 Wenyu Liu1,2†Ming Liu1,2†
Wenyu Liu1,2†Ming Liu1,2† Di Lu1,2Jiwei Wang1,2Zexin Cao1,2
Di Lu1,2Jiwei Wang1,2Zexin Cao1,2 Xuchen Liu1,2
Xuchen Liu1,2 Zichao Feng1,2Bin Huang1,2
Zichao Feng1,2Bin Huang1,2 Xinyu Wang1,2*
Xinyu Wang1,2*- 1Department of Neurosurgery, Qilu Hospital, Cheeloo College of Medicine, Shandong University and Institute of Brain and Brain-Inspired Science, Shandong University, Jinan, China
- 2Key Laboratory of Brain Function Remodeling, Qilu Hospital of Shandong University, Jinan, China
Background: Familial cerebral cavernous malformation (FCCM) is a vascular malformation disease closely linked to three identified genes: KRIT1/CCM1, MGC4607/CCM2 and PDCD10/CCM3. Over the past decade, a few cases of cerebral cavernous malformation (CCM) caused by different gene mutations have been reported in Chinese families. Herein, we introduce a Chinese family affected by FCCM due to a kind of KRIT1/CCM1 frameshift mutation. At the same time, a literature review was conducted to identify case reports of familial cerebral cavernous malformation.
Case presentation: The proband in the family in question demonstrated a series of clinical symptoms and features, including headache and bleeding. The proband was hospitalized for headache twice and, both times was examined under suspicion of CCM and received surgical treatment. Magnetic resonance imaging results showed that the proband had multiple intracranial vascular lesions, including on the brain, brainstem, and cerebellum. Genetic test results showed that the classic KRIT1 gene in the proband had a pathogenic mutation. The family members of the proband also showed typical cerebral cavernous malformation when considering clinical manifestations, magnetic resonance imaging findings and genetic test results.
Conclusions: We report a case of Chinese FCCM and its associated symptoms with CCM1-deletion mutations in China. Our findings deepen our understanding of CCM mutations and related phenotypes, the investigation results of this clinical experiment further show that the gene mutation form we reported plays an important role in human FCCM, and this trial investigation is beneficial for genetic counseling for CCM patients.
Introduction
Cerebral cavernous malformations (CCMs) are relatively rare vascular anomalies occurring in single or multiple sites (1) in the brain and spinal cord where small blood vessels are enlarged with irregular structures (2). A large autopsy study showed that the prevalence of CCM accounted for 0.1 to 0.5% of the total population (3), while other research has indicated that CCM influences ~10 to 20% of all cerebral vascular abnormalities (4). The disease can occur sporadically or by way of familial autosomal dominant inheritance without complete penetrance (5), with most cases having the former nature. The age of onset of CCM is typically 20 to 30 years of age or older. About 40% of CCM patients can be clinically asymptomatic, while others may suffer from repeated headaches, hemorrhagic strokes, seizures, or focal neurological deficits (6). Currently, magnetic resonance imaging (MRI) is commonly used to diagnose CCM (7), and the characteristic appearance of this phenomenon on MRI appears with concentrated mixed signal strengths and a hemosiderin-containing ring, and performs best on the inversion recovery sequence of T2-weighting and fluid attenuation. It is a complex undertaking to surgically remove multiple lesions at once, making the successful treatment of FCCM difficult (8).
Genetic locus heterogeneity has also been demonstrated by linkage mapping to additional loci on chromosomes 7p13–15 (CCM2, OMIM 603284) and 3q25.2–27 (CCM3, OMIM 603285) (9). To our knowledge, KRIT1/CCM1 encodes for the krev interaction trapped 1 (Krit1) protein. Mutations that occur mainly in CCM1 can cause the stop codon to terminate prematurely, resulting in truncation of the protein (10).
Reports of FCCM in the Chinese population are limited in number, and most FCCM families that have been reported are Mexican/Hispanic (11, 12). In this article, we describe a frame-shift CCM1 mutation in exon 14 leading to a premature stop codon in a Chinese family with incomplete penetrance of the disease, which presents further evidence for phenotypic heterogeneity.
Case Series
The proband was a 52-year-old male, diagnosed with intracranial cavernous hemangioma at the age of 30 years because of an intracranial hemorrhage. Four years ago, he had experienced an intracranial hemorrhage again in another cavernous hemangioma, and the lesions were removed by craniotomy on both occasions. He mentioned that several of his sisters had similar manifestations as he did. Considering the proband's family as one with FCCM, we therefore analyzed and herein report on the following familial case series.
Patients and Methods
Patients
We examined 11 members of a family of Chinese descent (Figure 1), with the help of the proband's son, and we established the familial pedigree. All participants signed written informed consent forms before blood sampling and genetic analysis, then we collected peripheral blood samples from members of the family. We checked the presence and location of central nervous system lesions by MRI. The study was approved by the Ethics Committee of Qilu Hospital of Shandong University.
