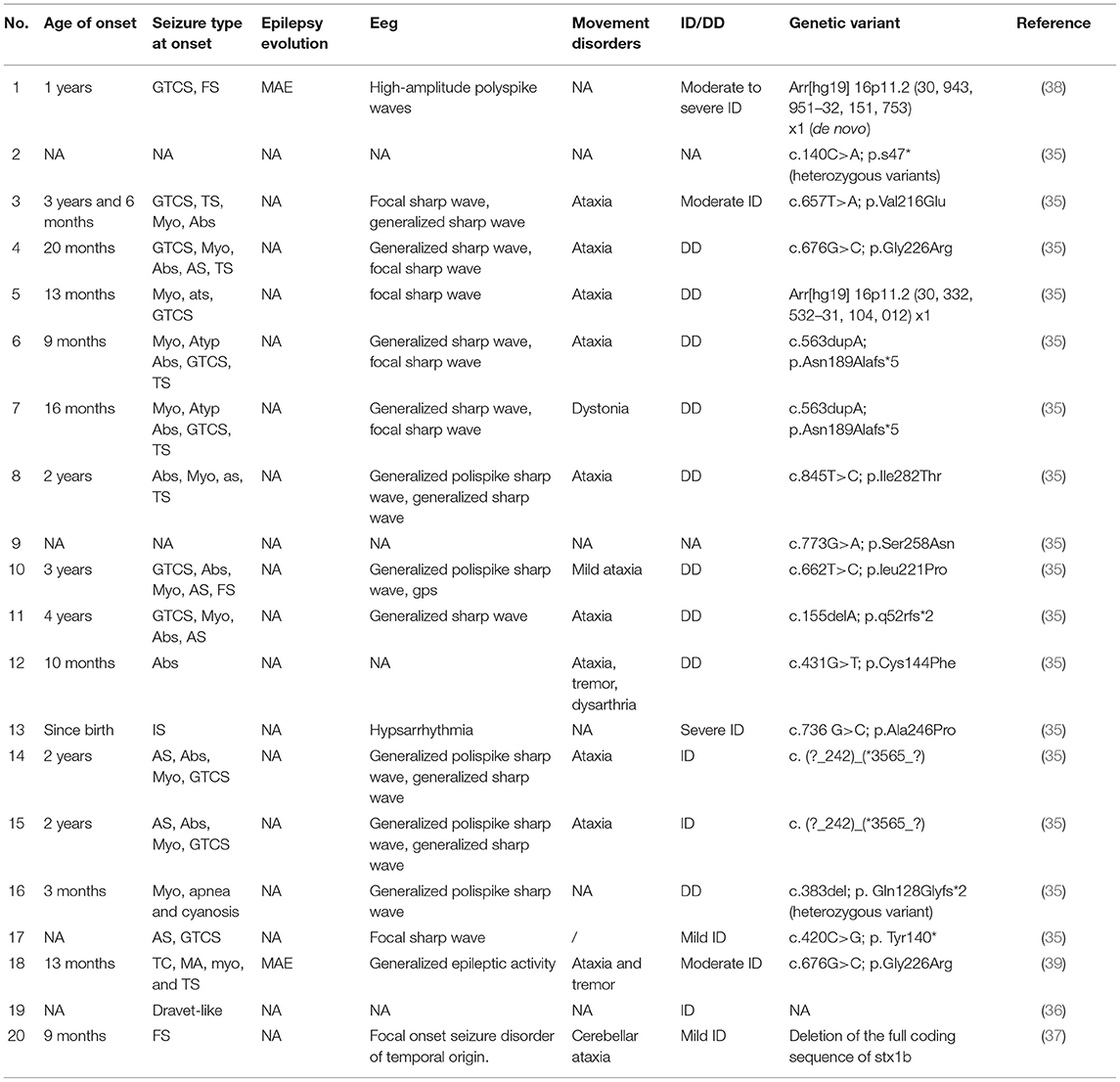
Table 1. Clinical features of STX1B-related patients.
The SNAP25 gene is mapped on chromosome 20 (20p12.2 region), and it encodes the synaptosomal-associated protein 25 kDa (Snap25) highly expressed in nerve and neuro-endocrine cells. Snap25 is a crucial component of the SNARE complex and, together with Stx1 and synaptobrevin-2, plays a crucial role in Ca2+-dependent exocytosis of the synaptic vesicles (14).
In mammals, due to the differential splicing of the SNAP25 gene, two different isoforms (Snap25a and Snap25b) are obtained, which differ only for nine amino acids. However, they show different expression and localization profiles in the various brain regions in humans and mice (43). The most crucial isoform in synaptic transmission is Snap25b, which is mainly expressed in the synapses of the central nervous system and the peripheral motor endplates, and it regulates the exocytosis of neurotransmitters. Given its fundamental role in nervous transmission, de novo SNAP25 variants are associated with various neurological disorders, such as epilepsy, movement disorders, and psychiatric conditions (39).
We reviewed the recent literature and found the description of 18 cases in which pathogenic variants of SNAP25 are associated with DEEs (15, 44–47). All clinical data are summarized in Table 2.
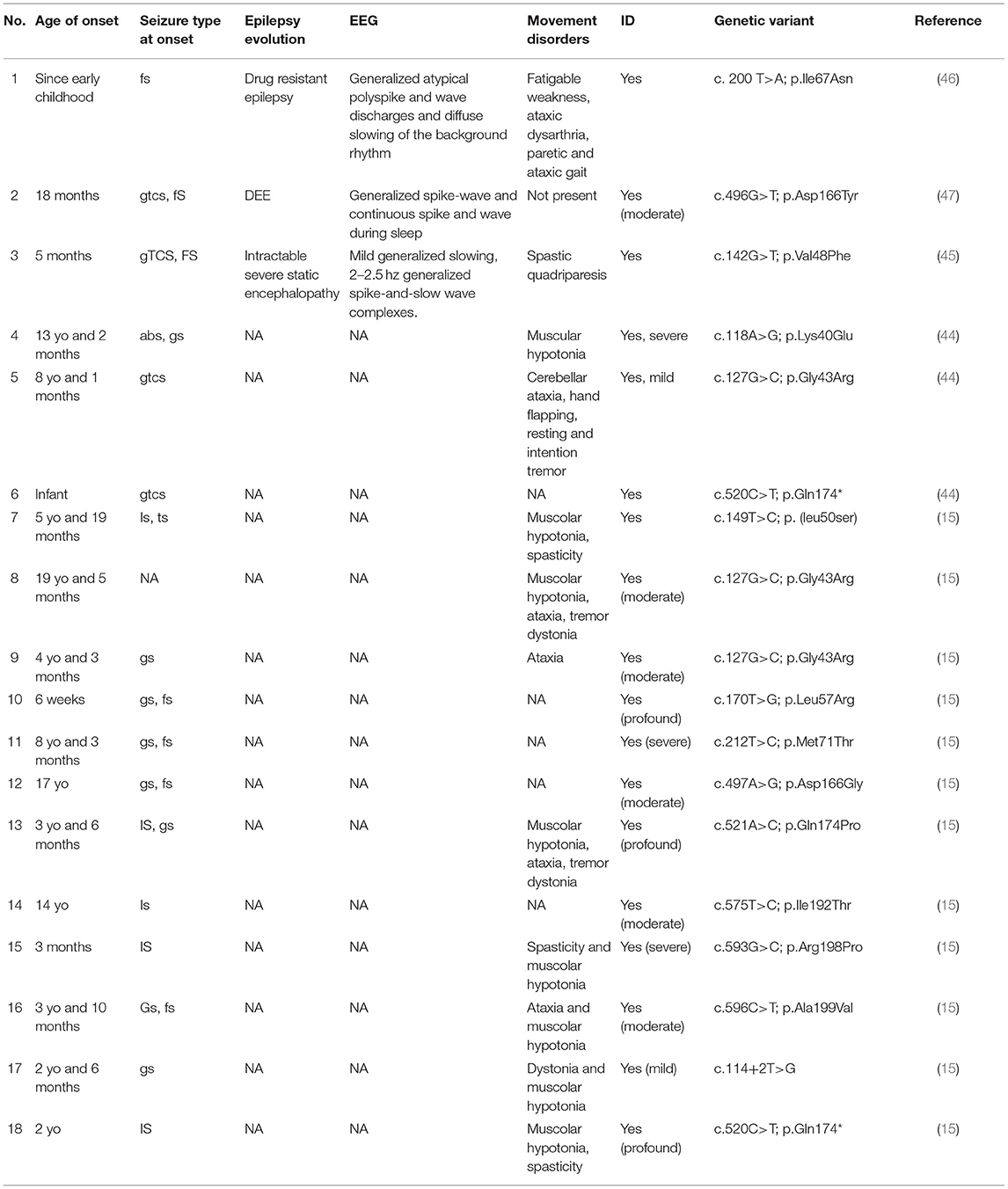
Table 2. Clinical features of SNAP25-related patients.
Most of the patients (13/18) showed seizures onset during the early childhood (ranging from 3 months to 8 years of age); two cases (2/18) presented with neonatal-onset epilepsy, while only three patients (3/18) showed epilepsy after the first decade of life (from 13 to 19 years of age). The semiology of the seizures appears to be heterogeneous: although most patients manifested generalized seizures (12/18), also focal seizures (5/18) and epileptic spasms (4/18) have been reported. In one case the description of the crisis was unavailable. Noteworthy, all patients (18/18) developed an intractable severe encephalopathy with moderate to severe ID. When available (3/18), the EEG showed multifocal abnormalities or generalized spike-and-slow wave complex, with a variable response to antiepileptic drugs (AEDs). Half of the patients (9/18) showed a negative MRI, and aspecific neuroradiological anomalies were reported only in three cases (3/18), such as leukoencephalopathy or brain volume loss. Frequent association with movement disorders, such as tremors, dystonia, muscular hypotonia, spasticity, and cerebellar ataxia, is described (13/18), and only in one patient (1/18) the absence of movement disorders was reported. Three patients (3/18) also showed behavioral disorders and autistic features. Response to AEDs was variable, and most of the patients (more than 50%) presented with frequent seizures despite being treated with several AEDs. A single patient showed a good response after therapy with valproic acid (VPA) and clonazepam, with a reduction of frequency and severity of seizures. Another case of highly drug-resistant epilepsy was treated with three-drug combinations and trials with a ketogenic diet and intravenous methylprednisolone. In some cases, information regarding the response to antiepileptic treatments were not reported (15, 44–47).
