 Filipp Maximilian Filippopulos
Filipp Maximilian Filippopulos Lutz Schnabel
Lutz Schnabel Konstanze Dunker
Konstanze Dunker Ralf Strobl
Ralf Strobl Doreen Huppert
Doreen Huppert
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 24 October 2022
Sec. Pediatric Neurology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.1016856
This article is part of the Research TopicVestibular Disorders in ChildrenView all 13 articles
Background: The main clinical presentation of episodic ataxias (EAs) consists of vertigo and dizziness attacks lasting for minutes to hours with widely varying accompanying symptoms. The differentiation of EA and episodic vertigo/dizziness syndromes in childhood and adolescence such as vestibular migraine (VM) and recurrent vertigo of childhood (RVC) can be challenging. Furthermore, only few prospective studies of children/adolescents with EA are available.
Objective: This study aims to characterize clinical and instrument-based findings in EA patients under 18 years of age, to delineate the clinical and therapeutic course in EA, and to present potentially new genetic mutations. Furthermore, the study aims to differentiate distinct characteristics between EA, VM, and RVC patients.
Methods: We prospectively collected clinical and instrument-based data of patients younger than 18 years, who presented at the German Center for Vertigo and Balance Disorders (DSGZ) at the LMU University Hospital in Munich with EA, VM, or RVC between January 2016 and December 2021. All patients underwent a comprehensive evaluation of neurological, ocular-motor, vestibular and cochlear function, including video-oculography with caloric testing, video head impulse test, vestibular evoked myogenic potentials, posturography, and gait analysis.
Results: Ten patients with EA, 15 with VM, and 15 with RVC were included. In EA the main symptoms were vertigo/dizziness attacks lasting between 5 min and 12 h. Common accompanying symptoms included walking difficulties, paleness, and speech difficulties. Six EA patients had a previously unknown gene mutation. In the interictal interval all EA patients showed distinct ocular-motor deficits. Significant differences between EA, VM, and RVC were found for accompanying symptoms such as speech disturbances and paleness, and for the trigger factor “physical activity”. Furthermore, in the interictal interval significant group differences were observed for different pathological nystagmus types, a saccadic smooth pursuit, and disturbed fixation suppression.
Conclusion: By combining clinical and ocular-motor characteristics we propose diagnostic criteria that can help to diagnose EA among children/adolescents and identify patients with EA even without distinct genetic findings. Nevertheless, broad genetic testing (e.g., next generation sequencing) in patients fulfilling the diagnostic criteria should be conducted to identify even rare or unknown genetic mutations for EA.
The diagnoses behind recurrent vertigo attacks in children and adolescents are manifold and often pose a major diagnostic challenge even for experienced clinicians. The most common diagnoses include vestibular migraine (VM) and the associated disorder of “Recurrent Vertigo of Childhood” (RVC), whereas central causes such as episodic ataxia (EA) are less frequent (1–3). The core features of these diseases may be very much alike and include recurrent attacks of vertigo (sensation of spinning of the environment)/dizziness (various sensations of body orientation and position), headaches, and different trigger-factors, as well as an inconclusive clinical examination, at least in early stages. Furthermore, overlap syndromes between e.g., migraine and EA (4, 5), epilepsy and EA (6), or progressive and episodic ataxias (e.g., spinocerebellar ataxia type 27 and EA 9) (7) have been described. Especially diseases with a rare occurrence such as EA are sometimes difficult to diagnose. Nevertheless, due to the direct therapeutic relevance (e.g., acetazolamide or 4-aminopyridine for EA, magnesium for VM), different prognosis (often progressive in EA, benign in RCV and VM), and impact on family planning (EA mostly has an autosomal dominant inheritance pattern), it is important not to miss such differential diagnoses of episodic vertigo syndromes.