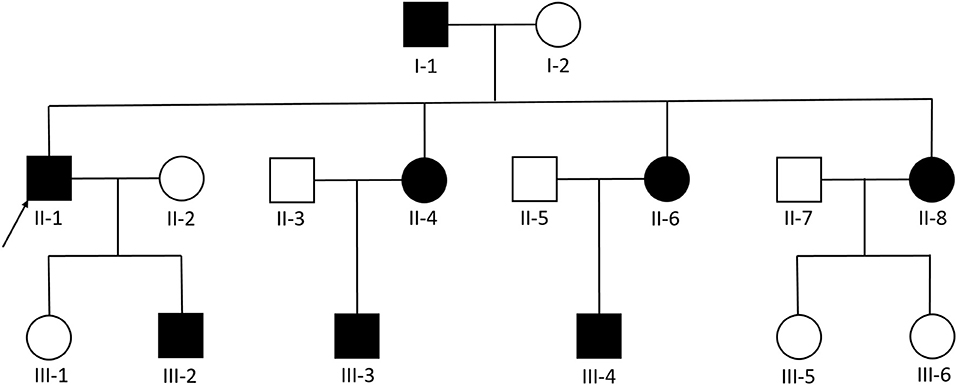
Figure 1. Pedigree diagram of the Chinese family with familial cerebral cavernous malformation, a heterozygous frameshift mutation described as c.1362_1363del (p.Gln455fs) was found in exon 14 of CCM1. The arrows designate the index patient (proband); filled squire = affected; unfilled square = unaffected.
Methods
Genomic DNA was extracted from the peripheral blood cells (II-1) of the proband, and we used polymerase chain reaction to amplify all coding exons of the three CCM genes (CCM1, CCM2, and CCM3). We used design primers that encompassed each exon and its respective exon intron-intron splice sites. The amplification primer was used as a sequencing primer, and the sequencing results were subjected to BLAST analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The primer sequences are listed in Table 1. The specific experimental steps were “Sample reception → DNA extraction → library construction (gDNA concentration detection and dilution → end repair and A base addition → adapter ligation and purification of ligation products → library fragment screening → PCR amplification and purification of amplified products → library hybridization and Capture → Product amplification and purification after capture) → On-machine sequencing (On-machine reagent information recording and preparation → Instrument cleaning → On-machine library preparation) → Bioinformatics analysis(details as follows).” First, we obtained fastq data from illumine sequencing machine (NovaSeq6000/HiSeq2500) by using Bcl2fastq (v2.0.1), so the acquired data could be processed by Trimmomatic (Version 0.36) to remove low-quality reads, bases, and trimming adaptors. We then cleaned the data and processed them with the Burrows–Wheeler Aligner (BWA) for alignment on a reference sequence of hg19, using the Genome Analysis Toolkit (GATK) for variant calling. Finally, the Variant Call format (VCF) files were analyzed using the ANNOVAR tool, which contains annotation databases, such as the 1000 genome databases, dbSNP database (dbSNP; http://www.ncbi.nlm.nih.gov/SNP), ClinVar database (ClinVar; http://www.ncbi.nlm.nih.gov/clinvar), the Polymorphism Phenotyping v2 database (Polyphen-2; http://genetics.bwh.harvard.edu/pph2), and the Sorting Intolerant from Tolerant database (SIFT; http://sift.jcvi.org). At the same time, the mutations found in the proband were searched for in the other family members.

Table 1. The primer sequence of this single gene sequencing.
Results
Clinical Investigation
A three-generation Chinese family with nine members affected by familial CCM was identified.
The proband was a 52-year-old male. His first symptom 30 years ago was a simple headache. He went to the local hospital for imaging examination and found multiple intracranial cavernous hemangioma with a small amount of bleeding (Unfortunately, the specific inspection method and lesion location are unknown). At that time, the cavernous hemangioma at the bleeding site was surgically removed. After the resection, he took Yangjiao granules for 2 to 3 years, and the pain symptoms basically disappeared. However, 4 years ago, he accidentally bumped his head at work and got a headache again without any other symptoms, another imaging examination revealed that a cavernous hemangioma in his skull had slight bleeding. He was resurgically removed the cavernous blood vessels at the bleeding site and recovered well after the operation, and except for numbness on the left face after a short period of time after the operation, he had no other discomfort, finally, the numbness on the left face gradually relieved on its own without any special treatment. The doctor considered that the postoperative symptom was the microbleeding caused by overwork and finally absorbed by itself, or the short-term sequelae caused by the operation, and recommended him to follow-up regularly. Since then, he has not had head symptoms again. The imaging examination of this trial shows multiple cavernous hemangioma in both cerebral hemispheres, as shown in Figure 2 (1–5). The proband's father was 76 years old. He hit his head at work when he was 65 years old and underwent craniocerebral imaging, and multiple cavernous hemangiomas were found in the brain (the specific location and manifestations of the lesions were unclear). So far, he has no obvious symptoms, and according to the doctor's recommendation, he has never given any special treatment to the intracranial cavernous hemangioma. Later, after 5 years of regular follow-up (Simple telephone follow-up), no obvious changes in the lesions were found. Due to his hunchback and some personal reasons, he did not have the latest MRI examination during this study, and the results of the previous cranial imaging examinations were not found. The daughter of the proband is 32 years old and has had no obvious symptoms so far, and her latest MRI examination does not reveal any abnormalities, and the son of the proband son has never had obvious head discomfort, but he once had multiple intracranial