 Jennifer Bauer
Jennifer Bauer Orrin Devinsky
Orrin Devinsky Markus Rothermel
Markus Rothermel Henner Koch
Henner Koch
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 06 January 2023
Sec. Epilepsy
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.1040648
This article is part of the Research Topic Forebrain Control of Breathing and Sudden Death in Epilepsy View all 10 articles
Epilepsy has a high prevalence and can severely impair quality of life and increase the risk of premature death. Sudden unexpected death in epilepsy (SUDEP) is the leading cause of death in drug-resistant epilepsy and most often results from respiratory and cardiac impairments due to brainstem dysfunction. Epileptic activity can spread widely, influencing neuronal activity in regions outside the epileptic network. The brainstem controls cardiorespiratory activity and arousal and reciprocally connects to cortical, diencephalic, and spinal cord areas. Epileptic activity can propagate trans-synaptically or via spreading depression (SD) to alter brainstem functions and cause cardiorespiratory dysfunction. The mechanisms by which seizures propagate to or otherwise impair brainstem function and trigger the cascading effects that cause SUDEP are poorly understood. We review insights from mouse models combined with new techniques to understand the pathophysiology of epilepsy and SUDEP. These techniques include in vivo, ex vivo, invasive and non-invasive methods in anesthetized and awake mice. Optogenetics combined with electrophysiological and optical manipulation and recording methods offer unique opportunities to study neuronal mechanisms under normal conditions, during and after non-fatal seizures, and in SUDEP. These combined approaches can advance our understanding of brainstem pathophysiology associated with seizures and SUDEP and may suggest strategies to prevent SUDEP.
Epilepsy affects ~0.75% of all people (1), or ~50 million people worldwide, with an incidence of 4–10/1000 people/year. Epileptic seizures result from abnormal hypersynchronous neuronal activity (2, 3). Most seizures arise from both hemispheres simultaneously (generalized) or from restricted regions in one or both hemispheres but can propagate widely (focal) (4, 5). Anti-seizure medicines (ASMs) prevent seizures for ~67% of patients, but many well-controlled patients experience cognitive and behavioral comorbid disorders and ASMs side effects. One-third of patients have drug-resistant epilepsy and often take multiple and high doses of ASMs with greater comorbidities, adverse effects, impairments of quality of life, and higher mortality (6–8).
Sudden unexpected death in epilepsy (SUDEP) is a witnessed or unwitnessed, non-drowning, and non-traumatic death in a person with epilepsy which often but not always follows a convulsive seizure. SUDEP excludes status epilepticus and cases where post-mortem examination or toxicology reveals another cause of death (9). SUDEP is the leading cause of death in drug-resistant epilepsy (DRE), with an incidence rate of 1–5 cases per 1000 patients per year (10, 11). SUDEP is the second leading neurological cause of lost years of life after stroke (12). Case-control studies reveal the following risk factors: generalized tonic-clonic seizures (GTCS) (any in the last year and further increased risk with ≥ 3/year), lack of adequate medication, nocturnal seizures, and lack of nocturnal supervision (13, 14). Many SUDEP cases are undetected or misclassified, suggesting the incidence is higher than reported (13, 15).
The few SUDEPs recorded on video with electroencephalographic and electrocardiographic data are biased toward more severe focal epilepsy cases admitted for presurgical evaluation with rapid ASMs reduction (16). By contrast, SUDEP affects the full spectrum of people with epilepsy (17), and results from epilepsy monitoring units cannot be generalized. The underlying mechanisms of SUDEP remain poorly defined. Most occur during sleep and follow convulsive seizures, with reduced brain activity and respiratory impairments commonly observed, although cardiac dysfunction can contribute (18–20). Postictal disruption of brainstem regulation of arousal, respiratory and cardiovascular functions is considered the common final pathway of death in SUDEP (9, 20–23). In animal models, arousal and cardiorespiratory dysfunctions can result from fast direct synaptic circuit mechanisms (24–27) and slower phenomenons like spreading depressions (SD) (28–31). How cortical seizure activity impairs brainstem functions postictally is a critical research issue. Understanding this pathophysiology will inform preventative and therapeutic strategies. We review potential mechanisms of seizure propagation and spread that might contribute to SUDEP, examine models used to study the mechanisms, and highlight advances in investigating complex network interaction in vivo in mouse models.
This section reviews the following questions: how does epileptic activity spread to the brainstem? Is this a rare event, or common but usually compensated for (and if so, how)?
Rodent, primate, and human brains orchestrate multiple areas to optimally assess internal and external conditions and determine behavioral outputs. This requires high connectivity, precise coordination, and balance between interacting cortical-subcortical networks. During cortical seizures, affected areas are directly impacted by aberrant excitation and inhibition. In addition, areas beyond the epileptic network can be severely disturbed by ictal spread to resonating areas. The brainstem receives projections from cortical and subcortical brain areas (32–34). During and after seizures, these connections can alter brainstem activity and potentially impair arousal and cardiorespiratory functions and contribute to SUDEP (32, 34–36). Understanding why some cortical seizures propagate to other cortical and subcortical areas and how this disrupts brainstem activity is a major challenge in SUDEP research. The brain regions involved in epileptic circuits - cerebral cortex (37), hippocampus (38), amygdala (39, 40), and thalamus (41) - are directly and indirectly connected to the brainstem and exert powerful influences over it. The brainstem and more rostral cerebral regions share strong reciprocal connections, complicating our understanding. We review new techniques to study network interaction involved in SUDEP in epileptic mouse models.
Epileptic seizures can be provoked by disrupting neuronal E/I balance by altering intrinsic properties, or by altering synaptic transmission and network stability causing hypersynchronous activity (42). The mechanisms underlying seizure propagation and termination are less well characterized. Focal seizures influence other brain areas via rapid axonal connections or spreading depression (SD), a slow propagating depolarization wave that inactivates neurons (25, 31, 35, 36). This slow ictal wavefront propagation corresponds to the gradual evolution of seizure symptoms, as in the Jacksonian sensory symptom march (43). The ictal wavefront may evoke a feedback loop to the seizure focus which triggers the clinical symptoms. Failure of feedforward inhibition supports epileptiform activity and seizure spread via this slow route in addition to classic synaptic pathways (44, 45). While SD contributes to symptoms of migraine and epilepsy, the mechanisms may be conserved or divergent (29, 46). The propagation rate of SD in migraine and epilepsy are similar, but their onset, duration, impacted brain regions and EEG changes can differ (47–50). Different SDs might exert distinct influences on brainstem function and SUDEP risk (29, 51). Debate persists whether this risk is primarily an ictal or post-ictal phenomenon. While the ictal seizure spreading into the brainstem might cause direct autonomic dysfunctions (36, 52, 53), the disturbance in the post-ictal period might substantially outlast the seizures. The post-ictal EEG suppression is viewed as a potential contributor but only a weak SUDEP predictor (54–56).
Mouse models of familial hemiplegic migraine with mutations in Cacna1a (57, 58), Atp1a2 (59) and Scn1a (60, 61) show increased mortality. In mice with Cacn1a variants, brainstem SD elicited by seizures can be fatal (31). Brainstem SD may directly impair cardiorespiratory function (21, 30, 31, 35, 40). In focal seizures, SD with seizure propagation may be restricted to cortical regions in most instances. SDs were directly triggered by high neuronal activity of focally induced seizures and prevented by applying tetrodotoxin (TTX; a potent sodium channel inhibitor) (62). The authors postulate that SD is an innate mammalian mechanism to prevent seizure propagation and generalization, and to induce seizure termination (62). However, if SDs reach brainstem autonomic centers, severe cardiorespiratory dysfunction may follow (31, 63).
Human studies used electrical stimulation or the time of seizure invasion to investigate cortical structures that alter breathing. These brain areas include the amygdala (32, 36, 64), the hippocampus head and body, anterior parahippocampal gyrus, and antero-mesial fusiform gyrus (65, 66). A pediatric study found apneas and seizure spread to the amygdala were strongly correlated (67), an adult study failed to replicate this (64). Electrical stimulation to the insula and left cingulate gyrus decreased cardiac output and induced cardiac asystole in epilepsy patients without effects on breathing (68, 69). However, electrical activation cannot precisely target specific neurons and circuits. Further, cortical and subcortical electrode coverage is limited. So the invasion of ictal activity to a region (e.g., amygdala) may be accompanied by spread to areas that were not sampled (e.g., hypothalamus, anterior cingulate, and orbitofrontal cortices). Also, correlating seizure invasion to apneas might reveal only some parts of the network involved in autonomic dysfunction. In animals and humans, physiological changes in subcortical areas (e.g., locus coeruleus) alter breathing (27, 70).
In addition to seizure invasion of cortical areas, altered connectivity between cortical areas and respiratory brainstem centers may be important (71). Functional magnetic resonance imaging (fMRI)-studies on epilepsy patients show reductions in resting-state functional connectivity and tissue loss in cortical, subcortical, and brainstem structures associated with impaired autonomic control and increased SUDEP risk (71–73). However, monitoring of patients who later died from SUDEP did not reveal a direct associated location or lateralization of the epileptogenic zone with their higher risk of death (16). Intracranial EEG recordings and stimulation studies implicate the amygdala, hippocampus, insular cortex, and seizure spread to the contralateral temporal lobe to correlate with ictal cardiorespiratory dysfunctions (32, 36, 53, 65, 67, 74). Thus, identifying the detailed and complex connectivity and the altered brain activity in regions controlling cardiorespiratory activity is crucial for SUDEP risk estimation.
Here, we review evidence of brainstem alterations (e.g., genetic or physiological/structural resulting from chronic epilepsy) associated with SUDEP risk. We discuss crucial brainstem areas generating and modulating autonomic rhythms, such as breathing, and discuss their potential role in SUDEP. The respiratory network flexibly adapts to environmental and metabolic changes while maintaining stability to guarantee effective gas exchange (75). This network integrates brainstem rhythm-generating nuclei with other central and peripheral neural regions (76). The brainstem respiratory network includes the parafacial respiratory group (pFRG), Bötzinger complex (BötC), pre-Bötzinger complex (pre-BötC), rostral ventral respiratory group (rVRG), and caudal VRG (cVRG). Pontine nuclei modulate respiratory activity via projections to medullary respiratory nuclei (34, 76, 77). The post-inspiratory complex (PiCo) provides excitatory input to generate post-inspiration patterns (78). Seizure-related effects on respiratory and cardiac brainstem centers can impair these functions and contribute to SUDEP (20, 21, 79, 80) (Figure 1). Cardiorespiratory dysfunction in SUDEP could result from the effects of higher cortical and limbic areas on brainstem function, direct brainstem alterations, or both, including descending and ascending circuitries (27, 38, 81) (Figure 2). Chronic alterations of respiratory control, such as reduced ventilatory responses to increased CO2 levels, occur in epilepsy patients (82).
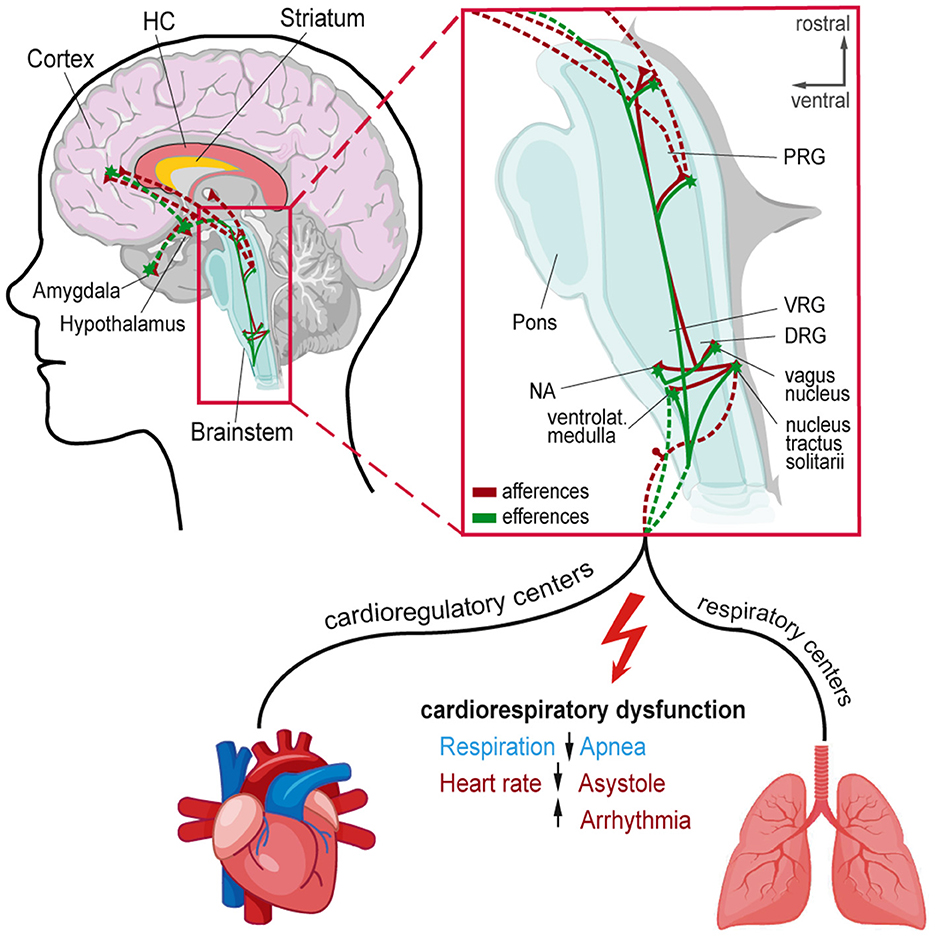
Figure 1. Afferent and efferent connections to central autonomic networks and proposed mechanisms involved in seizure-induced cardiorespiratory dysfunction leading to SUDEP. Cortical and subcortical regions involved in epileptic activity and cardiorespiratory modulation in the brainstem. Magnification shows connections and brainstem nuclei of cardioregulatory and respiratory centers that can be affected by seizure activity leading to cardiorespiratory dysfunction. DRG, Dorsal respiratory group; HC, Hippocampus; NA, Nucleus ambiguous; PRG, Pontine respiratory group; VRG, Ventral respiratory group.