The VAMP2 gene, mapped on chromosome 17 (17p13.1 region), encodes for Vamp2 (also called synaptobrevin-2) (16, 48). As mentioned, Vamp2 protein constitutes the v-SNARE, and results fundamental to driving synaptic transmission, which is also regulated by Ca2+ ions and other proteins (16). Pathogenic variants of VAMP2 gene are associated with neurodevelopmental disorders, such as visual impairment, hyperkinetic movements, autism spectrum disorder and epilepsy (16, 39). More severe neurological phenotypes are described in individuals with nonsynonymous mutations of VAMP2 (39). The importance of a correct mechanism of neuronal trafficking mediated by Vamp2 has been highlighted by a recent study evaluating the essential role of VAMP2 and DLG4 in the progression of epilepsy and behavioral disorders, in particular ADHD (49). In these conditions, the expression of Dlg4 and Vamp2 is downregulated, which determines abnormal neurotransmission, presumably the cause of these disorders (49).
To date, just three patients carrying pathogenic variants of VAMP2 associated with DEE are described (see Table 3) (16). In particular, Salpietro et al. (16) reported five individuals (from 2 months to 14 years of age) with de novo heterozygous mutations of the VAMP2 gene presenting with various neurodevelopmental phenotypes, such as epilepsy, hypotonia, ID, and autistic features. Two of these patients did not present epileptic manifestations, but they showed EEG anomalies (such as high-voltage delta activity, sharp-and-slow-wave complexes or only a disorganized EEG). Conversely, three patients showed seizures onset within the first months of life: one presented with focal seizures, another one reported generalized tonic-clonic seizures and focal seizures, the last one developed infantile spasms. EEGs showed disorganized activity, generalized and/or multifocal abnormalities, sharp wave-slow wave complexes, or other focal paroxysms. Language is always compromised (1/3) or absent (2/3), and all the patients presented with autistic features and variable motor stereotypies comparable to Rett syndrome (RTT) (3/3) (16). The patients showed highly drug-resistant epilepsy: they trialed several AEDs, such as VPA, vigabatrin, and lamotrigine. VPA has been proved the most beneficial in two individuals, and particularly one of them was reported as seizure-free since the age of 12 years, and his follow-up EEGs were normal (16).
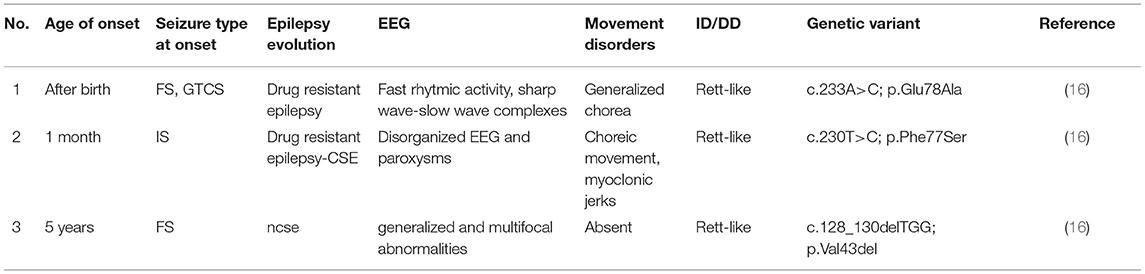
Table 3. Clinical features of VAMP2-related patients.
The CPLX1 gene is located on chromosome 4 (4p16.3), and it encodes for the complexin 1 (Cplx1). The complexin-family is a group of highly conserved cytosolic proteins expressed at the pre-synaptic terminal and interacting with the SNARE complex: Cplx1 and Cplx2 are the two paralogues mainly expressed in the CNS, the former being the most represented isoform, while Cplx3 and Cplx4 are predominantly identified at retinal ribbon synapses (13, 50, 51).
The exact function of the complexins has yet to be fully unraveled, but two mechanisms of action have been proposed. A controversial role as an inhibitor of the spontaneous release of neurotransmitters has been reported, with stronger evidence in invertebrates than in mammals; in fact, an increase in a spontaneous release of neurotransmitter was observed in cultured complexin-1/2 knockdown cortical neurons but not in complexin-1, −2 and double complexin-1/2 knockdown mice (13, 51–53). Secondly, a role as a Ca2+-triggered release promoter has been suggested, given the evidence showing a reduction in synaptic response amplitude after the action potential stimulation (51). Moreover, a reduction in both evoked and spontaneous release of glutamate is described in complexin-1/2/3 null cultured hippocampal neurons (13, 53). To date, the prominent hypothesis for the complexin mechanism of action is represented by its involvement in regulating the vesicle fusogenicity by lowering the energy barrier for primed vesicles to undergo either Ca2+-evoked or spontaneous fusion (13, 51).
To date, only five patients with DEEs associated with pathogenic variants CPLX1 have been described (50, 54), with epilepsy onset from 6 weeks to a maximum of 2 years of life. Infantile spasms (2/5) were the most frequent seizures type at onset, while a single patient showed myoclonic seizures (50). These three patients (3/5) developed myoclonic seizures, and EEG showed generalized epileptiform activity (50). However, all patients presented drug-resistant epilepsy. Conversely, Karaka et al. (54) reported two sisters that developed malignant migrating epilepsy and unspecified ID (54). No association with autistic features were reported in all the subjects. A single patient showed movement disorders and cerebral palsy (50). Brain MRI resulted normal in two patients (2/5), showed cortical atrophy in two patients (2/5), and in a single case (1/5) detected cerebellar abnormality (50). All data are summarized in Table 4.
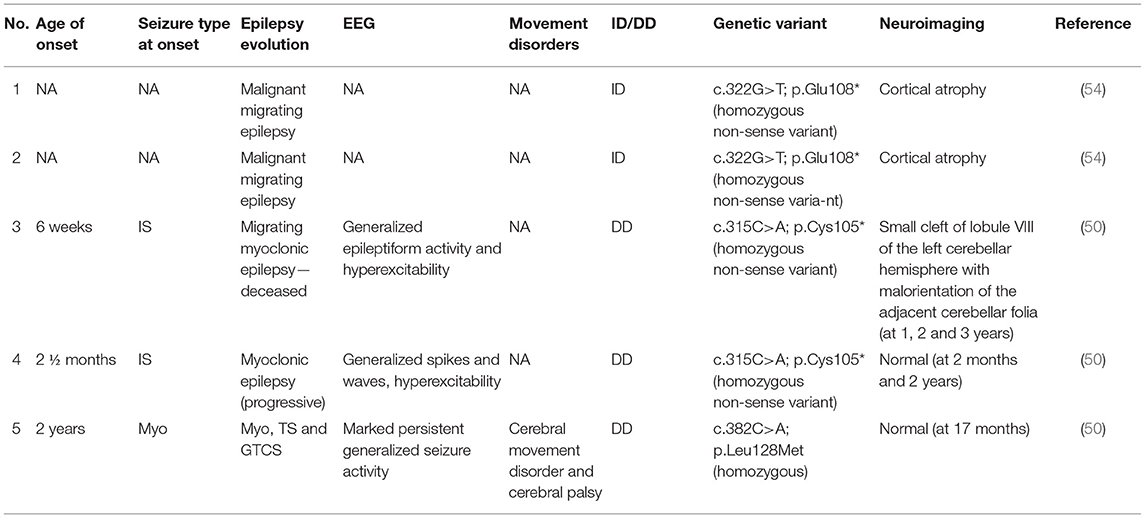
Table 4. Clinical features of CPLX1-related patients.
The STXBP1 (also known as MUNC18-1) is a gene located on chromosome 9 (9q34.11 region), which encodes the Syntaxin1a binding protein (Stxbp1), a protein of the SEC1 family that is essential for vesicles trafficking (19, 55). The Stxbp1 is a neural-specific binding protein that organizes the protein complexes that induce secretory vesicle exocytosis (56, 57). Notably, it is crucial to promote the conformation change in Stx1a, allowing the SNARE complex formation (22). This Stxbp1-Stx1a binding serves two purposes: firstly, when Stx1a is in a “closed” conformation, it interacts with the Habc domain and prevents the formation of ectopic and uncontrolled SNARE complexes in the synapses; secondly, when it binds the N-terminal peptide of the “open” Stx1a, Stxbp1 facilitates the synaptic vesicle priming and fusion, allowing the neurotransmitter release (58). It has also been demonstrated that Stxbp1 levels correlate with secretion capacity and synaptic strength, making this protein fundamental in synaptic transmission and maintenance of synaptic connections in adulthood (59–61).
Mutations of the STXBP1 gene determines an alteration in the elaborate mechanism of synaptic exocytosis, leading to an excitatory/inhibitory imbalance which can trigger an increased epileptic activity (60). Additionally, is ubiquitously expressed in the neuron. Stxbp1 also has non-synaptic functions: it regulates the post-Golgi transport of vesicles, it chaperons the α-synuclein, and it is a fundamental element in the development of the brain allowing neurite extension and radial migration of the cortical neurons (58).
Initially, the STXBP1 gene was associated with Ohtahara syndrome by Saitsu et al. (62). Since this discovery, the development and wide application of genetic testing helped recognize numerous new STXBP1-related severe early-onset DEEs (with a median onset of 6 weeks in 85% of the cases). The epileptic phenotypic spectrum includes West syndrome, Lennox-Gastaut syndrome, Dravet syndrome, early myoclonic encephalopathy, and several unclassified DEEs, with the former and the latter representing together half of STXBP1-related epilepsy (58, 63). Moreover, patients with disease-causing variants of STXBP1 have diverse phenotypes, including Rett-like syndrome and non-epileptic presentations, but they always show severe to profound DD/ID (58, 63, 64). In addition, an evolution of STXBP1-associated disease with the development of neurologic symptoms similar to early-onset parkinsonism has been recently pointed out (65).
Given the wide clinical spectrum of STXBP1-related diseases, a genotype-phenotype correlation is difficult to achieve: haploinsufficiency of the gene associated with a dominant-negative effect has been proposed as the primary pathogenic mechanism behind STXBP1 encephalopathy, potentially explaining the complex expression of pathogenic variants of this gene (64, 66). Previous reports showed an association between non-sense mutations and early onset DEEs, while missense pathogenic variants are correlated to more various clinical phenotypes (58). Interestingly, most of the pathogenic variants of STXBP1 described in the literature are heterozygous mutations, leading to the assumption that they were the only ones tolerated. However, two siblings presenting with Lennox-Gastaut syndrome were recently reported, carrying a homozygous missense mutation of STXBP1, causing a gain-of-function (64).
Regarding the treatment options for STXBP1-related DEEs, it is noteworthy that the patients carrying a pathogenic variant of STXBP1 showed seizures refractory to standard AEDs (58). Similarly to most of the neonatal-onset epileptic encephalopathies, Phenobarbital, VPA, vigabatrin, and levetiracetam are the most used drugs, though over half of the patients are treated with more than three AEDs, included ACTH and corticosteroids (58, 63, 67). Novel treatments such as trehalose, sorbitol, and 4-phenylbutyrate have been proven efficacious in restoring Stxbp1 protein levels in primary mouse neurons and C. elegans models: these compounds may reverse the deficit caused by the mutant protein and were also able to increase levels of wild-type Stxbp1 (58). Given the above, a clinical trial evaluating the safety and tolerability of Glycerol Phenylbutyrate in a small group of patients began in 2021 and is set to end in 2023 (ClinicalTrials.gov identifier: NCT04937062; accessed on 02 Jan2021).