cavernous hemangioma in a cranial MRI when he needed a physical examination due to work. Based on his clinical manifestations and imaging data, the doctor recommended no special treatment for hemangioma. He has not usually received MRI of the brain. The results of this imaging examination suggests that he has multiple cavernous hemangioma in the right cerebellar hemisphere and right prefrontal lobe, as shown in the 22–27 diagram of Figure 2. The eldest sister of the proband is 50 years old and, last year, experienced stutter and salivation when the right side of her head was tilted without an obvious cause, she therefore underwent an MRI examination, prompting the diagnosis of CCM. Following her diagnosis, she received conservative treatment at the local hospital and was observed carefully, she gradually recovered, and was discharged from the hospital. Now she is being followed up with regularly. Her latest MRI examination mainly reveals multiple cavernous hemangioma in the brain stem and bilateral cerebral hemispheres, as shown in the 6–10 diagram of Figure 2. The son of the proband's eldest sister has never had obvious head discomfort, nor has he undergone a cranial examination. His latest MRI examination revealed multiple cavernous hemangiomas in the right cerebellar hemisphere and right prefrontal lobe, as shown in the 28–31 diagram of Figure 2. The second sister of the proband is now 47 years old, and 2 years ago, she experienced blurred vision on the right side without any obvious cause, after conservative treatment, her vision improved, and she is currently taking vincamine orally and being followed up with regularly. Her latest MRI results mainly indicate multiple cavernous hemangiomas in the cerebellum, brain stem, and bilateral cerebral hemispheres, as shown in the 11–16 diagram of Figure 2. Her son is 28 years old, and experienced a seizure when he was 13 years old, which was stiff and convulsive, at the time, an imaging examination revealed CCM, then he was treated conservatively and taken carbamazepine orally. The drug was eventually stopped because his symptoms did not relapse, however, 2 years ago, he experienced another epileptic seizure with the same symptoms as the previous one, and an imaging examination revealed multiple cavernous hemangiomas in the brain, one of which was bleeding, so he underwent surgery and the cavernous hemangioma was removed. Since the operation, he has been taking sodium valproate sustained-release tablets regularly to prevent seizures. He experienced epilepsy three months after surgery and once in May 2019, but is now asymptomatic and is followed up with regularly. His latest cranial MRI examination reveals multiple cavernous hemangiomas in the brain stem and bilateral cerebral hemispheres, as shown in the 32–35 diagram of Figure 2. The third sister of the proband is 44 years old, at the age of 27 years, she had no obvious cause for general fatigue, and an imaging examination revealed multiple intracranial cavernous hemangiomas, one of which had ruptured and was bleeding. She underwent surgery to remove the cavernous hemangioma at the lesion, and now she walks with a slight limp, favoring the left half of her body. Her latest MRI examination reveals multiple cavernous hemangiomas in both cerebellar hemispheres, brainstem, bilateral thalamus, and bilateral cerebral hemispheres, as shown in the 17–21 diagram of Figure 2. The genetic analysis of the two daughters of the proband's third sister did not find the sequence of the mutant gene, and the previous MRI results of the brain did not indicate abnormalities, so MRI was not performed in this test. Table 2 shows the incidence, diagnosis, treatment, and prognosis of the family members. Representative MRIs of the family members with imaging changes can be found in Figure 2
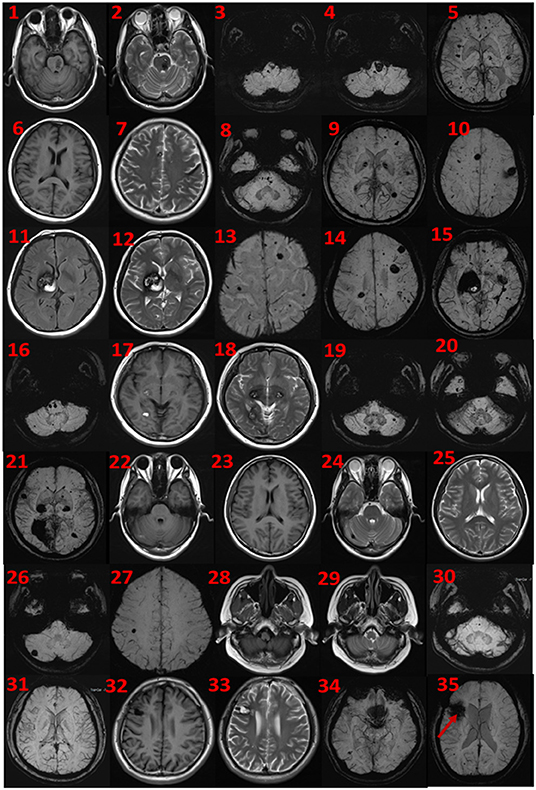
Figure 2. MRI findings. Cerebral MRI in subjects II-1 (1–5), II-4 (6–10), II-6 (11–16), II-8 (17–21), III-2 (22–27), III-3 (28–31), and iii-4 (32–35). 1–5 the MRI imaging showing there are multiple lesions in bilateral cerebellar hemispheres, brainstem area, left thalamus, and white matter areas in both cerebral hemispheres. 6–10 the MRI imaging showing there are multiple lesions in the brain stem and bilateral cerebral hemispheres. 11–16 the MRI imaging showing there are multiple lesions in the cerebellum, brainstem, and bilateral cerebral hemispheres, the large lesions are located in the right basal ganglia area, with a slight space–occupying effect. 17–21 the MRI imaging showing there are multiple lesions in bilateral cerebellar hemispheres, brainstem, bilateral thalamus, and bilateral cerebral hemispheres. 22–27 the MRI imaging showing there are lesions in the right cerebellar hemisphere and the right parietal lobe. 28–31 the MRI imaging showing there are multiple lesions on both sides of the brain, and the arrow points to the postoperative manifestations. 33–35 the MRI imaging showing there are multiple lesions in the brain stem and bilateral cerebral hemispheres.
