
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 23 January 2023
Sec. Multiple Sclerosis and Neuroimmunology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.1102484
This article is part of the Research TopicNeuroglial antibodies: from clinical associations to pathophysiological investigationsView all 8 articles
 Christopher M. Bartley1,2
Christopher M. Bartley1,2 Thomas T. Ngo1,2
Thomas T. Ngo1,2 Cathryn R. Cadwell1,3,4
Cathryn R. Cadwell1,3,4 Adil Harroud1,5
Adil Harroud1,5 Ryan D. Schubert1,5Bonny D. Alvarenga1,5Isobel A. Hawes1,5,6
Ryan D. Schubert1,5Bonny D. Alvarenga1,5Isobel A. Hawes1,5,6 Kelsey C. Zorn7
Kelsey C. Zorn7 Trung Hunyh1,5Lindsay H. Teliska8
Trung Hunyh1,5Lindsay H. Teliska8 Andrew F. Kung9
Andrew F. Kung9 Shailee Shah10,11Jeffrey M. Gelfand1,5
Shailee Shah10,11Jeffrey M. Gelfand1,5 Felicia C. Chow1,5
Felicia C. Chow1,5 Matthew N. Rasband8
Matthew N. Rasband8 Divyanshu Dubey12,13
Divyanshu Dubey12,13 Sean J. Pittock11,12,13Joseph L. DeRisi7,14
Sean J. Pittock11,12,13Joseph L. DeRisi7,14 Michael R. Wilson1,5*
Michael R. Wilson1,5* Samuel J. Pleasure1,5*
Samuel J. Pleasure1,5*Neuroinvasive infection is the most common cause of meningoencephalitis in people living with human immunodeficiency virus (HIV), but autoimmune etiologies have been reported. We present the case of a 51-year-old man living with HIV infection with steroid-responsive meningoencephalitis whose comprehensive pathogen testing was non-diagnostic. Subsequent tissue-based immunofluorescence with acute-phase cerebrospinal fluid revealed anti-neural antibodies localizing to the axon initial segment (AIS), the node of Ranvier (NoR), and the subpial space. Phage display immunoprecipitation sequencing identified ankyrinG (AnkG) as the leading candidate autoantigen. A synthetic blocking peptide encoding the PhIP-Seq-identified AnkG epitope neutralized CSF IgG binding to the AIS and NoR, thereby confirming a monoepitopic AnkG antibody response. However, subpial immunostaining persisted, indicating the presence of additional autoantibodies. Review of archival tissue-based staining identified candidate AnkG autoantibodies in a 60-year-old woman with metastatic ovarian cancer and seizures that were subsequently validated by cell-based assay. AnkG antibodies were not detected by tissue-based assay and/or PhIP-Seq in control CSF (N = 39), HIV CSF (N = 79), or other suspected and confirmed neuroinflammatory CSF cases (N = 1,236). Therefore, AnkG autoantibodies in CSF are rare but extend the catalog of AIS and NoR autoantibodies associated with neurological autoimmunity.
Immunocompromised individuals are vulnerable to neuroinvasive pathogens that can cause meningitis, encephalitis, and myelitis. Infections of the central nervous system (CNS) are a leading cause of death in persons with acquired immunodeficiency syndrome (AIDS) but are uncommon in those with well-controlled HIV. Yet, because CNS infection may still occur in well-controlled HIV (1, 2), comprehensive microbial testing and empiric antibiotics are warranted in patients with signs and symptoms consistent with CNS infection. However, ongoing neuroinflammation, despite comprehensive and persistently negative pathogen testing, presents a clinical dilemma.
Other causes of neuroinflammation in persons with well-controlled HIV include macrophage-mediated HIV encephalitis and CD8 encephalitis (3)—a hallmark of both of which is CNS HIV escape, in which HIV RNA can be detected in the CSF (4). Persons living with HIV are also at an increased risk for immune dysregulation (5, 6) and autoimmune disorders of the nervous system including acute and chronic inflammatory demyelinating polyneuropathy (7) (AIDP and CIDP, respectively).
Here, we present the case of a man with well-controlled HIV who developed steroid-responsive meningoencephalitis in the absence of HIV escape. Subsequent research studies identified a novel anti-ankyrinG (AnkG) antibody and a second antibody directed at a subpial antigen in his CSF. A search of archival neuroinflammatory cases identified an HIV-negative woman with a history of cancer and seizures who harbored the same anti-neuronal antibody but not the subpial antibody, indicating that AnkG may in rare cases be the target autoantigen in seronegative autoimmune neurological syndromes.
A 51-year-old man with well-controlled HIV (CD4+ count 791 cells/μL and undetectable viral load) on tenofovir alafenamide, emtricitabine, and dolutegravir presented to the emergency room with several weeks of headache, right-sided hearing loss, and memory impairment. He smoked one pack of cigarette per day and marijuana but did not use other drugs. He had lived in the San Francisco Bay Area for over 20 years and was an avid gardener. He did not have a history of incarceration, homelessness, or international travel (Figure 1A).
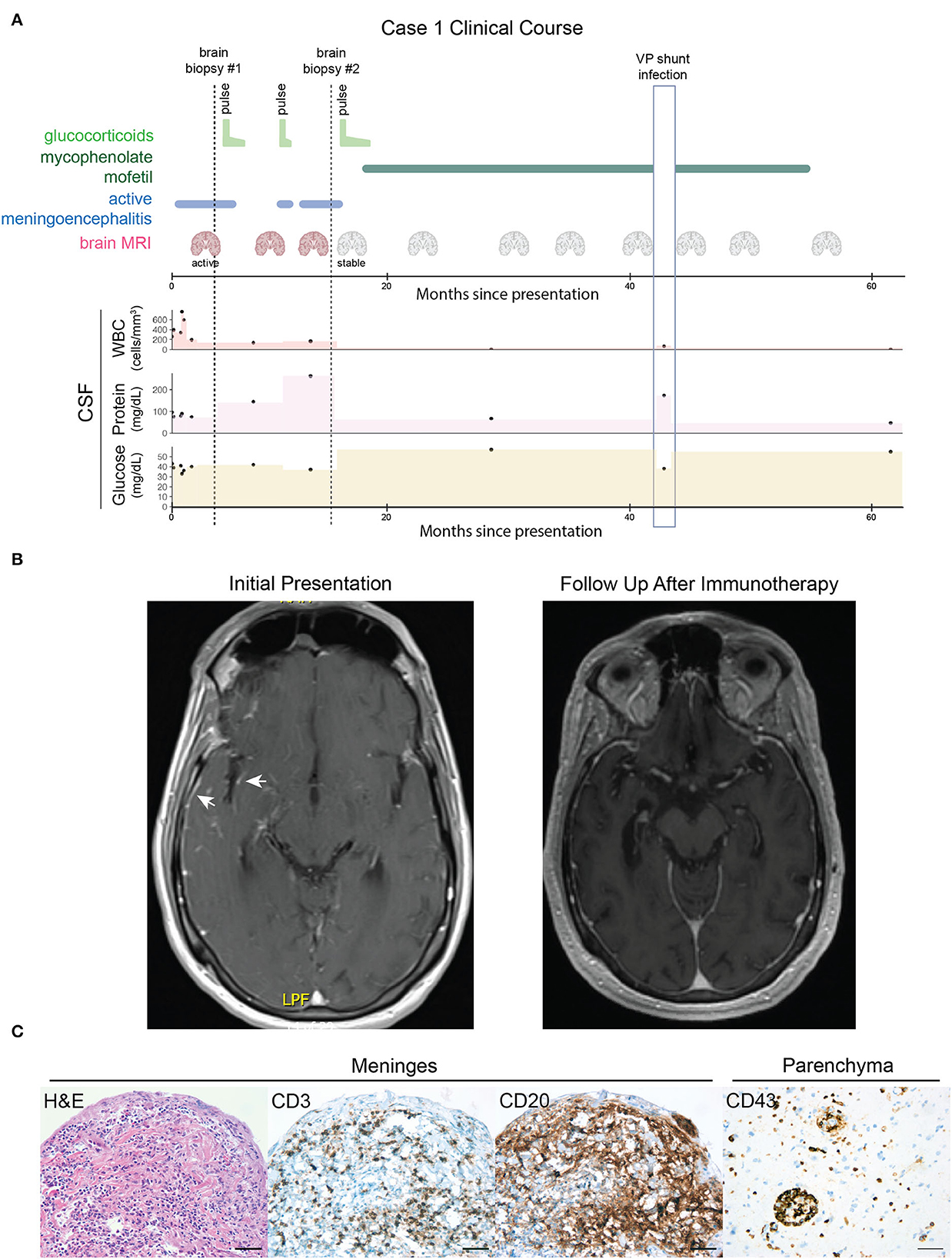
