 Luca Marsili
Luca Marsili Kevin R. Duque
Kevin R. Duque Rachel L. Bode1
Rachel L. Bode1 Marcelo A. Kauffman
Marcelo A. Kauffman Alberto J. Espay
Alberto J. Espay- 1James J. and Joan A. Gardner Center for Parkinson's Disease and Movement Disorders, Department of Neurology, University of Cincinnati, Cincinnati, OH, United States
- 2Consultorio y Laboratorio de Neurogenética, Centro Universitario de Neurología José María Ramos Mejía, Buenos Aires, Argentina
Long-read sequencing (LRS) technologies have been recently introduced to overcome intrinsic limitations of widely-used next-generation sequencing (NGS) technologies, namely the sequencing limited to short-read fragments (150–300 base pairs). Since its introduction, LRS has permitted many successes in unraveling hidden mutational mechanisms. One area in clinical neurology in need of rethinking as it applies to genetic mechanisms is essential tremor (ET). This disorder, among the most common in neurology, is a syndrome often exhibiting an autosomal dominant pattern of inheritance whose large phenotypic spectrum suggest a multitude of genetic etiologies. Exome sequencing has revealed the genetic etiology only in rare ET families (FUS, SORT1, SCN4A, NOS3, KCNS2, HAPLN4/BRAL2, and USP46). We hypothesize that a reason for this shortcoming may be non-classical genetic mechanism(s) underpinning ET, among them trinucleotide, tetranucleotide, or pentanucleotide repeat disorders. In support of this hypothesis, trinucleotide (e.g., GGC repeats in NOTCH2NLC) and pentanucleotide repeat disorders (e.g., ATTTC repeats in STARD7) have been revealed as pathogenic in patients with a past history of what has come to be referred to as “ET plus,” bilateral hand tremor associated with epilepsy and/or leukoencephalopathy. A systematic review of LRS in neurodegenerative disorders showed that 10 of the 22 (45%) genetic etiologies ascertained by LRS include tremor in their phenotypic spectrum, suggesting that future clinical applications of LRS for tremor disorders may uncover genetic subtypes of familial ET that have eluded NGS, particularly those with associated leukoencephalopathy or family history of epilepsy. LRS provides a pathway for potentially uncovering novel genes and genetic mechanisms, helping narrow the large proportion of “idiopathic” ET.
Introduction
Genetic factors contribute to various neurodegenerative diseases, as historically demonstrated by the study of selected families harboring specific pathogenic variants (1, 2). The development of next-generation sequencing (NGS) technologies during the last 15 years has accelerated the discovery of novel familial movement disorders and disease-causing variants in unusual sporadic phenotypes (2). However, despite the increase in genes and variants discovered in this group of disorders, routine NGS techniques have intrinsic limitations, mainly due to their ability to only sequence short-read length fragments (150–300 base pairs— bp) (3, 4) (Table 1). These limitations were among the motivations for the development of a new sequencing technology, the long-read sequencing (LRS).

Table 1. Main limitations of short-read next-generation sequencing (NGS) which may be overcome by long-read sequencing.
There are two main long-read platforms that have in common the ability to sequence in real-time single molecules of DNA, PacBio family (PacBio CCS) and Oxford Nanopore Technologies (ONT). Unlike the short-read NGS (mainly provided by Illumina platforms), LRS allows longer reads (over 10 kilo-base pair –Kbp in one single read) with high accuracy, thereby reducing the number of individual reads needed for complete coverage of individual genomic regions (4, 7, 8). Of note, long reads are single DNA molecules, and ONT reads can be ultra-long, even Mb-sized (9). LRS platforms offer the possibility of evaluating long reads of several kilobases, including highly repetitive regions, and significantly reduce GC errors. These advantages and new variant calling algorithms [namely, the algorithms used for the identification of variants from sequence data (10)] have recently allowed the detection and validation of variants which only a few years ago had eluded detection by short-read sequencing (8).
LRS technologies are currently used in several fields of neurology and have been the source of newly discovered pathogenic mutational mechanisms (4). Here we systematically review the main accomplishments of LRS technologies in the field of neurodegenerative disorders and focus on their potential utility on the syndrome of essential tremor (ET), where we envision an opportunity for their clinical application. In fact, the genetic etiology of some ET subtypes might be uncovered with LRS, particularly when other “pluses,” or atypical features, may be present, such as family history of epilepsy or associated white matter abnormalities on imaging. We will also discuss the main challenges of LRS technologies, and provide an overview of future applications of these exciting new methodologies in research. To facilitate understanding, we have created a glossary of key terms.
Long-Read Sequencing: From Limitations to Opportunities
Limitations of Next-Generation Sequencing
It has become clear that the human genome variability is caused by structural variations more commonly than previously thought (11). Structural variants (SVs) are currently defined as insertions, deletions, or inversions larger than 1 Kbp (This cutoff is arbitrary, and others may refer to mutations >50 bp as SVs) (12). Copy number variants (CNVs) refer to the number of copies of a particular gene present in the genome of an individual (13). The expanded use of sequencing techniques in clinical practice has increased, at least in part, the spectrum of both SVs and CNVs routinely identified. However, the information amenable to be obtained by classical short reads NGS platforms is still not sensitive enough for SVs and CNVs detection (11). Moreover, a clear limitation of short-read sequencing appears when regions of the genome harboring SVs, GC-rich regions, and repetitive sequences or sequences with many homologous elements (widespread repeats and segmental duplications) (3, 7) need to be mapped to reference assemblies (Table 1). Repetitive sequences of DNA are highly-prevalent across different species ranging from bacteria to humans, and they account for about 50% of the human genome (3).
Repeats Constitute a Huge Challenge for Sequence Alignment and Assembly Preparations
NGS techniques, due to their short read lengths, have the disadvantage of making these problems harder to solve (3). Hence, in the last 20 years, several technological developments have allowed increasing both the speed and the length of sequencing reads with the goal of reducing cycle times, accelerating sequence assembly, and enabling the accurate sequencing of repeat-rich areas of the genome (14–16). These discoveries have allowed a fast and relatively low-cost genome sequencing (16), facilitating the development of LRS, where the sequencing process is in real-time and polymerase chain reaction (PCR) amplification is no longer necessary (Figure 1). With the reading of longer fragments, LRS raises the accuracy in read mapping (3, 4) enhacing the odds for sequencing-generated discoveries where short read sequencing has previously failed (4, 7, 8, 17, 18).
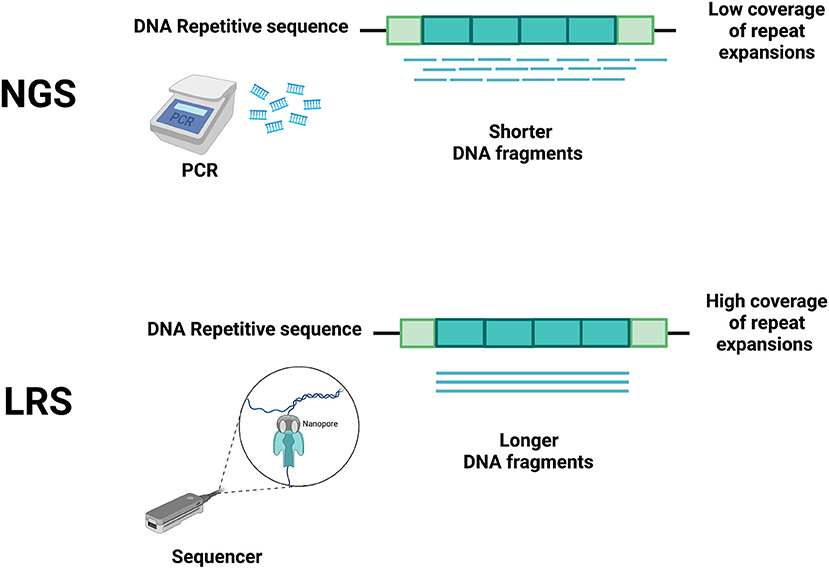
Figure 1. Main differences between short-read next-generation sequencing (NGS) and long-read sequencing (LRS) technologies. Short-read NGS (upper panel) uses PCR to obtain short DNA fragments that do not accurately cover the whole repeated DNA sequence, thus allowing many interpretation errors (e.g., including edge of repeat and flanking regions, spanning boundaries of repeats). The differing technology of LRS (lower panel) does not require PCR, allowing a more accurate coverage of the repeated DNA sequence. Created with BioRender.com.