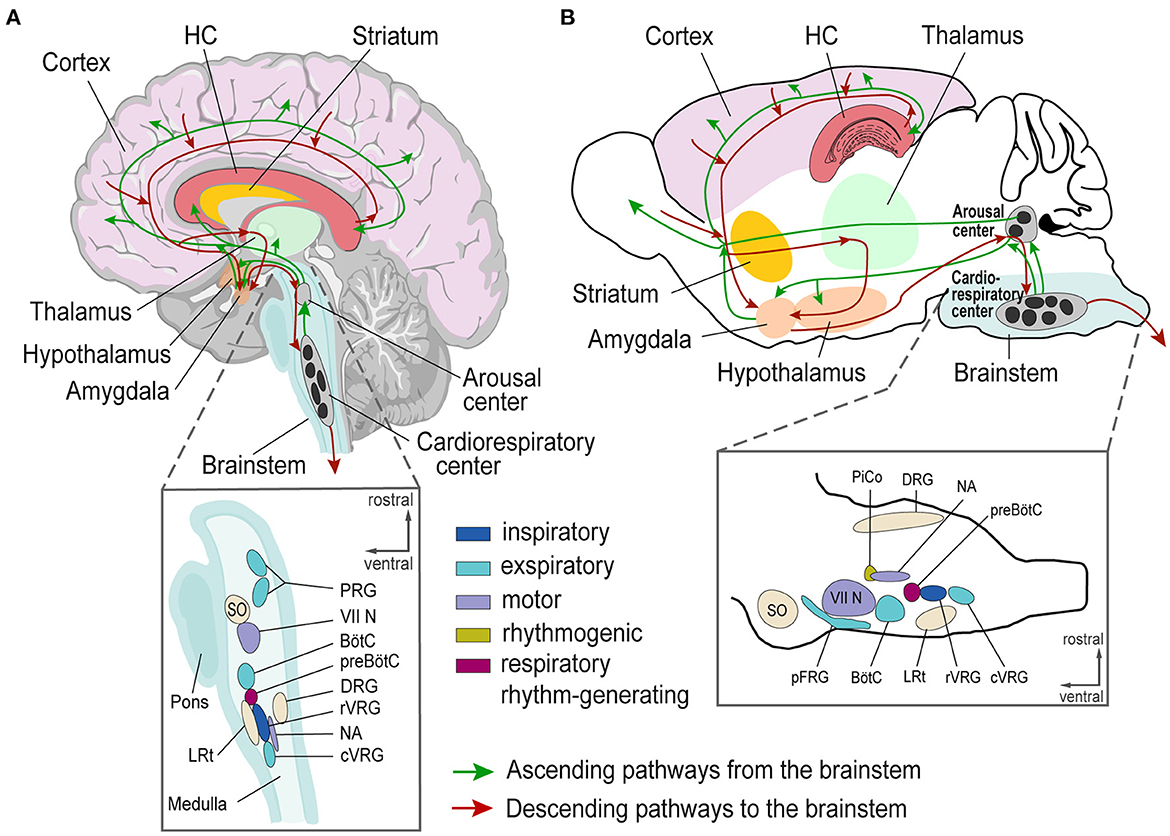
Figure 2. Schematic of brain regions involved in epileptic activity and cardiorespiratory regulation in humans (A) and mice (B). (A, B) Top, Human and mouse brain with depicted Cortex (pink), Hippocampus (red), Striatum (yellow), Thalamus (green), Hypothalamus (orange), and brainstem (blue) with ascending (green lines) and descending (red lines) projections. Bottom, Magnification of brainstem nuclei. VII N, facial nucleus; BötC, Bötzinger Complex; cVRG, caudal ventral respiratory group; DRG, Dorsal respiratory group; HC, Hippocampus; LRt, lateral reticular nucleus; NA, Nucleus ambiguus; pFRG, para-facial respiratory group; PRG, Pontine respiratory group; PiCo, Post-inspiratory Complex; preBötC, preBötzinger Complex; rVRG, rostral ventral respiratory group; SO, superior olive; VRG, Ventral respiratory group.
Epileptic seizures can directly alter heart rhythms and heart rate variability (HRV), which reflects balanced sympathetic and parasympathetic activity (83–86). High sympathetic tone and elevated levels of several neuropeptides can follow seizures (87, 88). Other seizure-induced acute changes include asystole, brady- and tachy-arrhythmias are most common with seizure foci in paralimbic and limbic cortices (69) and may contribute to SUDEP (23, 89, 90). Reduced HRV can result from voltage-gated sodium channel gene variants (91), and low-frequency HRV power is associated with SUDEP risk (92). Temporal lobe seizures may disturb arousal and vigilance networks (93).
A critical challenge is distinguishing indirect vs. direct effects on brainstem autonomic centers. For example, PreBötC dysfunction can result from mutations in ion channels (94–96) and mitochondrial genes (97), as well as transcription factors (98). In animal SUDEP models with Kcna1 and Scn1a mutations, the threshold to trigger brainstem SD is reduced (21). However, respiratory networks are state-dependent; neuromodulators influencing respiratory activity include norepinephrine, serotonin, acetylcholine, substance P, ATP, somatostatin, dopamine, endorphins, and adenosine (99). Several have been shown to be elevated during and following seizures and potentially could contribute to SUDEP (88, 100–102).
The brainstem is crucial for controlling cardiorespiratory autonomic function impairments likely contribute to sudden infant death syndrome (SIDS), the sudden and unexpected death of a seemingly healthy baby under age 1 year (103–105). There are striking similarities between SIDS, sudden unexplained death in childhood (SUDC), and SUDEP (106–109) with the exclusion of other causes, nocturnal occurrence in the prone position, and an unwitnessed death (106). Arousal can be triggered by increased CO2 (hypercapnia) and reduced oxygen levels (hypoxia), further preventing a build-up of end-tidal CO2 and restoration of normal oxygen levels (103). This arousal response is linked to breathing and is normally initiated with a sigh (augmented breath) (110–112). Sighs are generated in the PreBötC by the same rhythm-generating network crucial for eupnea and gasping (95, 113). In addition, several other areas such as the dorsal raphe nucleus, the nucleus tractus solitarius, the parabrachial nucleus, and the retrotrapezoid nucleus are involved in arousal (106). Seizures in the amygdala [bed nucleus of the stria terminalis (BNST)] can activate projections to the brainstem, disturbing structures like the parabrachial nucleus involved in arousal and respiratory function (40). The BNST is highly interconnected to cortical regions, the hippocampus, the hypothalamus, the midbrain, and other brainstem nuclei and may serve as an integrator of autonomic and neuroendocrine responses (40, 114–117). As discussed above, massive release of neuromodulators (e.g., norepinephrine, serotonin, and acetylcholine) can disturb arousal. Since hypoxia and hypercapnia trigger arousal and gasping, they are a focus of SIDS research. However, another vulnerability phase is reoxygenation after a hypoxia/hypercapnia. This phase includes post-hypoxic ventilatory depression (118, 119), which can occur after generalized tonic-clonic seizures and could be potentially prolonged in SUDEP. To dissect these mechanisms, modern experimental technology, including optogenetics and chemogenetics in animal models, as discussed below, is critical.
Future directions of SUDEP research seek to identify common molecular and cellular changes overlapping in several SUDEP animal models and potentially identify common changes in SIDS and SUDC models. While expression changes in RNA levels in brainstem areas of animals showing SD in cortical areas were detected (120), more investigations in epileptic animal models (genetic and induced) are needed to unravel molecular changes that participate in SUDEP.
Another potential SUDEP mechanism is direct ASM effects on brainstem function. ASM can reduce SUDEP risk by a reduction of seizure frequency and severity, thereby preventing seizure-induced impairment of brainstem autonomic centers. Under normal oxygen concentrations, mammals are eupneic, their robust respiratory network combines diverse synaptic and intrinsic signals in the respiratory network (99). During severe hypoxia, the respiratory network generates gasping (121, 122) through reduced mechanisms of rhythm generation (99, 113). During gasping, changes include reduced inhibition (123) and a switch to sodium-dependent intrinsic neuronal bursting securing rhythm generation (124–126). These altered rhythm-generating properties of the respiratory network alter the sensitivity to sodium channel-blocking ASMs and may interrupt the gasping response during seizure-induced postictal hypoxia (127). These direct brainstem effects may contribute to increased mortality associated with lamotrigine use observed in some studies (128). Moreover, during seizures, patients can experience repeated hypoxic episodes combined with increased norepinephrine and other neurotransmitter/modulator levels. This combination can destabilize PreBötC function (129) and may parallel secondary changes induced by the hypoxic conditions during and after cortical seizures.
More investigations of the brainstem function and gene expression of the brainstem areas controlling respiratory and cardiac functions are needed in epileptic mice to get a better understanding of the mechanisms underlying SUDEP. New insights into chronic brainstem changes could stem from novel techniques of brainstem transcriptome using single-cell RNA-Seq or spatial transcriptomics (130).
Model systems help to investigate the pathological interactions between brain regions that can result in a collapse of cardiorespiratory function and SUDEP. In vitro and in vivo models can study neuronal disease network mechanisms (131–133). In vitro models standardize experimental conditions, but oversimplify neuronal network function or whole organism interaction, which may be critical in SUDEP. In vivo models comprise diverse methods and model organisms (134–138). Brain areas involved in epileptic activity and cardiorespiratory regulation are similar in mice and humans (78, 139) (Figures 2A,B). Thus, mice can appropriately model human epilepsy. Modern techniques can target specific brain areas and predefined neuronal cell populations to decipher their role in epilepsy and SUDEP (140).
Epilepsy can result from diverse pathological processes, including trauma, stroke, tumors, infections, autoimmune disorders, and >150 genetic variants (141, 142). Epilepsy is often accompanied by comorbid disorders, including autism spectrum, cognitive, psychiatric, and hyperkinetic (143–148). The developmental and epileptic encephalopathies (DEEs) include a diverse spectrum of early-life epilepsies, often resulting from genetic disorders, and associated with developmental delays partly attributable to seizures and interictal epileptiform activity (149). Across these disorders, E/I imbalances occur in the amygdala, cortex, hippocampus, and other epileptogenic regions (150, 151). Complex and heterogeneous genetic mouse models recapitulate various human pathologies, offering insights into epilepsy and SUDEP and allowing controlled experiments on mechanisms by controlling for different confounds. Epilepsy mouse models are divided into induced and genetic models. In kindling models, stimulation (electrical, chemical, or acoustic) induces seizures, whereas in genetic models, gene mutations result in spontaneous seizures (136, 152). Mouse models can mimic focal and generalized epilepsies as well as post-traumatic epilepsy (153), temporal lobe epilepsy (TLE) (152), genetic variants (80, 154), and scores of rare genetic disorders (e.g., tuberous sclerosis complex, CDKL5, Rett Syndrome, Dravet Syndrome, FOXG1 syndrome, STXBP1 syndrome and many more) (Figure 3).
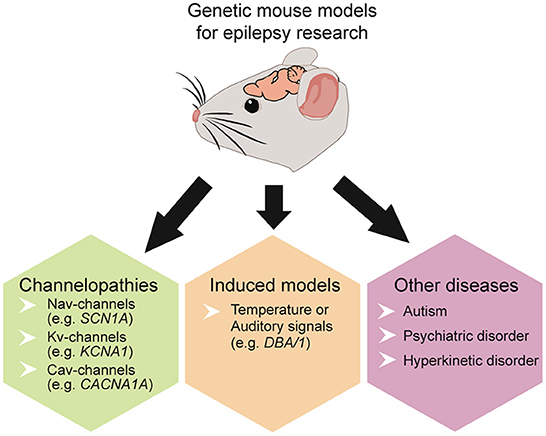
Figure 3. Overview of commonly used genetic mouse models to study epilepsy, SUDEP, and co-existing disorders.
Some genetically modified mouse lines model SUDEP with deadly seizure-induced cardiorespiratory abnormalities (39, 155). Many genetic models involve ion channels, including sodium voltage-gated channels (Nav) (154, 156–158), potassium voltage-gated channels (Kv) (28, 159, 160), or calcium voltage-gated channels (Cav) (30, 161) (Table 1). Some display spontaneous epileptic seizures (e.g., Scn1a, Scn1b, Kcna1, Kcnq1, Cacna1a, Shank3) (31, 143, 159, 162, 163) while others are susceptible to heat or audiogenic-induced seizures (e.g. Scn1a, Scn8a, DBA/1) (80, 164) (Figure 3).