Figure 1. Case 1, course of illness and clinical studies. (A) A graphical time course of events. Brain biopsies are indicated by vertical dotted lines. Periods of glucocorticoid and mycophenolate mofetil immunosuppressive therapy are indicated by light and dark green lines, respectively. Active meningoencephalitis is indicated by the blue line. MRI scans are indicated by the coronal brain icon (red = active meningoencephalitis and gray = stable/no enhancement or worsening of the encephalitic process). Black dots indicate clinical CSF values for white blood count (WBC, ref. ≤5 cells/μL), protein (ref. ≤60 mg/dL), and glucose levels (ref. ≥50 and ≤80 mg/dL). Colored areas between dots have been added to aid visual assessment. The vertical light blue outlined box indicates a period of acute illness secondary to a confirmed ventriculoperitoneal (VP) shunt infection resulting in a 2-month interruption in mycophenolate mofetil treatment and transient CSF abnormalities thought to be unrelated to the autoimmune meningoencephalitis. (B) Axial T1 post-gadolinium MRIs demonstrate that the right temporal lobe and insular leptomeningeal enhancement present at initial presentation (arrows, left image) had resolved at a follow-up appointment after immunosuppressive therapy (right image). (C) Histopathology findings: hematoxylin and eosin (H&E) staining of a biopsy of the meninges revealed a dense, mixed inflammatory infiltrate. Immunohistochemistry of the meninges identified scattered CD3+ T cells and scattered CD20+ B cells. A separate biopsy of the brain parenchyma revealed perivascular and intraparenchymal collections of CD43+ inflammatory cells. Scale bars = 50 μm.
In the emergency department, he had a witnessed seizure and an MRI of the brain showed swelling of the right cerebral cortex, with abnormal diffusion, T2/FLAIR hyperintensity, and patchy enhancement, and without abnormal susceptibility weighted imaging (Figure 1B). The imaging was consistent with meningoencephalitis, and serial lumbar punctures (LP) revealed a neutrophil-predominant pleocytosis (WBC: 260–756/μL with 12–70% neutrophils), mild hypoglycorrhachia (nadir 33 mg/dL), and elevated protein (76–95 mg/dL). Gram stain and bacterial culture in CSF were negative, as was cryptococcal antigen. He was started on empiric valacyclovir and broad-spectrum antibiotics.
After 3 weeks of inpatient antimicrobial treatment, the patient failed to improve. Comprehensive serological and cerebrospinal fluid (CSF) testing for infectious etiologies was non-diagnostic including serum toxoplasmosis IgG and CSF fungal cultures, microbial antigen and antibody testing, VDRL, universal bacterial, fungal, and mycobacterial PCR, and metagenomic next-generation sequencing (mNGS). Diagnostic biopsy of the leptomeninges overlying the right cerebrum revealed dense connective tissue with a mixed inflammatory infiltrate including granulocytes, and B and T cells (Figure 1C), suggestive of an infectious etiology. The underlying cortical brain tissue was also biopsied and showed no significant pathological abnormality. Histochemical stains for acid-fast bacillus (AFB), Grocott's methenamine silver stain (GMS), and period acid-Schiff (PAS) stains on the brain tissue were negative, arguing against mycobacterial or fungal infection and Whipple's disease. A Quantiferon Gold TB test was negative. However, given the insensitivity of CNS TB diagnostics, the lack of clinical improvement, and a persistent neutrophil-predominant CSF pleocytosis with mild hypoglycorrhachia, he was started on empiric therapy for TB meningoencephalitis with isoniazid, rifampin, ethambutol, and pyrazinamide and transferred to an outpatient rehabilitation facility.
One week after discharge (week 7), the patient presented to the emergency department with new-onset expressive aphasia, hyponatremia, and ataxia. EEG revealed right greater than left diffuse background slowing and disorganization without epileptiform activity. Repeat LP again showed a neutrophilic pleocytosis (WBC 194/μL with 50% neutrophils), protein 75 mg/dL, elevated IgG index (0.7, ref. ≤0.6), and five restricted oligoclonal bands (OCBs). Brain MRI showed persistent meningoencephalitis with new ventriculomegaly. A large volume LP was performed. The opening pressure was 25 cm and the patient regained his speech after 1 h later, had improved gait, and improved mental status. He proceeded to get a ventriculoperitoneal shunt and continued on anti-TB therapy.
A CSF autoimmune encephalopathy panel was reported as positive for NMDA receptor autoantibodies (1:8 tissue titer) as well as putatively non-specific additional staining on tissue. The whole body PET and scrotal ultrasound were negative. A repeat autoimmune encephalopathy panel was NMDA receptor antibody positive on cell-based assay but negative on reflex tissue staining. Because the clinical phenotype was markedly inconsistent with anti-NMDAR encephalitis, empiric anti-TB therapy was initially continued. Nonetheless, an autoimmune etiology was increasingly considered as a result of the repeatedly negative infectious workup and lack of improvement in antibiotic therapy. In this context, he received a 5-day course of methylprednisolone 1 gram daily on week 8 followed by a prednisone taper with dramatic improvement in his memory and language. Anti-TB therapy was ultimately discontinued after ~1 month of treatment.
Over the following months, attempts to taper off prednisone resulted in clinical and radiological deterioration. Surveillance brain MRIs demonstrated worsening of meningoencephalitis with a new focus on enhancement in the left amygdala. A second brain biopsy of glucocorticoids at month 10 showed severe granulomatous and suppurative meningoencephalitis with a perivascular and intraparenchymal lymphocytic infiltrate, diffuse astrogliosis, and microglial activation. The pattern of inflammation was again suspicious for mycobacterial infection; however, AFB and AFB-FITE stains and AFB culture of the brain tissue were negative, as were mycobacterial universal PCR and metagenomic next-generation sequencing.