Episodic ataxias are hereditary chanellopathies with symptoms mainly attributable to a cerebellar dysfunction, that are genetically and phenotypically heterogeneous (8), which further complicates the diagnostic approach. To date, nine phenotypes and genotypes of EA have been described (EA 1–9) (7, 9), with EA 2 being most common (10). EA 1 is typically caused by mutations of the potassium channel Kv1.1-encoding gene KCNA1 on chromosome 12q13 (11), and EA 2 by mutations in the CACNA1A gene on chromosome 19p13 which encodes the Cav2.1 subunit of the P/Q-type voltage-gated calcium channel (5). Although EAs usually have an autosomal-dominant inheritance pattern (7–9), spontaneous mutations have been described (4, 12), so that affected children/adolescents might not always have a positive family history, again complicating the correct diagnosis. The age of onset, disease characteristics, and symptom constellation during and between attacks is highly variable even in patients with the same gene mutation causing the EA (6, 10). No causative treatments are available for any EA syndrome, but there are some symptomatic treatment options available for EA 1 and 2 such as acetazolamide and 4-aminopyridine (10), that may have a significant impact on the patients' quality of life.
Since the knowledge of the clinical spectrum as well as typical instrument-based findings in patients with EA are the key for early diagnosis and specific treatment, we prospectively examined ten children/adolescents with episodic ataxia syndromes. We describe clinical and instrument-based findings, treatment response, and clinical course. Furthermore, to depict the most important features for diagnosing EA, we compared clinical and instrument-based findings between EA patients and patients with the most common episodic vertigo syndromes in children/adolescents, namely VM and RVC.
All children/adolescents younger than 18 years of age who presented at the German Center for Vertigo and Balance Disorders (DSGZ) at the LMU University Hospital in Munich between January 2016 and December 2021 were screened for inclusion. All patients with an episodic ataxia syndrome were included in the study. Furthermore, patients younger than 18 years with the final diagnosis of vestibular migraine (VM) or RVC according to the diagnostic criteria of the Bárány Society (13) were included in the study as comparative groups. All patients who were recruited before the publication of the diagnostic criteria in 2021 were reevaluated and only included for further analysis, if the diagnostic criteria for VM or RVC were fulfilled. Written informed consent was obtained from all participants included in the study.
Due to the lack of evidence on distinct clinical and instrument-based findings in patients with EA, an extensive, standardized work-up was designed to broadly evaluate clinical features and instrument-based findings these patients. The work-up was comprised to include most of the available neuro-otological examinations for the assessment of the peripheral and central vestibular system. Following examinations were performed:
A) Medical history (including duration and quality of symptoms, accompanying symptoms, trigger factors, family history, medication).
B) Clinical examination of the vestibular function including neuro-otological and neuro-ophthalmological examination (including head impulse test, test for spontaneous nystagmus, provocation nystagmus, positional nystagmus, skew deviation, smooth pursuit, saccades, gaze-holding, fixation suppression of the vestibulo-ocular reflex, hearing).
C) Neurological status (including motor, sensory, coordination, cranial nerve, cognitive function assessment).
D) Instrument-based assessment:
a. Video-oculography with caloric irrigation was conducted with cold (30°C) and worm (44°C) water on both sides to evaluate the low frequency function of the vestibular system
b. Video-Head-Impulse-Test (vHIT) was performed on a standard commercial v-HIT system to evaluate the high frequency function of the vestibular system
c. Ocular and cervical Vestibular-Evoked Myogenic Potentials (c/o VEMP's) were analyzed for the evaluation of utricular and saccular function respectively
d. Auditory-Evoked Potentials (AEP's) for the evaluation of the hearing function
e. Posturography and gait analysis was performed to measure body sway and gait patterns
f. Testing of subjective visual vertical, and fundus photography.
D) Clinical follow-up for patients with EA syndrome.
We report mean and standard deviation for continuous variables, and absolute frequency and the relative frequency as percentages for categorical variables. Group comparison is based on the chi-squared test for categorical data and on the likelihood ratio test for continuous variables.
Two-tailed p-values <5% were considered as statistically significant. As this is an exploratory study, no correction for multiple testing was done. R 4.1.2 was used for statistical analyses (14).
