 Ethiraj Ravindran1,2,3
Ethiraj Ravindran1,2,3 Noor Ullah4,5
Noor Ullah4,5 Shyamala Mani1,2,3Elaine Guo Yan Chew6,7
Shyamala Mani1,2,3Elaine Guo Yan Chew6,7 Moses Tandiono6,7
Moses Tandiono6,7 Jia Nee Foo6,7
Jia Nee Foo6,7 Chiea Chuen Khor6,8
Chiea Chuen Khor6,8 Angela M. Kaindl1,2,3*
Angela M. Kaindl1,2,3* Saima Siddiqi4*
Saima Siddiqi4*- 1Charité–Universitätsmedizin Berlin, Institute of Cell Biology and Neurobiology, Berlin, Germany
- 2Charité–Universitätsmedizin Berlin, Department of Pediatric Neurology, Berlin, Germany
- 3Charité–Universitätsmedizin Berlin, Center for Chronically Sick Children (Sozialpädiatrisches Zentrum, SPZ), Berlin, Germany
- 4Institute of Biomedical and Genetic Engineering (IBGE), Islamabad, Pakistan
- 5Khyber Medical University Institute of Paramedical Sciences (KMU IPMS), Peshawar, Pakistan
- 6Human Genetics, Genome Institute of Singapore, A*STAR, Singapore, Singapore
- 7Lee Kong Chian School of Medicine, Nanyang Technological University Singapore, Singapore, Singapore
- 8Singapore Eye Research Institute, Singapore, Singapore
RhoGTPase regulators play a key role in the development of the nervous system, and their dysfunction can result in brain malformation and associated disorders. Several guanine nucleotide exchange factors (GEF) have been linked to neurodevelopmental disorders. In line with this, ARHGEF17 has been recently linked as a risk gene to intracranial aneurysms. Here we report siblings of a consanguineous Pakistani family with biallelic variants in the ARHGEF17 gene associated with a neurodevelopmental disorder with intellectual disability, speech delay and motor dysfunction but not aneurysms. Cranial MRI performed in one patient revealed generalized brain atrophy with an enlarged ventricular system, thin corpus callosum and microcephaly. Whole exome sequencing followed by Sanger sequencing in two of the affected individuals revealed a homozygous missense variant (g.11:73021307, c.1624C>T (NM_014786.4), p.R542W) in the ARHGEF17 gene. This variant is in a highly conserved DCLK1 phosphorylation consensus site (I/L/V/F/M]RRXX[pS/pT][I/L/M/V/F) of the protein. Our report expands the phenotypic spectrum of ARHGEF17 variants from increased intracranial aneurysm risk to neurodevelopmental disease and thereby add ARHGEF17 to the list of GEF genes involved in neurodevelopmental disorders.
Introduction
Neurodevelopmental disorders encompass a broad range of symptoms such as developmental delay, intellectual disability (ID), motor disorders, attention deficit and autism spectrum disorders (1). Such disorders can be caused by a disruption of the array of spatially and temporally regulated gene products that orchestrate nervous system development (2, 3). An important group of proteins known to contribute to pre- and postnatal brain development are the regulators of Rho family of GTPases (4).
Guanine nucleotide exchange factors (GEFs) stimulate the exchange of GDP for GTP and when bound to GTP the small GTPases bind various effectors to influence processes such as cell cycle progression, cell survival, cytoskeleton organization, and vesicular and nuclear transport (5). Several GEFs have been reported to regulate these processes during brain development, and biallelic variants in Rho guanine nucleotide exchange factor (ARHGEF) genes have been associated with human neurodevelopmental disorders: midbrain-hindbrain malformation (ARHGEF2) (6), nonsyndromic intellectual disability (ARHGEF6) (7), epileptic encephalopathy (ARHGEF9) (8), peripheral demyelinating neuropathy (ARHGEF10) (9). Recently, ARHGEF17 has been reported as a risk gene for intracranial aneurysms (IA) (10). Here we report siblings of a consanguineous Pakistani family with biallelic variants in the ARHGEF17 gene associated with a neurodevelopmental disorder with intellectual disability, speech delay and motor dysfunction but not aneurysms.