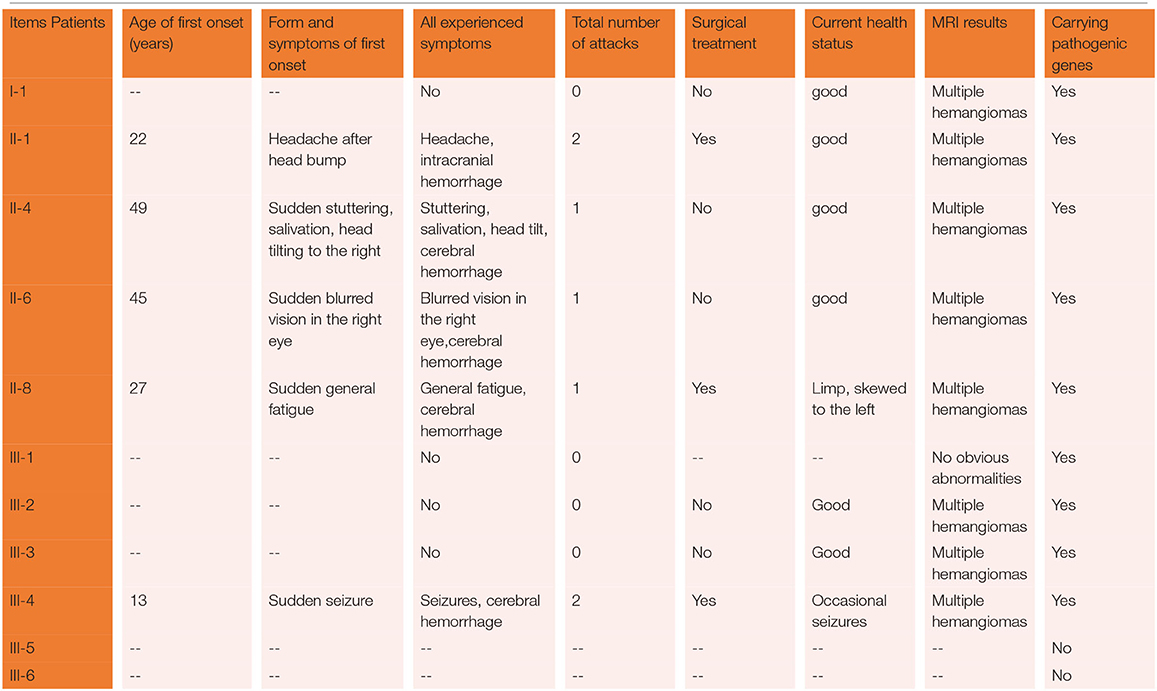
Table 2. The incidence, diagnosis, treatment, and prognosis of family members.
Genetic Analysis
We found a heterozygous deletion mutation (frameshift mutation) in the KRIT1/CCM1 gene of the proband, which we descrbied as c.1362_1363del (p.Gln455fs), in contrast, sequence analysis of CCM2 and CCM3 did not reveal any pathogenic mutations. At the same time, we also analyzed the KRIT1/CCM1 sequence of his father, son, sister and niece, and found the same mutation in all family members whose MRI scan showed cavernous hemangioma. The c.1362_ 1363del is a type of frame-shift mutation found in the patient's DNA (Figure 3), which alters the reading frame of the KRIT1/CCM1 gene, leading to a premature stop codon.
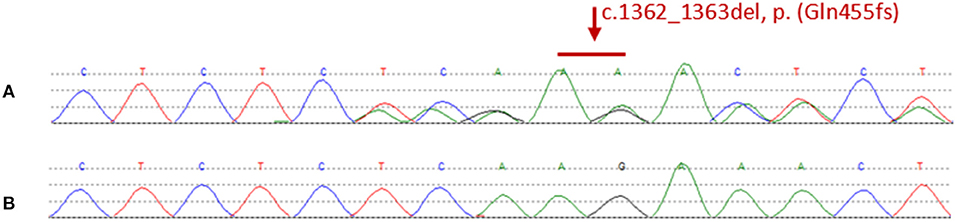
Figure 3. (A) Representative sequencing results from a patient in the family. The graph shows a “c.1362_1363del” deletion mutation of exon 14 in krit1. (B) Representative sequencing results from a normal member without mutation in the family.