Three weeks after the second biopsy, the patient was retreated with IV methylprednisolone followed by oral prednisone maintenance with subsequent clinical improvement over the next few months. Repeat imaging 3 months after the resumption of glucocorticoid treatment demonstrated near the total resolution of leptomeningeal enhancement and non-progression of his encephalitis. The patient was then transitioned empirically to mycophenolate mofetil and successfully weaned off prednisone without recurrence of his meningoencephalitis. He remained seizure-free on antiseizure therapy. Repeat CSF at 26 months post-presentation showed normal cellularity (3 WBC/μL with 87% lymphocytes and 17% monocytes), normal glucose (57 mg/dL), mildly elevated protein (67 mg/dL), persistent elevated IgG index (0.7 ref. ≤0.6), and two oligoclonal bands. MRI brain every 6 months showed stable multifocal T2/FLAIR cortical and white matter hyperintensities without associated enhancement. Following 3 years of immunosuppressive therapy, 4.5 years after his initial presentation, mycophenolate mofetil was stopped and the patient remained stable. Neurological assessment documented fixed sequelae consisting of dysarthria, executive dysfunction, and episodic memory impairment.
A 60-year-old woman with a history significant for smoking, non-insulin-dependent type II diabetes, renal cell carcinoma status post-left radical nephrectomy and node dissection and essential hypertension, and no family history of autoimmune neurologic or oncologic disease was diagnosed with stage IIIC metastatic ovarian cancer. She underwent bilateral salpingoopherectomy and supracervical hysterectomy, and carboplatin and taxol therapy. Two years later, in the context of recurrent ovarian cancer again undergoing carboplatin, taxol, and avastin therapy, she developed seizures with interictal abnormalities noted in the left frontotemporal region on EEG. Her MRI at the time had bilateral parieto-occipital FLAIR abnormalities without evidence of meningeal involvement. Her spinal fluid was notable for elevated protein. Her seizures were attributed to posterior reversible encephalopathy syndrome (PRES) in the setting of carboplatin use. Six years after her initial diagnosis, she had a recurrence of her ovarian cancer. She was placed on tamoxifen, and over the next 18 months, she was treated with topotecan and bevacizumab (15 cycles), paclitaxel (five cycles), gemcitabine (two doses), and doxorubicin. Eight years after her initial diagnosis, she again developed seizures characterized by unresponsiveness, rightward head and eye deviation, and right upper extremity tonic–clonic movements. EEGs at the time captured left temporal seizures and interictal abnormalities and were treated with levetiracetam. An MRI brain with contrast at the time showed non-specific ischemic changes without metastatic disease. Her spinal fluid was non-inflammatory, and autoantibody testing at the time was negative. Her CSF protein was elevated (100 mg/dL), and oligoclonal bands were not sent. Four months later, she was admitted to the hospital with neutropenic fever (absolute neutrophil count 0.3), mouth sores, and dysphagia, and treated for sepsis. Due to ongoing failure to thrive, the patient elected to pursue hospice and she subsequently died.
CSF from cases 1 and 2 was screened for autoantibodies by tissue-based immunofluorescence (TBIF). At a 1:100 dilution, acute-phase CSF from case 1 produced a pattern of immunoreactivity that localized to axon initial segment (AIS)-like structures throughout the cortex and node of Ranvier (NoR)-like structures in the optic tract. In contrast, case 1's convalescent CSF was not immunoreactive at a 1:100 dilution and stained only weekly at a 1:4 dilution, suggesting a decrease in the autoantibody titer following immunosuppressive therapy (Figure 2A).
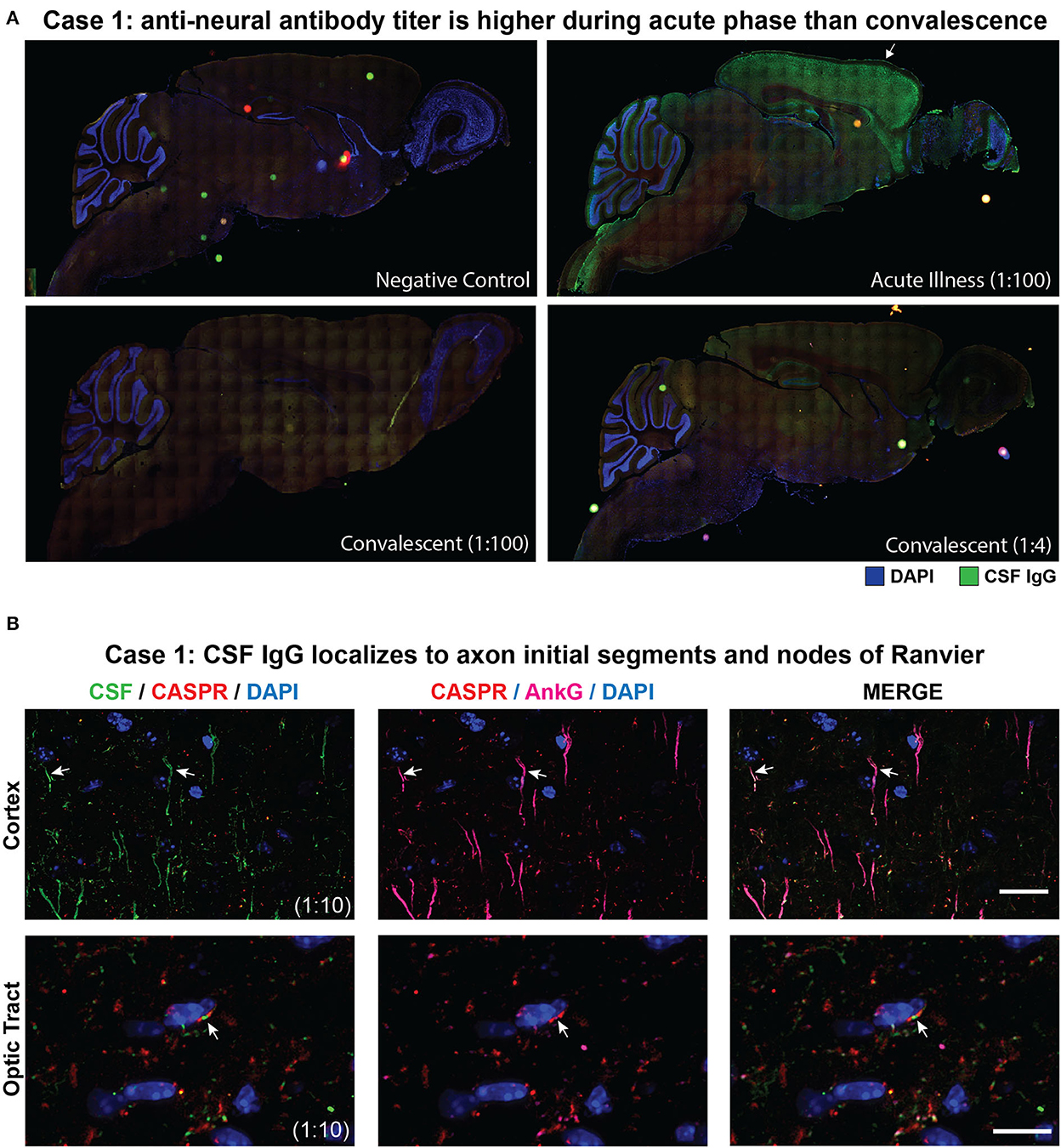
Figure 2. Tissue-based immunofluorescence (TBIF) assays. (A) Panoramic images of sagittal mouse brain TBIF with acute-phase CSF at a 1:100 dilution or convalescent CSF at a 1:4 dilution. (B) Colocalization of case 1 acute-phase CSF with AnkG at the AIS in the cortex (arrows, top row) and NoR in the optic tract (arrows, bottom row). Nodal CSF IgG localizes to the NoR delimited by paranodal CASPR (red). Scale bars = 10 μm.