ARHGEF17 is a member of the RhoGEF (Rho GTPase GEF) of the diffuse B-cell lymphoma (Dbl) family (11). Members of this large protein family regulate the GDP-GTP cycling of specific proteins through a catalytic Dbl homology (DH) domain and have a regulatory pleckstrin homology (PH) domain that binds to phosphatidylinositol lipids (12). In addition, ARHGEF17 contains a WD40 domain that is important for protein-protein interactions and an actin binding domain (ABD) that binds to actin thereby controlling intracellular localization and its activity. Thus, distinct domains and sequences of ARHGEF17 exert autoregulation of its activity in a spatio-temporal manner and control cellular processes implicated during brain development (13, 14).
In this study, we report novel biallelic ARHGEF17 variants and expand the phenotypic spectrum of ARHGEF17 variants from increased intracranial aneurysm risk to neurodevelopmental disease.
Subjects and methods
The human study was approved by the ethics committee of the Institute of Biomedical and genetic Engineering, Islamabad. The written informed consent was obtained for the molecular genetic analysis, the publication of clinical data, photos, and magnetic resonance images (MRI) from the index family. Blood samples were drawn from seven individuals from the affected family that included three affected (III.2, III.3, and III.4) and four unaffected (II.5, II.6, III.1 and III.5) individuals. Medical history but no clinical data were available from III.4 and III.6.
Genotyping
Genomic DNA was extracted using standard methods and samples were genotyped on the Illumina OmniExpress 24v1-0-A BeadChip array. We confirmed the reported familial relationships among genotyped samples using PLINK identity by descent (IBD) analysis (–genome) (15, 16). We then scanned the data for homozygous segments (>1 Mb) which are shared among affected individuals but not for the unaffected individuals. Homozygosity mapping was conducted using PLINK v1.07 using the default settings (15).
Whole exome sequencing
Targeted enrichment was performed on 1 μg of genomic DNA from two affected individuals (III.2 and III.3) using the Nimblegen SeqCap EZ Exome v3 kit and barcoded with four other samples for multiplexed 2 × 101 bp sequencing on a single lane of the Illumina HiSeq 2000 System. Each individual was sequenced to a mean coverage of 84.3–91.2 reads per target base, with 98% of the target exome covered by 10 or more reads. Reads were mapped using BWA v1.7. Variants were called using the GATK v2 Unified Genotyper following the recommended guidelines by GATK “Best practices for variant calling v3” (17). We used the following primer sequences for the confirmation of the identified ARHGEF17 variant through Sanger sequencing: 5'-AGGCACCTCTAGGGCATTG-3' and 5'-ACATCCCCTGCCCAGTC-3'.
Homozygosity mapping
Homozygous segments of >1 Mb in length accounted for 8.8–15.8% of the genome in all three affected individuals and their two non-affected siblings, confirming that these individuals are the offspring of a consanguineous union. We identified three large segments that were homozygous only in affected individuals (Chr4q11-q13.3 LOD 2.7, Chr11q13.4-q13.5 LOD 2.7 and Chr15q14-q21.2 LOD 2.7).