Long-Read Sequencing in Neurodegenerative Disorders
LRS has been used to successfully sequence patients with diseases associated with repeat expansions. It permitted the identification of pathogenic trinucleotide and pentanucleotide repeat expansion disorders, as the examples below illustrate.
The Example of Spinocerebellar Ataxia Type 10
Spinocerebellar ataxias (SCAs) are a heterogeneous group of conditions associated with ataxia (19). Some SCAs are caused by exonic CAG trinucleotide repeat expansions in selected coding regions, translating into long polyglutamine chains (20). However, non-coding repeat expansions and other conventional mutations may also cause other types of SCAs (4). As an example, SCA10 is caused by a noncoding ATTCT pentanucleotide repeat expansion located in intron 9 of the ATXN10 gene (21). SCA10 shows a wide range of phenotypic variability, which may include epileptic seizures (21).
From a technical standpoint, previous NGS studies of repeat expansions were based on amplicon sequencing. For disorders caused by large expansions such as SCA10, this approach is not feasible (22). Therefore, for an optimal analysis of these large intronic repetitions, it has been proposed to sequence DNA without using any preceding PCR amplification (23). For this purpose, a specific CRISPR/Cas9-based amplification-free target enrichment method (also known as No-Amp targeted sequencing) was created (24). Through the combination of LRS technologies matched with the no-Amp approach, it has become possible to identify interruption motifs, potentially acting as phenotypic modifiers, within the tandem repeat expansion locus in ATXN10 in patients presenting with variable phenotypes not always in the spectrum of SCA10, including a Parkinson's disease (PD) phenotype (25, 26). The hypothesis is that the interruptions present in the SCA10 repeat expansion may serve as a “phenotypic modifier” of the genotype-phenotype correlation between ATXN10 and parkinsonism (4, 26).
Targeted-LRS (T-LRS) can be obtained not only with the No-Amp sequencing, but also using PCR enrichment. The T-LRS allows to use LRS for selected, targeted genomic regions with computational method, thus permitting to select and sequence native DNA (18). This approach is called adaptive sampling T-LRS, and permits to accept or reject the DNA fragments for sequencing, based on pre-set target sequences with the advantage to be modified in real time (27, 28) (further in Discussion).
The Example of Huntington Disease
Huntington disease (HD) is caused by a CAG repeat expansion in the HTT gene located on chromosome 4 (29). The number of repeats inversely correlate with the disease severity and the age at onset (30). Hence, the precise calculation of the number of repeats has crucial diagnostic and prognostic values. To date, due to the intrinsic limitation of the PCR stutter and high GC content, NGS techniques are limited in the diagnostic accuracy of HD. For this purpose, the No-Amp targeted sequencing has been used to overcome the difficulties due to PCR stutter and to the high GC content, thus permitting a detailed sequence information and the assessment of somatic variability of repeated elements without the confounding errors related to the PCR stutter (7). In addition, LRS demonstrated the feasibility of rapidly genotyping and haplotype phasing to differentiate the genetic variability inherited either from paternal or maternal origins without familial genotype/haplotype data (31). In HD, this has led to determine the allele bearing both the pathogenic CAG expansion and SNPs 165 kb downstream, essential for personalized therapies.
The Example of Amyotrophic Lateral Sclerosis/Frontotemporal Dementia
A similar approach has been in amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD) associated with C9ORF72 gene variants (23, 32). ALS and FTD are considered a spectrum of disorders that may share some common genetic bases (27). In particular, the documentation of an expanded hexanucleotide repeat in the noncoding region of C9ORF72 gene on chromosome 9, has linked ALS with FTD genetic variants (33). Again, due to its high length and GC content, the hexanucleotide repeat expansion was difficult to sequence (32, 34, 35), but LRS technologies made it possible (4, 32).
The Example of Genetic Parkinsonism
LRS technologies have been useful in investigating two genetically-determined parkinsonisms, namely X-linked dystonia-parkinsonism (XDP) and GBA-related parkinsonism. XDP is a monogenic movement disorder presenting with both dystonia and parkinsonian features (36). The underlying genetic mechanisms of XDP have been thoroughly investigated using LRS techniques which, initially, narrowed the causal mutation to the TAF1 gene locus, thus hypothesizing an alternative splicing defect mechanism (Using PacBio, see also Section 4) (37). Later, thanks to the Nanopore LRS technologies (See also Section 4), it has been demonstrated that alternative splicing of TAF1 is not the molecular basis for this disease, thus marking a paradigm shift in the understanding of this condition and suggesting that the decrease in TAF1-messenger RNA is implicated in XDP pathogenesis (38). In GBA-related parkinsonism, heterozygous mutations in the GBA gene are considered strong risk factors for this condition. However the analysis of GBA is complicated by the nearby pseudogene which shares 96% exonic sequence homology to the GBA coding region (39). As noted above, LRS technologies have been useful in detecting disease-causing intronic and exonic variants with good haplotype phasing information (39, 40).
Systematic Review of the Literature on Long-Read Sequencing in Neurodegenerative Disorders
To better understand the impact of LRS technologies in neurodegenerative research, we performed a systematic review of the LRS literature focusing on new mutations discovered by LRS. We also searched for studies about LRS currently active or soon-to-be active in neurodegenerative diseases. We found 59 articles in PubMed and Cochrane, and only one record in ClinicalTrials.gov of a study not yet recruiting, aimed to analyze through LRS already existing biological samples from patients with specific mutations for neurodegenerative diseases. The main results of our systematic search (Table 2, Supplementary File) demonstrates the increasing impact LRS is making in the discovery of new pathogenic variants in previously unidentified genes and novel mutational mechanisms, especially those comprising repetitive and large structural variation. Importantly, 10 of the 22 (45%) genetic etiologies ascertained by LRS included tremor in their phenotypic spectrum.
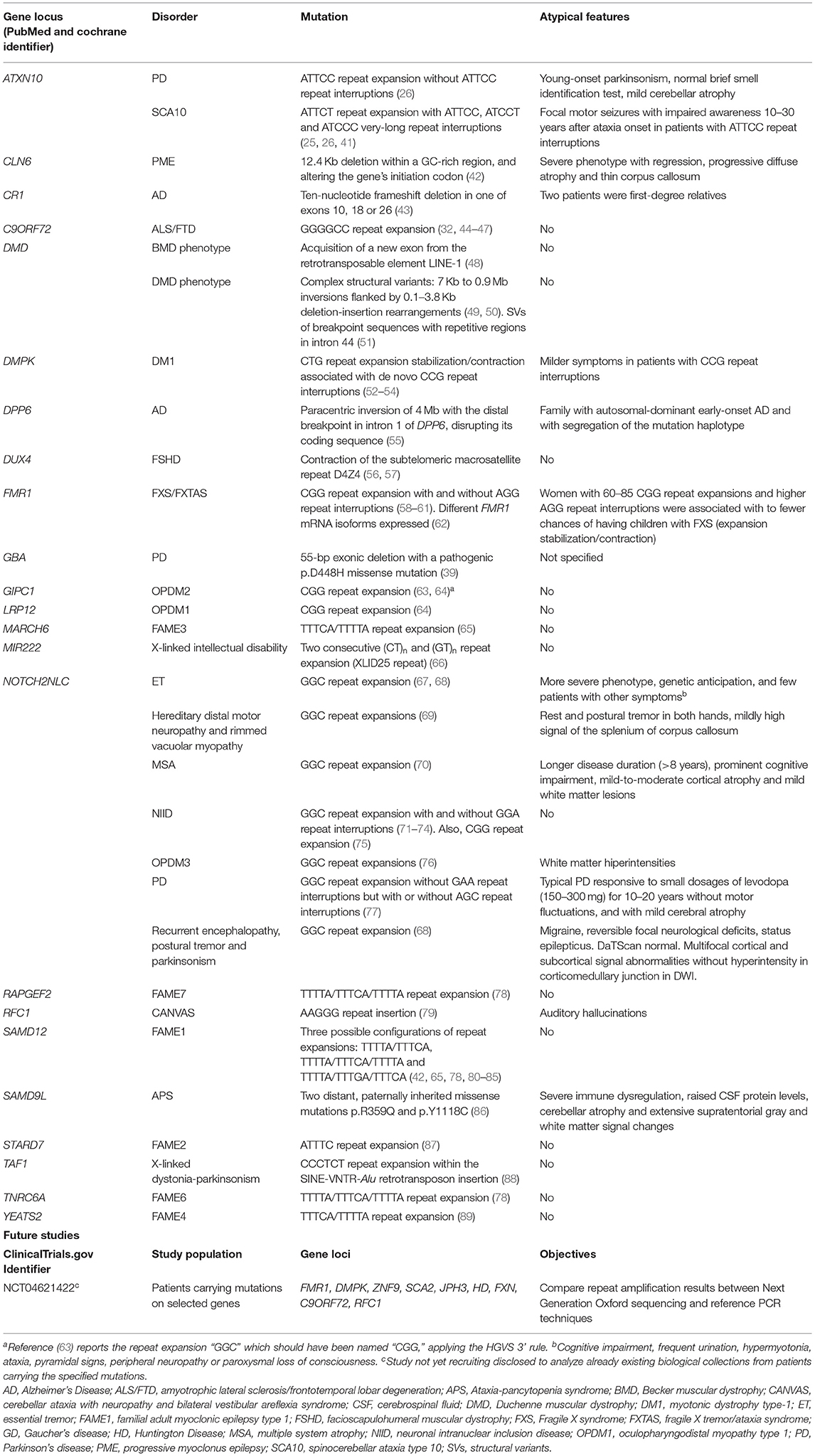
Table 2. Mutations previously hidden from standard genetic techniques but resolved after long-read sequencinga.