To verify immunoreactivity to the AIS and NoR, we immunostained mouse brain tissue with acute-phase CSF and commercial antibodies to the AIS and nodal marker AnkG and the paranodal marker protein contactin-associated protein 1 (CASPR). Case 1 CSF IgG colocalized with AnkG at the AIS and NoR, thereby confirming AIS-NoR immunoreactivity (Figure 2B). A review of TBIF results from a separate study revealed that none of 46 CSF samples from people living with HIV produced AIS/NoR immunostaining (8).
Case 2 was identified as part of a screen for TRIM46 autoimmunity (9). CSF was screened at a 1:4 dilution and produced a similar AIS and NoR-like pattern of immunoreactivity throughout the murine brain (Supplementary Figure 1A) but was negative for TRIM46 on the cell-based assay. Owing to limited sample volume, co-staining with commercial AnkG and CASPR antibodies was not performed, and the remaining CSF was reserved for candidate autoantigen discovery and validation.
We did not identify another case of anti-AIS/NoR immunoreactivity among the 39 control and 1,236 candidate CNS neuroinflammatory patients screened by TBIF in our laboratory.
To identify candidate anti-AIS/NoR autoantibodies, we screened both cases in technical replicate by phage display immunoprecipitation sequence (PhIP-Seq) (10). We restricted our analysis to AIS/NoR proteins (as defined by EMBL-EBI, see Supplementary methods) and normalized total protein enrichments to 4,215 control samples (comprised of beads only and healthy CSF and serum including replicates). Case 1 nominally enriched AnkG 2.4-fold above controls (Z-score of 0.6); however, case 2 failed to enrich any AIS/NoR proteins (Figure 3A).
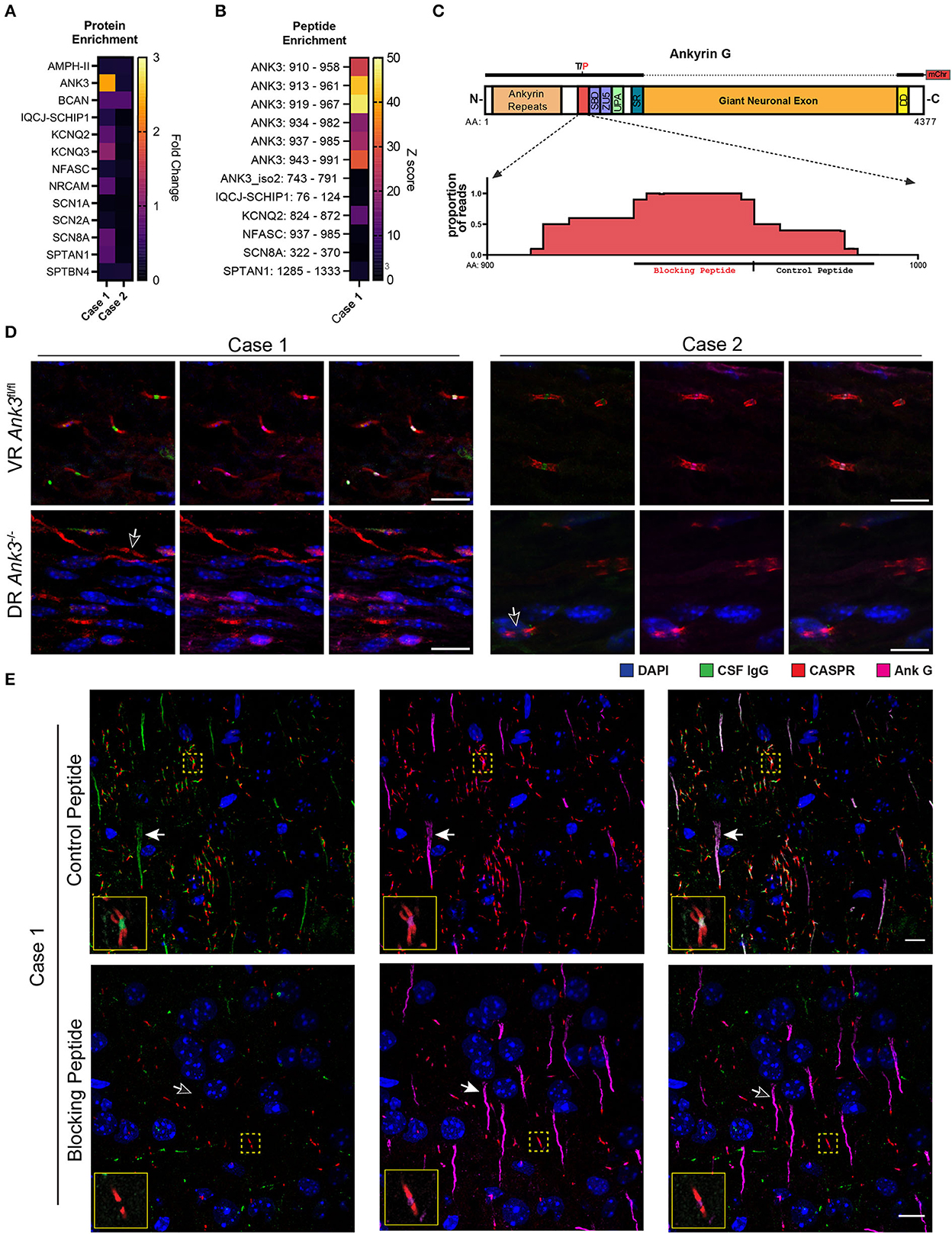
Figure 3. Validation of AnkG autoantibodies in the CSF of cases 1 and 2. (A) A heatmap of the fold change of PhIP-Seq AIS and NoR protein enrichments for cases 1 and 2 CSF relative to control PhIP-Seq runs (N = 4,216, comprised of beads only and healthy control CSF and serum). (B) Heatmap of case 1 acute-phase CSF AIS and NoR peptides enriched with ≥10-fold above controls (as above) in both technical replicates relative to the same controls as above. Individual peptides are annotated as “GENE: amino acid range” (e.g., ANK3: 910–958). Enrichment values are expressed as Z-scores. (C) Model of AnkG, its protein domains, and the case 1 target epitope. SBD, spectrin-binding domain (first ZU5 domain); UPA, UPA domain; SR, serine rich; DD, death domain. Above the model is a representation of the 270 kDa rat AnkG-mCherry construct used for the CBAs (T/P indicates that rats encode a proline residue instead of the threonine residue present in humans and mice). The dotted line indicates that the giant neuronal exon is absent in the 270 kDa construct. Below the model is a pileup graph of the position of residues encoded by AnkG-enriched peptides mapped to human AnkG (Uniprot.org Q12955-3). The relative positions of the synthetic blocking and control peptides used in TBIF experiments are indicated below the pileup map. (D) Cases 1 and 2 CSF immunostaining of AnkG-expressing ventral roots (VR-Ank3fl/fl) and AnkG-deficient dorsal roots (DR-Ank3−/−) from AdvillinCre/+ and Ank3flox/flox mice. The white outlined arrow indicates the absence of nodal immunostaining by CSF IgG (green). (E) Immunostaining of mouse cortex with case 1 CSF after preabsorption with the control peptide (top row) or blocking peptide (lower row). Filled arrows indicate the AIS, whereas the unfilled arrow indicates the absence of CSF IgG immunostaining of the AIS in the blocking peptide condition. The solid yellow outlined insets show the NoRs in the yellow dotted squares at higher magnification. Scale bar = 10 μm.