Between January 2016 and December 2021, 336 patients under the age of 18 were screened for inclusion. Of all screened children/adolescents, ataxia syndromes represented the fifth most common diagnosis following VM, functional dizziness, RVC, and orthostatic hypotension. A total of 18 children/adolescents with ataxia syndromes presented in the period mentioned, 10 of whom had an episodic ataxia syndrome and were included in the present study (see Figure 1). Furthermore, 15 age- and gender- matched children and adolescents with VM and RVC were included in the comparative groups.
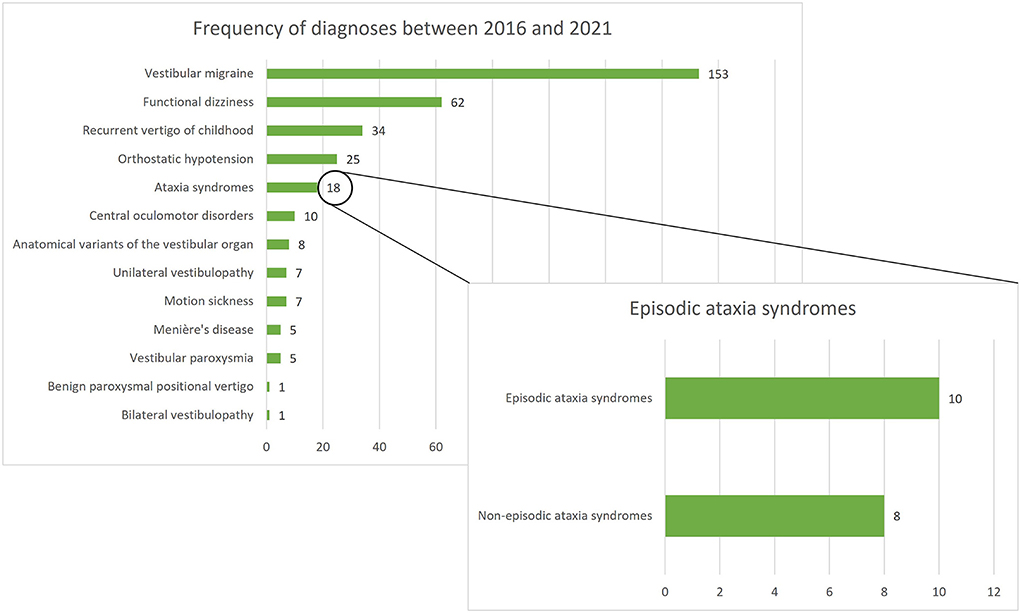
Figure 1. Frequency of diagnoses among 336 children/adolescents presenting at the German Center for Vertigo and Balance Disorders (DSGZ) between 2016 and 2021. Ataxia syndromes are the fifth most frequent diagnosis with episodic ataxias being the leading subgroup.
Of the 10 children/adolescents with EA, seven had a genetically confirmed EA 2, one had a genetically confirmed EA 1, one a genetically confirmed EA 9, and one child had a suspected EA 1. In the latter child, a mutation in the CACNA1A gene was ruled out; further testing for mutations e.g., in the KCNA1 gene was not possible due to insurance restrictions (Table 1). Nevertheless, the child was included in the EA group due to the distinct clinical and instrument-based findings (see Tables 2, 3), as well as due to the fact, that in a considerable amount of children and adolescents with EA, no mutation at all is found (8). Of the 10 children/adolescents five had a spontaneous mutation defined as no genetic marker in tested family members nor a positive family history (including the suspected EA 1 child). Six children/adolescents (five with EA 2, one with EA 1) had a new mutation, that has not been described in common gene databases such as the University of California Santa Cruz (UCSC) genome browser, the human gene mutation database (HGMD), ClinVar at the National Center for Biotechnology Information (NCBI), and global variome shared Leiden Open Variation Database (LOVD) (see Table 1).

Table 1. Genetic and clinical course characteristics of 10 children/adolescents with episodic ataxia.