Genetic analysis
For confirmation of causal mutation, we performed whole exome sequencing analysis in two affected siblings (III.2 and III.3) to identify a set of homozygous mutations that are shared between the two siblings within the homozygous segment. A total of 38,237 variants were found in these two individuals, out of which 26,042 were coding and 13,145 were nonsynonymous, frameshift or splice site variants in well-annotated transcripts. Of these, 902 were rare, either absent or present in < 1% of all populations in HapMap, 1000 genomes populations (17) and the NHLBI exome variant server (EVS) databases (URL: http://evs.gs.washington.edu/EVS/). Of these, 12 variants were homozygous in both affected siblings and six resided within the shared homozygous intervals on chromosomes 4, 11 and 15. Only two were predicted to be damaging: one in ARHGEF17 [Chr11:73021307, hg19/GRCh37, c.1624 C>T (NM_014786.4)] and one in Kinase insert domain receptor (KDR) [Chr4:55955592, hg19/GRCh37, c.3352 C>T (NM_002253.4)]. While both mutations were present in gnomAD South Asians, the KDR variant (allele frequency of 0.24%) is present at higher frequencies than the ARHGEF17 variant (allele frequency of 0.0098%). KDR has been shown to play an essential role in the regulation of angiogenesis, vascular development, vascular permeability, and embryonic hematopoiesis and heterozygous variants in KDR has been linked to Hemangioma (MIM# 602089). Since members of the ARHGEF family have been associated to neurodevelopmental disorders, we consider ARHGEF17 as a strong candidate for the phenotype observed in our index patients.
Results
Clinical presentation
We report four individuals of a consanguineous family of Pakistani descent with a neurodevelopmental disorder. Two affected individuals (III.2, III.3) were available for assessment. Two further individuals were reported to have been affected by a similar developmental disease but were deceased due to a tetanus infection (III.4) and severe head injury (III.6) at ages 20 and 7 years, respectively, and were not available for evaluation.
Proband 1 (P1, III.2) was a 24-year-old male born at term without complications with normal body weight and length (Figures 1A,B, Table 1). He had developmental delay, and later moderate-to-severe intellectual disability and a speech disorder was diagnosed. He began to speak at 3.5 years of age but could not communicate properly. He could understand only simple commands. He was able to walk at the age of 1 year. No dysmorphism or cranial nerve paralysis were noted on clinical examination. He had hypotonia. No