We hypothesized that spurious enrichment of non-target peptides could attenuate the contribution of individual target peptides to total protein enrichment values. Therefore, we filtered for AIS/NoR peptides that were enriched at least 10-fold in both technical replicates. For case 1, more than half of enriched peptides mapped to ankyrinG, six of which shared an epitope and had Z-scores ranging between 18 and 47 (Figure 3B). Multiple sequence alignment indicated that case 1's putative epitope lays within amino acids (AA) 937–958 (Figure 3C).
Because of case 1's HIV status, we reviewed PhIP-Seq data from 33 HIV CSF samples (N = 15 well-controlled and N = 18 untreated HIV) and found that neither AnkG nor epitope AnkG937−958 were enriched above negative controls (8). Furthermore, a BLAST search revealed that this epitope does not harbor sequence similarity to HIV arguing against molecular mimicry. Although NMDAR antibodies have been reported in anti-GFAP autoimmune meningoencephalitis (11), GFAP peptides were not observed in case 1's PhIP-Seq data despite being enriched by a commercial polyclonal antibody to GFAP as a positive control (data not shown).
In contrast to case 1, a peptide-level analysis of case 2 PhIP-Seq data failed to identify significant AIS/NoR peptide enrichments (data not shown) suggesting that case 2's autoantibodies recognize conformational or post-translationally modified epitopes that are not represented in our PhIP-Seq library.
Surprisingly, case 1 failed to bind rat-mCherry-AnkG (data not shown). However, a comparison of human, rat, and murine ankyrinG revealed that rat AnkG differs from human and murine AnkG by a single amino acid (proline in rat vs. threonine in human and mouse) in the center of the putative AnkG937−958 epitope (Figure 3C).
Based on the PhIP-Seq and TBIF findings, we hypothesized that case 2's target epitopes were conformational or post-translationally modified. We screened case 2's CSF against previously established AnkG and βIV spectrin variant Σ1 and Σ6 HEK 293T cell-based assays (12). We found that case 2 was strongly immunoreactive to overexpressed rat-mCherry-AnkG (Supplementary Figure 1B) but not βIV spectrin Σ1 or Σ6 (data not shown).
To confirm anti-AnkG antibodies in both cases, we immunostained conditional AnkG knockout mice with patient's CSF. The Advillin promoter is active in dorsal sensory neurons but inactive in ventral motor neurons (13). Therefore, we immunostained ventral root motor neurons (VR-Ank3flfl) and dorsal root sensory neurons (DR-Ank3−/−) from Advillin-Cre:Ank3fl/fl mice with CSF from cases 1 and 2. In both cases, CSF IgG localized to VR-Ank3flfl NoR but not DR-Ank3−/− NoR, thereby confirming that the staining was due to the presence of AnkG autoantibodies in both cases' CSF samples (Figure 3D).
We hypothesized that case 1's anti-AnkG antibody population was monoepitopic because AnkG PhIP-Seq-enriched peptides shared a common epitope. Although PhIP-Seq lacks conformational and post-translational epitopes, the negative cell-based assay using rat AnkG further suggested that case 1's autoantibody binding was restricted to amino acid 937–958.
To test for monoepitopic antibody binding, we commercially synthesized a peptide encoding case 1's putative epitope (blocking peptide: AnkG934−961) and a peptide encoding the adjacent C-terminal 25 AA as a non-epitope peptide control (control peptide: AnkG962−989) (Figure 3C).
In technical quadruplicate, we immunostained rodent brain tissue with acute-phase CSF without peptide, or after pre-incubation with control or blocking peptide in five molar excess. Preincubation of case 1 CSF with the blocking peptide, but not the control peptide, completely abrogated CSF IgG binding to both the AIS and NoR without affecting the binding of a commercial AnkG antibody (NeuroMab N106/36) that binds outside of the control and blocking epitopes (Figure 3E).
We observed that the AnkG blocking peptide abrogated AIS and NoR immunoreactivity but not the limited nuclear and subpial CSF IgG immunoreactivity (Figure 4). However, a review of the PhIP-Seq data failed to yield an obvious candidate antigen that would explain the subpial immunostaining.
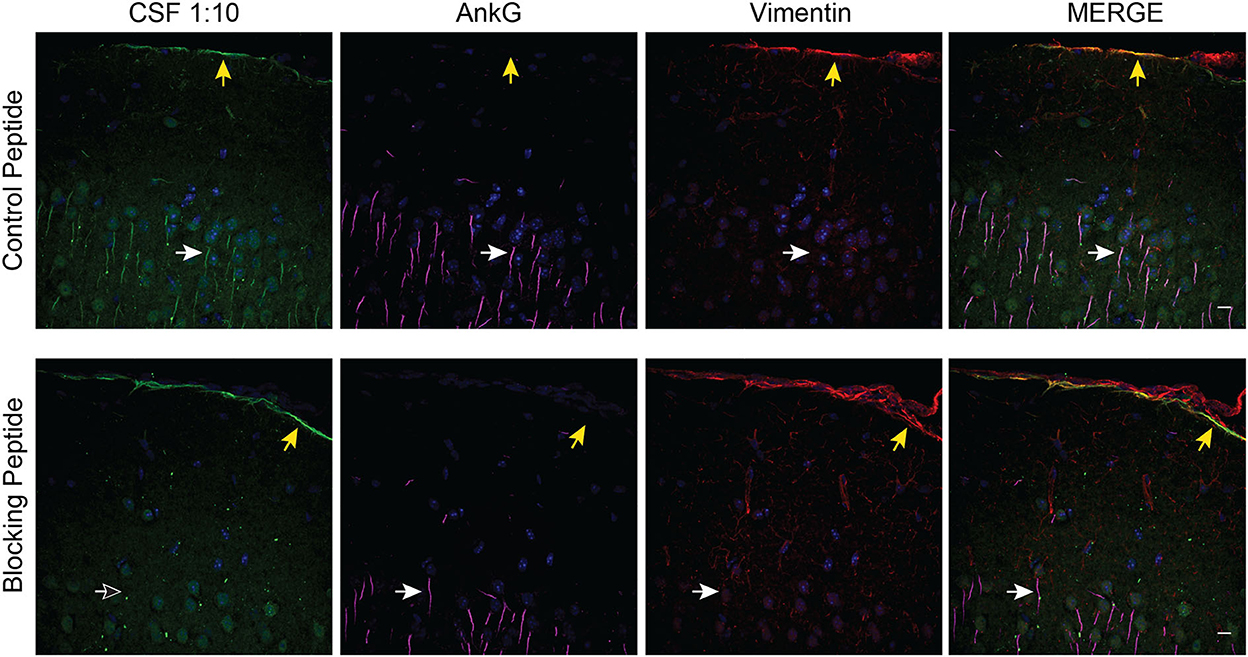
Figure 4. Identification of a subpial autoantibody. Case 1 CSF TBIF of murine cortex with control or blocking peptide. Tissue was co-stained with AnkG (magenta) and Vimentin (red) to identify the overlying pial tissue. Yellow arrows point to subpial CSF IgG. White arrows point to an AIS. The empty arrow in the lower left panel indicates the absence of AIS immunostaining. Scale bar = 10 μm.