Synaptic vesicle glycoproteins 2 (Sv2) are a transmembrane glycoprotein family located in the synaptic vesicles of neurons. Three isoforms of Sv2 are reported: Sv2a, 2b and 2c (68). The SV2A gene, mapped on chromosome 1 (1q21.2 region), encodes for Sv2a, a protein first described by Buckley and Kelly (69) that consists of 12 transmembrane domains and cytoplasmic N- and C-terminal sequences (70). It is widely expressed in GABAergic and glutamatergic neurons of the cerebral cortex, hippocampus and cerebellum, and it controls synaptic transmission through multiple mechanisms: Sv2a promotes the formation of the SNARE complex; also, it intervenes in both immediate synaptic vesicle release and Ca2+-dependent release as it interacts directly with synaptotagmin1 (Syt1). It has also been recognized as an ATP-binding site that would be involved in the process of vesicular priming (71, 72). It also represents the target site of Levetiracetam (LEV) (73). In literature, several studies on animal models showed how SV2A missense mutations cause an imbalance in GABAergic and glutamatergic transmission, leading to epilepsy (74–76).
To the best of our knowledge, only three cases of epilepsy associated with mutations in the SV2A gene have been reported in humans. Calame et al. (77) described a patient with epilepsy onset at 2 years of age. The administration of LEV caused a worsening of seizures and the development of a status epilepticus. Genetic investigations demonstrated a rare de novo variant in heterozygosity in SV2A. Subsequently, LEV was suspended, and the patient achieved good seizure control with VPA and a ketogenic diet. Developmental milestones in this patient are described as adequate, ruling out the hypothesis of a DEE.
On the contrary, Wang et al. (78) described a girl with a slight developmental delay and myoclonic seizures. The child was initially treated with levetiracetam, showing a deterioration of the clinical picture with the development of infantile spasms. Nevertheless, epileptic episodes disappeared after the suspension of levetiracetam and the administration of VPA and Clonazepam (78).
Only a case associated with drug-resistant epilepsy and DD/ID was reported by Serajee and Huq (79). This patient showed microcephaly, optic atrophy, epileptic spasms and myoclonus, with onset at 2 months of age (79). The clinical features of these two cases are summarized in Table 5.
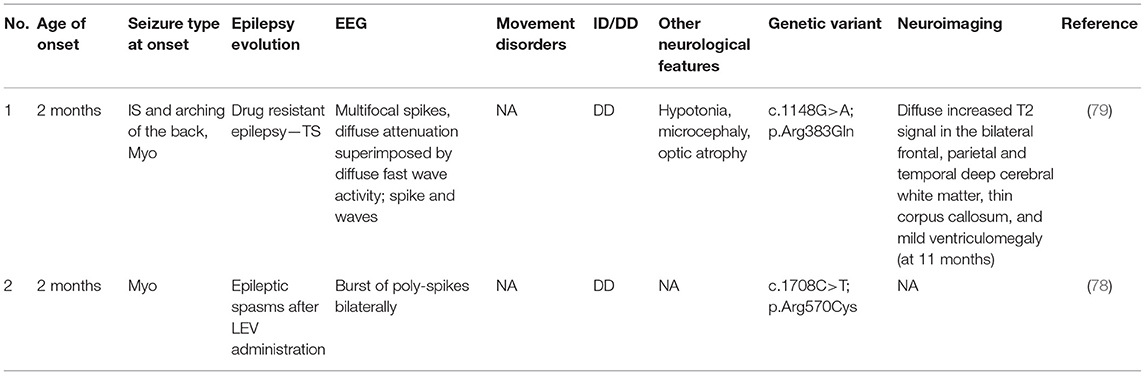
Table 5. Clinical features of SV2A-related patients.
The PRRT2 gene is located on chromosome 16 (16p11.2 region) and encodes for proline-rich transmembrane protein 2 (Prrt2), a pre-synaptic protein widely expressed in the cerebellum, the basal nuclei, and the neocortex (80, 81). Its expression is increased at major synaptogenesis stages, and it intervenes in modulating neuronal excitability. Two mechanisms are responsible for this process: Prrt2 regulates Nav1.2/1.6 currents (82) and contributes to controlling the vesicular trafficking and releasing the neurotransmitters. However, the mechanism of action of PRRT2 still appears controversial: according to Coleman et al. (83), the N-terminal portion of Prrt2 interacts directly with the SNARE complex reducing its formation, thus causing a decrease in the process of vesicular exocytosis (83). On the contrary, Valente et al. (81) proposed an alternative mechanism of action, according to which Prrt2 participates in the regulation of the Ca2+ sensing apparatus for the rapid synchronous release of synaptic vesicles by binding Snap25 and Syt1/2. Specifically, they observed a reduced neurotransmitter release in PRRT2-silenced primary neurons (81). Therefore, according to these evidences, in both cases Prrt2 influences the vesicular neurotransmitter release but with two apparently opposite effects (81, 83).
Pathogenic variants of PRRT2 are associated with a large spectrum of familial neurological disorders: PRRT2 is primarily associated with paroxysmal dyskinesia (PKD) but also hemiplegic migraine (HM), infantile convulsions and choreoathetosis (ICCA) and benign sporadic and familial seizures (BFIS) (84, 85).
Movement disorders are salient features of PRRT2-associated conditions, usually with onset in adulthood, and consisting principally in dystonia and choreoathetosis (84). The PKD chorea and dystonia episodes are typically brief (about 1 min) and can be triggered by prolonged exercise (86).
Regarding epilepsy, BIFS usually occurs within the first year of life and has a good response to drug therapy, especially with carbamazepine, phenobarbital, and VPA (87). Usually, seizures consist of brief episodes of psychomotor arrest, accompanied by generalized hypertonia, cyanosis, and limb jerks. In contrast, ICCA is characterized by the precocious onset of seizures and the subsequent development of movement disorders (paroxysmal dyskinesias), which generally occur at 5 years (86).
A genotype-phenotype correlation of PRRT2-related disorders is complicated, as it may be inferred by the wide phenotypic variability of diseases associated with the frameshift mutation c.649dupC (p.R217Pfs*8), which is reported as recurrent in the literature (82, 87, 88).
Generally, the neurological outcome is favorable (87), and there are no clear correlations between PRRT2 and epileptic encephalopathies (89).
Few cases of pathogenic variants of PRRT2 associated with clinical phenotypes comparable to DEEs were described: Pavone et al. (90) described a single patient with drug-resistant epilepsy, persistent electroencephalographic abnormalities, and severe DD. Djémié et al. (91) and Jafarpour and Desai (92) reported two cases of West Syndrome; Guerrero-López et al. (93) described a patient with epilepsy and severe ID. Moreover, severe conditions have been associated with homozygosity mutations of PRRT2 (94).
Although PRRT2 is known to be responsible for cognitive disorders (95), more studies are needed to assess the frequency of ID and psychiatric symptoms in PRRT2-associated syndromes (96).
NAPA and NAPB genes are mapped respectively on 19 (19q13.33 region) and 20 (20p11.21 region) chromosomes, and encoding for the soluble NSF Attachment Proteins Alpha (αSnap) and Beta (βSnap), ubiquitarious proteins with higher expression in the brain, βSnap only being expressed post-natally (97, 98). They are highly homologous to each other and represent essential components in the vesicular transport, the membrane fusion, and the release of neurotransmitters. In particular, they play a crucial role in dissociating and recycling the SNARE complex, making its components available for subsequent fusion reactions (98). The Snap proteins participate as a co-factor of the NSF ATPase during the SNARE complex disassembly, inducing increased levels of the free SNARE components (5, 98).
Given their role in neuronal regulation and brain development, variants of these genes may be associated with various neurological disorders, yet a clear link to developmental and epileptic encephalopathies has not been reported (97). However, a study conducted on βSnap-KOs mice demonstrated an epileptic phenotype with onset 11 days after birth, consistent with the developmental expression pattern of βSNAP: the mice developed severe recurrent epileptic seizures, occasionally leading to death (98).
To the best of our knowledge, solely four patients with DEEs associated with disease-causing variants of NAPB are described (29, 97, 99) (see Table 6). Clinical findings of the patient reported by Reuter et al. (29) was unavailable. All the other patients presented a very early onset of seizures (range from 2 to 6 months), and the most frequently reported type of seizures is clonic (3/4), but tonic seizures are also described in association with the clonic ones (97, 99). In the majority of the cases (3/4), the epilepsy evolution is a multifocal epileptic encephalopathy, and all the patients (4/4) presented a severe DD/ID (29, 97, 99). Neuroimaging was reported as normal in 2/4 subjects (2/4 are unavailable) (97, 99). One patient developed movement disorders such as axial and peripheral hypotonia, limb tremulousness and stereotypies (kicking, hand, wrist-twisting, and bringing to the midline) (99). No information about the therapeutic approach was reported.
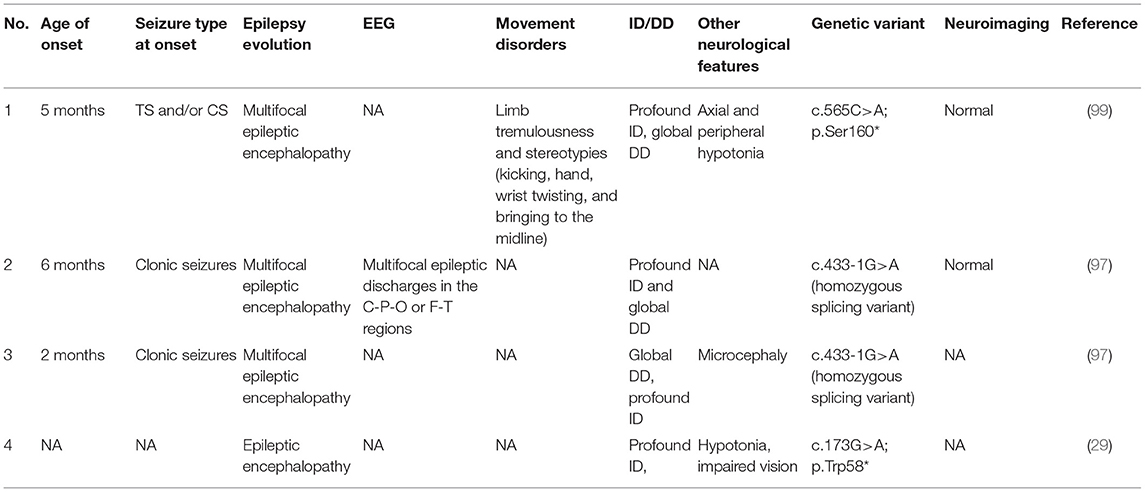
Table 6. Clinical features of NAPB-related patients.