Long-Read Sequencing in Tremor-Related Disorders
Long-Read Sequencing in Fragile X-Associated Tremor/Ataxia Syndrome
Some of the positive results of LRS technologies in the field of neurology have been accomplished on tremor-related disorders. Historically, the first application of LRS technologies in neurodegenerative disorders was on fragile X-associated tremor/ataxia syndrome (FXTAS) (58). FXTAS and fragile X syndrome (FXS) are both associated with a CGG trinucleotide repeat expansion on the FMR1 gene located on the X chromosome (90). Abnormal repeats in FMR1 in the so-called premutation range of 55–200 repeats express as FXTAS whereas more than 200 repeats manifests as FXS (4), markedly reducing the expression of FMR1. Despite the shared genetic background, these two conditions have discrepant phenotypes. FXTAS patients develop prominent tremor, ataxia, cognitive impairment, and peripheral neuropathy (with many patients carrying a diagnosis of ET or “ET plus”); instead, FXS patients mostly develop cognitive impairment (91). However, the premutation range of trinucleotide repeats is unstable between generations. With subsequent expansions, it may reach a full mutation, particularly during maternal transmission (92). Interestingly, LRS technologies have shown that the presence of specific interruption sequences (AGG) into the trinucleotide repeats may reduce this instability (92). Therefore, LRS technologies accurately assess the number of repetitions and the presence of interruption sequences, thus allowing a more precise estimation of expansion risk and opening the possibility of preimplantation genetic diagnosis (93, 94). This level of information only obtainable by LRS greatly inform early diagnosis and family counseling in these two X-linked conditions (4).
Long-Read Sequencing in Cortical Tremor—Familial Adult Myoclonic Epilepsies
Another interesting application of LRS technologies in the field of tremor-related disorders is on the familial adult myoclonic epilepsies (FAMEs) (75). FAME, also known as familial cortical myoclonic tremor with epilepsy (FCMTE) in Asia, belongs to the spectrum of the so-called benign adult familial myoclonic epilepsy (BAFME) (4, 75). These conditions are characterized by cortical tremor (phenomenologically tremor, electrophysiologically rhythmic myoclonus) and infrequent seizures with benign progression that segregate under an autosomal dominant familiar inheritance pattern (95, 96). As a result, many of these patients were considered within the spectrum of ET before their genetic etiology was unraveled (97). Although Mori et al. (98) mapped the disease locus to chromosome 8, through linkage analysis in 2011, no causative mutations were described until 2018 (75). Ishiura et al. (75) identified a pentanucleotide repeat expansion and some extra repeat sequences in a specific intron of the SAMD12 gene in a pool of Japanese families with BAFME1, using LRS technologies. Later, this same pattern was confirmed in a large family of Chinese patients using LRS technologies, thus achieving two important results: 1) The confirmation of previously published results on Japanese pedigrees; and 2) The confirmation that LRS is effective for diagnosing genetically determined diseases, particularly when NGS-based technologies have failed to identify a causative mutation (80). Interestingly, other pentanucleotide repeats were also found in genes different from the SAMD12, such as TNRC6 (BAFME6) and RAPGEF2 (BAFME7) (75). Subsequently, intronic repeat expansions have been found in the STARD7 (FAME2) and MARCH6 (FAME3) genes (although using short-read NGS and not LRS technologies) (65, 87). Finally, intronic expansions in YEATS2 were found as causal in BAFME4 (4, 89) (Table 2). In conclusion, similar tremor phenotypes have been caused by non-coding repeats, suggesting that in this class of disorders (BAFME/FAME/FCMTE) the presence of large repeats may underpin its pathogenesis to a greater extent than a single genomic locus per se.
Long-Read Sequencing in Essential Tremor
In the field of ET, Sun and colleagues (67) recently demonstrated using a cohort of 197 participants that a GGC repeat expansion in the NOTCH2NLC gene can manifest as ET (5.8% of cases studied). They also found that the abnormal repeat expansion correlated with the severity of the phenotype and with anticipation in subsequent generations (67). Of interest, NOTCH2NLC has also been previously identified as the same gene causing neuronal intranuclear inclusion disease (NIID) (75), a condition characterized by peripheral neuropathy and cognitive impairment, phenotypically unrelated to ET. Furthermore, GGC repeat expansions (>79 repeats) in the NOTCH2NLC gene have been described in patients classified as “benign PD” (e.g., treated with low levodopa doses), and no other clinical or imaging features of NIID, after several years of follow-up (77). Taken together, these results suggest that there are subsets of patients affected with tremor within the “ET,” “PD,” and even “dystonic tremor” (DT) spectrum, for which LRS may identify causative loci and pathogenic mechanisms. Of note, NOTCH2NLC is highly homologous to other four paralogs of the human genome, making it extremely difficult to obtain its proper sequence when short-read NGS technologies are used.
What is missing in the field of familial essential tremor? To date, the definition and classification of tremor-related disorders is based on their phenotypic manifestations (99). ET is a syndrome, often inherited in an autosomal dominant pattern (100, 101). Due to discrepancies between studies, the estimated rate of inheritance is highly variable, ranging from 20% to 90% (101). Rare genes have been found in selected ET families by exome sequencing (e.g., FUS, SORT1, SCN4A, NOS3, KCNS2, HAPLN4/BRAL2, USP46) (101). If classical genetic mechanisms were at play, not only would have other families with these genetic etiologies would have been reported but other genes might have been identified. Based on the recent successes of LRS technologies, and in line with precision medicine principles, a new molecular approach is required (102–105). Conditions traditionally grouped under such clinical categories as “ET,” “ET plus tremor,” “orthostatic tremor” (OT), “pseudo-OT,” and “indeterminate tremor” could be phenotypic expressions of several genetic etiologies (106). This hypothesis is supported by the abnormalities recently discovered in genes associated with “ET/ET plus” conditions, such as trinucleotide (e.g., NOTCH2NLC, FMR1) and pentanucleotide (e.g., ATTTC repeats) repeat disorders (100, 102, 107) (Table 2). For this purpose, based also on the results of previous studies conducted in patients with FXTAS, FAME, and ET, we believe it is recommended to apply LRS to cohorts of patients with the phenotype of “ET”, potentially including those with familial bilateral hand tremor and any associated conditions, particularly epilepsy, or even minor white matter abnormalities changes. This approach is expected to reveal the genetic underpinnings of a certain subset of individuals currently classified within the broad “ET” spectrum. In fact, familial ET-like disorders have classically been described as segregating in an autosomal dominant fashion, yet only repeat expansion mutations (e.g., NOTCH2NLC, FMR1, ATTTC repeats) have been ascertained as pathogenic. We hypothesize that a reason for this glaring shortcoming may be the non-classical nature of the genetic mechanism(s) underpinning ET, among them trinucleotide, tetranucleotide, or pentanucleotide repeat disorders.