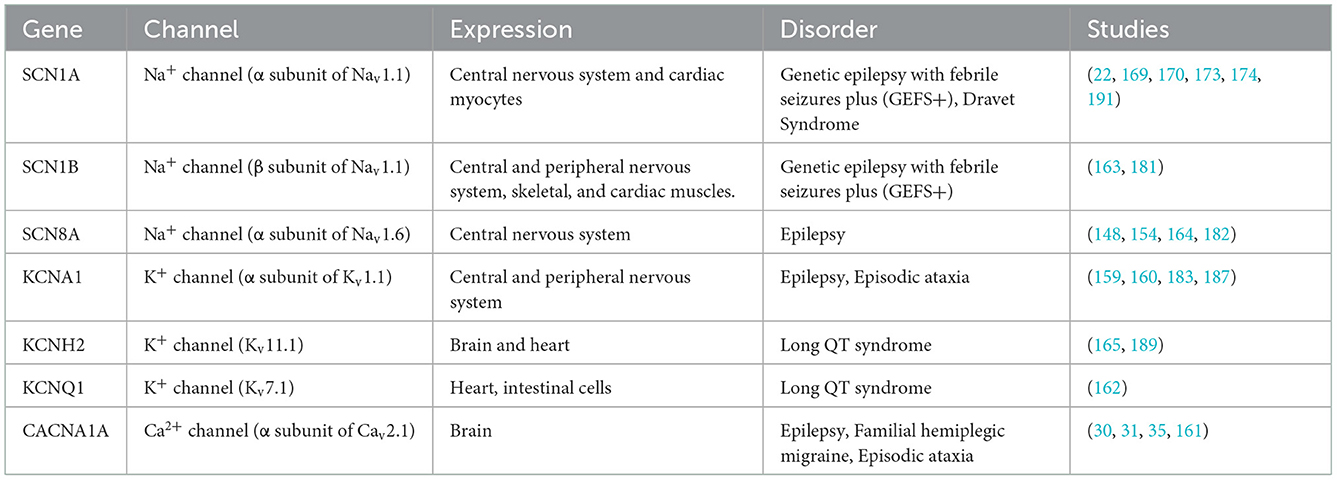
Table 1. Overview of common channelopathies in mouse models of epilepsy and SUDEP.
Commonly SUDEP mouse models carry mutations in the Nav (1.1, 1.6) and Kv (1.1, 7.1, 11.1) genes (159, 162, 165). Scn1a mutations alter the Navα1 subunit (Nav1.1) and Nav1.1 haploinsufficiency can cause Dravet Syndrome (DS). DS is a treatment-resistant early-onset epilepsy with 70-80% of cases due to Scn1a variants and high rates of SUDEP (9, 83, 166–169). Nav1.1 is expressed in inhibitory neurons. A loss of function decreases their excitability, increasing network excitability, altering action potential (AP) dynamics (170–173) and impairs thalamic glutamatergic and GABAergic function, disrupting thalamocortical networks and facilitating seizure generation (174, 175). Nav1.1 deficient mice recapitulate many aspects of human DS pathology including severe epilepsy, multiple neuropsychiatric comorbidities, and increased SUDEP risk (21, 22, 83, 173, 176–180). Other gene mutations (e.g., Scn1b and Scn8a) display similar symptoms (163, 164, 181, 182). Mice with mutations in genes encoding for Kv show cardiorespiratory failure including cardiac abnormalities and apnea observed in SUDEP (21, 183). Kv1.1-α1 subunits, encoded by the Kcna1 gene, are crucial for neuronal excitability and are broadly expressed in the cortex, hippocampus, cerebellum, and brainstem (184, 185). Kcna1 knockout mice display early-onset generalized tonic-clonic seizures, seizure-related death, and cardiorespiratory dysfunction (159, 186–188). These mice exhibit apneas, increased respiratory variability, and precede cardiac failure as risk factors for SUDEP (183, 187). Further Kv-channelopathies (e.g., Kcnh2 and Kcnq1) are susceptible to recurrent seizures and long QT syndrome (LQTS); i.e., arrhythmias and SUDEP (162, 189, 190).
Mutations in genes encoding for Nav1.1, Kv1.1, and Cav2.1 are moreover linked to brainstem seizures, medullary SD, and cortical seizures propagating to the brainstem causing cardiorespiratory arrest (21, 30, 31, 35). Thereby, local brainstem SD can elicit EEG suppression, apnea, bradycardia, and asystole, mimicking the involvement of SD in epileptic activity propagation and its relevance as SUDEP models.
Thus, a number of model systems and especially mouse models, are nowadays available for epilepsy and SUDEP research. In the direct context of SUDEP, models with Nav (1.1, 1.6) and Kv (1.1, 7.1, 11.1) mutations seem particularly promising. Of these, Scn1a models have been extensively studied and largely model the human SUDEP pathology and phenotypes well (152, 167, 191). Future studies need to extend to clinically and genetically characterized epilepsies to explore if common or distinct pathways of autonomic dysfunction mediate SUDEP.
To understand SUDEP mechanisms, we need models and techniques to represent and measure cortical seizure generation and propagation as well as cardiorespiratory function. In vivo techniques allow direct epileptic activity measurements and manipulations (192, 193) of complex circuitries and brain connections. Ex vivo recordings from targeted brain regions allow cellular processes to be investigated at high resolution. Ex vivo measurements like histological reconstructions, stainings, and spatial transcriptomics (180, 194) can reveal anatomical brain changes associated with epilepsy, which may be the cause or effect of epilepsy or an epiphenomenon of the underlying pathology.
Next, we will discuss recent advancements in methods to investigate in vivo and ex vivo models, including optogenetics, electrophysiology, imaging, and other measurements (Figure 4).
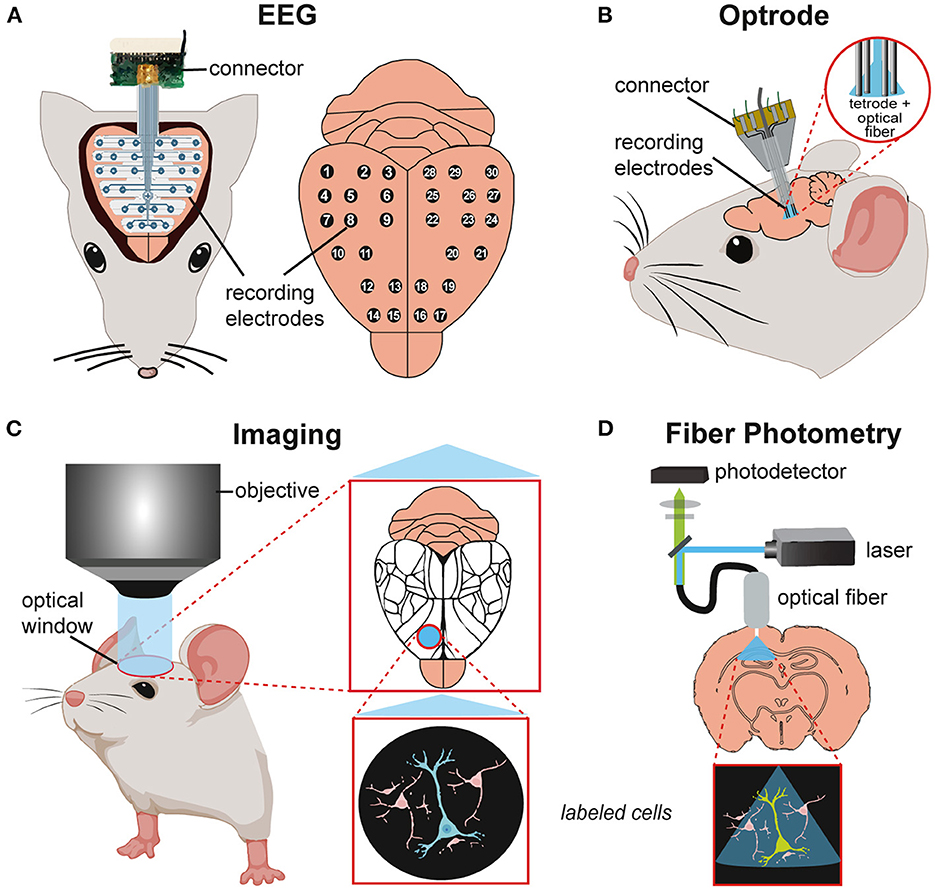
Figure 4. Schematics of in vivo techniques for studying neuronal activity in mouse models. (A) Electroencephalography (EEG) recording with implanted EEG probe and recording electrodes (30 channels) on the brain surface. (B) Implated optrode with integration of optical fiber and recording electrodes for simultaneous optical stimulation and electrophysiological recordings. (C) Imaging of the mouse brain through an optical window for visualizing brain areas (mapping, Allen Brain Atlas) or/and single-cell activity in the region of interest. (D) Fiber photometry of a target brain region with simultaneous optical stimulation (laser) and calcium imaging (photodetector) via an implanted fiber probe.
Optogenetics is a technique to study specific cells and their relations to brain functions and disorders (195). Optogenetics utilizes the expression of light-sensitive proteins (opsins) in brain areas or specific cells. Depending on the opsin used, targeted neurons can be activated or inhibited (or even both) using light stimulation to precisely control neuronal activity. Optogenetics can trigger or prevention of epileptic activity (196) with millisecond temporal precision, enabling the assessment of how specific firing patterns affect brain cells and networks (197). Optogenetics can be applied invasively and non-invasively (198) and can be combined with electrophysiological recordings and imaging techniques. Chemogenetics can selectively modulate cellular pathways using restricted artificial chemogenetic receptors [e.g. DREADDs (Designer Receptors Exclusively Activated by Designer Drugs)] delivered to specific neuronal populations. Instead of light stimulation, chemogenetics systemically injected or microinfused can activate ligands that excite or inhibit targeted neurons (199). Optogenetics can be combined with chemogenetics to manipulate neuronal activity with a high temporal and spatial resolution (200). In epilepsy animal models, these combined methods can identify and manipulate specific neuron populations, brain regions, and neuronal circuitries involved in epileptic activity (201).
Seizures and interictal epileptic discharges (IEDs) can be restricted to certain brain regions and networks. Electroencephalographic (EEG) recordings can localize brain regions giving rise to seizures and examine epilepsy-related neuronal activity changes across brain regions (202). Scalp EEG records changes in electrical potentials caused by ion flow across neural membranes, mainly at the brain's surface. It can detect the origin and propagation of epileptic activity throughout different brain regions at a macro scale (203, 204) (Figure 4A). Invasive methods include intracranial EEG (iEEG) using depth or subdural EEG recordings (ECoG) to study seizure onset and spread as well as SD and seizure propagation in SUDEP models at a higher spatiotemporal resolution (22, 30). Stereotaxically inserted multi-channel electrodes can record local field potentials (LFPs) and single-cell activity (204, 205). Modulation of neuronal activity via electrical stimulation with these electrodes is possible but is much less precise than optogenetic manipulation. For example, SDs can be induced by electrical, and optogenetic techniques (206) whereas electrophysiological and optical recording methods can assess their propagation and effects on other structures (207, 208).
Combined optogenetics and electrophysiology in vivo, using optical microelectrodes are called optrodes (209, 210) enabling a direct readout of manipulated cell activity. Here, a single microelectrode probe with integrated optical fiber can simultaneously record and transmit light to genetically modified, opsin-expressing cells. Optrodes can study neuronal circuit dynamics in awake-behaving animals (211, 212) (Figure 4B).
Imaging allows the visualization/mapping of cortical activity with high spatial and temporal resolution (213, 214). Voltage-sensitive dyes (VSDs) or genetically encoded calcium indicators (GECIs) react to direct or indirect (Ca2+) changes in neuronal activity. VSD imaging incorporates dyes into the cell membrane that signal membrane-potential differences as changes in fluorescence. VDS imaging can monitor synaptic transmission and propagation of cortical activity but has a low signal-to-noise ratio and lacks cellular specificity (215, 216). GECIs allow cell-specific targeting but have a slower temporal resolution. GECIs have been used to record population activity in wide-field calcium imaging experiments. Combined with two-photon imaging, GECIS can reveal activity dynamics of hundreds of individual neurons (217). Since the activity-dependent changes in calcium-sensitive proteins can be visualized in vivo over months, large-scale longitudinal functional studies can assess activity before, during, and after seizures and in a single animal (Figure 4C). Imaging techniques can visualize seizures in vivo at high temporal and cellular resolution (172, 218).
Fiber photometry can combine imaging and optogenetics using an implanted fiber-optic cannula to deliver excitation pulses and monitor activity-dependent fluorescence changes (219). This technique is ideal for deep brain recordings and can study calcium signals in distinct epileptic brain regions in freely moving mice (220) (Figure 4D). Fiber photometry can be used simultaneously with electrophysiological recordings to combine cell-type-specific imaging with high temporal-resolution spike recordings in freely behaving mice (221).
These methods have provided new insights into the role of brain regions and cell populations in epilepsy. Optogenetics combined with optical manipulation, and electrophysiological recordings revealed the key role of inhibitory GABAergic interneuron signaling in seizure generation and ictal propagation in epileptic mice (212, 222). Other studies addressing brainstem excitatory neurons showed a direct correlation to reduced subcortical activity during seizures (223). Together, these techniques provide new research opportunities on epilepsy networks and seizure dynamics over the whole brain.
Investigating SUDEP and cardiorespiratory dysfunctions requires additional recording techniques for in vivo monitoring of autonomic functions including breathing and heart rate. Several methods are available in the mouse (155, 182, 183, 224). Cardiac activity is typically recorded via electrocardiography (ECG) (225, 226). Methods can monitor breathing (227) including invasive (telemetry systems and intranasal cannulas) and non-invasive methods (movement sensors, restraining systems, plethysmographs) (Figure 5). The whole-chamber plethysmography approach offers a non-invasive method in freely, non-restrained animals (95, 97, 228). This technique allows recordings of breathing under hypoxia/hypoxemia conditions (low blood oxygen levels and insufficient oxygen supply) linked to SUDEP (30, 52, 229).