This case demonstrates the complexity of diagnosis and clinical management of people living with well-controlled HIV who present with neuroinflammatory syndromes. Although broad-based testing of blood, CSF, and brain tissue for infections was consistently negative, it is possible that a preceding CNS infection was the precipitant of this patient's autoimmune meningoencephalitis. Nonetheless, this case also highlights the need for continued autoantibody discovery in seronegative autoimmune CNS syndromes to decrease the time to treatment.
Despite negative infectious testing, case 1's low CSF glucose sustained concern for an undetected microorganism. Hypoglycorrhachia is common in infectious meningitis and is unlikely to be due to pleocytosis alone in aseptic meningitis except for specific non-infectious syndromes such as systemic lupus erythematosus (14), which case 1 did not have. Hypoglycorrhachia can also be observed in neurosarcoidosis, but the absence of sarcoidosis signs and symptoms on physical examination, laboratory studies, body imaging acutely, and longitudinal yearly followup were inconsistent with this diagnosis. Low CSF glucose has also been reported in autoimmune meningoencephalitis due to checkpoint inhibitor therapy (15–17) and GFAP meningoencephalitis (18)—the most common known autoantigen associated with autoimmune meningoencephalits (11). Although we did not test for GFAP antibodies by CBA, our case did not show GFAP-like staining by TBIF.
Although IgG class NMDAR autoantibodies were detected in the CSF of our patient, they were low titer and inconsistently detected on clinical testing; this, along with the lack of typical clinical features of anti-NMDAR encephalitis, led us to conclude that these represented false-positive anti-NMDAR testing, which is known to occur (19). NMDAR antibodies have also been observed in the setting of HIV escape (20), infectious meningoencephalitis (21, 22), and GFAP astrocytopathy (11). However, in these cases, as in ours, the phenotype was atypical for NMDAR encephalitis.
Instead, research testing identified a high-titer antibody to AnkG that decreased in titer during convalescence. AnkG is enriched in the AIS and NoR, subcellular structures with similar molecular profiles that are targeted by autoantibodies in diverse autoimmune neurological disorders including antibodies that bind to gangliosides (23), neurofascins (24, 25), CASPR (26), CASPR2 (27, 28), TRIM46 (9, 29), and βIV-spectrin (12). Grouping autoantibody-mediated neurological patients by target epitope may reveal unappreciated relationships between target epitope and clinical characteristics (30, 31). Because AnkG is an intracellular protein, anti-AnkG antibodies may not be directly pathogenic and may instead be reflective of autoreactive T-cell activity. However, both T- and B-cell infiltrates were detected in neuropathology.
Although intracellular, AnkG is a membrane-associated protein owing to its interaction with many AIS-enriched membrane proteins, an N-terminal palmitoyl moiety (32), and an adjacent solenoid ankyrin repeat domain that makes periodic contact with the plasma membrane (33). Using a synthetic blocking peptide, we were able to localize case 1's anti-AnkG epitope within amino acids 937–957, which lie in the linker region between the ankyrin repeat and the spectrin-binding domain (34). Replacement of AnkG's linker with the ankyrin B's linker region disrupts AnkG's association with the plasma membrane leading to an intracellular localization (35). As autoantibodies targeting other peripheral membrane proteins may be directly pathogenic (36, 37), it remains to be seen whether anti-AnkG937−957 antibodies directly alter AnkG function.
The AnkG937−957 blocking peptide also allowed us to identify a distinct population of subpial autoantibodies in the CSF of case 1. Although we were unable to identify the antigen, this subpial immune response may explain the different clinical phenotypes between case 1 and case 2. Alternatively, the difference may be because case 1 and case 2 target different AnkG target epitopes (30, 31) or anti-AnkG antibodies are rare epiphenomena of various neuroinflammatory processes.
Although AnkG autoantibodies have been reported in sera from healthy controls and individuals with Alzheimer's disease (38), these samples were not tested by TBIF, and the finding was not replicated (39). By TBIF, AIS-specific immunoreactivity has only been reported with CSF or serum from patients with TRIM46 (9, 29) or βIV-spectrin (12, 40) autoimmune neurological disorders. In contrast, we have not observed AIS immunostaining with CSF and serum from patients with systemic autoimmunity, cancer without neurological illness, or healthy controls (9).
Here, we report for the first time AnkG autoantibodies in the CSF of two patients with neurological illness. Because case 1 is living with HIV, we verified that AnkG autoantibodies were not secondary to his HIV status (8). Albeit extremely rare, these data suggest that, like myriad other antibodies targeting the AIS and NoR, AnkG autoantibodies are biomarkers of neurological autoimmunity.
Case 1 AnkG patient was inconsistently positive for NMDAR antibodies; therefore, we cannot rule out that this immune response contributed to his symptoms despite the atypical phenotype though that would be extremely unlikely clinically. It is possible that anti-AnkG occurred as a post-infectious autoimmune phenomenon in case 1, but the specific infection was not identified. Because CNS autoimmunity was not considered in case 2, she was not treated with immunotherapy. Moreover, both cases had different symptoms, suggesting that AnkG autoantibodies may not be biomarkers of a coherent neuroinflammatory syndrome. Furthermore, although we confirmed the presence of a distinct subpial antibody, we could not confirm its identity.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by University of California San Francisco Institutional Review Board (IRB) numbers 13-12236 and 15-18425, and by Mayo Clinic Institutional Review Board (IRB) numbers 08-006647 and 08-007846. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements. All procedures used in this study complied with federal guidelines and the institutional policies of the UCSF, Mayo Clinic, and Baylor College of Medicine Institutional Animal Care and Use Committees.
CB: conceptual design, experimental design, tumor histology, IHC, CBAs, PhIP-Seq analysis, data interpretation, and writing. TN: CBAs, IHC, confocal microscopy, and writing. CC: neuropathological studies and editing. AH: clinical data and writing and editing. BA and IH: PhIP-Seq assays. RS: PhIP-Seq and writing and editing. KZ: clinical data collection and patient sample collection. TH and MR: AnkyrinG-deficient mice and writing and editing. AK and JD: PhIP-Seq database design. SS, JG, FC, and SeP: clinical data and editing. DD: tissue-based assay, clinical data, and writing and editing. MW and SaP: conceptual design, experimental design, data interpretation, and writing. All authors contributed to the article and approved the submitted version.
Confocal microscopy with the CSU-W1 spinning disk was supported by the S10 Shared Instrumentation grant (1S10OD017993-01A1). Sequencing was performed at the UCSF CAT, supported by UCSF PBBR, RRP IMIA, and NIH 1S10OD028511-01 grants. This work was further supported by NIMH R01MH122471 (SPl, MW, KZ, and JD) and R25MH060482 (CB), NINDS K08NS096117 (MW), the Brain Research Foundation (SPl), the Westridge Foundation (MW), NINDS R25NS070680 (CC), NINDS K08NS126573 (CC), NINDS R35NS122073 (MR), the National Multiple Sclerosis Society Clinician Scientist Development Award FAN-1608- 25607 (RS), the Sandler Program for Breakthrough Biomedical Research (MW and SPl), the John A. Watson Scholar Program, UCSF (CB), the Hanna H. Gray Fellowship, the Howard Hughes Medical Institute (CB), the President's Postdoctoral Fellowship Program, the University of California (CB), and the Deeda Blair Research Initiative for Disorders of the Brain (CB).
We thank Delaine Larsen, Ph.D., Kari Herrington, Ph.D., and SoYeon Kim, Ph.D., of the University of California, San Francisco, Nikon Imaging Center, for their imaging support.