Technical Challenges
Studying ET and other tremor-related disorders with a LRS-based approach will come with technical and methodological challenges. A first consideration entails the methodological strategy to adopt for this purpose. A possible solution could be to perform a genetic linkage analysis using microsatellite markers (80) or single nucleotide polymorphisms (SNPs) (108). Afterward, NGS-based technologies, namely whole exome sequencing (WES) and whole genome sequencing (WGS) can be used to identify the disease locus of interest (80). Finally, even if a low coverage (~10× -15× ) might suffice for studies that combine both technologies, a higher coverage is desirable with both PacBio CCS and ONT platforms to identify causative mutations (80). After that, repeat-primed PCR can be then applied for the possible validation and confirmation of the identified repeat expansions and rapid screening of other subjects at risk (80). We know that the LRS technologies are accurate in providing coverage for the most difficult regions of the genome (Table 1). However, there are sequencing challenges to be overcome by LRS technologies, including the analysis of telomeric regions, due to their high variability and number of masked satellites, are poorly covered by all long- and short-read technologies (109). Despite a great improvement in the last years, ONT platforms seem still behind other platforms in terms of accuracy of sequencing and coverage (109) although this is rapidly changing because of continuous updates in chemistry and bioinformatic analysis of this platform (9) (Table 3). PacBio CCS platforms are also more accurate for searching specific interruption sequences (e.g., AGG) as demonstrated in FXTAS (92). In particular, the circular consensus sequencing (CCS) protocol allows long, high-fidelity reads with high accuracy (113). Hence, PacBio CCS platforms allow the utilization of smaller fragment sizes, with a global quality higher than Illumina (109). To complicate things further, improved analysis of Illumina short read data is now possible thanks to new algorithms like the ExpansionHunter and GangSTR (68, 114, 115). To the best of our knowledge, a systematic comparison of these short-read analyses to LRS is not available. However, the big advantage of ONT platforms is the lower and more competitive cost for their service, when compared to PacBio CCS platforms. Also, it is estimated that within a year, ONT platforms will significantly improve their accuracy and quality. This aspect should be considered when envisioning long-term research efforts in which data analysis will be undertaken years after biospecimen collection (when improvements in both technologies will be available). Both ONT and PacBio CCS are more suitable for DNA extracted from blood instead of saliva or other tissues, as compared with short-read NGS (we have personally observed that compared to blood-isolated DNA, saliva-isolated DNA shows lower yields and lower molecular weight, resulting in much smaller DNA fragment sizes), another aspect to consider when planning a study. The final challenge is related to the fact that both LRS technologies are less accurate when the source material come from shorter DNA samples previously stored for other purposes, because of the need of a dedicated extraction of the DNA in bigger fragment sizes.
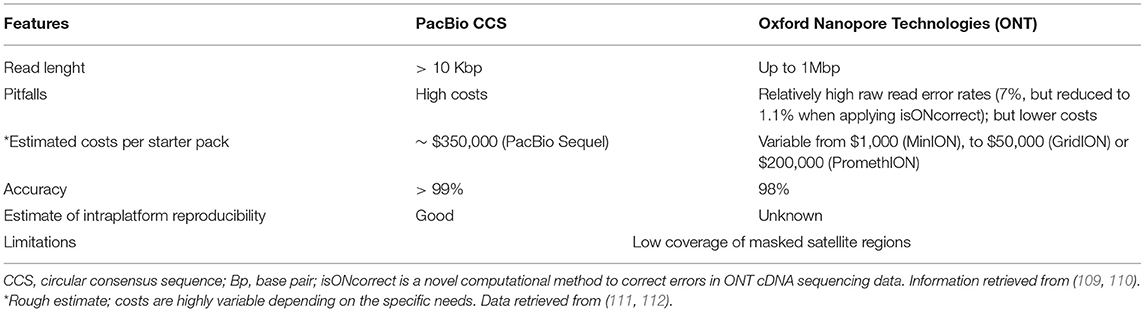
Table 3. Main features of the two long-read sequencing platforms: PacBio CCS and ONT.
Discussion
In the present review we have summarized the novel features of LRS genetic technologies, compared to short-read NGS as applied the field of ET and “ET plus.” The last decade has been a prolific era for the discovery of several genes (and related nucleotide repeat expansions) associated with tremor-related disorders and with other ostensibly unrelated phenotypes. Our systematic review confirmed the growth in new genes and genetic mechanisms discovered by LRS technologies, suggesting an underappreciated potential. Also, LRS technologies have revealed new insights about the relationship between coding and non-coding regions of our genome, thus improving the discovery of novel disease-causing variants. The discovery of these new variants must go in parallel with the development of the computational frameworks, thus bridging the gap between frontline research and practical efforts. In fact, the output of LRS-based studies clearly show that analysis of non-coding region is increasingly important in the regular workflows of clinicians determining the best approach for selected patients with suspected neurogenetic disorders (2).
The systematic application of LRS technologies in the field of tremor-related disorders will allow to determine the extent to which selected phenotypes are associated with repeat expansions. There is a high probability that the systematic application of LRS and WGS technologies will uncover novel repeat expansion disorders as potentially etiologic of “ET plus” and other tremor-related disorders. In the near future, LRS will be the first tier test for many tremor-related disorders and a serious alternative to short-read NGS (18).
The potential of LRS technologies may get us closer to elucidating the underlying genetic mechanisms of molecular subsets of ET on the road to precision medicine (102). While this approach can only be phenotype-driven at the outset, we envision a data driven, unbiased analytic approach as of higher hierarchical order in the future (116). This unique approach will allow a reverse biology-to-phenotype direction of research development in which clinical and imaging data are subordinate to genetic and biological signals of interest (116). A hypothesis free, causally- and data driven-based analyses, with an inclusive recruitment of patients with different phenotypes of neurodegenerative disorders, formed the foundations of the ongoing Cincinnati Cohort Biomarker Program (CCBP) study (116).
Promising results have come from a recent study conducted on over 3,000 inhabitants in Iceland, showing that LRS may improve the classification of SVs at the population level, when using a genome-wide non-targeted methodology (17). LRS can be useful for further large-scale efforts aimed at the exploration of different SVs unreachable with the NGS short reads sequencing approach (17). Another example was the findings of a common repeat expansion locus in ATXN10, associated with both ataxia (SCA10) and parkinsonism, two unrelated phenotypes (67, 77), suggesting that quite diverse phenotypes may share common genotypes. Another example of this successful strategy is that the GGC repeat expansion in the NOTCH2NLC gene may be associated with ET, “benign” idiopathic PD, and NIID, also three different phenotypes (67). Interestingly, genome-wide association studies have demonstrated an association between variants in the LINGO1 gene and familial ET (117). Recently, a CNV (tandem duplication) affecting the LINGO1 gene was found in a family with a predominantly dystonic tremor, using genome-wide SNP microarray analysis (118). Hence, LINGO1 gene may be involved in both ET and dystonic tremor. However, the LINGO1 risk variants are located in intronic regions, and the sequencing of LINGO1 exons in ET patients have failed to identify any pathogenic variants (118).These studies did not include LRS; if used, LRS could have helped in solving the problem and, perhaps, in identifying the repeats in the intronic regions. In sum, we are confident that a LRS-based approach will allow the discovery of the biological and genetic underpinnings of several neurodegenerative conditions, at least in the field of tremor-related disorders.
From a clinical standpoint, LRS is also useful to assign pathogenicity to a given detected variant especially when haplotypes can be assigned from long reads and not from short reads. We are aware that the genetic and phenotypic heterogeneity of neurogenetic diseases may force patients into a sort of “diagnostic odyssey” (104). A possible meaningful approach could be using a targeted gene panel sequencing combined with WES/WGS. From a previous combined experience in our centers (in Buenos Aires, Argentina and Cincinnati, USA), focusing on the usefulness of reinterpreting variants previously classified as of “uncertain significance,” about 30% of those variants were reclassified as pathogenic (104) using this approach. The use of NGS has allowed the identification of both germline and mosaic pathogenic variants, expanding the diagnostic yield (104). These results demonstrated the high clinical impact of periodic reanalyses of undetermined variants in clinical neurology. A major challenge is that the generation of LRS data for a thorough genome-wide analysis of these cases of “uncertain significance” is extremely expensive (18). For this purpose, the adaptive sampling T-LRS is a promising tool to broaden the clinical use of LRS with a cost comparable to the current one for the short-read WGS and with high sensitivity and specificity, although larger prospective studies are needed to confirm these results (18). We can assume that, in the future, the WG-LRS could easily replace almost every other genetic tests currently used in the clinical field (18) (Figure 2).

Figure 2. Traditional genetic workup and possibilities derived from long-read sequencing. The traditional (current) genetic workup is not diagnostic in about 50% of cases; other 20% of unresolved cases are represented by variants of uncertain significance. LRS (when coupled with whole genome sequencing— WGS, namely LR-WGS) has the potential to diagnose up to 70% of currently unresolved cases. The remaining 25% of cases are diagnosed using targeted genetic panel sequencing (TGPS) and whole exome sequencing (WES), and 5% of cases are diagnosed by WGS. Data derived from Salinas et al. (104) and Frésard et al. (119). Created with BioRender.com.