Discussion
Cavernous hemangioma is a type of hemangioma that usually occurs in young adults and rarely occurs in older adults (13). The incidence of this condition in men and women appears to be equal, although, in some reports, women with cavernous hemangioma exhibit a higher bleeding rate (14). In the current family, five patients had bleeding, including two men and three women, which is different from the findings of other studies. There are two known manifestations of intracranial cavernous hemangioma: one is sporadic, usually with isolated lesions, and the other is a familial disease, which usually has multiple lesions and is inherited in an autosomal dominant nature with incomplete penetrance. In most cases, CCMs are located inside the brain, and 40% of patients have no obvious symptoms, while about 60% will experience generalized or focal seizures, focal neurological deficits, headaches and other symptoms of intracranial hemorrhage. The risk of symptomatic bleeding varies depending upon the location of the cavernoma, and is generally greater for lesions deeper in the brain (4). The risk of bleeding recurrence in each patient every year increases from 5 to 60% (15). Therefore, it is necessary to treat lesions early on after the first seizure or bleeding event. Among the cases of intracranial cavernous hemangioma caused by CCM1, CCM2, and CCM3 gene mutations, only reports of CCM1 mutations accompanied by cutaneous capillary—venous malformations have been published (16), and there is no cavernous hemangioma patient with skin venous—capillary malformations in the family detailed in this report. According to reports, the coexistence of brain CCM and spine CCM is very rare (17), and there was no such patient in the family involved in this report.
MRI is the most sensitive method for detecting intracranial CCM lesions. According to the results of MRI examination, CCM can be divided into four types (18). Type 1 lesions show high-intensity signals on both T1 and T2 sequences. Type 2 lesions are characterized by a mixed signal of high and low signals surrounded by low signal edges and are the most common CCM lesions. Type 3 lesions show low signals on both T1 and T2 sequences. Finally, type 4 lesions are low-signal lesions that can only be detected in radio-echo sequences. One of the main characteristics of both symptomatic and asymptomatic mutation carriers is whether there are multiple lesions on the brain MRI scan (7). Among the family members involved in this report, the brain MRI of mutation carriers also showed multiple lesions. When there are multiple lesions, gradient-echo sequences are more sensitive than turbo spin-echo (TSE) sequences; however, on TSE and gradient echo MRI sequences, some mutation carriers may not show any lesions (19). Therefore, it is necessary to use systematic gradient-echo sequences to detect patients suspected of having CCM. In patients with hemorrhagic metastases, the diagnosis may become difficult; moreover, if the peripheral edema causes a contrast enhancement effect after bleeding, rapid recurring bleeding occurs, or the lesions increase, the diagnosis will be more difficult. If there is doubt about a diagnosis, surgical removal of the lesions can be considered. For an uncertain or doubtful diagnosis, a biopsy can be performed by surgically removing the lesion. However, due to the risk of bleeding, a biopsy is usually not recommended.
Our findings demonstrate that not only incomplete clinical but also radiological penetrance precludes the use of cerebral MRI to firmly establish a non-carrier status even in adults and using highly sensitive gradient-echo sequences. In this case series, we found that we cannot rely solely on cranial MRI to determine whether a person is a non-mutant carrier, even in adults or using highly sensitive gradient echo sequences because of the clinical and radiological incomplete appearance. According to prior reports, based on the results of MRI examinations, about 75% of patients with significantly isolated CCM will have a parent with neuroradiological changes (8). Therefore, we infer that only those family members who carry familial cavernous hemangioma mutant genes require surveillance and medical management and intervention for symptoms and lesions.
In the past decade, the incidence of familial cavernomas hemangioma in Hispanic American (20) and Chinese families (21) has been higher than that in other populations, and existing clinical reports are mainly concentrated on these two groups of people.
In this study, we found a case of FCCM through a proband from a Chinese family. Patients from this family showed a series of clinical symptoms and features, including seizures, language disorders, dyskinesia, and other symptoms resulting from multiple brain lesions and bleeding. In this case, a heterozygous frame-shift mutation, which has been reported by several times in other countries in the past (22–25), was found in exon 14 (c.1362_1363del) of CCM1. This single-stranded deletion was predicted to cause coding disorder of amino acid Gln at position 455, leading to premature appearance of stop codons, making the encoded protein truncated and losing normal function.
The CCM1 gene with 16 coding exons encodes the Krit1 protein (containing 736 amino acids, which has three ankyrin domains and a FERM domain). Mutations in CCM1 all generate premature stop codons (26). So far, more than 90 different CCM1 mutations have been reported (27). It is speculated that these mutations in the CCM1 gene can lead to abnormal mRNA, which in turn leads to non-functional proteins (28). Familial CCM mutations are inherited in an autosomal dominant manner, of which the penetrance is incomplete at both the clinical and neuroradiological levels (29). Therefore, clinically asymptomatic “carrier” individuals with a disease-causing mutation should be monitored for lesions by MRI (30). In Japan, a recent survey showed genomic differences between different ethnic groups (31), where the ratio of CCM1, CCM2, and CCM3 mutation rates may reflect the unique genomic characteristics of the Japanese population (32).