head circumference data was available at birth. At the age of 12 years, the proband experienced frequent falls and gradually lost the ability to walk. He could not lift his extremities and became restricted to his bed. He was able to swallow food but could not eat or drink without any support. No gastrostomy was performed. P1 had progressive loss of muscle strength with muscle weakness and wasting, but no joint contractures. Tendon reflexes were brisk, no pathological reflexes were present. There were no coordination problems, and no tremor. Pes equinus was noted. He lost the ability to communicate at the age of 12 years. No visual or hearing impairments were noted. Echocardiogram did not show any abnormality, particularly no sign of cardiomyopathy. Microcephaly was observed with an occipitofrontal head circumference (OFC) of 53.3 cm [ < 3rd centile, < −2 standard deviations (SD)] at the age of 22 years. Postnatal OFC values were not available. MRI findings revealed generalized brain atrophy, accentuated at the temporal lobes, with incomplete opercularization, enlarged ventricular system and thinned corpus callosum (consequence of the parenchymal atrophy), chronic ischemic changes periventricular white matter surrounding occipital horns, and thickened skull bone (Figure 1C).
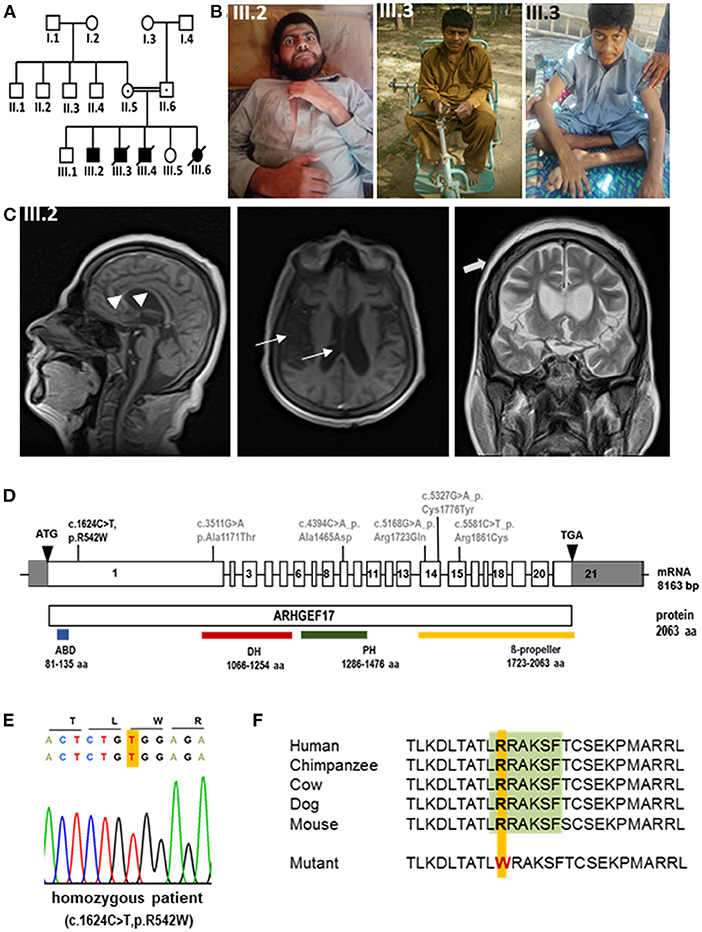
Figure 1. Phenotype and genotype of index patients with ARHGEF17 mutation (A) Pedigree of index family indicating affected patients (III.2, III.3). (B) Photomicrographs of III.2 and III.3. (C) MRI images of III.2 revealed general atrophy/hypoplasia of the brain parenchyma, enlarged ventricular system (thin arrows) and thin, but complete corpus callosum (arrow heads), chronic ischemic changes periventricular white matter surrounding occipital horns, moderate cerebral atrophy and thickened scull bone (thick arrows). (D) Schematic representation of ARHGEF17 cDNA with 21 exons and the wild-type ARHGEF17 protein with the identified variants of our index family (black) and previously reported homozygous variants (gray). A homozygous exchange of a single base C to T in exon 1 of the ARHGEF17 (c.1624C>T (NM_014786.4), p.R542W (NP_055601.2) was identified and confirmed through Sanger sequencing. (E) Electropherogram traces depicting the exchange of C to T at position 1624 of the ARHGEF17 gene. (F) The site of mutation is located in DCLK1 consensus site [LRRAKSF (green)] of ARHGEF17 and it is highly conserved across species.
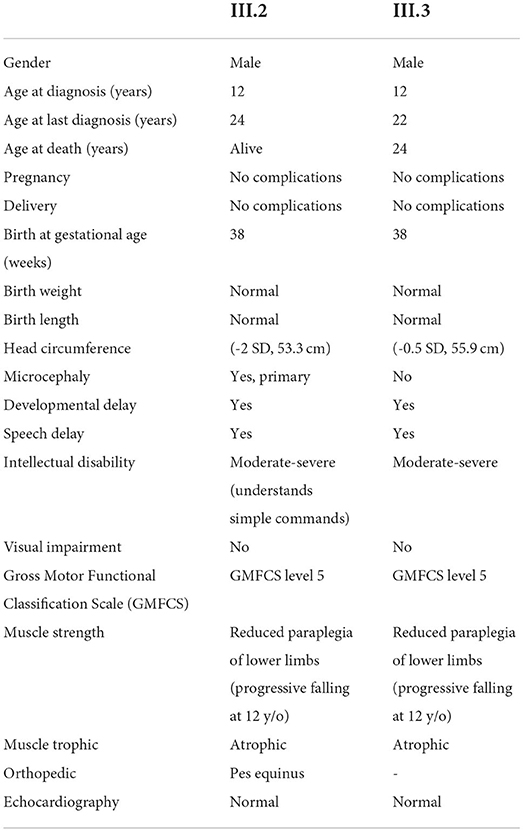
Table 1. Clinical phenotype of index patients with biallelic ARHGEF17 variant c.1624C>T (NM_014786.4).