The dynamin (Dmn) is a GTPase involved in vesicular transport and clathrin-dependent endocytosis (100). There are three different isoforms of the protein, called Dnm 1, 2 and 3; variant 1 is the most expressed in neurons (101). Dnm1 is encoded by the DNM1 gene located in 9q34.11, and its levels increase in parallel with synaptogenesis, particularly during the post-natal phase (102, 103). The process of phosphorylation/dephosphorylation of Dnm1, mediated by Cdk5 and Ca2+-dependent calmodulin, respectively, regulates the activation of Dnm1 by facilitating its interaction with other proteins involved in endocytosis. For this reason, pathogenic variants of DNM1 can cause an impairment of endocytosis of synaptic vesicles with consequent impact on vesicle recycling and synaptic function (104). To date, it has been clarified that DNM1 pathogenic variants can be associated with Lennox-Gastaut syndrome and infantile spasms (105, 106).
Our review of the literature has led to the identification of 33 cases with pathogenic variants of DMN1, but only 30 of these are associated with epilepsy and some degree of ID (100, 103, 106–114). Brereton et al. (115) described some cases presenting a milder phenotype, characterized by autistic symptoms and ID without seizure. Clinical features of patients with DNM1-related DEEs are summarized in Table 7.
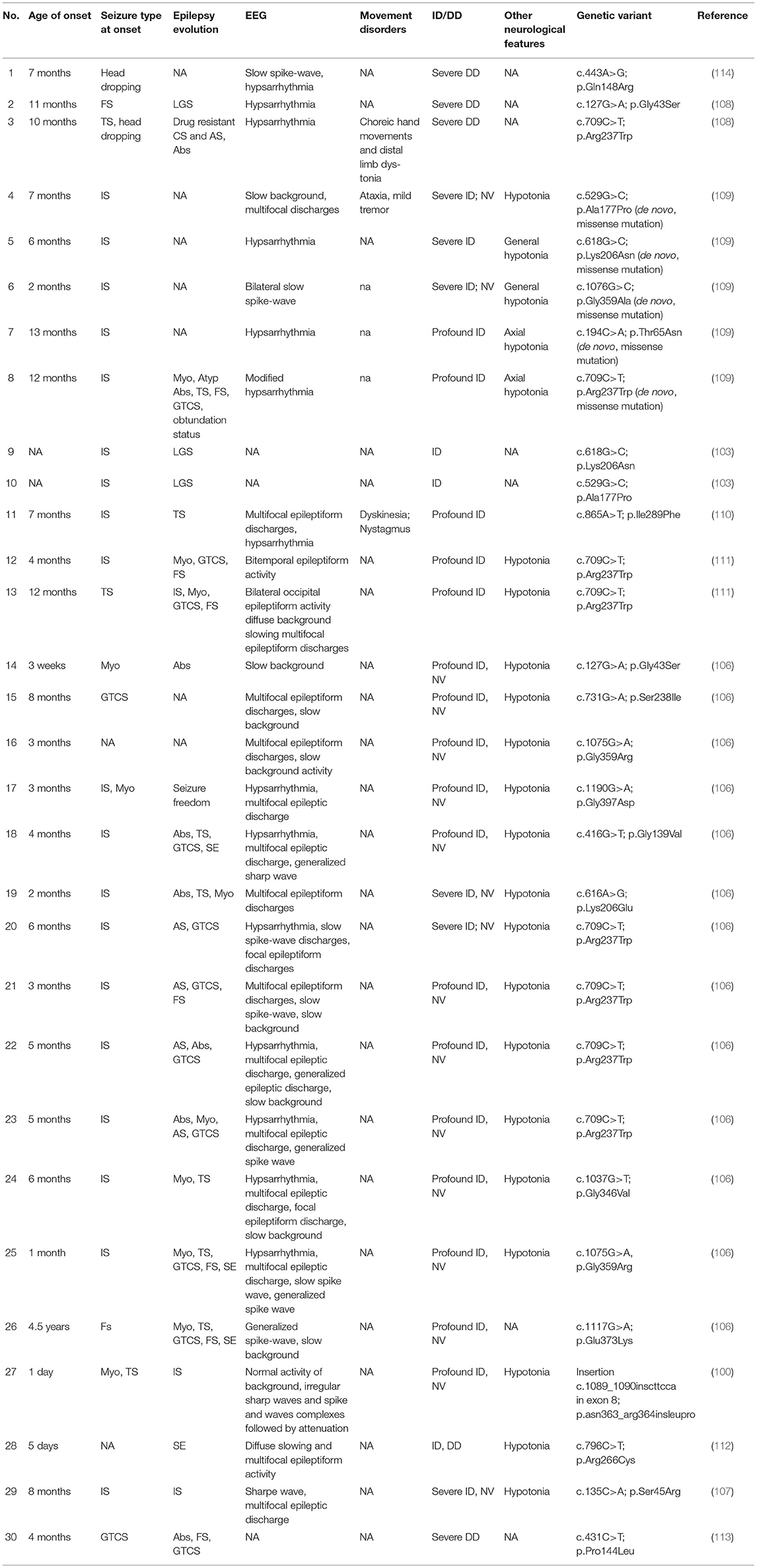
Table 7. Clinical features of DNM1-related patients.
The onset of seizures occurred in almost all cases within the first year of life, and the most frequently reported seizures type is represented by infantile spasms (19/30). Noteworthy is the case of a patient who did not report further epileptic episodes despite presenting an onset with infantile spasms in the first year of life, developing a neurological impairment with profound ID (106).
All patients presented with severe/profound ID, in many cases (17/30) associated with the absence of verbal communication (100, 106, 107, 109). Also, two-thirds of the cases (22/30) showed deep axial and/or diffuse hypotonia and severe involvement of motor skills (100, 106–109, 111, 112).
It is noteworthy mentioning that the genetic variant c.709C>T (p.Arg237Trp) was found in 8 cases out of 30 (27% of patients identified) and that c.1075G>A (p.Gly359Arg) was reported twice (106, 108, 109, 111).
Data regarding the therapy was not available in 7/30 of the patient. Only four patients (4/30) were seizure-free, and in particular, two of them have achieved the absence of seizures following the ketogenic diet (106, 109, 114). In addition, another one obtained the absence of epileptic episodes for a long time following a ketogenic diet, although epilepsy recurred later (109).
The ZFYVE20 gene, mapped on chromosome 3 (3p25 region), encodes for Rabenosyn-5 (Rbsn-5), a large, highly conserved, multidomain protein, which is ubiquitously expressed in mammalian cells. Rbsn-5 is involved in receptor-mediated endocytosis and neurotransmitter recycling (116, 117). Specifically, Rbsn-5 main function is to regulate the intracellular route of internalized neurotransmitters receptors, facilitating their recycling to the plasma membrane through direct interaction with regulatory proteins and lipids, such as the endocytic GTPases Rab4 and Rab5, and phosphatidylinositol-3 phosphate (116–118).
We reviewed recent literature and identified a single case in which a pathogenic variant of ZFYVE20 is associated with a form of DEE (116). A girl with a homozygous missense mutation of ZFYVE20, detected by whole-exome sequencing, showed a severe drug-resistant epileptic encephalopathy and ID (116). She presented seizures onset at 5 months of life with infantile spasms and had marked hypotonus, with the impossibility of sitting and walking independently. The patient also presented facial dysmorphisms, microcephaly, macrocytosis and megaloblastoid erythropoiesis (116). She gained poor seizures control with several anticonvulsive drugs (VPA, Phenobarbital, Levetiracetam, Lamotrigine). At 14 months, a ketogenic diet was started with a report of improvement, and she was clinically seizure-free at 6.5 years of age (116).
The TBC1D24 gene, located on chromosome 16 (16p13.3 region), encodes for Tbc1d24, a highly conserved 553 amino acid protein, which consists of two domains, the Tre2/Bub2/Cdc16 domain (TBC) and a TBC/Lysin Motif Domain/Catalytic (TLDc) domain (119). The TBC domain is involved in the regulation of synaptic traffic, while the function of the TLDc domain is less known, but it appears to be involved in oxidative stress processes (119).
Tbc1d24 is involved in vesicle trafficking in the brain, neuronal migration, and somatic cellular development (120, 121). In particular, in the pre-synaptic terminal, it acts as a selective GTPase activating protein for the GTPase Rab35, which allows the endosomal sorting of synaptic vesicle proteins and the replacement of damaged components (122). Moreover, Falace et al. (120) demonstrated that Tbc1d24 binds the GTPase Arf6, proving in mouse models the role of TBC1D24 in regulating neuronal migration.
Pathogenic variants of this gene are associated with heterogeneous clinical manifestations, including non-syndromic hearing loss and drug-resistant epilepsy, cerebellar alterations, alternating hemiplegia, and symptoms of neurodegeneration (119).
Epilepsy phenotypes associated with pathogenic variants of TBC1D24 include familial infantile myoclonic epilepsy, epilepsy of infancy with migrating focal seizures (EIMFS) and DOORS (deafness, onychodystrophy, osteodystrophy, intellectual disabilities, and seizures) syndrome (123–125).
In the recent literature, we found the description of 30 cases in which mutations of TBC1D24 are associated with DEEs (107, 123–131). All clinical features are summarized in Table 8.
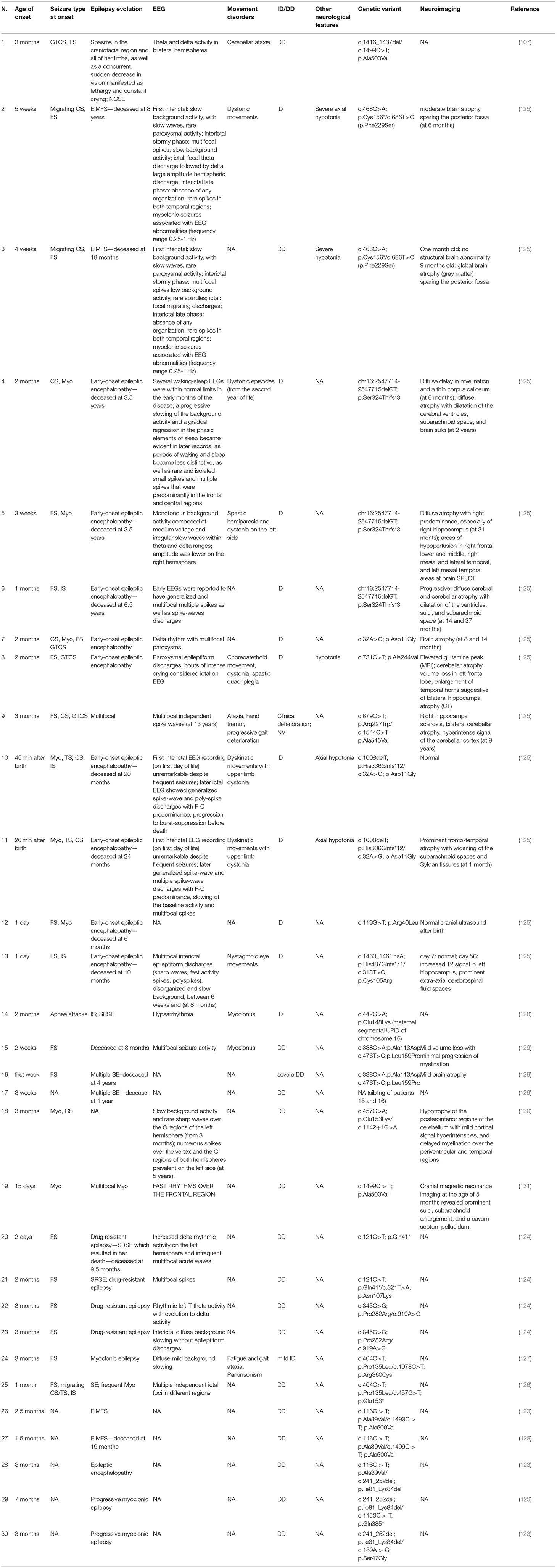
Table 8. Clinical features of TBC1D24-related patients.