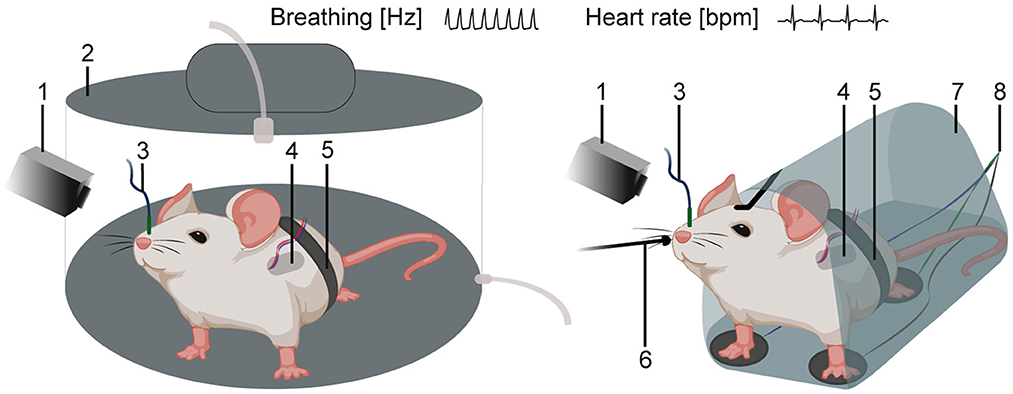
Figure 5. Schematics of in vivo techniques for the recording of cardiac and respiratory activity. Mouse in a whole-chamber plethysmograph (left) or in a restraining system (right) with additional recording methods. (1) video monitoring and infrared camera, (2) whole-chamber plethysmography, (3) intranasal cannula, (4) telemetry system, (5) movement sensor, (6) external temperature probe (thermocouple), (7) restraining system, and (8) 3-point ECG.
Although in vivo methods provide insights into the network mechanisms, ex vivo studies offer more focused investigations of cellular changes. Histological reconstructions and stainings of brain regions can follow in vivo experiments to verify transgene expressions and precisely localize implanted electrodes or optical fibers (30, 180, 230). Brain slice preparations containing cortical, hippocampal, or brainstem microcircuits allow single-cell recordings or small network analysis to gain insights into pathophysiology (78, 95, 231, 232). Spatial transcriptomics can map the organization and connectivity of distinct genetically defined cell types (194, 233). In epilepsy research, this can provide a deeper exploration of disease mechanisms and pathogenic changes in the spatial organization and molecular signaling networks (234).
Thus, combining different techniques can provide a greater definition of the dysfunctions associated with epileptic activity and its interplay with autonomic functions on different levels to identify possible biomarkers for epilepsy, seizure onset, and SUDEP (202, 235).
Possible therapeutic approaches could be based on electrical or optical stimulation of specific brain areas to “rebalance” their E/I activity and maintain cardiorespiratory function during and after seizures (198, 236). Electrical stimulation in patients to map epileptic zones can inhibit or enhance respiration (237). Optogenetic neuronal activation has been shown to suppress seizure-induced respiratory arrest and exert an anticonvulsant effect in a SUDEP mouse model (238). Further, ex vivo methods might provide opportunities for new molecular targets and drug screening (233, 234, 239). However, these invasive approaches will require far more refinement for their potential benefits to exceed their definite risks.
SUDEP is the leading epilepsy-related cause of death, affecting all age groups and epilepsy severities. SUDEP mechanisms are poorly understood but are critical for preventive and therapeutic strategies. Although ASMs can control seizures in most patients, they do not alter long-term prognosis or cure epilepsy (240). Further, their side effects can be severe (241, 242). 30% of the patients with ASM-resistant epilepsy suffer ongoing seizures and experience an increased SUDEP risk. Medications/treatments that prevent seizures in those that are currently uncontrolled with minimal side effects are desperately needed. Understanding SUDEP mechanisms in more detail is a desperate need.
Epilepsy mouse models with ion channel mutations mimic human epilepsies (176) and are critical in translational neuroscience research (243). They offer possibilities to investigate the link between genetic alterations and their underlying neurobiological mechanisms in much greater detail compared to humans. Translation of basic animal research to human epilepsy is exemplified by SCN1A-mice whose response to ASM has enabled the development of FDA-approved medications and gene therapy trials (191). Translational research with new molecular targets for anti-epileptogenic and anti-seizure research can empower novel drug discoveries and identify potential biomarkers for early diagnoses and more effective treatments (235, 243, 244).
Cardiorespiratory inhibition following epileptic seizures may be the common final mechanism of SUDEP. Cardiorespiratory dysfunctions from cortical or subcortical epileptic activity propagating to brainstem regions could cause SUDEP (21, 30, 31). SD might be directly involved in SUDEP-related seizure spread to the brainstem (29). Mouse models combining technological advances allow precise investigations of the brain networks implicated in SUDEP (235). These brain areas may provide new targets for interventions to prevent SUDEP.
In mice, invasive methods such as optical or electrical stimulations can manipulate neuronal networks (198, 245) whereas neurostimulation-based techniques can also be applied to epilepsy patients. Acute and chronic deep brain stimulation (DBS), as well as vagus nerve stimulation (VNS), are epilepsy therapies (236, 246–248). Combining neurostimulation and ASM may be more effective in controlling seizures than either alone (249).
There remains a critical need to better understand the mechanisms of epilepsy and SUDEP. Mouse models combined with precise methods are an important tools to assess these mechanisms and translate this knowledge into preventive and therapeutic strategies.
JB, OD, MR, and HK reviewed the literature and wrote this review article. All authors contributed to the article and approved the submitted version.
This study was supported by the Deutsche Forschungsgemeinschaft [DFG, German Research Foundation, 250583768/RO4046/2-1 and /2-2 (MR), 445965029/RO4046/5-1 (MR), 466488864/RO4046/6-1 and 7-1 (MR), 368482240/GRK2416 (MR)], 263938822/KO-4877/2-1 and the Interdisciplinary Center for Clinical Research within the faculty of Medicine at the RWTH Aachen University Grant IZKF TN1-7 532007 (MR) and FACES (Finding a Cure for Epilepsy and Seizures) (HK).
Figures were partly created with Biorender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Beghi E., Giussani G., Nichols E., Abd-Allah F., Abdela J., Abdelalim A.L., et al (2019). Global, regional, and national burden of epilepsy, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18, 357–375. doi: 10.1016/S1474-4422(18)30454-X
2. Beghi E. The epidemiology of epilepsy. Neuroepidemiology. (2020) 54:185–91. doi: 10.1159/000503831
3. Fisher RS, Van Emde Boas W, Blume W, Elger C, Genton P, Lee P, et al. Epileptic Seizures and Epilepsy: Definitions Proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia. (2005) 46:470–2. doi: 10.1111/j.0013-9580.2005.66104.x
4. Cuello Oderiz C, von Ellenrieder N, Dubeau F, Eisenberg A, Gotman J, Hall J, et al. Association of cortical stimulation-induced seizure with surgical outcome in patients with focal drug-resistant epilepsy. JAMA Neurol. (2019) 76:1070–78. doi: 10.1001/jamaneurol.2019.1464
5. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the international league against epilepsy: position paper of the ILAE commission for classification and terminology. Epilepsia. (2017) 58:522–30. doi: 10.1111/epi.13670
6. Berto P. Quality of life in patients with epilepsy and impact of treatments. Pharmacoeconomics. (2002) 20:1039–59. doi: 10.2165/00019053-200220150-00002
7. Kwan, P., and Brodie, M. (2000). Early identification of refractory epilepsy. N Engl J Med., 342, 314–319. doi: 10.1056/NEJM200002033420503
8. Sveinsson O, Andersson T, Carlsson S, Tomson T. The incidence of SUDEP: A natiowide population-based cohort study. Neurology. (2017) 89:170–7. doi: 10.1212/WNL.0000000000004094
9. Devinsky O, Hesdorffer DC, Thurman DJ, Lhatoo S, Richerson G. Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol. (2016) 15:1075–88. doi: 10.1016/S1474-4422(16)30158-2
10. Surges R, Thijs RD, Tan HL, Sander JW. Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nat Rev Neurol. (2009) 5:492–504. doi: 10.1038/nrneurol.2009.118
11. Tomson T, Walczak T, Sillanpaa M, Sander J. Sudden unexpected death in epilepsy: a review of incidenceand risk factors. Epilepsia. (2005) 46:54–61. doi: 10.1111/j.1528-1167.2005.00411.x
12. Thurman DJ, Hesdorffer DC, French JA. Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia. (2014) 55:1479–85. doi: 10.1111/epi.12666
13. Sveinsson O, Andersson T, Mattsson P, Carlsson S, Tomson T. Pharmacologic treatment and SUDEP risk: A nationwide, population-based, case-control study. Neurology. (2020) 95:e2509–18. doi: 10.1212/WNL.0000000000010874
14. van der Lende M, Hesdorffer DC, Sander JW, Thijs RD. Nocturnal supervision and SUDEP risk at different epilepsy care settings. Neurology. (2018) 91:e1508–18. doi: 10.1212/WNL.0000000000006356
15. Devinsky O, Friedman D, Cheng JY, Moffatt E, Kim A, Tseng ZH, et al. Underestimation of sudden deaths among patients with seizures and epilepsy. Neurolgy. (2017) 89:886–92. doi: 10.1212/WNL.0000000000004292
16. Nei M, Ho RT, Abou-Khalil BW, Drislane FW, Liporace J, Romeo A, et al. EEG and ECG in sudden unexplained death in epilepsy. Epilepsia. (2004) 45:338–45. doi: 10.1111/j.0013-9580.2004.05503.x
17. Verducci C, Hussain F, Donner E, Moseley BD, Buchhalter J, Hesdorffer D, et al. SUDEP in the North American SUDEP registry: the full spectrum of epilepsies. Neurology. (2019) 93:e227–36. doi: 10.1212/WNL.0000000000007778
18. Blum A. Respiratory physiology of seizures. J Clin Neurophysiol. (2009) 26:309–15. doi: 10.1097/WNP.0b013e3181b7f14d
19. Lamberts RJ, Thijs RD, Laffan A, Langan Y, Sander JW. Sudden unexpected death in epilepsy: people with nocturnal seizures may be at highest risk. Epilepsia. (2012) 53:253–7. doi: 10.1111/j.1528-1167.2011.03360.x
20. Ryvlin P, Nashef L, Lhatoo SD, Bateman LM, Bird J, Bleasel A, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurology. (2013) 12:966–77. doi: 10.1016/S1474-4422(13)70214-X
21. Aiba I, Noebels JL. Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med. (2015) 7:246. doi: 10.1126/scitranslmed.aaa4050
22. Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. (2013) 123:1798–808. doi: 10.1172/JCI66220
23. Leung H, Kwan P, Elger CE. Finding the missing link between ictal bradyarrhythmia, ictal asystole, and sudden unexpected death in epilepsy. Epilepsy Behav. (2006) 9:19–30. doi: 10.1016/j.yebeh.2006.05.009
24. Derera ID, Delisle BP, Smith BN. Functional neuroplasticity in the nucleus tractus solitarius and increased risk of sudden death in mice with acquired temporal lobe epilepsy. eNeuro. (2017) 4:5. doi: 10.1523/ENEURO.0319-17.2017
25. Lertwittayanon W, Devinsky O, Carlen PL. Cardiorespiratory depression from brainstem seizure activity in freely moving rats. Neurobiol Dis. (2020) 134:104628. doi: 10.1016/j.nbd.2019.104628
26. Pilar M, DeFelipe J. Altered synaptic circuitry in the human temporal neocortex removed from epileptic patients. Exp Brain Res. (1997) 114:1–10. doi: 10.1007/PL00005608
27. Yackle K, Schwarz LA, Kam K, Sorokin JM, Huguenard JR, Feldman JL, et al. Breathing control center neurons that promote arousal in mice. Science. (2017) 355:1411–5. doi: 10.1126/science.aai7984
28. Aiba I, Noebels JL. Kcnq2/Kv7.2 controls the threshold and bi-hemispheric symmetry of cortical spreading depolarization. Brain. (2021) 144:2863–78. doi: 10.1093/brain/awab141
29. Cozzolino O, Marchese M, Trovato F, Pracucci E, Ratto GM, Buzzi MG, et al. Understanding spreading depression from headache to sudden unexpected death. Front Neurol. (2018) 9:19. doi: 10.3389/fneur.2018.00019
30. Jansen NA, Schenke M, Voskuyl RA, Thijs RD, van den Maagdenberg A, Tolner EA, et al. Apnea associated with brainstem seizures in Cacna1a (S218L) mice is caused by medullary spreading depolarization. J Neurosci. (2019) 39:9633–44. doi: 10.1523/JNEUROSCI.1713-19.2019
31. Loonen ICM, Jansen NA, Cain SM, Schenke M, Voskuyl RA, Yung AC, et al. Brainstem spreading depolarization and cortical dynamics during fatal seizures in Cacna1a S218L mice. Brain. (2019) 142:412–25. doi: 10.1093/brain/awy325
32. Dlouhy BJ, Gehlbach BK, Kreple CJ, Kawasaki H, Oya H, Buzza C, et al. Breathing inhibited when seizures spread to the amygdala and upon amygdala stimulation. J Neurosci. (2015) 35:10281–9. doi: 10.1523/JNEUROSCI.0888-15.2015
33. Herrero JL, Khuvis S, Yeagle E, Cerf M, Mehta AD. Breathing above the brain stem: volitional control and attentional modulation in humans. J Neurophysiol. (2018) 119:145–59. doi: 10.1152/jn.00551.2017
34. Yang CF, Feldman JL. Efferent projections of excitatory and inhibitory preBotzinger Complex neurons. J Comp Neurol. (2018) 526:1389–402. doi: 10.1002/cne.24415
35. Cain SM, Bernier LP, ZhangY, Yung AC, Kass J, Bohnet B, et al. Hyperexcitable superior colliculus and fatal brainstem spreading depolarization in a model of sudden unexpected death in epilepsy. Brain Commun. (2022) 4:fcac006. doi: 10.1093/braincomms/fcac006