CB owns shares in NowRx Inc. JG has received unrelated clinical trial support through UCSF from Roche/Genentech and Vigil Neurosciences, personal fees for consulting from Biogen, and personal fees for medical-legal consulting. FC has received personal fees for medical-legal consulting. SS has received personal compensation for serving on medical advisory boards for Alexion Pharmaceuticals, and Horizon Therapeutics. DD has patents pending for KLHL11-IgG, LUZP4-IgG, and cavin-4-IgG as markers of neurological autoimmunity. DD has also received funding from the US Department of Defense (DOD) (CA210208). DD has consulted for UCB, Immunovant, Argenx, and Astellas. All compensation for consulting activities is paid directly to Mayo Clinic. SeP has received personal compensation for serving as a consultant for Genentech, Sage Therapeutics, and Astellas. He's received personal compensation for serving on scientific advisory boards or data safety monitoring boards for F. Hoffman-LaRoche AG, Genentech, and UCB. His institution has received compensation for serving as a consultant for Astellas, Alexion, and Viela Bio/MedImmune. All compensation is paid to Mayo Clinic. He has received research support from Alexion, Viela Bio/MedImmune, Roche/Genentech. He has a patent, Patent# 8,889,102 (Application#12-678350, Neuromyelitis Optica Autoantibodies as a Marker for Neoplasia)—issued; a patent, Patent# 9,891,219B2 (Application#12-573942, Methods for Treating Neuromyelitis Optica (NMO) by Administration of Eculizumab to an individual that is Aquaporin-4 (AQP4)-IgG Autoantibody positive)—issued. JD has received grants from the Chan Zuckerberg Biohub, personal fees from the Public Health Company and from Allen & Company. MW has received unrelated grants from Roche/Genentech and Novartis as well as speaking honoraria from Novartis, Takeda, WebMD and Genentech.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.1102484/full#supplementary-material
Supplementary Figure 1. Identification and validation of anti-AnkG antibodies in case 2 CSF by TBIF and CBA. (A) TBIF of murine cortex with CSF from case 2 shows AIS-like structures in the cortex (arrow), hippocampus, and the cerebellum (ml, molecular layer; gl, granule cell layer) and NoR-like structures in the midbrain. (B) 270 kDa rat AnkG-mCherry CBA. HEK 293T cells were transfected with AnkG-mCherry, fixed, permeabilized, and stained with CSF at a 1:100 dilution and DAPI. Scale bar = 10 μm.
1. Lynch C, Green S, Lewis N, Megson C, Parsons H. Neurologic symptomatic cryptococcal antigenaemia in a patient with well-controlled human immunodeficiency virus. Clin Infect Dis. (2019) 70:1521–2. doi: 10.1093/cid/ciz658
2. Yoon Ha, Riska PF, Jain R, Morales C, Pirofski L-a. Unexpected case of cryptococcal meningoencephalitis in a patient with long-standing well-controlled HIV infection. Med Mycol Case Rep. (2021) 32:14–6. doi: 10.1016/j.mmcr.2021.01.003
3. Johnson TP, Nath A. Biotypes of HIV-associated neurocognitive disorders based on viral and immune pathogenesis. Curr Opin Infect Dis. (2022) 35:223–30. doi: 10.1097/QCO.0000000000000825
4. Winston A, Antinori A, Cinque P, Fox HS, Gisslen M, Henrich TJ, et al. Defining cerebrospinal fluid HIV RNA escape: editorial review AIDS. Aids. (2019) 33(Suppl. 2:S107–11. doi: 10.1097/QAD.0000000000002252
5. Lederman MM, Funderburg NT, Sekaly RP, Klatt NR, Hunt PW. Residual immune dysregulation syndrome in treated HIV infection. Adv Immunol. (2013) 119:51–83. doi: 10.1016/B978-0-12-407707-2.00002-3
6. Genet P, Sutton L, Chaoui D, Jijakli AA, Gerbe J, Masse V, et al. Prevalence of monoclonal gammopathy in HIV patients in 2014. J Int AIDS Soc. (2014) 17(4 Suppl. 3):19649. doi: 10.7448/IAS.17.4.19649
7. Verma A. Epidemiology and clinical features of HIV-1 associated neuropathies. J Peripher Nerv Syst. (2001) 6:8–13. doi: 10.1046/j.1529-8027.2001.006001008.x
8. Hawes IA, Browne W, Wapinarski A, Dandekar R, Bartley CM, Sowa G, et al. Unbiased serology reveals autoimmunity and HIV antibody signatures in HIV CNS Escape. MedRxivorg. (2022). doi: 10.1101/2022.12.13.22283439
9. Valencia-Sanchez C, Knight AM, Hammami MB, Guo Y, Mills JR, Kryzer TJ, et al. Characterisation of TRIM46 autoantibody-associated paraneoplastic neurological syndrome. J Neurol Neurosurg Psychiatry. (2021) 93:196–200. doi: 10.1136/jnnp-2021-326656
10. O'Donovan B, Mandel-Brehm C, Vazquez SE, Liu J, Parent AV, Anderson MS, et al. High-resolution epitope mapping of anti-Hu and anti-Yo autoimmunity by programmable phage display. Brain Commun. (2020) 2:fcaa059. doi: 10.1093/braincomms/fcaa059
11. Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. (2016) 73:1297–307. doi: 10.1001/jamaneurol.2016.2549
12. Bartley CM, Ngo TT, Alvarenga BD, Kung AF, Teliska LH, Sy M, et al. βIV-spectrin autoantibodies in 2 individuals with neuropathy of possible paraneoplastic origin: a case series. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e1188. doi: 10.1212/NXI.0000000000001188
13. Ho TS, Zollinger DR, Chang KJ, Xu M, Cooper EC, Stankewich MC, et al. A hierarchy of ankyrin-spectrin complexes clusters sodium channels at nodes of Ranvier. Nat Neurosci. (2014) 17:1664–72. doi: 10.1038/nn.3859
14. Baud MO, Vitt JR, Robbins NM, Wabl R, Wilson MR, Chow FC, et al. Pleocytosis is not fully responsible for low CSF glucose in meningitis. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e425. doi: 10.1212/NXI.0000000000000425
15. Larkin J, Chmielowski B, Lao CD, Hodi FS, Sharfman W, Weber J, et al. Neurologic serious adverse events associated with nivolumab plus ipilimumab or nivolumab alone in advanced melanoma, including a case series of encephalitis. Oncologist. (2017) 22:709–18. doi: 10.1634/theoncologist.2016-0487
16. Stein MK, Summers BB, Wong CA, Box HL, Cleveland KO. Meningoencephalitis following ipilimumab administration in metastatic melanoma. Am J Med Sci. (2015) 350:512–3. doi: 10.1097/MAJ.0000000000000584
17. Quach HT, Robbins CJ, Balko JM, Chiu CY, Miller S, Wilson MR, et al. Severe epididymo-orchitis and encephalitis complicating anti-PD-1 therapy. Oncologist. (2019) 24:872–6. doi: 10.1634/theoncologist.2018-0722