All patients presented a history of early onset of seizures, ranging from 20 minutes after birth to 8 months of life: in the majority of the patients (28/30), the onset was within 3 months of life, and, among them, 6/30 individuals developed seizures within the first week (107, 123–131).
The most commonly reported type of seizures is migrating focal ones (17/30), followed by migrating clonic ones (9/30), myoclonic ones (9/30), generalized tonic-clonic ones (4/30), epileptic spasm (4/30), tonic ones (3/30), and apnea attacks (1/30) (107, 123–126, 128–131). In six patients, the description of the seizures was not available (123, 129).
All patients developed a drug-resistant epileptic encephalopathy: five patients (5/30) showed epilepsy of infancy with migrating focal seizures (EIMFS) (123, 125, 126). In two cases, an evolution in progressive myoclonic epilepsy has been described, poorly controlled by drug therapy and often triggered by fever (123).
A non-convulsive super-refractory status epilepticus treated with midazolam, ketamine, and pentobarbital was reported in two cases (124). One of these patients was also treated with hypothermia and died at 9 years (124). Another patient presented with super-refractory status epilepticus treated with midazolam or thiamylal (128). Moreover, the patient described by Li et al. (107) presented with non-convulsive status epilepticus treated with intravenous injection of diazepam, while the patient reported by Lozano et al. (129) developed multiple status epilepticus and died at 1 year of age.
Overall, 13 patients (13/30) died at a very young age (ranging from 3 months to 9.5 years of age) (123–125, 129).
The EEGs did not show a typical pattern, and various abnormalities were described: focal epileptiform discharges in different regions of cerebral hemispheres were reported in seven cases (7/30) (124–126), while 8/30 patients showed generalized or and/or multifocal discharges (125). A diffuse background slowing was described in nine patients (9/30), despite the presence or absence of focal/multifocal discharges (124, 125, 127), while 3/30 initially presented EEGs within the normal limits (125). Lately, 5/30 patients showed a total absence of organization (125). One patient (1/33) developed a burst-suppression pattern (125) and one (1/30) presented with hypsarrhythmia at 4 months of age (128). In 8/30 cases, EEG data was not available (123, 125, 129).
All of them showed DD/ID, and in many cases, the expressive language was inadequate or dysarthric (107, 123–126, 128–131).
In one case, a young woman with an early onset of myoclonic epilepsy showed movement disorders (1/30) with cerebellar ataxia and fatigue, parkinsonism and symptoms of psychosis with hallucinations and depression (127). Other reported movement disorders were Dystonic movements (6/30), cerebellar ataxia (2/30), dyskinetic movements (2/30), non-epileptic myoclonus (2/30), spastic hemiparesis (1/30), choreoatethoid movements (1/30), spastic quadriplegia (1/30), nystagmoid eye movements (1/30) (107, 124–126, 128, 129). In 7/30 cases, the description of movement disorders and other additional features was not available (123, 130, 131).
Most of the patients (29/30) developed an epileptic encephalopathy with frequent seizures non-responsive to various therapeutic approaches and ketogenic diet therapy (107, 123–125, 128–131). Fang et al. (126) described a single patient that experienced >50 % seizure reduction after diazepam treatment. The ketogenic diet was adopted in two cases (123, 124), causing a reduction of seizures and cognitive improvement after a year and a half of diet in one patient (123). In one case, a description of the therapeutic response was not available (127).
DEEs are a heterogeneous group of conditions with onset in infancy or early childhood, in which the epileptic activity significantly interferes with the development, determining severe DD/ID and other neuropsychiatric disorders (9, 10). These diseases may be caused by an alteration of the synapse, the fundamental unit of signal transmission in the nervous system (1, 3). Numerous genes contribute to the synaptic transmission's proper functioning, and alterations of this complex mechanism may result in synaptopathy (1, 8).
We reviewed the literature, focusing on those genes involved in the correct operation of the pre-synaptic terminal, and analyzed the clinical features of 119 patients that showed a clinical presentation resembling a DEE.
A valid genotype-phenotype correlation is difficult to deduce especially in those reports including only few patients with DEEs (50, 79, 116). On the contrary, when many patients with pathogenic variants of the same gene present with different phenotypes, a clear correlation is difficult to achieve: this is even truer in those cases in which the same mutation is related to various clinical presentations (58, 63, 64, 82, 87, 88). This phenotypic heterogeneity has been long studied, and it is related to several factors intervening during the development, including epigenetic factors, timing and location of physiological gene expression and modifier genes (9).
Nevertheless, some noteworthy features may be underlined regarding the clinical phenotypes of DEEs related to the genes we reviewed, which can lead the clinician to a genetic suspect.
For instance, patients with VAMP2 pathogenic variants showed a neurodevelopmental disorder characterized by ID, central visual impairment, movement disorders, epilepsy or electroencephalographic abnormalities, autistic features, and loss of purposeful hand movements resembling Rett syndrome (16).
STX1B-related DEE must be hypothesized when a patient presents with myoclonic seizures at onset (35, 40–42). Subsequently, these patients may manifest ataxia (35, 37, 39).
Patients carrying DNM1 mutations may present with infantile spasms in more than a half of the cases, usually associated with axial and/or diffuse hypotonia with severe impairment of motor skills (100, 106–109, 111, 112).
Almost the totality of the patients with a pathogenic variant of TBC1D24 present the first seizure within 3 months after birth, with severe progression frequently leading to death within the first decade of life (107, 123–131). Although EIMFS was reported as a typical epileptic phenotype related to TBC1D24, in our review, we could identify 5/30 (16%) patients that developed this type of DEE (123–126). Instead, noteworthy mentioning is the association with status epilepticus (107, 124, 128, 129).
Usually, patients with SNAP25-DEEs show seizure's onset after 2 years of age, with generalized seizures and a frequent association to movement disorders (15, 44–47), while NAPB-associated DEEs are characterized by a high frequency of clonic seizures and an evolution in multifocal epileptic encephalopathy (97, 99).
Overall, when a patient presents with some degree of DD, an early epilepsy onset with drug-resistant seizures, possibly associated with a movement disorder, the suspect of a synaptopathy must be taken into account when approaching the differential diagnosis.
Concerning the therapeutic approaches, an individually-tailored treatment is desirable to intervene directly on the altered mechanism determining the DEE, improving the seizure control and the developmental outcome. However, in most severe epilepsies, a gene-specific therapy is not available, and the treatment options are represented by the usual AEDs, that do not address the underlying causative mechanism (9). Our literature review regarding DEEs related to genes involved in pre-synaptic mechanisms confirms these data: most patients showed a bad prognosis with highly drug-resistant seizures, despite the multiple therapeutic combinations. Moreover, literature data suggested the efficacy of the ketogenic diet in part of the patients with DEEs related to DNM1, TBC1D24, and ZFYVE2O. In particular, two subjects with pathogenic variants of DNM1 achieved the absence of seizures, and another one obtained the absence of epileptic episodes for a long time, although epilepsy recurred later (106, 109). The ketogenic diet was also considered as a valid therapeutic approach for seizure control in two cases related to TBC1D24, and especially one of these patients showed a decrease in the frequency of seizures and cognitive improvement after a year and a half of diet (123, 124). Finally, the girl with a disease-causing variant of ZFYVE2O, after several AEDs trials, started a ketogenic diet, obtaining a gradual improvement and seizure freedom at 6.5 years of age (116).
This work's limitation is the paucity of a complete description of the patients, making it difficult to obtain homogenous information and, therefore, to deduce a clear genotype-phenotype correlation. A more detailed clinical description of the patients may be desirable to improve the genotype-phenotype correlation and better guide the choice of the genetic testing, allowing to obtain an early diagnosis and to develop individually-tailored therapies.
AN and GD conceived planned and supervised the study. GS, GV, and MS wrote the first draft of the manuscript and prepared the tables. GS prepared the figure. AB, GA, and VS helped supervise the project. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abs, Absence; AS, atonic seizure; Atyp Abs, Atypical Absence; CS, clonic seizures; CSE, convulsive status epilepticus; DD, developmental delay; EIMFS, Epilepsy of Infancy with Migrating Focal Seizures; FS, Focal Seizure; GTCS, generalized tonic clonic seizure; ID, intellectual disability; IS, Infantile Spasm; LGS, Lennox Gastaut Syndrome; MA, myoclonic atonic seizure; MAE, myclonic atonic epilepsy; Myo, Myclonic seizure; NA, not available; NCSE, non-convulsive status epilepticus; NV, non-verbal; SE, Status Epilepticus; TS, Tonic Seizure.