In sum, in the last decade the broad application of NGS and LRS technologies has helped interpret a large proportion of rare non-coding, missense, “uncertain significance,” and structural variants in the genome in several neurodegenerative diseases. We envision a great potential for the application of LRS in ET and “ET plus” disorders. This approach will help with the diagnosis, classification, and possibly also design of disease-specific treatment of patients with different neurogenetic neurodegenerative disorders. The development of LRS technologies are moving us closer to the desired reversed genotype-to-phenotype paradigm (2). This shift has the potential to usher a systems biology model of precision medicine for patients with neurodegenerative diseases.
Author Contributions
LM: conception, organization, execution of the study design, writing of the first draft, and review and critique. KD: organization, execution of the study design, review, and critique. RB: execution of the study design, review, and critique. MK: conception, organization, of the study design, review, and critique. AE: conception, organization of the study design, review, critique, and final approval of the submitted version. All authors contributed to subsequent drafts and approved the submitted version.
Conflict of Interest
LM has received honoraria from the International Association of Parkinsonism and Related Disorders (IAPRD) Society for social media and web support. MK is an employee of the CONICET and has received grant support from the Ministry of Science and Technology of Argentina and the Ministry of Health of Buenos Aires. AE has received grant support from the NIH and the Michael J Fox Foundation; personal compensation as a consult-ant/scientific advisory board member for Abbvie, Neuroderm, Neurocrine, Amneal, Adamas, Acadia, Acorda, Kyowa Kirin, Sunovion, Lundbeck, and USWorldMeds; publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer; and honoraria from USWorldMeds, Acadia, and Sunovion. He is cofounder of REGAIN Therapeutics, owner of a provisional patent on compositions and methods for treatment and/or prophylaxis of proteinopathies.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.821189/full#supplementary-material
References
1. MacDonald ME, Barnes G, Srinidhi J, Duyao MP, Ambrose CM, Myers RH, et al. Gametic but not somatic instability of CAG repeat length in Huntington's disease. J Med Genet. (1993) 30:982–6. doi: 10.1136/jmg.30.12.982
2. Sturchio A, Marsili L, Mahajan A, Grimberg MB, Kauffman MA, Espay AJ. How have advances in genetic technology modified movement disorder nosology? Eur J Neurol. (2020) 27:1461–70. doi: 10.1111/ene.14294
3. Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. (2011) 13:36–46. doi: 10.1038/nrg3117
4. Su Y, Fan L, Shi C, Wang T, Zheng H, Luo H, et al. Deciphering neurodegenerative diseases using long-read sequencing. Neurology. (2021) 97:423–33. doi: 10.1212/WNL.0000000000012466
5. Nattestad M, Goodwin S, Ng K, Baslan T, Sedlazeck FJ, Rescheneder P, et al. Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line. Genome Res. (2018) 28:1126–35. doi: 10.1101/gr.231100.117
6. Silvestre-Ryan J, Holmes I. Pair consensus decoding improves accuracy of neural network basecallers for nanopore sequencing. Genome Biol. (2021) 22:38. doi: 10.1186/s13059-020-02255-1
7. Mantere T, Kersten S, Hoischen A. Long-Read sequencing emerging in medical genetics. Front Genet. (2019) 10:426. doi: 10.3389/fgene.2019.00426
8. De Coster W, Weissensteiner MH, Sedlazeck FJ. Towards population-scale long-read sequencing. Nat Rev Genet. (2021) 22:572–87. doi: 10.1038/s41576-021-00367-3
9. Lin B, Hui J, Mao H. Nanopore technology and its applications in gene sequencing. Biosensors(Basel). (2021) 11:214. doi: 10.3390/bios11070214
10. EMBL-EBI WGC, Hinxton, Cambridgeshire, CB10, 1SD, UK. Human Genetic Variation. An Introduction. (2021). Available online at: https://www.ebi.ac.uk/training/online/courses/human-genetic-variation-introduction/variant-identification-and-analysis/.
11. Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. (2011) 12:363–76. doi: 10.1038/nrg2958
12. Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. (2006) 7:85–97. doi: 10.1038/nrg1767
13. Thapar A, Cooper M. Copy number variation: what is it and what has it told us about child psychiatric disorders? J Am Acad Child Adolesc Psychiatry. (2013) 52:772–4. doi: 10.1016/j.jaac.2013.05.013
14. Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Characterization of individual polynucleotide molecules using a membrane channel. Proc Natl Acad Sci U S A. (1996) 93:13770–3. doi: 10.1073/pnas.93.24.13770
15. Howorka S, Cheley S, Bayley H. Sequence-specific detection of individual DNA strands using engineered nanopores. Nat Biotechnol. (2001) 19:636–9. doi: 10.1038/90236
16. Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, et al. Real-time DNA sequencing from single polymerase molecules. Science. (2009) 323:133–8. doi: 10.1126/science.1162986
17. Beyter D, Ingimundardottir H, Oddsson A, Eggertsson HP, Bjornsson E, Jonsson H, et al. Long-read sequencing of 3,622 icelanders provides insight into the role of structural variants in human diseases and other traits. Nat Genet. (2021) 53:779–86. doi: 10.1038/s41588-021-00865-4
18. Miller DE, Sulovari A, Wang T, Loucks H, Hoekzema K, Munson KM, et al. Targeted long-read sequencing identifies missing disease-causing variation. Am J Hum Genet. (2021) 108:1436–49. doi: 10.1016/j.ajhg.2021.06.006
19. Orr HT, Chung MY, Banfi S, Kwiatkowski TJ. Jr., Servadio A, Beaudet AL, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. (1993) 4:221–6. doi: 10.1038/ng0793-221
20. Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primers. (2019) 5:24. doi: 10.1038/s41572-019-0074-3
21. Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. (2000) 26:191–4. doi: 10.1038/79911
22. Laver TW, Caswell RC, Moore KA, Poschmann J, Johnson MB, Owens MM, et al. Pitfalls of haplotype phasing from amplicon-based long-read sequencing. Sci Rep. (2016) 6:21746. doi: 10.1038/srep21746
23. Höijer I, Tsai YC, Clark TA, Kotturi P, Dahl N, Stattin EL, et al. Detailed analysis of HTT repeat elements in human blood using targeted amplification-free long-read sequencing. Hum Mutat. (2018) 39:1262–72. doi: 10.1002/humu.23580
24. Tsai Y-C, Greenberg D, Powell J, Höijer I, Ameur A, Strahl M, et al. Amplification-free, CRISPR-Cas9 targeted enrichment and SMRT sequencing of repeat-expansion disease causative genomic regions. BioRxiv. (2017):203919. doi: 10.1101/203919
25. McFarland KN, Liu J, Landrian I, Godiska R, Shanker S, Yu F, et al. SMRT sequencing of long tandem nucleotide repeats in SCA10 reveals unique insight of repeat expansion structure. PLoS ONE. (2015) 10:e0135906. doi: 10.1371/journal.pone.0135906
26. Schüle B, McFarland KN, Lee K, Tsai YC, Nguyen KD, Sun C, et al. Parkinson's disease associated with pure ATXN10 repeat expansion. NPJ Parkinsons Dis. (2017) 3:27. doi: 10.1038/s41531-017-0029-x
27. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. (2006) 314:130–3. doi: 10.1126/science.1134108
28. Loose M, Malla S, Stout M. Real-time selective sequencing using nanopore technology. Nat Methods. (2016) 13:751–4. doi: 10.1038/nmeth.3930
29. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. the Huntington's disease collaborative research group. Cell. (1993) 72:971–83. doi: 10.1016/0092-8674[93]90585-E
30. Losekoot M, van Belzen MJ, Seneca S, Bauer P, Stenhouse SA, Barton DE. EMQN/CMGS best practice guidelines for the molecular genetic testing of Huntington disease. Eur J Hum Genet. (2013) 21:480–6. doi: 10.1038/ejhg.2012.200
31. Svrzikapa N, Longo KA, Prasad N, Boyanapalli R, Brown JM, Dorset D, et al. Investigational assay for haplotype phasing of the huntingtin gene. Mol Ther Methods Clin Dev. (2020) 19:162–73. doi: 10.1016/j.omtm.2020.09.003
32. Ebbert MTW, Farrugia SL, Sens JP, Jansen-West K, Gendron TF, Prudencio M, et al. Long-read sequencing across the C9orf72 “GGGGCC” repeat expansion: implications for clinical use and genetic discovery efforts in human disease. Mol Neurodegener. (2018) 13:46. doi: 10.1186/s13024-018-0274-4
33. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
34. Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EM, Parkinson G, et al. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep. (2012) 2:1016. doi: 10.1038/srep01016
35. Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. (2014) 507:195–200. doi: 10.1038/nature13124