The form of genetic mutation discovered in this study is a type of CCM1 mutation which traditionally leads to the premature generation of stop codons, resulting in truncated proteins. Several reports on FCCM caused by CCM1 mutations in China have published in recent years, as shown in Table 3. One point mutation (c.2835CT, p.Q698X) in exon 19 caused the stop codon (Q698X) to be generated prematurely, which resulted in a truncated Krit1 protein (11). Another point mutation (c.1298CG, p.S430X) in exon 14 caused the stop codon to be generated prematurely, which produced another truncated form of the Krit1 protein (12). A deletion mutation of the AT frameshift at NTs 1292 and 1293 in exon 13 destroyed the Krit1 protein-encoding mechanism (21). The splicing mutation of the GTA deletion of the intron 9/exon 10 receptor splice site of the CCM1 gene produced a premature stop codon at the 23rd amino acid, which led to the truncation of the Krit1 protein (33). A deletion mutation (c.1197delCAAA) in exon 12 led to an early stop codon (TGA) at NT 1,228, which produced a truncated Krit1 protein of only 409 amino acids (28). One T—deletion mutation in exon 14 (c.1396delT) caused a premature stop codon in the second half of the CCM1 gene, resulting in a Krit1 protein of only 493 amino acids in length (34). A heterozygous T deletion in exon 15 (c.1542delT) of the 16 coding exons (exons 5–20) led to an early stop codon, producing a truncated Krit1 protein of 526 amino acids (35). Detection of DNA using exome - capture sequencing technology revealed a c.1159G>T transition in exon 14 of the CCM1 gene, which led to premature termination at codon 387 (p. E387*) (36). A T—insertion mutation in exon 18 (c.1896_1897insT) caused a frameshift at NTs 1896 and 1897, which destroyed the coding of Krit1 protein (37). A novel heterozygous nonsense NT transition (c.1864C>T; p.Gln622X) in exon 17 of the CCM1/KRIT1 gene was predicted to trigger a premature stop codon (TAG) at NTs 1864 to 1866 to generate a truncated Krit1 of 621 amino acids in length (30). DNA sequencing analysis of the proband disclosed a novel heterozygous deletion mutation (c.1919delT; p.Phe640SerfsX21) in exon 17 of the CCM1 gene. This mutation led to a frameshift and caused a premature termination codon to generate a truncated Krit1 of 659 amino acids (38). Finally, a deletion frameshift mutation, c.1635delA (p.Thr545fsTer6), in the CCM1 gene, produced a truncated protein lacking 191 (546–736) amino acids at the C-terminal of the Krit1 protein (27).

Table 3. Reports of 12 Chinese articles on FCCM in the past 10 years.
In summary, we report a case series of FCCM in a Chinese family with a frame-shift mutation of CCM1 that has been previously reported at abroad, and we report the corresponding classical genetic characteristics, clinical characteristics and imaging characteristics. Our findings further deepen the understanding of CCM mutations and the associated clinical phenotypes. Moreover, our findings is beneficial knowledge to support genetic counseling of FCCM patients.
Conclusions
The study demonstrates Chinese FCCM with the KRIT1/CCM1 insertion mutation, here in the course of this trial, we have collected three generations of families with this mutation, and then we perfected the clinical symptoms and signs caused by this mutation of the gene, as well as the corresponding imaging changes. The results of this trial investigation are very consistent with the classic FCCM in terms of clinical manifestations, imaging manifestations, or genetic manifestations, which deepen the understanding of CCM mutations and the associated clinical phenotypes. Compared with previous reports, the investigation results of this clinical experiment further show that the gene mutation form we reported plays an important role in human familial cavernous hemangioma. Moreover, it will be of significance in genetic counseling for CCM.
Data Availability Statement
The datasets presented in this article are not readily available due to ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethical Committee of Qilu Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
XW and BH: study design. WL and ML: data collection. WL, ML, JW, DL, ZC, XL, and ZF: statistical analysis. WL, ML, JW, DL, and ZC: data interpretation. WL, ML, JW, DL, ZC, and XL: manuscript preparation. XW, BH, and JW: literature search. XW: funds collection. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the family members for their cooperation. Moreover, the authors thank the JinYu Medical Testing Center for technical assistance and the doctors in the imaging department of Qilu Hospital for the help with the laboratory results. The study was supported by the Ethical Committee of Qilu Hospital of Shandong University and the Natural Science Foundation of Shandong Province.
References
1. Leblanc G, Golanov E, Awad I, Young W. Biology of vascular malformations of the brain. Stroke. (2009) 40:E694–702. doi: 10.1161/STROKEAHA.109.563692
2. Uebelhoer M, Boon L, Vikkula M. Vascular anomalies: from genetics toward models for therapeutic trials. Cold Spring Harbor Perspect Med. (2012) 2:8. doi: 10.1101/cshperspect.a009688
3. Otten P, Pizzolato G, Rilliet B, Berney J. [131 Cases of Cavernous Angioma (Cavernomas) of the CNS, discovered by retrospective analysis of 24,535 autopsies]. Neuro-Chirurgie. (1989) 35:128–31.