Proband 2 (P2, III.3) was a 22-year-old male born at term with normal birth weight and length (Figures 1A,B, Table 1). He had developmental delay, and later moderate-to-severe intellectual disability and a speech disorder were diagnosed. He began to speak at 3.5 years of age but could not communicate properly. He was able to walk by 1 year of age. Clinical examination showed no dysmorphism or cranial nerve paralysis. Similar to his brother, frequent falls started at the age of 12 years, he developed progressive loss of muscle strength with muscle weakness and wasting. He could not lift his extremities and lost the ability to walk, and became wheelchair bound. He was able to swallow food but could not eat or drink without any support. No gastrostomy was performed. He became wheelchair bound. He could not communicate at the age of diagnosis. No visual or hearing impairments were noted. Echocardiogram did not show any abnormality, particularly no signs of cardiomyopathy. No microcephaly was observed (55.9 cm, −0.5 SD). No MRI was performed. He died at the age of 24 years due to unknown cause in an episode of a febrile illness and abdominal pain.
Genetic findings
Through whole exome sequencing, we identified the homozygous exchange of a single base C to T in exon 1 of the ARHGEF17 gene (Chr 11:73021307, hg19/GRCh37, c.1624C>T, NM_014786.4; p.R542W, NP_055601.2) in two affected siblings (III.2 and III.3) (Figure 1D) and confirmed the mutation by Sanger sequencing (Figure 1E). The identified variant lies in a protein region highly conserved across species (Figure 1F). The variant is disease-causative in nature, as predicted by Mutation Taster and SIFT (https://www.mutationtaster.org/, http://genetics.bwh.harvard.edu/pph2/, https://sift.bii.a-star.edu.sg).
Discussion
Here we describe the novel biallelic variant c.1624C>T in the ARHGEF17 gene (NM_014786.4) to be associated with a neurodevelopmental disorder with intellectual disability, speech disorder and progressive motor dysfunction. ARHGEF17 is highly expressed in the brain throughout development but not in skeletal muscle according to the EMBL-EBI expression atlas (https://www.ebi.ac.uk/gxa/home; ensg00000110237) (11). Given this expression pattern of ARHGEF17, muscle weakness and wasting in light of brisk reflexes observed in index patients may result from a central nervous system effect rather than a neuropathy or myopathy. Unfortunately, no electrophysiology or muscle biopsy data was available to further discriminate the cause of progressive motor dysfunction.
A previous report had identified five variants in the ARHGEF17 gene in individuals with intracranial aneurysms (IA) and classified the gene as a genetic risk factor for IA (10). All the reported variants affected the C-terminus and mapped to the known functional DH, PH and WD40 domains (Figure 1D). Morpholino-based knockdown of Arhgef17 in zebrafish (Danio rerio) led to intracranial hemorrhage and erythrocyte extravasation, further supporting the conclusion that ARHGEF17 mutations are a risk factor for IA (10). The current identified variant of the index patients resides in the N-terminus and results in an amino acid change of arginine to tryptophan at position 542. This results in the disruption of a DCLK1 (doublecortin-like kinase 1) phosphorylation consensus motif I/L/V/F/M]RRXX[pS/pT][I/L/M/V/F. Sequence alignment showed that the DCLK1 consensus site in ARHGEF17, LRRAKSF, is highly conserved across species, including H. sapiens, P. troglodytes, M. mulatta, C. lupus, B. taurus, M. musculus, R. norvegicus, and G. gallus. Intriguingly, the N-terminus of ARHGEF17 is present neither in Danio rerio nor Xenopus tropicalis, suggesting that the additional functions were acquired by the N-terminus during evolution. DCLK1 is a protein very similar to doublecortin (DCX) that has been implicated in neural development including cortical malformation in humans (lissencephaly, subcortical laminal heterotopia) (18, 19). DCLK1 plays an important role in controlling mitosis and spindle organization as well as in neuronal cell fate (19) and suggests a mechanism by which the novel ARHGEF17 variant described here could result in a neurodevelopmental disorder. In line with this, a heterozygous variant in the DCLK1 gene has been reported in an individual with abnormalities in the musculoskeletal and nervous system in the DECIPHER database (https://www.deciphergenomics.org/gene/DCLK1/ddd-research-variant-overlap).