36. Nobis WP, Gonzalez Otarula KA, Templer JW, Gerard EE, VanHaerents S, Lane G, et al. The effect of seizure spread to the amygdala on respiration and onset of ictal central apnea. J Neurosurg. (2019) 132:1313–23. doi: 10.3171/2019.1.JNS183157
37. Jarre G, Altwegg-Boussac T, Williams MS, Studer F, Chipaux M, David O, et al. Building up absence seizures in the somatosensory cortex: from network to cellular epileptogenic processes. Cereb Cortex. (2017) 27:4607–23. doi: 10.1093/cercor/bhx174
38. Karalis N, Sirota A. Breathing coordinates cortico-hippocampal dynamics in mice during offline states. Nat Commun. (2022) 13:467. doi: 10.1038/s41467-022-28090-5
39. Marincovich A, Bravo E, Dlouhy B, Richerson GB. Amygdala lesions reduce seizure-induced respiratory arrest in DBA/1 mice. Epilepsy Behav. (2021) 121:106440. doi: 10.1016/j.yebeh.2019.07.041
40. Yan WW, Xia M, Chiang J, Levitt A, Hawkins N, Kearney J, et al. Enhanced synaptic transmission in the extended amygdala and altered excitability in an extended amygdala to brainstem circuit in a dravet syndrome mouse model. eNeuro. (2021) 8:3. doi: 10.1523/ENEURO.0306-20.2021
41. Kostopoulus G. Involvement of the thalamocortical system in epileptic loss of consciousness. Epilepsia. (2001) 42:13–9. doi: 10.1046/j.1528-1157.2001.042suppl.3013.x
42. Bonansco C, Fuenzalida M. Plasticity of hippocampal excitatory-inhibitory balance: missing the synaptic control in the epileptic brain. Neural Plast. (2016) 2016:8607038. doi: 10.1155/2016/8607038
43. Liou JY, Smith EH, Bateman LM, Bruce SL, McKhann GM, Goodman RR, et al. A model for focal seizure onset, propagation, evolution, and progression. Elife. (2020) 9: 50927. doi: 10.7554/eLife.50927
44. Eissa TL, Dijkstra K, Brune C, Emerson RG, van Putten M, Goodman RR, et al. Cross-scale effects of neural interactions during human neocortical seizure activity. Proc Natl Acad Sci U S A. (2017) 114:10761–6. doi: 10.1073/pnas.1702490114
45. Schevon CA, Weiss SA, McKhann GJr, Goodman RR, Yuste R, Emerson RG, et al. Evidence of an inhibitory restraint of seizure activity in humans. Nat Commun. (2012) 3:1060. doi: 10.1038/ncomms2056
46. Chever O, Zerimech S, Scalmani P, Lemaire L, Pizzamiglio L, Loucif A, et al. Initiation of migraine-related cortical spreading depolarization by hyperactivity of GABAergic neurons and NaV1, 1 Channels. J Clin Invest. (2021) 131:21. doi: 10.1172/JCI142203
47. Bigal ME, Lipton RB, Cohen J, Silberstein SD. Epilepsy and migraine. Epilepsy Behav. (2003) 4:S13–24. doi: 10.1016/j.yebeh.2003.07.003
48. Charles AC, Baca SM. Cortical spreading depression and migraine. Nat Rev Neurol. (2013) 9:637–44. doi: 10.1038/nrneurol.2013.192
49. Rho YI, Kim SH, Kang HC, Lee YJ, Kim YO, Kim SK, et al. EEG Characteristics and diagnostic implications in childhood headache: a multi-center study. Front Neurol. (2020) 11:569486. doi: 10.3389/fneur.2020.569486
50. Rogawski MA. Migraine and epilepsy-shared mechanisms within the family of episodic disorders. Jasper's Basic Mech Epilepsies. (2012) 51:1–5. doi: 10.1093/med/9780199746545.003.0073
51. Chong CD, Plasencia JD, Frakes DH, Schwedt TJ. Structural alterations of the brainstem in migraine. Neuroimage Clin. (2017) 13:223–7. doi: 10.1016/j.nicl.2016.10.023
52. Bateman LM, Li CS, Seyal M. Ictal hypoxemia in localization-related epilepsy: analysis of incidence, severity and risk factors. Brain. (2008) 131:3239–45. doi: 10.1093/brain/awn277
53. Seyal M, Bateman LM. Ictal apnea linked to contralateral spread of temporal lobe seizures: Intracranial EEG recordings in refractory temporal lobe epilepsy. Epilepsia. (2009) 50:2557–62. doi: 10.1111/j.1528-1167.2009.02245.x
54. Kang JY, Rabiei AH, Myint L, Nei M. Equivocal significance of post-ictal generalized EEG suppression as a marker of SUDEP risk. Seizure. (2017) 48:28–32. doi: 10.1016/j.seizure.2017.03.017
55. Seyal M, Bateman LM, Li CS. Impact of periictal interventions on respiratory dysfunction, postictal EEG suppression, and postictal immobility. Epilepsia. (2013) 54:377–82. doi: 10.1111/j.1528-1167.2012.03691.x
56. Surges R, Strzelczyk A, Scott CA, Walker MC, Sander JW. Postictal generalized electroencephalographic suppression is associated with generalized seizures. Epilepsy Behav. (2011) 21:271–4. doi: 10.1016/j.yebeh.2011.04.008
57. van den Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T, et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. (2004) 41:701–10. doi: 10.1016/S0896-6273(04)00085-6
58. van den Maagdenberg AM, Pizzorusso T, Kaja S, Terpolilli N, Shapovalova M, Hoebeek FE, et al. High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann Neurol. (2010) 67:85–98. doi: 10.1002/ana.21815
59. Leo L, Gherardini L, Barone V, Fusco D, Pietrobon M, et al. Increased susceptibility to cortical spreading depression in the mouse model of familial hemiplegic migraine type 2. PLoS Genet. (2011) 7:e1002129. doi: 10.1371/journal.pgen.1002129
60. Auffenberg E, Hedrich UB, Barbieri R, Miely D, Groschup B, Wuttke TV, et al. Hyperexcitable interneurons trigger cortical spreading depression in an Scn1a migraine model. J Clin Invest. (2021) 131:21. doi: 10.1172/JCI142202
61. Jansen NA, Dehghani A, Linssen MML, Breukel C, Tolner EA, van den Maagdenberg A, et al. First FHM3 mouse model shows spontaneous cortical spreading depolarizations. Ann Clin Transl Neurol. (2020) 7:132–8. doi: 10.1002/acn3.50971
62. Tamim I, Chung DY, Morais D, Loonen AL, Qin ICM, Misra T, et al. Spreading depression as an innate antiseizure mechanism. Nat Commun. (2021) 12:2206. doi: 10.1038/s41467-021-22464-x
63. Noebels JLMD. Brainstem spreading depolarization: rapid descent into the shadow of SUDEP. Brain. (2019) 142:231–3. doi: 10.1093/brain/awy356
64. Park K, Kanth K, Bajwa S, Girgis F, Shahlaie K, Seyal M, et al. Seizure-related apneas have an inconsistent linkage to amygdala seizure spread. Epilepsia. (2020) 61:1253–60. doi: 10.1111/epi.16518
65. Lacuey N, Hampson JP, Harper RM, Miller JP, Lhatoo S. Limbic and paralimbic structures driving ictal central apnea. Neurology. (2019) 92:e655–69. doi: 10.1212/WNL.0000000000006920
66. Lacuey N, Zonjy B, Londono L, Lhatoo SD. Amygdala and hippocampus are symptomatogenic zones for central apneic seizures. Neurology. (2017) 88:701–5. doi: 10.1212/WNL.0000000000003613
67. Rhone AE, Kovach CK, Harmata GI, Sullivan AW, Tranel D, Ciliberto MA, et al. A human amygdala site that inhibits respiration and elicits apnea in pediatric epilepsy. JCI Insight. (2020) 5:134852. doi: 10.1172/jci.insight.134852
68. Leung H, Schindler K, Kwan P, Elger C. Asystole induced by electrical stimulation of the left cingulate gyrus. Epileptic Disord. (2007) 9:77–81. doi: 10.1684/epd.2007.0054
69. Sanchez-Larsen A, Principe A, Ley M, Navarro-Cuartero J, Rocamora R. Characterization of the insular role in cardiac function through intracranial electrical stimulation of the human insula. Ann Neurol. (2021) 89:1172–80. doi: 10.1002/ana.26074
70. Doi A, Ramirez JM. State-dependent interactions between excitatory neuromodulators in the neuronal control of breathing. J Neurosci. (2010) 30:8251–62. doi: 10.1523/JNEUROSCI.5361-09.2010
71. Allen LA, Harper RM, Lhatoo S, Lemieux L, Diehl B. Neuroimaging of sudden unexpected death in epilepsy (SUDEP): insights from structural and resting-state functional MRI studies. Front Neurol. (2019) 10:185. doi: 10.3389/fneur.2019.00185
72. Mueller SG, Nei M, Bateman LM, Knowlton R, Laxer KD, Friedman D, et al. Brainstem network disruption: a pathway to sudden unexplained death in epilepsy? Hum Brain Mapp. (2018) 39:4820–30. doi: 10.1002/hbm.24325
73. Tang Y, Chen Q, Yu X, Xia W, Luo C, Huang X, et al. A resting-state functional connectivity study in patients at high risk for sudden unexpected death in epilepsy. Epilepsy Behav. (2014) 41:33–8. doi: 10.1016/j.yebeh.2014.08.140
74. Afif A, Minotti L, Kahane P, Hoffmann D. Anatomofunctional organization of the insular cortex: a study using intracerebral electrical stimulation in epileptic patients. Epilepsia. (2010) 51:2305–15. doi: 10.1111/j.1528-1167.2010.02755.x
75. Ramirez JM, Baertsch N. Defining the Rhythmogenic Elements of Mammalian Breathing. Physiology. (2018) 33:302–16. doi: 10.1152/physiol.00025.2018
76. Garcia AJ, Zanella S, Koch H, Doi A, Ramirez JM. Chapter 3–networks within networks: the neuronal control of breathing. Prog Brain Res. (2011) 188:31–50. doi: 10.1016/B978-0-444-53825-3.00008-5
77. Dutschmann M, Dick TE. Pontine mechanisms of respiratory control. Compr Physiol. (2012) 2:2443–69. doi: 10.1002/cphy.c100015
78. Anderson TM, Garcia AJ, Baertsch NA, Pollak J, Bloom JC, Wei AD, et al. A novel excitatory network for the control of breathing. Nature. (2016) 536:76–80. doi: 10.1038/nature18944
79. Manolis TA, Manolis AA, Melita H, Manolis AS. Sudden unexpected death in epilepsy: The neuro-cardio-respiratory connection. Seizure. (2019) 64:65–73. doi: 10.1016/j.seizure.2018.12.007
80. Schilling WP, McGrath MK, Yang T, Glazebrook PA, Faingold CL, Kunze DL, et al. Simultaneous cardiac and respiratory inhibition during seizure precedes death in the DBA/1 audiogenic mouse model of SUDEP. PLoS One. (2019) 14:e0223468. doi: 10.1371/journal.pone.0223468
81. Yang CF, Kim EJ, Callaway EM, Feldman JL. Monosynaptic Projections to Excitatory and Inhibitory preBotzinger Complex Neurons. Front Neuroanat. (2020) 14:58. doi: 10.3389/fnana.2020.00058
82. Sainju RK, Dragon DN, Winnike HB, Nashelsky MB, Granner MA, Gehlbach BK, et al. Ventilatory response to CO2 in patients with epilepsy. Epilepsia. (2019) 60:508–17. doi: 10.1111/epi.14660
83. Kalume F. Sudden unexpected death in Dravet syndrome: respiratory and other physiological dysfunctions. Respir Physiol Neurobiol. (2013) 189:324–8. doi: 10.1016/j.resp.2013.06.026
84. Massey CA, Sowers LP, Dlouhy BJ, Richerson GB. Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol. (2014) 10:271–82. doi: 10.1038/nrneurol.2014.64
85. Menuet C, Connelly AA, Bassi JK, Melo MR, Le S, Kamar J, et al. PreBotzinger complex neurons drive respiratory modulation of blood pressure and heart rate. Elife. (2020) 9. doi: 10.7554/eLife.57288.sa2
86. Moridani MK, Farhadi H. Heart rate variability as a biomarker for epilepsy seizure prediction. Bratisl Lek Listy. (2017) 118:3–8. doi: 10.4149/BLL_2017_001
87. Hampel KG, Jahanbekam A, Elger CE, Surges R. Seizure-related modulation of systemic arterial blood pressure in focal epilepsy. Epilepsia. (2016) 57:1709–18. doi: 10.1111/epi.13504
88. Patodia S, Somani A, Thom M. Review: Neuropathology findings in autonomic brain regions in SUDEP and future research directions. Auton Neurosci. (2021) 235:102862. doi: 10.1016/j.autneu.2021.102862
89. Barot N, Nei M. Autonomic aspects of sudden unexpected death in epilepsy (SUDEP). Clin Auton Res. (2019) 29:151–60. doi: 10.1007/s10286-018-0576-1
90. van der Lende M, Surges R, Sander JW, Thijs RD. Cardiac arrhythmias during or after epileptic seizures. J Neurol Neurosurg Psychiatry. (2016) 87:69–74. doi: 10.1016/j.autneu.2015.07.066
91. Myers KA, Bello-Espinosa LE, Symonds JD, Zuberi SM, Clegg R, Sadleir LG, et al. Heart rate variability in epilepsy: a potential biomarker of sudden unexpected death in epilepsy risk. Epilepsia. (2018) 59:1372–80. doi: 10.1111/epi.14438
92. Sivathamboo S, Friedman D, Laze J, Nightscales R, Chen Z, Kuhlmann L, et al. Association of short-term heart rate variability and sudden unexpected death in epilepsy. Neurology. (2021) 97:e2357–67. doi: 10.1212/WNL.0000000000012946
93. Englot DJ, Morgan VL, Chang C. Impaired vigilance networks in temporal lobe epilepsy: Mechanisms and clinical implications. Epilepsia. (2020) 61:189–202. doi: 10.1111/epi.16423
94. Buehler PK, Bleiler D, Tegtmeier I, Heitzmann D, Both C, Georgieff M, et al. Abnormal respiration under hyperoxia in TASK-1/3 potassium channel double knockout mice. Respir Physiol Neurobiol. (2017) 244:17–25. doi: 10.1016/j.resp.2017.06.009
95. Koch H, Zanella S, Elsen GE, Smith L, Doi A, Garcia AJ, et al. Stable respiratory activity requires both P/Q-type and N-type voltage-gated calcium channels. J Neurosci. (2013) 33:3633–45. doi: 10.1523/JNEUROSCI.6390-11.2013
96. Lu B, Su Y, Das S, Liu J, Xia J, Ren D, et al. The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell. (2007) 129:371–83. doi: 10.1016/j.cell.2007.02.041