4. Kondziolka D, Lunsford L, Kestle J. The natural history of cerebral cavernous malformations. J Neurosurg. (1995) 83:820–4. doi: 10.3171/jns.1995.83.5.0820
5. Han G, Ma L, Qiao H, Han L, Wu Q, Li Q. CCM2A novel missense variant caused cerebral cavernous malformations in a Chinese family. Front Neurosci. (2020) 14:604350. doi: 10.3389/fnins.2020.604350
6. Choquet H, Nelson J, Pawlikowska L, McCulloch C, Akers A, Baca B, et al. Association of cardiovascular risk factors with disease severity in cerebral cavernous malformation type 1 subjects with the common hispanic mutation. Cerebrovascular Dis. (2014) 37:57–3. doi: 10.1159/000356839
7. Akers A, Al-Shahi Salman R, A Awad I, Dahlem K, Flemming K, Hart B, et al. Synopsis of guidelines for the clinical management of cerebral cavernous malformations: consensus recommendations based on systematic literature review by the angioma alliance scientific advisory board clinical experts panel. Neurosurgery. (2017) 80:665–80. doi: 10.1093/neuros/nyx091
8. Labauge P, Laberge S, Brunereau L, Levy C, Tournier-Lasserve E. Hereditary cerebral cavernous angiomas: clinical and genetic features in 57 French families. Société Française de Neurochirurgie. (1998) 352:1892–7. doi: 10.1016/S0140-6736(98)03011-6
9. Liquori C, Berg M, Siegel A, Huang E, Zawistowski J, Stoffer T, et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genetics. (2003) 73:1459–64. doi: 10.1086/380314
10. Béraud-Dufour S, Gautier R, Albiges-Rizo C, Chardin P, Faurobert E. Krit 1 interactions with microtubules and membranes are regulated by Rap1 and integrin cytoplasmic domain associated protein-1. The FEBS J. (2007) 274:5518–32. doi: 10.1111/j.1742-4658.2007.06068.x
11. Chen D, Lipe H, Qin Z, Bird T. Cerebral cavernous malformation: novel mutation in a Chinese family and evidence for heterogeneity. J Neurologic Sci. (2002) 196:91–6. doi: 10.1016/S0022-510X(02)00031-X
12. Xu Y, Zhao J, Wu B, Zhong H, Wang S, Heng W. [A Novel Krit-1 Mutation in Han Family With Cerebral Cavernous Malformation]. Zhonghua Bing li xue za zhi = Chinese J Pathol. (2003) 32:220–5.
13. Zabramski J, Wascher T, Spetzler R, Johnson B, Golfinos J, Drayer B, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg. (1994) 80:422–32. doi: 10.3171/jns.1994.80.3.0422
14. Moriarity J, Wetzel M, Clatterbuck R, Javedan S, Sheppard J, Hoenig-Rigamonti K, et al. The natural history of cavernous malformations: a prospective study of 68 patients. Neurosurgery. (1999) 44:1166–71; Discussion 72–3. doi: 10.1227/00006123-199906000-00003
15. Willie J, Malcolm J, Stern M, Lowder L, Neill S, Cabaniss B, et al. Safety and effectiveness of stereotactic laser ablation for epileptogenic cerebral cavernous malformations. Epilepsia. (2019) 60:220–32. doi: 10.1111/epi.14634
16. Sirvente J, Enjolras O, Wassef M, Tournier-Lasserve E, Labauge P. Frequency and phenotypes of cutaneous vascular malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. J Euro Acad Dermatol Venereol JEADV. (2009) 23:1066–72. doi: 10.1111/j.1468-3083.2009.03263.x
17. Toldo I, Drigo P, Mammi I, Marini V, Carollo C. Vertebral and spinal cavernous angiomas associated with familial cerebral cavernous malformation. Surgic Neurol. (2009) 71:167–71. doi: 10.1016/j.surneu.2007.07.067
18. Sabayan B, Lineback C, Viswanathan A, Leslie-Mazwi T, Shaibani A. Central nervous system vascular malformations: a clinical review. Annal Clinic Translat Neurol. (2021) 8:504–22. doi: 10.1002/acn3.51277
19. Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M, et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Annal Neurol. (2006) 60:550–6. doi: 10.1002/ana.20947
20. Rigamonti D, Hadley M, Drayer B, Johnson P, Hoenig-Rigamonti K, Knight J, et al. Cerebral cavernous malformations. Incid Famil Occurren New Engl J Med. (1988) 319:343–7. doi: 10.1056/NEJM198808113190605
21. Mao Y, Zhao Y, Zhou L, Huang C, Shou X, Gong J, et al. A Novel gene mutation (1292 deletion) in a Chinese family with cerebral cavernous malformations. Neurosurgery. (2005) 56:1149–53; Discussion −53.