Considering the severity of the phenotype in the index patients during development and with age and the indirect cause of death of three patients might suggests that ARHGEF17 is implicated in neurodevelopmental as well as in neurodegenerative disorder. ARHGEF17 has been linked to intracranial aneurysm which is in line with its higher expression levels in blood vessels and maintenance of the integrity of blood vessels (11). In support of this, studies have shown the importance of vascular functions during early stages of brain development and cognitive functioning throughout adulthood (20–22). Vascular dysfunctions have been associated to various neurodevelopmental (autism spectrum disorders, microcephaly, schizophrenia) and neurodegenerative disorders (Alzheimer's disease, Parkinson's disease, multiple sclerosis) (20–22).
In conclusion, we have identified a new variant in ARHGEF17 in individuals with a neurodevelopmental disorder. Identification of additional families and functional experiments are necessary to delineate the role of ARHGEF17 in brain and musculoskeletal development. Our report expands the phenotypic spectrum of ARHGEF17-associated human disorders and adds ARHGEF17 to the list of GEF genes linked to neurodevelopmental diseases.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Institute of Biomedical and Genetic Engineering, Islamabad. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
SS was responsible for project conception. AK analyzed and interpreted patient's clinical and genetic data. NU and SS contributed clinical samples by recruiting subjects, gathering patient history, clinical information, and written informed consents. EC, MT, JF, and CK performed WES and bioinformatics data analysis. EC, MT, JF, and SS performed Sanger sequencing and segregation analysis. SM interpreted the genetic data and performed bioinformatic analysis. SM, ER, and AK drafted the manuscript that was revised and accepted by all coauthors. All authors contributed to the article and approved the submitted version.
Funding
We acknowledge the funding resources for this study: The genetic analysis was supported by the Agency for Science, Technology and Research, Singapore (to CK) and a Singapore National Research Foundation Fellowship (NRF-NRFF2016-03; to JF). Family and clinical data collection was supported by departmental grant (IBGE) (SS). Further funding includes the German Research Foundation (SFB665, SFB1315, FOR3004, AK), the Sonnenfeld Stiftung (AK), the Berlin Institute of Health (BIH, CRG1, AK) and the Charité (AK, ER, and SM). This study makes use of data generated by the DECIPHER community available on the DECIPHER webpage (www.deciphergenomics.org).
Acknowledgments
The authors thank the index family for their participation in this study. We acknowledge Anna Tietze for the analysis of cranial MR images.
Conflict of interest
Author CK was employed by Singapore Eye Research Institute.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Morris-Rosendahl DJ, Crocq MA. Neurodevelopmental disorders-the history and future of a diagnostic concept. Dialogues Clin Neurosci. (2020) 22:65–72. doi: 10.31887/DCNS.2020.22.1/macrocq
2. Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, et al. Transcriptional landscape of the prenatal human brain. Nature. (2014) 508:199–206. doi: 10.1038/nature13185
3. Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, et al. Spatio-temporal transcriptome of the human brain. Nature. (2011) 478:483–9. doi: 10.1038/nature10523
4. Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. (2005) 19:1–49. doi: 10.1101/gad.1256405
5. Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. (2002) 16:1587–609. doi: 10.1101/gad.1003302