18. Kapadia RK, Ney DE, Hannan M, Farley M, Pastula DM, Piquet al. Glial fibrillary acidic protein (GFAP) associated autoimmune meningoencephalitis in a patient receiving nivolumab. J Neuroimmunol. (2020) 344:577259. doi: 10.1016/j.jneuroim.2020.577259
19. Knudtzen FC, Nilsson AC, Skarphedinsson S, Blaabjerg M. False-positive anti-NMDA receptor antibodies in severe case of Lyme neuroborreliosis. Neurol Sci. (2020) 41:197–9. doi: 10.1007/s10072-019-04004-0
20. Moloney PB, Hutchinson S, Heskin J, Mulcahy F, Langan Y, Conlon NP, et al. Possible N-methyl-D-aspartate receptor antibody-mediated encephalitis in the setting of HIV cerebrospinal fluid escape. J Neurol. (2020) 267:1348–52. doi: 10.1007/s00415-019-09693-3
21. Xiaoli C, Qun W, Jing L, Huan Y, Si C. Concurrent tuberculous meningoencephalitis and anti-NMDAR encephalitis: a case report. Front Neurol. (2022) 13:870607. doi: 10.3389/fneur.2022.870607
22. Issa N, Martin C, Dulau C, Camou F. Severe anti-GFAP meningo-encephalomyelitis following viral infection. Multiple Scler Relat Disord. (2020) 45:102448. doi: 10.1016/j.msard.2020.102448
23. Hafer-Macko C, Hsieh ST, Li CY, Ho TW, Sheikh K, Cornblath DR, et al. Acute motor axonal neuropathy: an antibody-mediated attack on axolemma. Ann Neurol. (1996) 40:635–44. doi: 10.1002/ana.410400414
24. Devaux JJ, Odaka M, Yuki N. Nodal proteins are target antigens in Guillain-Barré syndrome. J Peripher Nerv Syst. (2012) 17:62–71. doi: 10.1111/j.1529-8027.2012.00372.x
25. Prüss H, Schwab JM, Derst C, Görtzen A, Veh RW. Neurofascin as target of autoantibodies in Guillain-Barre syndrome. Brain. (2011) 134(Pt 5):e173. doi: 10.1093/brain/awq372
26. Doppler K, Appeltshauser L, Villmann C, Martin C, Peles E, Krämer HH, et al. Auto-antibodies to contactin-associated protein 1 (Caspr) in two patients with painful inflammatory neuropathy. Brain. (2016) 139(Pt 10):2617–30. doi: 10.1093/brain/aww189
27. Seery N, Butzkueven H, O'Brien TJ, Monif M. Contemporary advances in antibody-mediated encephalitis: anti-LGI1 and anti-Caspr2 antibody (Ab)-mediated encephalitides. Autoimmun Rev. (2022) 21:103074. doi: 10.1016/j.autrev.2022.103074
28. Irani SR, Pettingill P, Kleopa KA, Schiza N, Waters P, Mazia C, et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol. (2012) 72:241–55. doi: 10.1002/ana.23577
29. van Coevorden-Hameete MH, van Beuningen SFB, Perrenoud M, Will LM, Hulsenboom E, Demonet J-F, et al. Antibodies to TRIM46 are associated with paraneoplastic neurological syndromes. Ann Clin Transl Neurol. (2017) 4:680–6. doi: 10.1002/acn3.396
30. Liimatainen S, Honnorat J, Pittock SJ, McKeon A, Manto M, Radtke JR, et al. GAD65 autoantibody characteristics in patients with co-occurring type 1 diabetes and epilepsy may help identify underlying epilepsy etiologies. Orphanet J Rare Dis. (2018) 13:55. doi: 10.1186/s13023-018-0787-5
31. Sinmaz N, Nguyen T, Tea F, Dale RC, Brilot F. Mapping autoantigen epitopes: molecular insights into autoantibody-associated disorders of the nervous system. J Neuroinflamm. (2016) 13:219. doi: 10.1186/s12974-016-0678-4
32. He M, Jenkins P, Bennett V. Cysteine 70 of ankyrin-G is S-palmitoylated and is required for function of ankyrin-G in membrane domain assembly. J Biol Chem. (2012) 287:43995–4005. doi: 10.1074/jbc.M112.417501
33. Fujiwara Y, Kondo HX, Shirota M, Kobayashi M, Takeshita K, Nakagawa A, et al. Structural basis for the membrane association of ankyrinG via palmitoylation. Sci Rep. (2016) 6:23981. doi: 10.1038/srep23981
34. Kordeli E, Lambert S, Bennett V. AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem. (1995) 270:2352–9. doi: 10.1074/jbc.270.5.2352
35. He M, Tseng WC, Bennett V. A single divergent exon inhibits ankyrin-B association with the plasma membrane. J Biol Chem. (2013) 288:14769–79. doi: 10.1074/jbc.M113.465328
36. Manto M, Mitoma H, Hampe CS. Anti-GAD antibodies and the cerebellum: where do we stand? Cerebellum. (2019) 18:153–6. doi: 10.1007/s12311-018-0986-6
37. Rocchi A, Sacchetti S, De Fusco A, Giovedi S, Parisi B, Cesca F, Höltje M, Ruprecht K, Ahnert-Hilger G, Benfenati F. Autoantibodies to synapsin I sequestrate synapsin I and alter synaptic function. Cell Death Dis. (2019) 10:864. doi: 10.1038/s41419-019-2106-z
38. Santuccione AC, Merlini M, Shetty A, Tackenberg C, Bali J, Ferretti MT, et al. Active vaccination with ankyrin G reduces β-amyloid pathology in APP transgenic mice. Mol Psychiatry. (2013) 18:358–68. doi: 10.1038/mp.2012.70
39. Segundo-Acosta PS, Montero-Calle A, Jernbom-Falk A, Alonso-Navarro M, Pin E, Andersson E, et al. Multiomics profiling of Alzheimer's disease serum for the identification of autoantibody biomarkers. J Proteome Res. (2021) 20:5115–30. doi: 10.1021/acs.jproteome.1c00630
Keywords: meningoencephalitis, axon initial segment (AIS), node of Ranvier, human immunodeficiency virus (HIV), ankyrinG, ANK3, autoantibody, autoimmune
Citation: Bartley CM, Ngo TT, Cadwell CR, Harroud A, Schubert RD, Alvarenga BD, Hawes IA, Zorn KC, Hunyh T, Teliska LH, Kung AF, Shah S, Gelfand JM, Chow FC, Rasband MN, Dubey D, Pittock SJ, DeRisi JL, Wilson MR and Pleasure SJ (2023) Dual ankyrinG and subpial autoantibodies in a man with well-controlled HIV infection with steroid-responsive meningoencephalitis: A case report. Front. Neurol. 13:1102484. doi: 10.3389/fneur.2022.1102484
Received: 18 November 2022; Accepted: 16 December 2022;
Published: 23 January 2023.
Edited by:
Sergio Muñiz-Castrillo, Stanford Center for Sleep Sciences and Medicine, United StatesReviewed by:
Tracey Cho, The University of Iowa, United StatesCopyright © 2023 Bartley, Ngo, Cadwell, Harroud, Schubert, Alvarenga, Hawes, Zorn, Hunyh, Teliska, Kung, Shah, Gelfand, Chow, Rasband, Dubey, Pittock, DeRisi, Wilson and Pleasure. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael R. Wilson,  bWljaGFlbC53aWxzb25AdWNzZi5lZHU=; Samuel J. Pleasure, c2FtdWVsLnBsZWFzdXJlQHVjc2YuZWR1
bWljaGFlbC53aWxzb25AdWNzZi5lZHU=; Samuel J. Pleasure, c2FtdWVsLnBsZWFzdXJlQHVjc2YuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.