97. Quintana A, Zanella S, Koch H, Kruse SE, Lee D, Ramirez JM, et al. Fatal breathing dysfunction in a mouse model of Leigh syndrome. J Clin Invest. (2012) 122:2359–68. doi: 10.1172/JCI62923
98. Gallego J, Dauger S. PHOX2B mutations and ventilatory control. Respir Physiol Neurobiol. (2008) 164:49–54. doi: 10.1016/j.resp.2008.07.003
99. Ramirez JM, Doi A, Garcia AJ, Elsen FP, Koch H, Wei AD. The cellular building blocks of breathing. Compr Physiol. (2012) 2:2683–731. doi: 10.1002/cphy.c110033
100. Murugesan A, Rani MRS, Hampson J, Zonjy B, Lacuey N, Faingold CL, et al. Serum serotonin levels in patients with epileptic seizures. Epilepsia. (2018) 59:e91–7. doi: 10.1111/epi.14198
101. Nass RD, Motloch LJ, Paar V, Lichtenauer M, Baumann J, Zur B, et al. Blood markers of cardiac stress after generalized convulsive seizures. Epilepsia. (2019) 60:201–10. doi: 10.1111/epi.14637
102. Simon RP, Aminhoff MJ, Benowitz NL. Changes in plasma catecholamines after tonic-clonic seizures. Neurology. (1984) 34:255–7. doi: 10.1212/WNL.34.2.255
103. Garcia AJ, Koschnitzky JE, Ramirez JM. The physiological determinants of sudden infant death syndrome. Respir Physiol Neurobiol. (2013) 189:288–300. doi: 10.1016/j.resp.2013.05.032
104. McCulloch K, Brouillette RT, Guzzetta AJ, Hunt CE. Arousal responses in near-miss sudden infant death syndrome and in normal infants. J Pediatr. (1982) 101:911–7. doi: 10.1016/S0022-3476(82)80009-7
105. Sawaguchi T, Kato I, Franco P, Sottiaux M, Kadhim H, Shimizu S, et al. Apnea, glial apoptosis and neuronal plasticity in the arousal pathway of victims of SIDS. Forensic Sci Int. (2005) 149:205–17. doi: 10.1016/j.forsciint.2004.10.015
106. Buchanan GF. Impaired CO2-induced arousal in SIDS and SUDEP. Trends Neurosci. (2019) 42:242–50. doi: 10.1016/j.tins.2019.02.002
107. Crandall LG, Lee JH, Stainman R, Friedman D, Devinsky O. Potential role of febrile seizures and other risk factors associated with sudden deaths in children. JAMA Netw Open. (2019) 2:e192739. doi: 10.1001/jamanetworkopen.2019.2739
108. Hesdorffer DC, Crandall LA, Friedman D, Devinsky O. Sudden unexplained death in childhood: A comparison of cases with and without a febrile seizure history. Epilepsia. (2015) 56:1294–300. doi: 10.1111/epi.13066
109. Richerson GB, Buchanan GF. The serotonin axis: Shared mechanisms in seizures, depression, and SUDEP. Epilepsia. (2011) 52:28–38. doi: 10.1111/j.1528-1167.2010.02908.x
110. Lijowska AS, Reed NW, Chiodini BA, Thach BT. Sequential arousal and airway-defensive behavior of infants in asphyxial sleep environments. J Appl Physiol. (1997) 83:219–28. doi: 10.1152/jappl.1997.83.1.219
111. McNamara F, Wulbrand H, Thach BT. Characteristics of the infant arousal response. J Appl Physiol. (1982) 85:2314–21. doi: 10.1152/jappl.1998.85.6.2314
112. Ramirez JM. The integrative role of the sigh in psychology, physiology, pathology, and neurobiology. Prog Brain Res. (2014) 209:91–129. doi: 10.1016/B978-0-444-63274-6.00006-0
113. Lieske SP, Thoby-Brisson M, Telgkamp P, Ramirez JM. Reconfiguration of the neural network controlling multiple breathing patterns: eupnea, sighs and gasps. Nat Neurosci. (2000) 3:600–7. doi: 10.1038/75776
114. Cassell MD, Freedman LJ, Shi C. The intrinsic organization of the central extended amygdala. Ann N Y Acad Sci. (1999) 877:217–41. doi: 10.1111/j.1749-6632.1999.tb09270.x
115. Dong HW, Swanson LW. Projections from bed nuclei of the stria terminalis, dorsomedial nucleus: implications for cerebral hemisphere integration of neuroendocrine, autonomic, and drinking responses. J Comp Neurol. (2006) 494:75–107. doi: 10.1002/cne.20790
116. Pollak Dorocic I, Furth D, Xuan Y, Johansson Y, Pozzi L, Silberberg G, et al. A whole-brain atlas of inputs to serotonergic neurons of the dorsal and median raphe nuclei. Neuron. (2014) 83:663–78. doi: 10.1016/j.neuron.2014.07.002
117. Reichard RA, Subramanian S, Desta MT, Sura T, Becker ML, Ghobadi CW, et al. Abundant collateralization of temporal lobe projections to the accumbens, bed nucleus of stria terminalis, central amygdala and lateral septum. Brain Struct Funct. (2017) 222:1971–88. doi: 10.1007/s00429-016-1321-y
118. Garcia AJ, Rotem-Kohavi N, Doi A, Ramirez JM. Post-hypoxic recovery of respiratory rhythm generation is gender dependent. PLoS ONE. (2013) 8:e60695. doi: 10.1371/journal.pone.0060695
119. Poets CF, Meny RG, Chobanian MR, Bobofiglo RE. Gasping and other cardiorespiratory patterns during sudden infant deaths. Pediatr Res. (1999) 45:350–4. doi: 10.1203/00006450-199903000-00010
120. Sintas C, Fernandez-Castillo N, Vila-Pueyo M, Pozo-Rosich P, Macaya A, Cormand B, et al. Transcriptomic changes in rat cortex and brainstem after cortical spreading depression with or without pretreatment with migraine prophylactic drugs. J Pain. (2017) 18:366–75. doi: 10.1016/j.jpain.2016.11.007
121. Paton JF, Abdala AP, Koizumi H, Smith JC, St-John WM. Respiratory rhythm generation during gasping depends on persistent sodium current. Nat Neurosci. (2006) 9:311–3. doi: 10.1038/nn1650
122. Telgkamp P, Ramirez JM. Differential responses of respiratory nuclei to anoxia in rhythmic brain stem slices of mice. J Neurophysiol. (1999) 82:2163–70. doi: 10.1152/jn.1999.82.5.2163
123. Schmidt C, Bellingham MC, Richter DW. Adenosinergic modulation of respiratory neurones and hypoxic responses in the anaesthetized cat. J Physiol. (1995) 483:769–81. doi: 10.1113/jphysiol.1995.sp020621
124. Koch H, Caughie C, Elsen FP, Doi A, Garcia AJ, Zanella S, Ramirez JM. Prostaglandin E2 differentially modulates the central control of eupnoea, sighs and gasping in mice. J Physiol. (2015) 593:305–19. doi: 10.1113/jphysiol.2014.279794
125. Pena F, Aguileta MA. Effects of riluzole and flufenamic acid on eupnea and gasping of neonatal mice in vivo. Neurosci Lett. (2007) 415:288–93. doi: 10.1016/j.neulet.2007.01.032
126. Pena F, Parkis MA, Tryba AK, Ramirez JM. Differential contribution of pacemaker properties to the generation of respiratory rhythms during normoxia and hypoxia. Neuron. (2004) 43:105–17. doi: 10.1016/j.neuron.2004.06.023
127. Layer N, Brandes J, Luhrs PJ, Wuttke TV, Koch H. The effect of lamotrigine and other antiepileptic drugs on respiratory rhythm generation in the pre-Botzinger complex. Epilepsia. (2021) 62:2790–803. doi: 10.1111/epi.17066
128. Christensen J, Trabjerg BB, Dreier JW. Cardiac morbidity and mortality associated with the use of lamotrigine. Epilepsia. (2022) 63:2371–80. doi: 10.1111/epi.17339
129. Zanella S, Doi A, Garcia AJ, Elsen F, Kirsch S, Wei AD, Ramirez JM. When norepinephrine becomes a driver of breathing irregularities: how intermittent hypoxia fundamentally alters the modulatory response of the respiratory network. J Neurosci. (2014) 34:36–50. doi: 10.1523/JNEUROSCI.3644-12.2014
130. Bruxel EM, Bruno DCF, Do Canto AM, Geraldis JC, Godoi AB, Martin M, et al. Multi-omics in mesial temporal lobe epilepsy with hippocampal sclerosis: Clues into the underlying mechanisms leading to disease. Seizure. (2021) 90:34–50. doi: 10.1016/j.seizure.2021.03.002
131. Grainger AI, King MC, Nagel DA, Parri HR, Coleman MD, Hill EJ, et al. In vitro models for seizure-liability testing using induced pluripotent stem cells. Front Neurosci. (2018) 12:590. doi: 10.3389/fnins.2018.00590
132. Raimondo JV, Heinemann U, Curtis D, Goodkin M, Dulla, HP, et al. Methodological standards for in vitro models of epilepsy and epileptic seizures. A TASK1-WG4 report of the AES/ILAE translational task force of the ILAE. Epilepsia. (2017) 58:40–52. doi: 10.1111/epi.13901
133. Ziobro JM, Deshpande LS, Delorenzo RJ. An organotypic hippocampal slice culture model of excitotoxic injury induced spontaneous recurrent epileptiform discharges. Brain Res. (2011) 1371:110–20. doi: 10.1016/j.brainres.2010.11.065
134. Baraban S. Emerging epilepsy models: insights from mice, flies, worms and fish. Curr Opin Neurol. (2007) 20:164–8. doi: 10.1097/WCO.0b013e328042bae0
135. Buchsbaum IY, Cappello S. Neuronal migration in the CNS during development and disease: insights from in vivo and in vitro models. Development. (2019) 146:163766. doi: 10.1242/dev.163766
136. Kandratavicius L, Balista PA, Lopes-Aguiar C, Ruggiero RN, Umeoka EH, Garcia-Cairasco N, et al. Animal models of epilepsy: use and limitations. Neuropsychiatr Dis Treat. (2014) 10:1693–705. doi: 10.2147/NDT.S50371
137. Opitz A, Falchier A, Linn GS, Milham MP, Schroeder CE. Limitations of ex vivo measurements for in vivo neuroscience. Proc Natl Acad Sci U S A. (2017) 114:5243–6. doi: 10.1073/pnas.1617024114
138. Stewart AM, Braubach O, Spitsbergen J, Gerlai R, Kalueff AV. Zebrafish models for translational neuroscience research: from tank to bedside. Trends Neurosci. (2014) 37:264–78. doi: 10.1016/j.tins.2014.02.011
139. Schwarzacher SW, Rub U, Deller T. Neuroanatomical characteristics of the human pre-Botzinger complex and its involvement in neurodegenerative brainstem diseases. Brain. (2011) 134:24–35. doi: 10.1093/brain/awq327
140. Bentley JN, Chestek C, Stacey WC, Patil PG. Optogenetics in epilepsy. Neurosurg Focus. (2013) 34:E4. doi: 10.3171/2013.3.FOCUS1364
141. Helbig I, Ellis CA. Personalized medicine in genetic epilepsies - possibilities, challenges, and new frontiers. Neuropharmacology. (2020) 172:107970. doi: 10.1016/j.neuropharm.2020.107970
142. Helbig I, Tayoun AA. Understanding Genotypes and Phenotypes in Epileptic Encephalopathies. Mol Syndromol. (2016) 7:172–81. doi: 10.1159/000448530
143. Han K, Holder JL, Schaaf CP, Lu H, Chen H, Kang H, et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature. (2013) 503:72–7. doi: 10.1038/nature12630
144. Jin C, Zhang Y, Kim S, Kim Y, Lee Y, Han K, et al. Spontaneous seizure and partial lethality of juvenile Shank3-overexpressing mice in C57BL/6 J background. Mol Brain. (2018) 11:57. doi: 10.1186/s13041-018-0403-6
145. Lee Y, Zhang Y, Kim S, Han K. Excitatory and inhibitory synaptic dysfunction in mania: an emerging hypothesis from animal model studies. Exp Mol Med. (2018) 50:1–11. doi: 10.1038/s12276-018-0187-x
146. Lewis ML, Kesler M, Candy SA, Rho JM, Pittman QJ. Comorbid epilepsy in autism spectrum disorder: Implications of postnatal inflammation for brain excitability. Epilepsia. (2018) 59:1316–26. doi: 10.1111/epi.14440
147. Seiffert S, Pendziwiat M, Bierhals T, Goel H, Schwarz N, van der Ven A, et al. Modulating effects of FGF12 variants on NaV1.2 and NaV1.6 being associated with developmental and epileptic encephalopathy and Autism spectrum disorder: a case series. EBioMedicine. (2022) 83:104234. doi: 10.1016/j.ebiom.2022.104234
148. Wong JC, Grieco SF, Dutt K, Chen L, Thelin JT, Inglis GAS, et al. Autistic-like behavior, spontaneous seizures, and increased neuronal excitability in a Scn8a mouse model. Neuropsychopharmacology. (2021) 46:2011–20. doi: 10.1038/s41386-021-00985-9
149. Guerrini R, Conti V, Mantegazza M, Balestrini S, Galanopoulou AS, Benfenati F, et al. Developmental and epileptic encephalopathies: from genetic heterogeneity to phenotypic continuum. Physiol Rev. (2023) 103:433–513. doi: 10.1152/physrev.00063.2021
150. Colmers PLW, Maguire J. Network dysfunction in comorbid psychiatric illnesses and epilepsy. Epilepsy Curr. (2020) 20:205–10. doi: 10.1177/1535759720934787
151. Weston CSE. Four Social Brain Regions, Their Dysfunctions, and Sequelae, Extensively Explain Autism Spectrum Disorder Symptomatology. Brain Sci. (2019) 9:130. doi: 10.3390/brainsci9060130
152. Marshall GF, Gonzalez-Sulser A, Abbott CM. Modelling epilepsy in the mouse: challenges and solutions. Dis Model Mech. (2021) 14:47449. doi: 10.1242/dmm.047449
153. Di Sapia R, Moro F, Montanarella M, Iori V, Micotti E, Tolomeo D, et al. In-depth characterization of a mouse model of post-traumatic epilepsy for biomarker and drug discovery. Acta Neuropathol Commun. (2021) 9:76. doi: 10.1186/s40478-021-01165-y
154. Meisler MH, Kearney J, Escayg A, MacDonald BT, Sprunger LK. Sodium channels and neurological disease: insights from Scn8a mutations in the mouse. Neuroscientist. (2001) 7:136–45. doi: 10.1177/107385840100700208
155. Goldman AM, Buchanan G, Aiba I, Noebels JL. SUDEP Animal Models. Models Seizures Epilepsy. (2017) 1:1007–18. doi: 10.1016/B978-0-12-804066-9.00070-5
156. Catterall WA. Sodium Channel Mutations and Epilepsy in Jasper's Basic Mechanisms of the Epilepsies. Bethesda, MD: National Center for Biotechnology Information (2012).