6. Ravindran E, Hu H, Yuzwa SA, Hernandez-Miranda LR, Kraemer N, Ninnemann O, et al. Homozygous ARHGEF2 mutation causes intellectual disability and midbrain-hindbrain malformation. PLoS Genet. (2017) 13:e1006746. doi: 10.1371/journal.pgen.1006746
7. Kutsche K, Yntema H, Brandt A, Jantke I, Nothwang HG, Orth U, et al. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat Genet. (2000) 26:247–50. doi: 10.1038/80002
8. Harvey K, Duguid IC, Alldred MJ, Beatty SE, Ward H, Keep NH, et al. The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J Neurosci. (2004) 24:5816–26. doi: 10.1523/JNEUROSCI.1184-04.2004
9. Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer-Grumbach M, Kwon JM, et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet. (2003) 72:722–7. doi: 10.1086/367847
10. Yang X, Li J, Fang Y, Zhang Z, Jin D, Chen X, et al. Rho Guanine Nucleotide Exchange Factor ARHGEF17 Is a Risk Gene for Intracranial Aneurysms. Circ Genom Precis Med. (2018) 11:e002099. doi: 10.1161/CIRCGEN.117.002099
11. Rumenapp U, Freichel-Blomquist A, Wittinghofer B, Jakobs KH, Wieland T. A mammalian Rho-specific guanine-nucleotide exchange factor (p164-RhoGEF) without a pleckstrin homology domain. Biochem J. (2002) 366(Pt 3):721–8. doi: 10.1042/bj20020654
12. Mitin N, Rossman KL, Der CJ. Identification of a novel actin-binding domain within the Rho guanine nucleotide exchange factor TEM4. PLoS ONE. (2012) 7:e41876. doi: 10.1371/journal.pone.0041876
13. Lutz S, Mohl M, Rauch J, Weber P, Wieland T. RhoGEF17, a Rho-specific guanine nucleotide exchange factor activated by phosphorylation via cyclic GMP-dependent kinase Ialpha. Cell Signal. (2013) 25:630–8. doi: 10.1016/j.cellsig.2012.11.016
14. Isokane M, Walter T, Mahen R, Nijmeijer B, Hériché JK, Miura K, et al. ARHGEF17 is an essential spindle assembly checkpoint factor that targets Mps1 to kinetochores. J Cell Biol. (2016) 212:647–59. doi: 10.1083/jcb.201408089
15. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. (2007) 81:559–75. doi: 10.1086/519795
16. Sobel E, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet. (1996) 58:1323–37.
17. Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. (2012) 491:56–65. doi: 10.1038/nature11632
18. Deuel TA, Liu JS, Corbo JC, Yoo SY, Rorke-Adams LB, Walsh CA. Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. (2006) 49:41–53. doi: 10.1016/j.neuron.2005.10.038
19. Shu T, Tseng HC, Sapir T, Stern P, Zhou Y, Sanada K, et al. Doublecortin-like kinase controls neurogenesis by regulating mitotic spindles and M phase progression. Neuron. (2006) 49:25–39. doi: 10.1016/j.neuron.2005.10.039
20. Kalailingam P, Wang KQ, Toh XR, Nguyen TQ, Chandrakanthan M, Hasan Z, et al. Deficiency of MFSD7c results in microcephaly-associated vasculopathy in Fowler syndrome. J Clin Invest. (2020) 130:4081–93. doi: 10.1172/JCI136727
21. Ouellette J, Lacoste B. From neurodevelopmental to neurodegenerative disorders: the vascular continuum. Front Aging Neurosci. (2021) 13:749026. doi: 10.3389/fnagi.2021.749026
Keywords: ARHGEF17, neurodevelopmental disorder, microcephaly, motor dysfunction, missense mutation
Citation: Ravindran E, Ullah N, Mani S, Chew EGY, Tandiono M, Foo JN, Khor CC, Kaindl AM and Siddiqi S (2022) Case report: Expanding the phenotype of ARHGEF17 mutations from increased intracranial aneurysm risk to a neurodevelopmental disease. Front. Neurol. 13:1017654. doi: 10.3389/fneur.2022.1017654
Received: 12 August 2022; Accepted: 30 September 2022;
Published: 20 October 2022.
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Jehan Suleiman, Tawam Hospital, United Arab EmiratesTanzila Mukhtar, University of California, San Francisco, United States
Copyright © 2022 Ravindran, Ullah, Mani, Chew, Tandiono, Foo, Khor, Kaindl and Siddiqi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela M. Kaindl, YW5nZWxhLmthaW5kbEBjaGFyaXRlLmRl; Saima Siddiqi, c2FpbWFzaWRkaXFpMkBnbWFpbC5jb20=