1. Lepeta K, Lourenco MV, Schweitzer BC, Martino Adami PV, Banerjee P, Catuara-Solarz S, et al. Synaptopathies: synaptic dysfunction in neurological disorders - a review from students to students. J Neurochem. (2016) 138:785–805. doi: 10.1111/jnc.13713
2. Cortès-Saladelafont E, Tristán-Noguero A, Artuch R, Altafaj X, Bayès A, García-Cazorla A. Diseases of the synaptic vesicle: a potential new group of neurometabolic disorders affecting neurotransmission. Semin Pediatr Neurol. (2016) 23:306–20. doi: 10.1016/j.spen.2016.11.005
3. Tristán-Noguero A, García-Cazorla À. Synaptic metabolism: a new approach to inborn errors of neurotransmission. J Inherit Metab Dis. (2018) 41:1065–75. doi: 10.1007/s10545-018-0235-7
4. Körber C, Kuner T. Molecular machines regulating the release probability of synaptic vesicles at the active zone. Front Synaptic Neurosci. (2016) 8:5. doi: 10.3389/fnsyn.2016.00005
5. Rizzoli SO. Synaptic vesicle recycling: steps and principles. EMBO J. (2014) 33:788–822. doi: 10.1002/embj.201386357
6. Syková E, Nicholson C. Diffusion in brain extracellular space. Physiol Rev. (2008) 88:1277–340. doi: 10.1152/physrev.00027.2007
7. Li JY, Plomann M, Brundin P. Huntington's disease: a synaptopathy?. Trends Mol Med. (2003) 9:414–20. doi: 10.1016/j.molmed.2003.08.006
8. Luo J, Norris RH, Gordon SL, Nithianantharajah J. Neurodevelopmental synaptopathies: insights from behaviour in rodent models of synapse gene mutations. Progress Neuro Psychopharmacol Biol Psychiatry. (2018) 84(Pt B):424–39. doi: 10.1016/j.pnpbp.2017.12.001
9. McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. (2016) 15:304–16. doi: 10.1016/S1474-4422(15)00250-1
10. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia (2017) 58:512–21. doi: 10.1111/epi.13709
11. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. (2001) 68:1327–32. doi: 10.1086/320609
12. Saifee O, Wei L, Nonet ML. The Caenorhabditis elegans unc-64 locus encodes a syntaxin that interacts genetically with synaptobrevin. Mol Biol Cell. (1998) 9:1235–52. doi: 10.1091/mbc.9.6.1235
13. Melland H, Carr EM, Gordon SL. Disorders of synaptic vesicle fusion machinery. J Neurochem. (2021) 157:130–64. doi: 10.1111/jnc.15181
14. Fukuda H, Imagawa E, Hamanaka K, Fujita A, Mitsuhashi S, Miyatake S, et al. A novel missense SNAP25b mutation in two affected siblings from an Israeli family showing seizures and cerebellar ataxia. J Hum Genet. (2018) 63:673–6. doi: 10.1038/s10038-018-0421-3
15. Klöckner C, Sticht H, Zacher P, Popp B, Babcock HE, Bakker DP, et al. De novo variants in SNAP25 cause an early-onset developmental and epileptic encephalopathy. Genet Med. (2021) 23:653–60. doi: 10.1038/s41436-020-01020-w
16. Salpietro V, Malintan NT, Llano-Rivas I, Spaeth CG, Efthymiou S, Striano P, et al. Mutations in the neuronal vesicular SNARE VAMP2 affect synaptic membrane fusion and impair human neurodevelopment. Am J Hum Genet. (2019) 104:721–30. doi: 10.1016/j.ajhg.2019.02.016
17. Rizo J, Xu J. The synaptic vesicle release machinery. Annu Rev Biophys. (2015) 44:339–67. doi: 10.1146/annurev-biophys-060414-034057
18. Fernandez I, Ubach J, Dulubova I, Zhang X, Südhof TC, Rizo J. Three-dimensional structure of an evolutionarily conserved N-terminal domain of syntaxin 1A. Cell. (1998) 94:841–9. doi: 10.1016/S0092-8674(00)81742-0
19. Misura KM, Scheller RH, Weis WI. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. (2000) 404:355–62. doi: 10.1038/35006120
20. Zhou P, Pang ZP, Yang X, Zhang Y, Rosenmund C, Bacaj T, et al. Syntaxin-1 N-peptide and Habc-domain perform distinct essential functions in synaptic vesicle fusion. EMBO J. (2013) 32:159–71. doi: 10.1038/emboj.2012.307
21. Verhage M, Sørensen JB. SNAREopathies: diversity in mechanisms and symptoms. Neuron. (2020) 107:22–37. doi: 10.1016/j.neuron.2020.05.036
22. Gerber SH, Rah JC, Min SW, Liu X, de Wit H, Dulubova I, et al. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science. (2008) 321:1507–10. doi: 10.1126/science.1163174
23. Lee S, Shin J, Jung Y, Son H, Shin J, Jeong C, et al. Munc18-1 induces conformational changes of syntaxin-1 in multiple intermediates for SNARE assembly. Sci Rep. (2020) 10:11623. doi: 10.1038/s41598-020-68476-3
24. Bennett MK, Calakos N, Scheller RH. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. (1992) 257:255–9. doi: 10.1126/science.1321498
25. Mishima T, Fujiwara T, Sanada M, Kofuji T, Kanai-Azuma M, Akagawa K. Syntaxin 1B, but not syntaxin 1A, is necessary for the regulation of synaptic vesicle exocytosis and of the readily releasable pool at central synapses. PLoS ONE. (2014) 9:e90004. doi: 10.1371/journal.pone.0090004
27. Gao MC, Bellugi U, Dai L, Mills DL, Sobel EM, Lange K, et al. Intelligence in Williams Syndrome is related to STX1A, which encodes a component of the presynaptic SNARE complex. PLoS ONE. (2010) 5:e10292. doi: 10.1371/journal.pone.0010292
28. Tropeano M, Wöber-Bingöl C, Karwautz A, Wagner G, Vassos E, Campos-de-Sousa S, et al. Association analysis of STX1A gene variants in common forms of migraine. Cephalalgia. (2012) 32:203–12. doi: 10.1177/0333102411433300
29. Reuter MS, Tawamie H, Buchert R, Hosny Gebril O, Froukh T, Thiel C, et al. Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiatry. (2017) 74:293–9. doi: 10.1001/jamapsychiatry.2016.3798
30. Yu YX, Shen L, Xia P, Tang YW, Bao L, Pei G. Syntaxin 1A promotes the endocytic sorting of EAAC1 leading to inhibition of glutamate transport. J Cell Sci. (2006) 119(Pt 18):3776–87. doi: 10.1242/jcs.03151
31. Fili O, Michaelevski I, Bledi Y, Chikvashvili D, Singer-Lahat D, Boshwitz H, et al. Direct interaction of a brain voltage-gated K+ channel with syntaxin 1A: functional impact on channel gating. J Neurosci. (2001) 21:1964–74. doi: 10.1523/JNEUROSCI.21-06-01964.2001
32. Baghel R, Grover S, Kaur H, Jajodia A, Parween S, Sinha J, et al. Synergistic association of STX1A and VAMP2 with cryptogenic epilepsy in North Indian population. Brain Behav. (2016) 6:e00490. doi: 10.1002/brb3.490
33. Smirnova T, Miniou P, Viegas-Pequignot E, Mallet J. Assignment of the human syntaxin 1B gene (STX) to chromosome 16p11.2 by fluorescence in situ hybridization. Genomics. (1996) 36:551–3. doi: 10.1006/geno.1996.0506
34. Wu YJ, Tejero R, Arancillo M, Vardar G, Korotkova T, Kintscher M, et al. Syntaxin 1B is important for mouse postnatal survival and proper synaptic function at the mouse neuromuscular junctions. J Neurophysiol. (2015) 114:2404–17. doi: 10.1152/jn.00577.2015
35. Wolking S, May P, Mei D, Møller RS, Balestrini S, Helbig KL, et al. Clinical spectrum of STX1B-related epileptic disorders. Neurology. (2019) 92:e1238–49. doi: 10.1212/WNL.0000000000007089
36. Liu YH, Cheng YT, Tsai MH, Chou IJ, Hung PC, Hsieh MY, et al. Genetics and clinical correlation of Dravet syndrome and its mimics - experience of a tertiary center in Taiwan. Pediatr Neonatol. (2021) 62:550–8. doi: 10.1016/j.pedneo.2021.05.022
37. Borlot F, de Almeida BI, Combe SL, Andrade DM, Filloux FM, Myers KA. Clinical utility of multigene panel testing in adults with epilepsy and intellectual disability. Epilepsia. (2019) 60:1661–9. doi: 10.1111/epi.16273
38. Vlaskamp DR, Rump P, Callenbach PM, Vos YJ, Sikkema-Raddatz B, van Ravenswaaij-Arts CM, et al. Haploinsufficiency of the STX1B gene is associated with myoclonic astatic epilepsy. European journal of paediatric neurology: EJPN. (2016) 20:489–92. doi: 10.1016/j.ejpn.2015.12.014
39. Tang S, Addis L, Smith A, Topp SD, Pendziwiat M, Mei D, et al. Phenotypic and genetic spectrum of epilepsy with myoclonic atonic seizures. Epilepsia. (2020) 61:995–1007. doi: 10.1111/epi.16508
40. Lerche H, Weber YG, Baier H, Jurkat-Rott K, Kraus de Camargo O, Ludolph AC, et al. (2001). Generalized epilepsy with febrile seizures plus: further heterogeneity in a large family. Neurology, 57, 1191–1198. doi: 10.1212/WNL.57.7.1191
41. Weber YG, Jacob M, Weber G, Lerche H. A BFIS-like syndrome with late onset and febrile seizures: suggestive linkage to chromosome 16p11.2-16q12.1. Epilepsia. (2008) 49:1959–64. doi: 10.1111/j.1528-1167.2008.01646.x
42. Mishima T, Fujiwara T, Kofuji T, Saito A, Terao Y, Akagawa K. Syntaxin 1B regulates synaptic GABA release and extracellular GABA concentration, and is associated with temperature-dependent seizures. J Neurochem. (2021) 156:604–13. doi: 10.1111/jnc.15159
43. Prescott GR, Chamberlain LH. Regional and developmental brain expression patterns of SNAP25 splice variants. BMC Neurosci. (2011) 12:35. doi: 10.1186/1471-2202-12-35
44. Heyne HO, Singh T, Stamberger H, Abou Jamra R, Caglayan H, Craiu D, et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat Genet. (2018) 50:1048–53. doi: 10.1038/s41588-018-0143-7
45. Rohena L, Neidich J, Truitt Cho M, Gonzalez KD, Tang S, Devinsky O, et al. Mutation in SNAP25 as a novel genetic cause of epilepsy and intellectual disability. Rare Dis. (2013) 1:e26314. doi: 10.4161/rdis.26314
46. Shen XM, Selcen D, Brengman J, Engel AG. Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology. (2014) 83:2247–55. doi: 10.1212/WNL.0000000000001079
47. Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet. (2017) 101:664–85. doi: 10.1016/j.ajhg.2017.09.008