36. Lee LV, Pascasio FM, Fuentes FD, Viterbo GH. Torsion dystonia in Panay, Philippines. Adv Neurol. (1976) 14:137–51.
37. Aneichyk T, Hendriks WT, Yadav R, Shin D, Gao D, Vaine CA, et al. Dissecting the causal mechanism of X-linked dystonia-parkinsonism by integrating genome and transcriptome assembly. Cell. (2018) 172:897–909.e21. doi: 10.1016/j.cell.2018.02.011
38. Capponi S, Stöffler N, Penney EB, Grütz K, Nizamuddin S, Vermunt MW, et al. Dissection of TAF1 neuronal splicing and implications for neurodegeneration in X-linked dystonia-parkinsonism. Brain Commun. (2021) 3:fcab253. doi: 10.1093/braincomms/fcab253
39. Leija-Salazar M, Sedlazeck FJ, Toffoli M, Mullin S, Mokretar K, Athanasopoulou M, et al. Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol Genet Genomic Med. (2019) 7:e564. doi: 10.1002/mgg3.564
40. Graham OEE, Pitcher TL, Liau Y, Miller AL, Dalrymple-Alford JC, Anderson TJ, et al. Nanopore sequencing of the glucocerebrosidase (GBA) gene in a New Zealand Parkinson's disease cohort. Parkinsonism Relat Disord. (2020) 70:36–41. doi: 10.1016/j.parkreldis.2019.11.022
41. Hashem V, Tiwari A, Bewick B, Teive HAG, Moscovich M, Schüele B, et al. Pulse-Field capillary electrophoresis of repeat-primed PCR amplicons for analysis of large repeats in spinocerebellar ataxia type 10. PLoS ONE. (2020) 15:e0228789. doi: 10.1371/journal.pone.0228789
42. Mizuguchi T, Suzuki T, Abe C, Umemura A, Tokunaga K, Kawai Y, et al. A 12-kb structural variation in progressive myoclonic epilepsy was newly identified by long-read whole-genome sequencing. J Hum Genet. (2019) 64:359–68. doi: 10.1038/s10038-019-0569-5
43. Ebbert MTW, Jensen TD, Jansen-West K, Sens JP, Reddy JS, Ridge PG, et al. Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome Biol. (2019) 20:97. doi: 10.1186/s13059-019-1707-2
44. Suh E, Grando K, Van Deerlin VM. Validation of a long-read PCR assay for sensitive detection and sizing of C9orf72 hexanucleotide repeat expansions. J Mol Diagn. (2018) 20:871–82. doi: 10.1016/j.jmoldx.2018.07.001
45. Bram E, Javanmardi K, Nicholson K, Culp K, Thibert JR, Kemppainen J, et al. Comprehensive genotyping of the C9orf72 hexanucleotide repeat region in 2095 ALS samples from the NINDS collection using a two-mode, long-read PCR assay. Amyotroph Lateral Scler Frontotemporal Degener. (2019) 20:107–14. doi: 10.1080/21678421.2018.1522353
46. DeJesus-Hernandez M, Aleff RA, Jackson JL, Finch NA, Baker MC, Gendron TF, et al. Long-read targeted sequencing uncovers clinicopathological associations for C9orf72-linked diseases. Brain. (2021) 144:1082–8. doi: 10.1093/brain/awab006
47. Wallace AD, Sasani TA, Swanier J, Gates BL, Greenland J, Pedersen BS, et al. CaBagE: a Cas9-based background elimination strategy for targeted, long-read DNA sequencing. PLoS ONE. (2021) 16:e0241253. doi: 10.1371/journal.pone.0241253
48. Gonçalves A, Oliveira J, Coelho T, Taipa R, Melo-Pires M, Sousa M, et al. Exonization of an intronic LINE-1 element causing becker muscular dystrophy as a novel mutational mechanism in dystrophin gene. Genes (Basel). (2017) 8:253. doi: 10.3390/genes8100253
49. Xie Z, Sun C, Liu Y, Yu M, Zheng Y, Meng L, et al. Practical approach to the genetic diagnosis of unsolved dystrophinopathies: a stepwise strategy in the genomic era. J Med Genet. (2021) 58:743–51. doi: 10.1136/jmedgenet-2020-107113
50. Xie Z, Sun C, Zhang S, Liu Y, Yu M, Zheng Y, et al. Long-read whole-genome sequencing for the genetic diagnosis of dystrophinopathies. Ann Clin Transl Neurol. (2020) 7:2041–6. doi: 10.1002/acn3.51201
51. Geng C, Tong Y, Zhang S, Ling C, Wu X, Wang D, et al. Sequence and structure characteristics of 22 deletion breakpoints in intron 44 of the DMD gene based on long-read sequencing. Front Genet. (2021) 12:638220. doi: 10.3389/fgene.2021.638220
52. Cumming SA, Hamilton MJ, Robb Y, Gregory H, McWilliam C, Cooper A, et al. De novo repeat interruptions are associated with reduced somatic instability and mild or absent clinical features in myotonic dystrophy type 1. Eur J Hum Genet. (2018) 26:1635–47. doi: 10.1038/s41431-018-0156-9
53. Dastidar S, Ardui S, Singh K, Majumdar D, Nair N, Fu Y, et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. (2018) 46:8275–98. doi: 10.1093/nar/gky548
54. Mangin A, de Pontual L, Tsai YC, Monteil L, Nizon M, Boisseau P, et al. Robust detection of somatic mosaicism and repeat interruptions by long-read targeted sequencing in myotonic dystrophy type 1. Int J Mol Sci. (2021) 22:2616. doi: 10.3390/ijms22052616
55. Cacace R, Heeman B, Van Mossevelde S, De Roeck A, Hoogmartens J, De Rijk P, et al. Loss of DPP6 in neurodegenerative dementia: a genetic player in the dysfunction of neuronal excitability. Acta Neuropathol. (2019) 137:901–18. doi: 10.1007/s00401-019-01976-3
56. Morioka MS, Kitazume M, Osaki K, Wood J, Tanaka Y. Filling in the gap of human chromosome 4: single molecule real time sequencing of macrosatellite repeats in the facioscapulohumeral muscular dystrophy locus. PLoS ONE. (2016) 11:e0151963. doi: 10.1371/journal.pone.0151963
57. Mitsuhashi S, Nakagawa S, Takahashi Ueda M, Imanishi T, Frith MC, Mitsuhashi H. Nanopore-based single molecule sequencing of the D4Z4 array responsible for facioscapulohumeral muscular dystrophy. Sci Rep. (2017) 7:14789. doi: 10.1038/s41598-017-13712-6
58. Loomis EW, Eid JS, Peluso P, Yin J, Hickey L, Rank D, et al. Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene. Genome Res. (2013) 23:121–8. doi: 10.1101/gr.141705.112
59. Pham TT, Yin J, Eid JS, Adams E, Lam R, Turner SW, et al. Single-locus enrichment without amplification for sequencing and direct detection of epigenetic modifications. Mol Genet Genomics. (2016) 291:1491–504. doi: 10.1007/s00438-016-1167-2
60. Tseng E, Tang HT, AlOlaby RR, Hickey L, Tassone F. Altered expression of the FMR1 splicing variants landscape in premutation carriers. Biochim Biophys Acta Gene Regul Mech. (2017) 1860:1117–26. doi: 10.1016/j.bbagrm.2017.08.007
61. Ardui S, Race V, de Ravel T, Van Esch H, Devriendt K, Matthijs G, et al. Detecting AGG interruptions in females with a FMR1 premutation by long-read single-molecule sequencing: a 1 year clinical experience. Front Genet. (2018) 9:150. doi: 10.3389/fgene.2018.00150
62. Pretto DI, Eid JS, Yrigollen CM, Tang HT, Loomis EW, Raske C, et al. Differential increases of specific FMR1 mRNA isoforms in premutation carriers. J Med Genet. (2015) 52:42–52. doi: 10.1136/jmedgenet-2014-102593
63. Deng J, Yu J, Li P, Luan X, Cao L, Zhao J, et al. Expansion of GGC Repeat in GIPC1 Is Associated with oculopharyngodistal myopathy. Am J Hum Genet. (2020) 106:793–804. doi: 10.1016/j.ajhg.2020.04.011