157. Mantegazza M, Cestele S, Catterall WA. Sodium channelopathies of skeletal muscle and brain. Physiol Rev. (2021) 101:1633–89. doi: 10.1152/physrev.00025.2020
158. Meisler MH, Hill SF, Yu W. Sodium channelopathies in neurodevelopmental disorders. Nat Rev Neurosci. (2021) 22:152–66. doi: 10.1038/s41583-020-00418-4
159. Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. (2010) 30:5167–75. doi: 10.1523/JNEUROSCI.5591-09.2010
160. Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, et al. Deletion of the KV1.1 potassium channel causes epilepsy in mice. Neuron. (1998) 20:809–19. doi: 10.1016/S0896-6273(00)81018-1
161. Felix R. Insights from mouse models of absence epilepsy into Ca2+ channel physiology and disease etiology. Cell Mol Neurobiol. (2002) 22:103–20. doi: 10.1023/A:1019807719343
162. Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL, et al. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. (2009) 1:6. doi: 10.1126/scitranslmed.3000289
163. Yuan Y, O'Malley HA, Smaldino MA, Bouza AA, Hull JM, Isom LL, et al. Delayed maturation of GABAergic signaling in the Scn1a and Scn1b mouse models of Dravet Syndrome. Sci Rep. (2019) 9:6210. doi: 10.1038/s41598-019-42191-0
164. Wengert ER, Wenker IC, Wagner EL, Wagley PK, Gaykema RP, Shin JB, et al. Adrenergic mechanisms of audiogenic seizure-induced death in a mouse model of SCN8A encephalopathy. Front Neurosci. (2021) 15:581048. doi: 10.3389/fnins.2021.581048
165. Zamorano-Leon JJ, Yanez R, Jaime G, Rodriguez-Sierra P, Calatrava-Ledrado L, Alvarez-Granada RR, et al. KCNH2 gene mutation: a potential link between epilepsy and long QT-2 syndrome. J Neurogenet. (2012) 26:382–6. doi: 10.3109/01677063.2012.674993
166. Auerbach DS, Jones J, Clawson BC, Offord J, Lenk GM, Ogiwara I, et al. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One. (2013) 8:e77843. doi: 10.1371/journal.pone.0077843
167. Brunklaus A, Zuberi SM. Dravet syndrome–from epileptic encephalopathy to channelopathy. Epilepsia. (2014) 55:979–84. doi: 10.1111/epi.12652
168. Connolly MB. Dravet syndrome: diagnosis and long-term course. Can J Neurol Sci. (2016) 43:S3–8. doi: 10.1017/cjn.2016.243
169. Kim Y, Bravo E, Thirnbeck CK, Smith-Mellecker LA, Kim SH, Gehlbach BK, et al. Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest. (2018) 128:1141–53. doi: 10.1172/JCI94999
170. Hedrich UB, Liautard C, Kirschenbaum D, Pofahl M, Lavigne J, Liu Y, et al. Impaired action potential initiation in GABAergic interneurons causes hyperexcitable networks in an epileptic mouse model carrying a human Na(V)1.1 mutation. J Neurosci. (2014) 34:14874–89. doi: 10.1523/JNEUROSCI.0721-14.2014
171. Layer N, Sonnenberg L, Pardo Gonzalez E, Benda J, Hedrich UBS, Lerche H, et al. Dravet Variant SCN1A (A1783V) Impairs Interneuron Firing Predominantly by Altered Channel Activation. Front Cell Neurosci. (2021) 15:754530. doi: 10.3389/fncel.2021.754530
172. Tran CH, Vaiana M, Nakuci J, Somarowthu A, Goff KM, Goldstein N, et al. Interneuron desynchronization precedes seizures in a mouse model of dravet syndrome. J Neurosci. (2020) 40:2764–75. doi: 10.1523/JNEUROSCI.2370-19.2020
173. Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. (2006) 9:1142–9. doi: 10.1038/nn1754
174. Lange d. e., Gunning I. M., Sonsma B., van Gemert A. C. M., van Kempen L., Verbeek M., et al. (2019). Outcomes and comorbidities of SCN1A-related seizure disorders. Epilepsy Behav 90, 252–259. doi: 10.1016/j.yebeh.2018.09.041
175. Studtmann C, Ladislav M, Topolski MA, Safari M, Swanger SA. NaV1.1 haploinsufficiency impairs glutamatergic and GABAergic neuron function in the thalamus. Neurobiol Dis. (2022) 167:105672. doi: 10.1016/j.nbd.2022.105672
176. Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC, Potter GB, et al. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A. (2012) 109:14646–51. doi: 10.1073/pnas.1211591109
177. Gataullina S, Dulac O. From genotype to phenotype in Dravet disease. Seizure. (2017) 44:58–64. doi: 10.1016/j.seizure.2016.10.014
178. Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, et al. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature. (2012) 489:385–90. doi: 10.1038/nature11356
179. Kuo FS, Cleary CM, LoTurco JJ, Chen X, Mulkey DK. Disordered breathing in a mouse model of Dravet syndrome. Elife. (2019) 8:22. doi: 10.7554/eLife.43387.022
180. Ricobaraza A, Mora-Jimenez L, Puerta E, Sanchez-Carpintero R, Mingorance A, Artieda J, et al. Epilepsy and neuropsychiatric comorbidities in mice carrying a recurrent Dravet syndrome SCN1A missense mutation. Sci Rep. (2019) 9:14172. doi: 10.1038/s41598-019-50627-w
181. Chen C, Westenbroek RE, Xu X, Edwards CA, Sorenson DR, Chen Y, et al. Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci. (2004) 24:4030–42. doi: 10.1523/JNEUROSCI.4139-03.2004
182. Wenker IC, Blizzard EA, Wagley PK, Patel MK. Peri-ictal autonomic control of cardiac function and seizure-induced death. Front Neurosci. (2021) 15:795145. doi: 10.3389/fnins.2021.795145
183. Dhaibar H, Gautier NM, Chernyshev OY, Dominic P, Glasscock E. Cardiorespiratory profiling reveals primary breathing dysfunction in Kcna1-null mice: Implications for sudden unexpected death in epilepsy. Neurobiol Dis. (2019) 127:502–11. doi: 10.1016/j.nbd.2019.04.006
184. Guan D, Lee JC, Higgs MH, Spain WJ, Foehring RC. Functional roles of Kv1 channels in neocortical pyramidal neurons. J Neurophysiol. (2007) 97:1931–40. doi: 10.1152/jn.00933.2006
185. Wang H, Kunkel DD, Schwartzkroin PA, Tempel BL. Localization of Kv1.1 and Kv1.2. Two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J Neurosci. (1994) 14:4588–99. doi: 10.1523/JNEUROSCI.14-08-04588.1994
186. Moore BM, Jerry Jou C, Tatalovic M, Kaufman ES, Kline DD, Kunze DL, et al. The Kv1.1 null mouse a model of sudden unexpected death in epilepsy (SUDEP). Epilepsia. (2014) 55:1808–16. doi: 10.1111/epi.12793
187. Simeone KA, Hallgren J, Bockman CS, Aggarwal A, Kansal V, Netzel L, et al. Respiratory dysfunction progresses with age in Kcna1-null mice, a model of sudden unexpected death in epilepsy. Epilepsia. (2018) 59:345–57. doi: 10.1111/epi.13971
188. Thouta S, Zhang Y, Garcia E, Snutch TP. Kv1.1 channels mediate network excitability and feed-forward inhibition in local amygdala circuits. Sci Rep. (2021) 11:15180. doi: 10.1038/s41598-021-94633-3
189. Partemi S, Cestele S, Pezzella M, Campuzano O, Paravidino R, Pascali VL, et al. Loss-of-function KCNH2 mutation in a family with long QT syndrome, epilepsy, and sudden death. Epilepsia. (2013) 54:e112–116. doi: 10.1111/epi.12259
190. Tiron C, Campuzano O, Perez-Serra A, Mademont I, Coll M, Allegue C, et al. Further evidence of the association between LQT syndrome and epilepsy in a family with KCNQ1 pathogenic variant. Seizure. (2015) 25:65–7. doi: 10.1016/j.seizure.2015.01.003
191. Hawkins NA, Anderson LL, Gertler TS, Laux L, George AL, Kearney JA. Screening of conventional anticonvulsants in a genetic mouse model of epilepsy. Ann Clin Transl Neurol. (2017) 4:326–39. doi: 10.1002/acn3.413
192. Chauvette S, Soltani S, Seigneur J, Timofeev I. In vivo models of cortical acquired epilepsy. J Neurosci Methods. (2016) 260:185–201. doi: 10.1016/j.jneumeth.2015.08.030
193. Rubio C, Rubio-Osornio M, Retana-Marquez S, Veronica Custodio ML, Paz C. In vivo experimental models of epilepsy. Cent Nerv Syst Agents Med Chem. (2010) 10:298–309. doi: 10.2174/187152410793429746
194. Ortiz C, Carlen M, Meletis K. Spatial transcriptomics: molecular maps of the mammalian brain. Annu Rev Neurosci. (2021) 44:547–62. doi: 10.1146/annurev-neuro-100520-082639
195. Boyden E. Optogenetics and the future of neuroscience. Nat. Neurosci. (2015) 18;1200–1. doi: 10.1038/nn.4094
196. Shimoda Y, Beppu K, Ikoma Y, Morizawa YM, Zuguchi S, Hino U, et al. Optogenetic stimulus-triggered acquisition of seizure resistance. Neurobiol Dis. (2022) 163:105602. doi: 10.1016/j.nbd.2021.105602
197. Mahmoudi P, Veladi H, Pakdel F. Optogenetics, tools and applications in neurobiology. J Med Signals Sens. (2017) 7:71–9. doi: 10.4103/2228-7477.205506
198. Pouliopoulos AN, Murillo MF, Noel RL, Batts AJ, Ji R, Kwon N, et al. Non-invasive optogenetics with ultrasound-mediated gene delivery and red-light excitation. Brain Stimul. (2022) 15:927–41. doi: 10.1016/j.brs.2022.06.007
199. Campbell EJ, Marchant NJ. The use of chemogenetics in behavioural neuroscience: receptor variants, targeting approaches and caveats. Br J Pharmacol. (2018) 175:994–1003. doi: 10.1111/bph.14146
200. Berglund K, Tung JK, Higashikubo B, Gross RE, Moore CI, Hochgeschwender U, et al. Combined optogenetic and chemogenetic control of neurons. Methods Mol Biol. (2016) 1408:207–25. doi: 10.1007/978-1-4939-3512-3_14
201. Cela E, Sjöström PJ. Novel optogenetic approaches in epilepsy research. Front Neurosci. (2019) 13:947. doi: 10.3389/fnins.2019.00947
202. Staba RJ, Stead M, Worrell GA. Electrophysiological biomarkers of epilepsy. Neurotherapeutics. (2014) 11:334–46. doi: 10.1007/s13311-014-0259-0
203. Choi JH, Koch KP, Poppendieck W, Lee M, Shin HS. High resolution electroencephalography in freely moving mice. J Neurophysiol. (2010) 104:1825–34. doi: 10.1152/jn.00188.2010
204. Sheybani L, Birot G, Contestabile A, Seeck M, Kiss JZ, Schaller K, et al. Electrophysiological evidence for the development of a self-sustained large-scale epileptic network in the kainate mouse model of temporal lobe epilepsy. J Neurosci. (2018) 38:3776–91. doi: 10.1523/JNEUROSCI.2193-17.2018
205. Dai Y, Song Y, Xie J, Xu S, Li X, He E, et al. In vivo microelectrode arrays for detecting multi-region epileptic activities in the hippocampus in the latent period of rat model of temporal lobe epilepsy. Micromachines. (2021) 12:6. doi: 10.3390/mi12060659