48. Zoraqi GK, Paradisi S, Falbo V, Taruscio D. Genomic organization and assignment of VAMP2 to 17p12 by FISH. Cytogenet Cell Genet. (2000) 89:199–203. doi: 10.1159/000015612
49. Xi XJ, Tang JH, Zhang BB, Xiao X, Hu XY, Wan Y, et al. Dlg4 and Vamp2 are involved in comorbid epilepsy and attention-deficit hyperactivity disorder: a microarray data study. Epilepsy Behav. (2020) 110:107192. doi: 10.1016/j.yebeh.2020.107192
50. Redler S, Strom TM, Wieland T, Cremer K, Engels H, Distelmaier F, et al. Variants in CPLX1 in two families with autosomal-recessive severe infantile myoclonic epilepsy and ID. Eur J Hum Genet. (2017) 25:889–93. doi: 10.1038/ejhg.2017.52
51. Trimbuch T, Rosenmund C. Should I stop or should I go? The role of complexin in neurotransmitter release. Nat Rev Neurosci. (2016) 17:118–25. doi: 10.1038/nrn.2015.16
52. Maximov A, Tang J, Yang X, Pang ZP, Südhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. (2009) 323:516–21. doi: 10.1126/science.1166505
53. Reim K, Mansour M, Varoqueaux F, McMahon HT, Südhof TC, Brose N, et al. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. (2001) 104:71–81. doi: 10.1016/S0092-8674(01)00192-1
54. Karaca E, Harel T, Pehlivan D, Jhangiani SN, Gambin T, Coban Akdemir Z, et al. Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron. (2015) 88:499–513. doi: 10.1016/j.neuron.2015.09.048
55. Swanson DA, Steel JM, Valle D. Identification and characterization of the human ortholog of rat STXBP1, a protein implicated in vesicle trafficking and neurotransmitter release. Genomics. (1998) 48:373–6. doi: 10.1006/geno.1997.5202
56. Pevsner J, Hsu SC, Scheller RH. n-Sec1: a neural-specific syntaxin-binding protein. Proc Natl Acad Sci USA. (1994) 91:1445–9. doi: 10.1073/pnas.91.4.1445
57. Toonen RF, Verhage M. Munc18-1 in secretion: lonely Munc joins SNARE team and takes control. Trends Neurosci. (2007) 30:564–72. doi: 10.1016/j.tins.2007.08.008
58. Abramov D, Guiberson N, Burré J. STXBP1 encephalopathies: clinical spectrum, disease mechanisms, and therapeutic strategies. J Neurochem. (2021) 157:165–78. doi: 10.1111/jnc.15120
59. Toonen RF, Kochubey O, de Wit H, Gulyas-Kovacs A, Konijnenburg B, Sørensen JB, et al. Dissecting docking and tethering of secretory vesicles at the target membrane. EMBO J. (2006) 25:3725–37. doi: 10.1038/sj.emboj.7601256
60. Al Mehdi K, Fouad B, Zouhair E, Boutaina B, Yassine N, Chaimaa A, et al. Molecular modelling and dynamics study of nsSNP in STXBP1 gene in early infantile epileptic encephalopathy disease. Biomed Res Int. (2019) 2019:4872101. doi: 10.1155/2019/4872101
61. Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science (New York, NY). (2000) 287:864–9. doi: 10.1126/science.287.5454.864
62. Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. (2008) 40:782–8. doi: 10.1038/ng.150
63. Stamberger H, Nikanorova M, Willemsen MH, Accorsi P, Angriman M, Baier H, et al. STXBP1 encephalopathy: a neurodevelopmental disorder including epilepsy. Neurology. (2016) 86:954–62. doi: 10.1212/WNL.0000000000002457
64. Lammertse H, van Berkel AA, Iacomino M, Toonen RF, Striano P, Gambardella A, et al. Homozygous STXBP1 variant causes encephalopathy and gain-of-function in synaptic transmission. Brain. (2020) 143:441–51. doi: 10.1093/brain/awz391
65. Lanoue V, Chai YJ, Brouillet JZ, Weckhuysen S, Palmer EE, Collins BM, et al. STXBP1 encephalopathy: connecting neurodevelopmental disorders with α-synucleinopathies? Neurology. (2019) 93:114–23. doi: 10.1212/WNL.0000000000007786
66. Kovacevic J, Maroteaux G, Schut D, Loos M, Dubey M, Pitsch J, et al. Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy. Brain. (2018) 141:1350–74. doi: 10.1093/brain/awy046
67. Di Rosa G, Dicanio D, Nicotera AG, Mondello P, Cannavò L, Gitto E. Efficacy of intravenous hydrocortisone treatment in refractory neonatal seizures: a report on three cases. Brain Sci. (2020) 10:885. doi: 10.3390/brainsci10110885
68. Vanoye-Carlo A, Gómez-Lira G. Differential expression of SV2A in hippocampal glutamatergic and GABAergic terminals during postnatal development. Brain Res. (2019) 1715:73–83. doi: 10.1016/j.brainres.2019.03.021
69. Buckley K, Kelly RB. Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J Cell Biol. (1985) 100:1284–94. doi: 10.1083/jcb.100.4.1284
70. Bajjalieh SM, Peterson K, Shinghal R, Scheller RH. SV2, a brain synaptic vesicle protein homologous to bacterial transporters. Science (New York, NY). (1992) 257:1271–3. doi: 10.1126/science.1519064
71. Ohno Y, Tokudome K. Therapeutic role of synaptic vesicle glycoprotein 2A (SV2A) in modulating epileptogenesis. CNS Neurol Disord Drug Targets. (2017) 16:463–71. doi: 10.2174/1871527316666170404115027
72. Yao J, Nowack A, Kensel-Hammes P, Gardner RG, Bajjalieh SM. Cotrafficking of SV2 and synaptotagmin at the synapse. J Neurosci. (2010) 30:5569–78. doi: 10.1523/JNEUROSCI.4781-09.2010
73. Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci USA. (2004) 101:9861–6. doi: 10.1073/pnas.0308208101
74. Harper CB, Small C, Davenport EC, Low DW, Smillie KJ, Martínez-Mármol R, et al. An epilepsy-associated SV2A mutation disrupts synaptotagmin-1 expression and activity-dependent trafficking. J Neurosci. (2020) 40:4586–95. doi: 10.1523/JNEUROSCI.0210-20.2020
75. Tokudome K, Okumura T, Terada R, Shimizu S, Kunisawa N, Mashimo T, et al. A missense mutation of the gene encoding Synaptic Vesicle Glycoprotein 2A (SV2A) confers seizure susceptibility by disrupting amygdalar synaptic GABA release. Front Pharmacol. (2016) 7:210. doi: 10.3389/fphar.2016.00210
76. Crowder KM, Gunther JM, Jones TA, Hale BD, Zhang HZ, Peterson MR, et al. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc Natl Acad Sci USA. (1999) 96:15268–73. doi: 10.1073/pnas.96.26.15268
77. Calame DG, Herman I, Riviello JJ. A de novo heterozygous rare variant in SV2A causes epilepsy and levetiracetam-induced drug-resistant status epilepticus. Epilepsy Behav Rep. (2021) 15:100425. doi: 10.1016/j.ebr.2020.100425
78. Wang D, Zhou Q, Ren L, Lin Y, Gao L, Du J, et al. Levetiracetam-induced a new seizure type in a girl with a novel SV2A gene mutation. Clin Neurol Neurosurg. (2019) 181:64–6. doi: 10.1016/j.clineuro.2019.03.020
79. Serajee FJ, Huq AM. Homozygous mutation in synaptic vesicle glycoprotein 2A gene results in intractable epilepsy, involuntary movements, microcephaly, and developmental and growth retardation. Pediatr Neurol. (2015) 52:642–6.e1. doi: 10.1016/j.pediatrneurol.2015.02.011
80. Valtorta F, Benfenati F, Zara F, Meldolesi J. PRRT2: from paroxysmal disorders to regulation of synaptic function. Trends Neurosci. (2016) 39:668–79. doi: 10.1016/j.tins.2016.08.005
81. Valente P, Castroflorio E, Rossi P, Fadda M, Sterlini B, Cervigni RI, et al. PRRT2 is a key component of the Ca(2+)-dependent neurotransmitter release machinery. Cell Rep. (2016) 15:117–31. doi: 10.1016/j.celrep.2016.03.005
82. Fruscione F, Valente P, Sterlini B, Romei A, Baldassari S, Fadda M, et al. PRRT2 controls neuronal excitability by negatively modulating Na+ channel 1.2/1.6 activity. Brain. (2018) 141:1000–16. doi: 10.1093/brain/awy051
83. Coleman J, Jouannot O, Ramakrishnan SK, Zanetti MN, Wang J, Salpietro V, et al. PRRT2 regulates synaptic fusion by directly modulating SNARE complex assembly. Cell Rep. (2018) 22:820–31. doi: 10.1016/j.celrep.2017.12.056
84. Döring JH, Saffari A, Bast T, Brockmann K, Ehrhardt L, Fazeli W, et al. The phenotypic spectrum of PRRT2-associated paroxysmal neurologic disorders in childhood. Biomedicines. (2020) 8:456. doi: 10.3390/biomedicines8110456
85. Yang L, You C, Qiu S, Yang X, Li Y, Liu F, et al. Novel and de novo point and large microdeletion mutation in PRRT2-related epilepsy. Brain Behav. (2020) 10:e01597. doi: 10.1002/brb3.1597
86. Erro R, Bhatia KP, Espay AJ, Striano P. The epileptic and nonepileptic spectrum of paroxysmal dyskinesias: channelopathies, synaptopathies, and transportopathies. Mov Disord. (2017) 32:310–8. doi: 10.1002/mds.26901
87. Ebrahimi-Fakhari D, Saffari A, Westenberger A, Klein C. The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain. (2015) 138(Pt 12):3476–95. doi: 10.1093/brain/awv317
88. Balagura G, Riva A, Marchese F, Iacomino M, Madia F, Giacomini T, et al. Clinical spectrum and genotype-phenotype correlations in PRRT2 Italian patients. Eur J Paediatr Neurol. (2020) 28:193–7. doi: 10.1016/j.ejpn.2020.06.005
89. Heron SE, Ong YS, Yendle SC, McMahon JM, Berkovic SF, Scheffer IE, et al. Mutations in PRRT2 are not a common cause of infantile epileptic encephalopathies. Epilepsia. (2013) 54:e86–9. doi: 10.1111/epi.12167
90. Pavone P, Corsello G, Cho SY, Pappalardo XG, Ruggieri M, Marino SD, et al. PRRT2 gene variant in a child with dysmorphic features, congenital microcephaly, and severe epileptic seizures: genotype-phenotype correlation? Ital J Pediatr. (2019) 45:159. doi: 10.1186/s13052-019-0755-2
91. Djémié T, Weckhuysen S, Holmgren P, Hardies K, Van Dyck T, Hendrickx R, et al. PRRT2 mutations: exploring the phenotypical boundaries. J Neurol Neurosurg Psychiatry. (2014) 85:462–5. doi: 10.1136/jnnp-2013-305122