64. Xi J, Wang X, Yue D, Dou T, Wu Q, Lu J, et al. 5' UTR CGG repeat expansion in GIPC1 is associated with oculopharyngodistal myopathy. Brain. (2021) 144:601–14. doi: 10.1093/brain/awaa426
65. Florian RT, Kraft F, Leitão E, Kaya S, Klebe S, Magnin E, et al. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with familial adult myoclonic epilepsy type 3. Nat Commun. (2019) 10:4919. doi: 10.1038/s41467-019-12763-9
66. Zablotskaya A, Van Esch H, Verstrepen KJ, Froyen G, Vermeesch JR. Mapping the landscape of tandem repeat variability by targeted long read single molecule sequencing in familial X-linked intellectual disability. BMC Med Genomics. (2018) 11:123. doi: 10.1186/s12920-018-0446-7
67. Sun QY, Xu Q, Tian Y, Hu ZM, Qin LX, Yang JX, et al. Expansion of GGC repeat in the human-specific NOTCH2NLC gene is associated with essential tremor. Brain. (2020) 143:222–33. doi: 10.1093/brain/awz372
68. Yau WY, Vandrovcova J, Sullivan R, Chen Z, Zecchinelli A, Cilia R, et al. Low Prevalence of NOTCH2NLC GGC repeat expansion in white patients with movement disorders. Mov Disord. (2021) 36:251–5. doi: 10.1002/mds.28302
69. Yu J, Luan XH Yu M, Zhang W, Lv H, Cao L, et al. GGC repeat expansions in NOTCH2NLC causing a phenotype of distal motor neuropathy and myopathy. Ann Clin Transl Neurol. (2021) 8:1330–42. doi: 10.1002/acn3.51371
70. Fang P, Yu Y, Yao S, Chen S, Zhu M, Chen Y, et al. Repeat expansion scanning of the NOTCH2NLC gene in patients with multiple system atrophy. Ann Clin Transl Neurol. (2020) 7:517–26. doi: 10.1002/acn3.51021
71. Deng J, Gu M, Miao Y, Yao S, Zhu M, Fang P, et al. Long-read sequencing identified repeat expansions in the 5'UTR of the NOTCH2NLC gene from Chinese patients with neuronal intranuclear inclusion disease. J Med Genet. (2019) 56:758–64. doi: 10.1136/jmedgenet-2019-106268
72. Sone J, Mitsuhashi S, Fujita A, Mizuguchi T, Hamanaka K, Mori K, et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet. (2019) 51:1215–21. doi: 10.1038/s41588-019-0459-y
73. Tian Y, Wang JL, Huang W, Zeng S, Jiao B, Liu Z, et al. Expansion of Human-Specific GGC Repeat in neuronal intranuclear inclusion disease-related disorders. Am J Hum Genet. (2019) 105:166–76. doi: 10.1016/j.ajhg.2019.05.013
74. Chen Z, Xu Z, Cheng Q, Tan YJ, Ong HL, Zhao Y, et al. Phenotypic bases of NOTCH2NLC GGC expansion positive neuronal intranuclear inclusion disease in a Southeast Asian cohort. Clin Genet. (2020) 98:274–81. doi: 10.1111/cge.13802
75. Ishiura H, Shibata S, Yoshimura J, Suzuki Y, Qu W, Doi K, et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet. (2019) 51:1222–32. doi: 10.1038/s41588-019-0458-z
76. Yu J, Deng J, Guo X, Shan J, Luan X, Cao L, et al. The GGC repeat expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy type 3. Brain. (2021) 144:1819–32. doi: 10.1093/brain/awab077
77. Ma D, Tan YJ, Ng ASL, Ong HL, Sim W, Lim WK, et al. Association of NOTCH2NLC repeat expansions with parkinson disease. JAMA Neurol. (2020) 77:1559–63. doi: 10.1001/jamaneurol.2020.3023
78. Ishiura H, Doi K, Mitsui J, Yoshimura J, Matsukawa MK, Fujiyama A, et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat Genet. (2018) 50:581–90. doi: 10.1038/s41588-018-0067-2
79. Nakamura H, Doi H, Mitsuhashi S, Miyatake S, Katoh K, Frith MC, et al. Long-read sequencing identifies the pathogenic nucleotide repeat expansion in RFC1 in a Japanese case of CANVAS. J Hum Genet. (2020) 65:475–80. doi: 10.1038/s10038-020-0733-y
80. Zeng S, Zhang MY, Wang XJ, Hu ZM, Li JC, Li N, et al. Long-read sequencing identified intronic repeat expansions in SAMD12 from Chinese pedigrees affected with familial cortical myoclonic tremor with epilepsy. J Med Genet. (2019) 56:265–70. doi: 10.1136/jmedgenet-2018-105484
81. Cen Z, Chen Y, Yang D, Zhu Q, Chen S, Chen X, et al. Intronic (TTTGA)(n) insertion in SAMD12 also causes familial cortical myoclonic tremor with epilepsy. Mov Disord. (2019) 34:1571–6. doi: 10.1002/mds.27832
82. Mizuguchi T, Toyota T, Adachi H, Miyake N, Matsumoto N, Miyatake S. Detecting a long insertion variant in SAMD12 by SMRT sequencing: implications of long-read whole-genome sequencing for repeat expansion diseases. J Hum Genet. (2019) 64:191–7. doi: 10.1038/s10038-018-0551-7
83. Pan S, Li X, Li L, Lin H, Wang D, Zhang X, et al. Comprehensive genetic, clinical and electrophysiological studies of familial cortical myoclonic tremor with epilepsy 1 highlight the role of gene configurations. Seizure. (2021) 87:69–74. doi: 10.1016/j.seizure.2021.02.026
84. Yeetong P, Chunharas C, Pongpanich M, Bennett MF, Srichomthong C, Pasutharnchat N, et al. Founder effect of the TTTCA repeat insertions in SAMD12 causing BAFME1. Eur J Hum Genet. (2021) 29:343–8. doi: 10.1038/s41431-020-00729-1
85. Zhou Y, Sood R, Wang Q, Carrington B, Park M, Young AC, et al. Clinical and genomic analysis of a large Chinese family with familial cortical myoclonic tremor with epilepsy and SAMD12 intronic repeat expansion. Epilepsia Open. (2021) 6:102–11. doi: 10.1002/epi4.12450
86. Bowden R, Davies RW, Heger A, Pagnamenta AT, de Cesare M, Oikkonen LE, et al. Sequencing of human genomes with nanopore technology. Nat Commun. (2019) 10:1869. doi: 10.1038/s41467-019-09637-5
87. Corbett MA, Kroes T, Veneziano L, Bennett MF, Florian R, Schneider AL, et al. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat Commun. (2019) 10:4920. doi: 10.1038/s41467-019-12671-y
88. Reyes CJ, Laabs BH, Schaake S, Lüth T, Ardicoglu R, Rakovic A, et al. Brain regional differences in hexanucleotide repeat length in X-linked dystonia-parkinsonism using nanopore sequencing. Neurol Genet. (2021) 7:e608. doi: 10.1212/NXG.0000000000000608
89. Yeetong P, Pongpanich M, Srichomthong C, Assawapitaksakul A, Shotelersuk V, Tantirukdham N, et al. TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain. (2019) 142:3360–6. doi: 10.1093/brain/awz267
90. Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, Brunberg JA, et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. (2003) 72:869–78. doi: 10.1086/374321
91. Hagerman RJ, Hagerman PJ. Fragile X syndrome: a model of gene-brain-behavior relationships. Mol Genet Metab. (2001) 74:89–97. doi: 10.1006/mgme.2001.3225
92. Yrigollen CM, Durbin-Johnson B, Gane L, Nelson DL, Hagerman R, Hagerman PJ, et al. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet Med. (2012) 14:729–36. doi: 10.1038/gim.2012.34
93. Apessos A, Abou-Sleiman PM, Harper JC, Delhanty JD. Preimplantation genetic diagnosis of the fragile X syndrome by use of linked polymorphic markers. Prenat Diagn. (2001) 21:504–11. doi: 10.1002/pd.111
94. Biancalana V, Glaeser D, McQuaid S, Steinbach P. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. Eur J Hum Genet. (2015) 23:417–25. doi: 10.1038/ejhg.2014.185