22. Ortiz L, Costa A, Bellido M, Solano F, García-Moreno J, Gamero M, et al. Study of cerebral cavernous malformation in Spain and Portugal: high prevalence of a 14 bp deletion in exon 5 of MGC4607 (CCM2 Gene). J Neurol. (2007) 254:322–6. doi: 10.1007/s00415-006-0359-9
23. Riant F, Cecillon M, Saugier-Veber P, Tournier-Lasserve E. CCM Molecular screening in a diagnosis context: novel unclassified variants leading to abnormal splicing and importance of large deletions. Neurogenetics. (2013) 14:133–41. doi: 10.1007/s10048-013-0362-0
24. Mondéjar R, Solano F, Rubio R, Delgado M, Pérez-Sempere A, González-Meneses A, et al. Mutation prevalence of cerebral cavernous malformation genes in Spanish patients. PloS one. (2014) 9:E86286. doi: 10.1371/journal.pone.0086286
25. Hirota K, Akagawa H, Kikuchi A, Oka H, Hino A, Mitsuyama T, et al. KRIT1 mutations in three japanese pedigrees with hereditary cavernous malformation. Hum Genome Variat. (2016) 3:16032. doi: 10.1038/hgv.2016.32
26. Lanfranconi S, Ronchi D, Ahmed N, Civelli V, Basilico P, Bresolin N, et al. A novel CCM1 mutation associated with multiple cerebral and vertebral cavernous malformations. BMC Neurol. (2014) 14:158. doi: 10.1186/s12883-014-0158-3
27. Zhang F, Xue Y, Zhang F, Wei X, Zhou Z, Ma Z, et al. Identification of a novel CCM1 frameshift mutation in a chinese han family with multiple cerebral cavernous malformations. Front Neurosci. (2020) 14:525986. doi: 10.3389/fnins.2020.525986
28. Zhao Y, Xie L, Li P, Song J, Qu T, Fan W, et al. A Novel CCM1 gene mutation causes cerebral cavernous malformation in a Chinese family. J Clinic Neurosci Offic J Neurosurgic Soc Australasia. (2011) 18:61–5. doi: 10.1016/j.jocn.2010.04.051
29. Antognelli C, Trapani E, Delle Monache S, Perrelli A, Daga M, Pizzimenti S, et al. KRIT1 Loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: implication for cerebral cavernous malformation disease. Free Radic Biol Med. (2018) 115:202–18. doi: 10.1016/j.freeradbiomed.2017.11.014
30. Yang C, Nicholas V, Zhao J, Wu B, Zhong H, Li Y, et al. A Novel CCM1/KRIT1 Heterozygous Nonsense Mutation (c. 1864C>T) associated with familial cerebral cavernous malformation: a genetic insight from an 8-year continuous observational study. J Mol Neurosci. (2017) 61:511–23. doi: 10.1007/s12031-017-0893-1
31. Li J, Yang T, Wang L, Yan H, Zhang Y, Guo Y, et al. Whole genome distribution and ethnic differentiation of copy number variation in caucasian and Asian populations. PloS one. (2009) 4:E7958. doi: 10.1371/journal.pone.0007958
32. Tsutsumi S, Ogino I, Miyajima M, Ikeda T, Shindo N, Yasumoto Y, et al. Genomic causes of multiple cerebral cavernous malformations in a Japanese population. J Clinic Neurosci Offic J Neurosurgic Soc Australasia. (2013) 20:667–9. doi: 10.1016/j.jocn.2012.05.041
33. Ji B, Qin W, Sun T, Feng G, He L, Wang Y. A Novel Deletion Mutation in CCM1 Gene (Krit1) Is Detected in a Chinese Family With Cerebral Cavernous Malformations. Yi Chuan xue bao = Acta Genetica Sinica. (2006) 33:105–10. doi: 10.1016/S0379-4172(06)60028-0
34. Wang X, Liu X, Lee N, Liu Q, Li W, Han T, et al. Features of a Chinese family with cerebral cavernous malformation induced by a novel CCM1 gene mutation. Chinese Med J. (2013) 126:3427−32.
35. Zhu H, Guo Y, Feng X, Zhang R, Zhou C, Li G, et al. Familial cerebral cavernous angiomas: clinical and genetic features in a Chinese family with a frame-shift mutation in the CCM1 Gene (Krit1). J Mol Neurosci: MN. (2014) 54:790–5. doi: 10.1007/s12031-014-0415-3
36. Mao C, Yang J, Zhang S, Luo H, Song B, Liu Y, et al. Exome capture sequencing identifies a novel CCM1 Mutation in a Chinese family with multiple cerebral cavernous malformations. Int J Neurosci. (2016) 126:1071–6. doi: 10.3109/00207454.2015.1118628
37. Wang H, Pan Y, Zhang Z, Li X, Xu Z, Suo Y, et al. A novel KRIT1/CCM1 gene insertion mutation associated with cerebral cavernous malformations in a Chinese family. J Mol Neurosci: MN. (2017) 61:221–6. doi: 10.1007/s12031-017-0881-5
Keywords: familial cerebral cavernous hemangioma, KRIT1/CCM1 gene, case report, literature review, Chinese family
Citation: Liu W, Liu M, Lu D, Wang J, Cao Z, Liu X, Feng Z, Huang B and Wang X (2022) A Chinese Family With Cerebral Cavernous Malformation Caused by a Frameshift Mutation of the CCM1 Gene: A Case Report and Review of the Literature. Front. Neurol. 13:795514. doi: 10.3389/fneur.2022.795514
Received: 15 October 2021; Accepted: 10 January 2022;
Published: 04 April 2022.
Edited by:
Dario Ronchi, University of Milan, ItalyReviewed by:
Lorenzo Pavone, University of Catania, ItalyJun Zhang, Texas Tech University Health Sciences Center, United States
Copyright © 2022 Liu, Liu, Lu, Wang, Cao, Liu, Feng, Huang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinyu Wang, d2FuZ3hpbnl1QHNkdS5lZHUuY24=
†These authors have contributed equally to this work