92. Jafarpour S, Desai J. Infantile spasms associated with a pathogenic PRRT2 variant. Pediatr Neurol. (2021) 115:41. doi: 10.1016/j.pediatrneurol.2020.10.010
93. Guerrero-López R, Ortega-Moreno L, Giráldez BG, Alarcón-Morcillo C, Sánchez-Martín G, Nieto-Barrera M, et al. Atypical course in individuals from Spanish families with benign familial infantile seizures and mutations in the PRRT2 gene. Epilepsy Res. (2014) 108:1274–8. doi: 10.1016/j.eplepsyres.2014.06.011
94. Labate A, Tarantino P, Viri M, Mumoli L, Gagliardi M, Romeo A, et al. Homozygous c.649dupC mutation in PRRT2 worsens the BFIS/PKD phenotype with mental retardation, episodic ataxia, and absences. Epilepsia. (2012) 53:e196–9. doi: 10.1111/epi.12009
95. Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. (2011) 478:57–63. doi: 10.1038/nature10423
96. Nobile C, Striano P. PRRT2: a major cause of infantile epilepsy and other paroxysmal disorders of childhood. Prog Brain Res. (2014) 213:141–58. doi: 10.1016/B978-0-444-63326-2.00008-9
97. Zhao X, Wang Y, Cai A, Mei S, Liu N, Kong X. A novel NAPB splicing mutation identified by Trio-based exome sequencing is associated with early-onset epileptic encephalopathy. Eur J Med Genet. (2021) 64:104101. doi: 10.1016/j.ejmg.2020.104101
98. Burgalossi A, Jung S, Meyer G, Jockusch WJ, Jahn O, Taschenberger H, et al. SNARE protein recycling by αSNAP and βSNAP supports synaptic vesicle priming. Neuron. (2010) 68:473–87. doi: 10.1016/j.neuron.2010.09.019
99. Conroy J, Allen NM, Gorman KM, Shahwan A, Ennis S, Lynch SA, et al. NAPB - a novel SNARE-associated protein for early-onset epileptic encephalopathy. Clin Genet. (2016) 89:E1–3. doi: 10.1111/cge.12648
100. Kolnikova M, Skopkova M, Ilencikova D, Foltan T, Payerova J, Danis D, et al. DNM1 encephalopathy - atypical phenotype with hypomyelination due to a novel de novo variant in the DNM1 gene. Seizure. (2018) 56:31–3. doi: 10.1016/j.seizure.2018.01.020
101. Cao H, Garcia F, McNiven MA. Differential distribution of dynamin isoforms in mammalian cells. Mol Biol Cell. (1998) 9:2595–609. doi: 10.1091/mbc.9.9.2595
102. Ferguson SM, Brasnjo G, Hayashi M, Wölfel M, Collesi C, Giovedi S, et al. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science. (2007) 316:570–4. doi: 10.1126/science.1140621
103. Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, et al. (2013). De novo mutations in epileptic encephalopathies. Nature, 501(7466), 217–221. doi: 10.1038/nature12439
104. Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol. (2012) 13:75–88. doi: 10.1038/nrm3266
105. Mastrangelo M. Lennox-gastaut syndrome: a state of the art review. Neuropediatrics. (2017) 48:143–51. doi: 10.1055/s-0037-1601324
106. von Spiczak S, Helbig KL, Shinde DN, Huether R, Pendziwiat M, Lourenço C, et al. DNM1 encephalopathy: a new disease of vesicle fission. Neurology. (2017) 89:385–94. doi: 10.1212/WNL.0000000000004152
107. Li H, Fang F, Xu M, Liu Z, Zhou J, Wang X, et al. Clinical assessments and EEG analyses of encephalopathies associated with dynamin-1 mutation. Front Pharmacol. (2019) 10:1454. doi: 10.3389/fphar.2019.01454
108. Nakashima M, Kouga T, Lourenço CM, Shiina M, Goto T, Tsurusaki Y, et al. De novo DNM1 mutations in two cases of epileptic encephalopathy. Epilepsia. (2016) 57:e18–23. doi: 10.1111/epi.13257
109. EuroEPINOMICS-RES Consortium Epilepsy Phenome/Genome Project and Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet. (2014) 95:360–70. doi: 10.1016/j.ajhg.2014.08.013
110. Allen NM, Conroy J, Shahwan A, Lynch B, Correa RG, Pena SD, et al. Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia. (2016) 57:e12–7. doi: 10.1111/epi.13250
111. Epi4K Consortium. De novo Mutations in SLC1A2 and CACNA1A Are important causes of epileptic encephalopathies. Am J Hum Genet. (2016) 99:287–98. doi: 10.1016/j.ajhg.2016.06.003
112. Lazzara A, Asghar S, Zacharia T, Byler D. DNM1 Mutation in a child associated with progressive bilateral mesial temporal sclerosis. Clin Case Rep. (2018) 6:2037–9. doi: 10.1002/ccr3.1793
113. Fung CW, Kwong AK, Wong VC. Gene panel analysis for nonsyndromic cryptogenic neonatal/infantile epileptic encephalopathy. Epilepsia Open. (2017) 2:236–43. doi: 10.1002/epi4.12055
114. Deng XL, Yin F, Zhang CL, Ma YP, He F, Wu LW, et al. Dynamin-1-related infantile spasms: a case report and review of literature. Zhonghua Er Ke Za Zhi. (2016) 54:856–9. doi: 10.3760/cma.j.issn.0578-1310.2016.11.014
115. Brereton E, Fassi E, Araujo GC, Dodd J, Telegrafi A, Pathak SJ, et al. Mutations in the PH Domain of DNM1 are associated with a nonepileptic phenotype characterized by developmental delay and neurobehavioral abnormalities. Mol Genet Genomic Med. (2018) 6:294–300. doi: 10.1002/mgg3.362
116. Stockler S, Corvera S, Lambright D, Fogarty K, Nosova E, Leonard D, et al. Single point mutation in Rabenosyn-5 in a female with intractable seizures and evidence of defective endocytotic trafficking. Orphanet J Rare Dis. (2014) 9:141. doi: 10.1186/s13023-014-0141-5
117. Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. (2004) 27:509–47. doi: 10.1146/annurev.neuro.26.041002.131412
118. Nielsen E, Christoforidis S, Uttenweiler-Joseph S, Miaczynska M, Dewitte F, Wilm M, et al. Rabenosyn-5, a novel Rab5 effector, is complexed with hVPS45 and recruited to endosomes through a FYVE finger domain. J Cell Biol. (2000) 151:601–12. doi: 10.1083/jcb.151.3.601
119. Lüthy K, Mei D, Fischer B, De Fusco M, Swerts J, Paesmans J, et al. TBC1D24-TLDc-related epilepsy exercise-induced dystonia: rescue by antioxidants in a disease model. Brain. (2019) 142:2319–35. doi: 10.1093/brain/awz175
120. Falace A, Buhler E, Fadda M, Watrin F, Lippiello P, Pallesi-Pocachard E, et al. TBC1D24 regulates neuronal migration and maturation through modulation of the ARF6-dependent pathway. Proc Natl Acad Sci USA. (2014) 111:2337–42. doi: 10.1073/pnas.1316294111
121. Kim Nguyen NT, Ohbayashi N, Kanaho Y, Funakoshi Y. TBC1D24 regulates recycling of clathrin-independent cargo proteins mediated by tubular recycling endosomes. Biochem Biophys Res Commun. (2020) 528:220–6. doi: 10.1016/j.bbrc.2020.05.007
122. Fassio A, Fadda M, Benfenati F. Molecular machines determining the fate of endocytosed synaptic vesicles in nerve terminals. Front Synaptic Neurosci. (2016) 8:10. doi: 10.3389/fnsyn.2016.00010
123. Zhang J, Chen J, Zeng Q, Zhang L, Tian X, Yang X, et al. Infantile epilepsy with multifocal myoclonus caused by TBC1D24 mutations. Seizure. (2019) 69:228–34. doi: 10.1016/j.seizure.2019.05.010
124. Appavu B, Guido-Estrada N, Lindstrom K, Grebe T, Kerrigan JF, Troester M. Electroclinical phenotypes and outcomes in TBC1D24-related epilepsy. Epileptic Disord. (2016) 18:324–8. doi: 10.1684/epd.2016.0849
125. Balestrini S, Milh M, Castiglioni C, Lüthy K, Finelli MJ, Verstreken P, et al. TBC1D24 genotype-phenotype correlation: epilepsies and other neurologic features. Neurology. (2016) 87:77–85. doi: 10.1212/WNL.0000000000002807
126. Fang ZX, Xie LL, Yan LS, Lin H, Pan YN, Liu BK, et al. Clinical and genetic characteristics of epilepsy of infancy with migrating focal seizures in Chinese children. Epilepsy Res. (2021) 174:106669. doi: 10.1016/j.eplepsyres.2021.106669
127. Banuelos E, Ramsey K, Belnap N, Krishnan M, Balak C, Szelinger S, et al. Case Report: Novel mutations in TBC1D24 are associated with autosomal dominant tonic-clonic and myoclonic epilepsy and recessive Parkinsonism, psychosis, and intellectual disability. F1000Res. (2017) 6:553. doi: 10.12688/f1000research.10588.1
128. Nakashima M, Negishi Y, Hori I, Hattori A, Saitoh S, Saitsu H. A case of early-onset epileptic encephalopathy with a homozygous TBC1D24 variant caused by uniparental isodisomy. Am J Med Genet Part A. (2019) 179:645–9. doi: 10.1002/ajmg.a.61056
129. Lozano R, Herman K, Rothfuss M, Rieger H, Bayrak-Toydemir P, Aprile D, et al. Clinical intrafamilial variability in lethal familial neonatal seizure disorder caused by TBC1D24 mutations. Am J Med Genet Part A. (2016) 170:3207–14. doi: 10.1002/ajmg.a.37933
130. Salemi M, Cali' F, Giambirtone M, Elia M, Romano C. TBC1D24 gene mRNA expression in a boy with early infantile epileptic encephalopathy-16. Acta Neurol Belgica. (2020) 120:381–3. doi: 10.1007/s13760-017-0818-3
Keywords: synaptopathy, developmental and epileptic encephalopathy (DEE), pre-synaptic mechanisms, drug resistant epilepsy, intellectual disability (ID), SNAREopathies
Citation: Spoto G, Valentini G, Saia MC, Butera A, Amore G, Salpietro V, Nicotera AG and Di Rosa G (2022) Synaptopathies in Developmental and Epileptic Encephalopathies: A Focus on Pre-synaptic Dysfunction. Front. Neurol. 13:826211. doi: 10.3389/fneur.2022.826211
Received: 30 November 2021; Accepted: 24 January 2022;
Published: 08 March 2022.
Edited by:
Kette D. Valente, Universidade de São Paulo, BrazilReviewed by:
Fabrizia Claudia Guarnieri, National Research Council (CNR), ItalyCopyright © 2022 Spoto, Valentini, Saia, Butera, Amore, Salpietro, Nicotera and Di Rosa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincenzo Salpietro, di5zYWxwaWV0cm9AdWNsLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.