95. Uyama E, Fu YH, Ptácek LJ. Familial adult myoclonic epilepsy (FAME). Adv Neurol. (2005) 95:281–8.
96. Latorre A, Rocchi L, Magrinelli F, Mulroy E, Berardelli A, Rothwell JC, et al. Unravelling the enigma of cortical tremor and other forms of cortical myoclonus. Brain. (2020) 143:2653–63. doi: 10.1093/brain/awaa129
97. Okuma Y, Shimo Y, Shimura H, Hatori K, Hattori T, Tanaka S, et al. Familial cortical tremor with epilepsy: an under-recognized familial tremor. Clin Neurol Neurosurg. (1998) 100:75–8. doi: 10.1016/S0303-8467(98)00003-1
98. Mori S, Nakamura M, Yasuda T, Ueno S, Kaneko S, Sano A. Remapping and mutation analysis of benign adult familial myoclonic epilepsy in a Japanese pedigree. J Hum Genet. (2011) 56:742–7. doi: 10.1038/jhg.2011.93
99. Bhatia KP, Bain P, Bajaj N, Elble RJ, Hallett M, Louis ED, et al. Consensus Statement on the classification of tremors from the task force on tremor of the international parkinson and movement disorder society Mov Disord. (2018) 33:75–87. doi: 10.1002/mds.27121
100. Haubenberger D, Hallett M. Essential tremor. N Engl J Med. (2018) 378:1802–10. doi: 10.1056/NEJMcp1707928
101. Hopfner F, Helmich RC. The etiology of essential tremor: genes versus environment. Parkinsonism Relat Disord. (2018) 46(Suppl. 1):S92–6. doi: 10.1016/j.parkreldis.2017.07.014
102. Espay AJ, Brundin P, Lang AE. Precision medicine for disease modification in parkinson disease. Nat Rev Neurol. (2017) 13:119–26. doi: 10.1038/nrneurol.2016.196
103. Fasano A, Lang AE, Espay AJ. What is “essential” about essential tremor? a diagnostic placeholder. Mov Disord. (2018) 33:58–61. doi: 10.1002/mds.27288
104. Salinas V, Vega P, Marsili L, Pérez-Maturo J, Martínez N, Zavala L, et al. The odyssey of complex neurogenetic disorders: from undetermined to positive. Am J Med Genet C Semin Med Genet. (2020) 184:876–84. doi: 10.1002/ajmg.c.31848
105. Mahajan A, Gupta P, Jacobs J, Marsili L, Sturchio A, Jinnah HA, et al. Impaired saccade adaptation in tremor-dominant cervical dystonia-evidence for maladaptive cerebellum. Cerebellum. (2020). doi: 10.1007/s12311-020-01104-y
106. Espay AJ, Lang AE, Erro R, Merola A, Fasano A, Berardelli A, et al. Essential pitfalls in “essential” tremor. Mov Disord. (2017) 32:325–31. doi: 10.1002/mds.26919
107. Kevelam SH, Steenweg ME, Srivastava S, Helman G, Naidu S, Schiffmann R, et al. Update on leukodystrophies: a historical perspective and adapted definition. Neuropediatrics. (2016) 47:349–54. doi: 10.1055/s-0036-1588020
108. Zhang S, Liang F, Lei C, Wu J, Fu J, Yang Q, et al. Long-read sequencing and haplotype linkage analysis enabled preimplantation genetic testing for patients carrying pathogenic inversions. J Med Genet. (2019) 56:741–9. doi: 10.1136/jmedgenet-2018-105976
109. Foox J, Tighe SW, Nicolet CM, Zook JM, Byrska-Bishop M, Clarke WE, et al. Performance assessment of DNA sequencing platforms in the ABRF next-generation sequencing study. Nat Biotechnol. (2021) 39:1129–40. doi: 10.1038/s41587-021-01049-5
110. Sahlin K, Medvedev P. Error correction enables use of Oxford Nanopore technology for reference-free transcriptome analysis. Nat Commun. (2021) 12:2. doi: 10.1038/s41467-020-20340-8
111. Cambridge Uo. Long Read Sequencing: Ready for the Clinic? (2022). Available online at: https://www.phgfoundation.org/briefing/long-read-sequencing-ready-for-clinic.
113. Wenger AM, Peluso P, Rowell WJ, Chang PC, Hall RJ, Concepcion GT, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol. (2019) 37:1155–62. doi: 10.1038/s41587-019-0217-9
114. Rajan-Babu IS, Peng JJ, Chiu R, Li C, Mohajeri A, Dolzhenko E, et al. Genome-wide sequencing as a first-tier screening test for short tandem repeat expansions. Genome Med. (2021) 13:126. doi: 10.1186/s13073-021-00932-9
115. Dolzhenko E, Bennett MF, Richmond PA, Trost B, Chen S, van Vugt J, et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol. (2020) 21:102. doi: 10.1186/s13059-020-02017-z
116. Sturchio A, Marsili L, Vizcarra JA, Dwivedi AK, Kauffman MA, Duker AP, et al. Phenotype-Agnostic molecular subtyping of neurodegenerative disorders: the Cincinnati Cohort Biomarker Program (CCBP). Front Aging Neurosci. (2020) 12:553635. doi: 10.3389/fnagi.2020.553635
117. Stefansson H, Steinberg S, Petursson H, Gustafsson O, Gudjonsdottir IH, Jonsdottir GA, et al. Variant in the sequence of the LINGO1 gene confers risk of essential tremor. Nat Genet. (2009) 41:277–9. doi: 10.1038/ng.299
118. Alakbarzade V, Iype T, Chioza BA, Singh R, Harlalka GV, Hardy H, et al. Copy number variation of LINGO1 in familial dystonic tremor. Neurol Genet. (2019) 5:e307. doi: 10.1212/NXG.0000000000000307
119. Frésard L, Montgomery SB. Diagnosing rare diseases after the exome. Cold Spring Harb Mol Case Stud. (2018) 4. doi: 10.1101/mcs.a003392
Glossary
• Read mapping = Read mapping consists of the process of aligning the reads to a reference genome. The mapper uses as input a reference genome and a set of reads. The aim is to align each read in the set of reads on the specific reference genome.
• Amplicon sequencing = Amplicon Sequencing represents a targeted approach that enables researchers to analyze selected genetic variation(s) in specific genomic regions. The sequencing of PCR products (e.g., amplicons) allows variant identification and characterization. However, this approach is subject to inherent PCR biases.
• CRISPR/Cas9-based amplification-free target enrichment method (also known as No-Amp targeted sequencing) = CRISPR/Cas9 is a revolutionary gene editing tool of short-guide RNAs to target gene-modifying enzymes to specific DNA sequences. LRS is able to target genomic regions such as long repeats, without the need of PCR, e.g. avoiding PCR biases. This method has been called No-Amp targeted sequencing by Pac Bio. However, it can be used with both LRS platforms.
• PCR bias = It consists of artifacts introduced by PCR that skew the distribution of PCR products due to an unequal allelic amplification efficiency.
• PCR stutter = Stutters are common artifacts of amplification of the short-tandem repeat (STR) regions occurring when using polymerase chain reaction (PCR). They consist of strands of one or more motifs shorter or longer than the parental allele.
• Haplotype phasing = Haplotype phasing, or estimation in human genetics, refers to the process of statistical estimation of haplotypes from genotype data. The most common scenario arises when genotypes are collected at a set of polymorphic sites from a group of individuals. LRS allows for direct haplotype phasing, without statistical inference.
• Satellite DNA = DNA containing several tandem (e.g., not inverted) repeats of a given short basic repeating unit. Satellite DNA is located near the centromeres and telomeres of chromosomes (masked satellites).
Keywords: long-read sequencing, whole-genome sequencing, genomics, tremor, movement disorders
Citation: Marsili L, Duque KR, Bode RL, Kauffman MA and Espay AJ (2022) Uncovering Essential Tremor Genetics: The Promise of Long-Read Sequencing. Front. Neurol. 13:821189. doi: 10.3389/fneur.2022.821189
Received: 23 November 2021; Accepted: 25 February 2022;
Published: 23 March 2022.
Edited by:
Harvey Checkoway, University of California, San Diego, United StatesReviewed by:
Christos Proukakis, University College London, United KingdomJoanne Trinh, University of Lübeck, Germany
Copyright © 2022 Marsili, Duque, Bode, Kauffman and Espay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alberto J. Espay, YWxiZXJ0by5lc3BheUB1Yy5lZHU=