206. Houben T, Loonen IC, Baca SM, Schenke M, Meijer JH, Ferrari MD, et al. Optogenetic induction of cortical spreading depression in anesthetized and freely behaving mice. J Cereb Blood Flow Metab. (2017) 37:1641–55. doi: 10.1177/0271678X16645113
207. Chung DY, Sugimoto K, Fischer P, Bohm M, Takizawa T, Sadeghian H, et al. Real-time non-invasive in vivo visible light detection of cortical spreading depolarizations in mice. J Neurosci Methods. (2018) 309:143–6. doi: 10.1016/j.jneumeth.2018.09.001
208. Smith JM, Bradley DP, James MF, Huang CL. Physiological studies of cortical spreading depression. Biol Rev Camb Philos Soc. (2006) 81:457–81. doi: 10.1017/S1464793106007081
209. Dufour S, Koninck Y. Optrodes for combined optogenetics and electrophysiology in live animals. Neurophotonics. (2015) 2:1–14. doi: 10.1117/1.NPh.2.3.031205
210. Tian H, Xu K, Zou L, Fang Y. Multimodal neural probes for combined optogenetics and electrophysiology. iScience. (2022) 25:103612. doi: 10.1016/j.isci.2021.103612
211. Anikeeva P, Andalman AS, Witten I, Warden M, Goshen I, Grosenick L, et al. Optetrode: a multichannel readout for optogenetic control in freely moving mice. Nat Neurosci. (2011) 15:163–70. doi: 10.1038/nn.2992
212. Lu Y, Zhong C, Wang L, Wei P, He W, Huang K, et al. Optogenetic dissection of ictal propagation in the hippocampal-entorhinal cortex structures. Nat Commun. (2016) 7:10962. doi: 10.1038/ncomms10962
213. Lim DH, Ledue J, Mohajerani MH, Vanni MP, Murphy TH. Optogenetic approaches for functional mouse brain mapping. Front Neurosci. (2013) 7:54. doi: 10.3389/fnins.2013.00054
214. Xiao D, Forys BJ, Vanni MP, Murphy TH. MesoNet allows automated scaling and segmentation of mouse mesoscale cortical maps using machine learning. Nat Commun. (2021) 12:5992. doi: 10.1038/s41467-021-26255-2
215. Fehervari TD, Okazaki Y, Sawai H, Yagi T. In vivo voltage-sensitive dye study of lateral spreading of cortical activity in mouse primary visual cortex induced by a current impulse. PLoS ONE. (2015) 10:e0133853. doi: 10.1371/journal.pone.0133853
216. Goldberg EM, Coulter DA. Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat Rev Neurosci. (2013) 14:337–49. doi: 10.1038/nrn3482
217. Oh J, Lee C, Kaang BK. Imaging and analysis of genetically encoded calcium indicators linking neural circuits and behaviors. Korean J Physiol Pharmacol. (2019) 23:237–49. doi: 10.4196/kjpp.2019.23.4.237
218. Somarowthu A, Goff KM, Goldberg EM. Two-photon calcium imaging of seizures in awake, head-fixed mice. Cell Calcium. (2021) 96:102380. doi: 10.1016/j.ceca.2021.102380
219. Li L, Tang Y, Sun L, Rahman K, Huang K, Xu W, et al. In vivo fiber photometry of neural activity in response to optogenetically manipulated inputs in freely moving mice. J Innov Opt Health Sci. (2017) 10:5. doi: 10.1142/S1793545817430015
220. Dong X, Zhang X, Wang F, Liu N, Liu A, Li Y, et al. Simultaneous calcium recordings of hippocampal CA1 and primary motor cortex M1 and their relations to behavioral activities in freely moving epileptic mice. Exp Brain Res. (2020) 238:1479–88. doi: 10.1007/s00221-020-05815-w
221. Patel AA, McAlinden N, Mathieson K, Sakata S. Simultaneous electrophysiology and fiber photometry in freely behaving mice. Front Neurosci. (2020) 14:148. doi: 10.3389/fnins.2020.00148
222. Levesque M, Wang S, Etter G, Williams S, Avoli M. Bilateral optogenetic activation of inhibitory cells favors ictogenesis. Neurobiol Dis. (2022) 171:105794. doi: 10.1016/j.nbd.2022.105794
223. Furman M, Zhan Q, McCafferty C, Lerner BA, Motelow JE, Meng J, et al. Optogenetic stimulation of cholinergic brainstem neurons during focal limbic seizures: Effects on cortical physiology. Epilepsia. (2015) 56:e198–202. doi: 10.1111/epi.13220
224. Liu S, Han S. Simultaneous recording of breathing and neural activity in awake behaving mice. STAR Protoc. (2022) 3:101412. doi: 10.1016/j.xpro.2022.101412
225. Ho D, Zhao X, Gao S, Hong C, Vatner DE, Vatner SF, et al. Heart rate and electrocardiography monitoring in mice. Curr Protoc Mouse Biol. (2011) 1:123–39. doi: 10.1002/9780470942390.mo100159
226. Mongue-Din H, Salmon A, Fiszman MY, Fromes Y. Non-invasive restrained ECG recording in conscious small rodents: a new tool for cardiac electrical activity investigation. Pflugers Arch. (2007) 454:165–71. doi: 10.1007/s00424-006-0197-8
227. Grimaud J, Murthy VN. How to monitor breathing in laboratory rodents: a review of the current methods. J Neurophysiol. (2018) 120:624–32. doi: 10.1152/jn.00708.2017
228. Viemari JC, Roux JC, Tryba AK, Saywell V, Burnet H, Pena F, et al. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. (2005) 25:11521–30. doi: 10.1523/JNEUROSCI.4373-05.2005
229. Park KJ, Sharma G, Kennedy JD, Seyal M. Potentially high-risk cardiac arrhythmias with focal to bilateral tonic-clonic seizures and generalized tonic-clonic seizures are associated with the duration of periictal hypoxemia. Epilepsia. (2017) 58:2164–71. doi: 10.1111/epi.13934
230. Medinaceli Quintela R, Bauer J, Wallhorn L, Le K, Brunert D, Rothermel M, et al. Dynamic impairment of olfactory behavior and signaling mediated by an olfactory corticofugal system. J Neurosci. (2020) 40:7269–85. doi: 10.1523/JNEUROSCI.2667-19.2020
231. Kaneko K, Currin CB, Goff KM, Wengert ER, Somarowthu A, Vogels TP, et al. Developmentally regulated impairment of parvalbumin interneuron synaptic transmission in an experimental model of Dravet syndrome. Cell Rep. (2022) 38:110580. doi: 10.1016/j.celrep.2022.110580
232. Valassina N, Brusco S, Salamone A, Serra L, Luoni M, Giannelli S, et al. Scn1a gene reactivation after symptom onset rescues pathological phenotypes in a mouse model of Dravet syndrome. Nat Commun. (2022) 13:161. doi: 10.1038/s41467-021-27837-w
233. Lein E, Borm LE, Linnarsson S. The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science. (2017) 358:64–9. doi: 10.1126/science.aan6827
234. Leitner DF, Mills JD, Pires G, Faustin A, Drummond E, Kanshin E, et al. Proteomics and Transcriptomics of the hippocampus and cortex in SUDEP and high-risk SUDEP Patients. Neurology. (2021) 96:e2639–52. doi: 10.1212/WNL.0000000000011999
235. Hutson TN, Rezaei F, Gautier NM, Indumathy J, Glasscock E, Iasemidis L, et al. Directed Connectivity Analysis of the Neuro-Cardio- and Respiratory Systems Reveals Novel Biomarkers of Susceptibility to SUDEP. IEEE Open J Eng Med Biol. (2020) 1:301–11. doi: 10.1109/OJEMB.2020.3036544
236. Boon P, Raedt R, Herdt DE, Wyckhuys VT, Vonck K. Electrical stimulation for the treatment of epilepsy. Neurotherapeutics. (2009) 6:218–27. doi: 10.1016/j.nurt.2008.12.003
237. Chaitanya G, Hampson JP, Toth E, Hupp NJ, Hampson JS, Mosher JC, et al. Limbic and paralimbic respiratory modulation: from inhibition to enhancement. Epilepsia. (2022) 63:1799–811. doi: 10.1111/epi.17244
238. Zhang H, Zhao H, Zeng C, Van Dort C, Faingold CL, Taylor NE, et al. Optogenetic activation of 5-HT neurons in the dorsal raphe suppresses seizure-induced respiratory arrest and produces anticonvulsant effect in the DBA/1 mouse SUDEP model. Neurobiol Dis. (2018) 110:47–58. doi: 10.1016/j.nbd.2017.11.003
239. Magalhaes DM, Pereira N, Rombo DM, Beltrao-Cavacas C, Sebastiao AM, Valente CA, et al. Ex vivo model of epilepsy in organotypic slices-a new tool for drug screening. J Neuroinflammation. (2018) 15:203. doi: 10.1186/s12974-018-1225-2
240. Chen Z, Brodie MJ, Liew D, Kwan P. Treatment outcomes in patients with newly diagnosed epilepsy treated with established and new antiepileptic drugs: a 30-year longitudinal cohort study. JAMA Neurol. (2018) 75:279–86. doi: 10.1001/jamaneurol.2017.3949
241. Luoni C, Bisulli F, Canevini MP, Sarro D, Fattore G, et al. Determinants of health-related quality of life in pharmacoresistant epilepsy: results from a large multicenter study of consecutively enrolled patients using validated quantitative assessments. Epilepsia. (2011) 52:2181–91. doi: 10.1111/j.1528-1167.2011.03325.x
242. Ortinski P, Meador KJ. Cognitive side effects of antiepileptic drugs. Epilepsy Behav. (2004) 5:S60–65. doi: 10.1016/j.yebeh.2003.11.008
243. Löscher W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. European Journal of Epilepsy. (2011) 20:359–68. doi: 10.1016/j.seizure.2011.01.003
244. Engel J, Pitkanen A, Loeb JA, Dudek FE, Bertram EH, Cole AJ, et al. Epilepsy biomarkers. Epilepsia. (2013) 54:61–9. doi: 10.1111/epi.12299
245. Panuccio G, Colombi I, Chiappalone M. Recording and modulation of epileptiform activity in rodent brain slices coupled to microelectrode arrays. J Vis Exp. (2018) 135:57548. doi: 10.3791/57548
246. Ben-Menachem E. Vagus-nerve stimulation for the treatment of epilepsy. Lancet Neurol. (2002) 1:477–82. doi: 10.1016/S1474-4422(02)00220-X
247. Han CL, Hu W, Stead M, Zhang T, Zhang JG, Worrell GA, et al. Electrical stimulation of hippocampus for the treatment of refractory temporal lobe epilepsy. Brain Res Bull. (2014) 109:13–21. doi: 10.1016/j.brainresbull.2014.08.007
248. Rodenkirch C, Carmel JB, Wang Q. Rapid effects of vagus nerve stimulation on sensory processing through activation of neuromodulatory systems. Front Neurosci. (2022) 16:922424. doi: 10.3389/fnins.2022.922424
Keywords: epilepsy, SUDEP, brainstem, mouse models, cardiorespiratory activity
Citation: Bauer J, Devinsky O, Rothermel M and Koch H (2023) Autonomic dysfunction in epilepsy mouse models with implications for SUDEP research. Front. Neurol. 13:1040648. doi: 10.3389/fneur.2022.1040648
Received: 09 September 2022; Accepted: 12 December 2022;
Published: 06 January 2023.
Edited by:
Brian James Dlouhy, The University of Iowa, United StatesReviewed by:
Irina Oane, Bucharest University Emergency Hospital, RomaniaCopyright © 2023 Bauer, Devinsky, Rothermel and Koch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Henner Koch,  aGtvY2hAdWthYWNoZW4uZGU=
aGtvY2hAdWthYWNoZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.