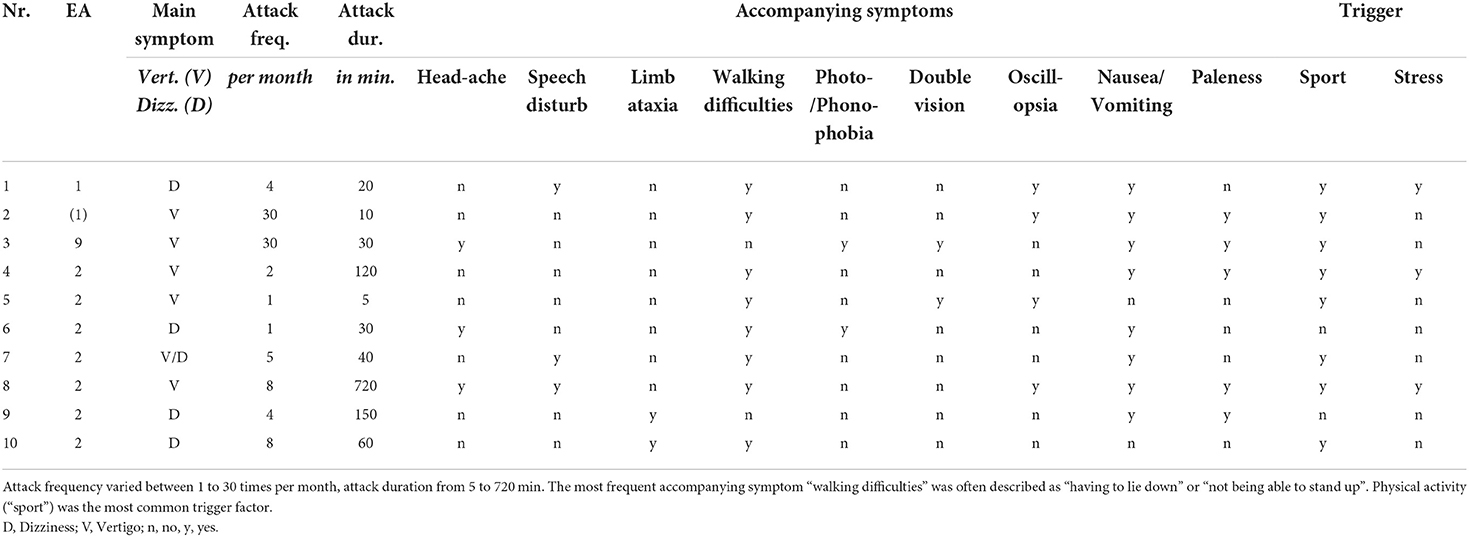
Table 2. Detailed clinical characteristics of the vertigo/dizziness attacks of children and adolescents with episodic ataxia.
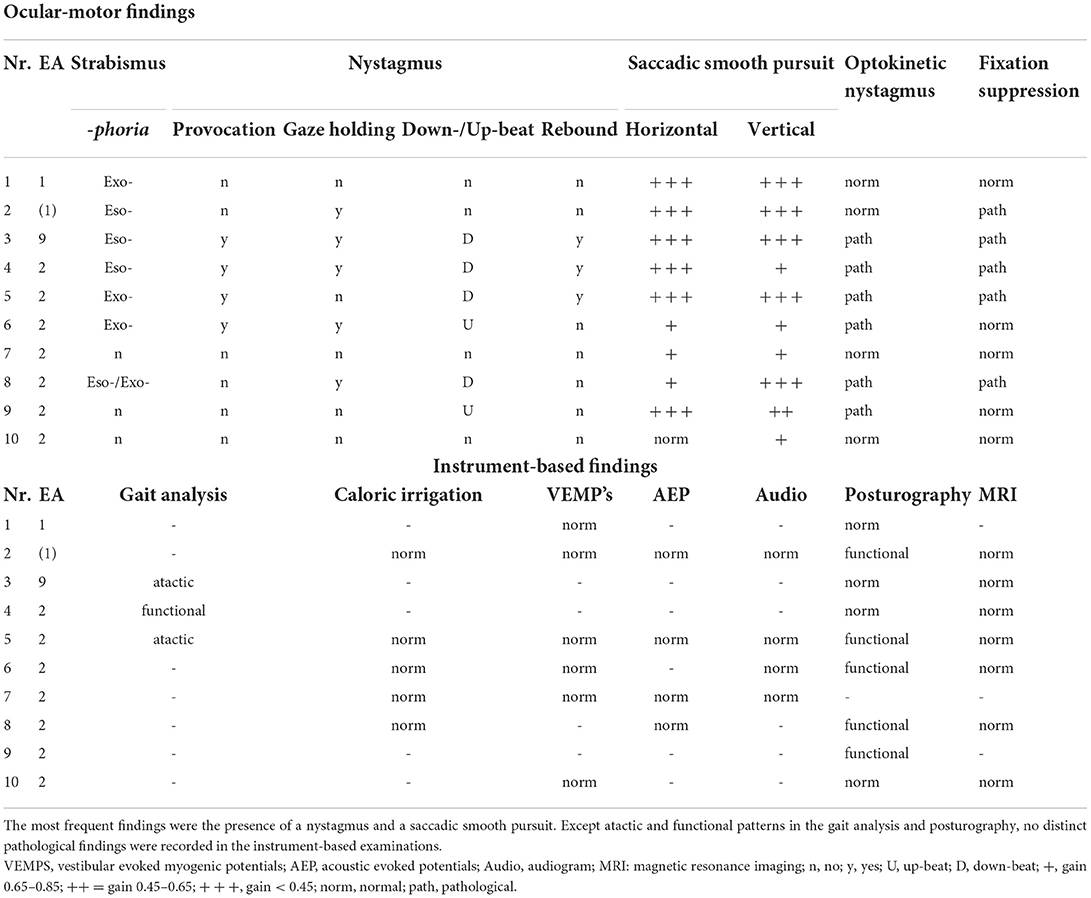
Table 3. Ocular-motor and instrument-based findings in the attack-free (interictal) interval in children and adolescents with episodic ataxia.
The mean age at symptom presentation in children/adolescents with EA was 7.4 ± 4.3 years. The earliest symptom manifestation was in the ninth month in one child with EA 2 and the latest at the age of 12 years and 6 months, also in an adolescent with EA 2.
Attack frequency varied between daily attacks (in EA 9 and suspected EA 1) and attacks once a month (mean ± sd = 9.3 ± 11.2). The Scale for the Assessment and Rating of Ataxia (SARA) in the attack-free interval was increased in three children/adolescents (for details see Table 1).
In a vertigo/dizziness attack, the most common accompanying symptoms were “nausea/vomiting” (80%) and experiencing “walking difficulties” (80%), which were often described as the “inability to walk” and “having to lie down” until the attack is over. Furthermore, “paleness” (50%) and “oscillopsia” (40%) were commonly described. The most frequent trigger for an attack was physical activity (“sport”, 80%), followed by psychosocial stress (30%), while two children/adolescents (20%) did not report any trigger factors. Detailed findings are listed in Tables 2, 3.
The broad ocular-motor evaluation showed in all patients with EA an impaired smooth pursuit, in most cases with a medium to highly reduced smooth pursuit gain at 0.1 and 0.2 Hz (Hertz). Furthermore, 70% of the patients had a pathological finding in the nystagmus examination (see Table 3). The optokinetic nystagmus was impaired in 60% of patients and fixation suppression in 50%. Overall, 70% of patients had more than one ocular-motor abnormality. No pathological findings were detected in additional instrument-based examinations such as caloric irrigation, VEMPS, AEPs, and audiogram.
The mean follow up of the patients with EA was 2.7 years. Seven children/adolescents received treatment with at least one medication, two declined treatment and one with genetically not confirmed EA 1 was not offered any specific medication. Medical treatment was administered for at least 6 weeks and then changed or discontinued if no therapeutic effect was noted. Initial treatment was chosen according to the side-effect profile of each substance and according to patient/parents' wishes; dosage was adapted to weight for acetazolamide (8–30 mg/kg), 4-aminopyridine was restricted to a maximum of 2 doses of 10 mg per day. In four cases acetazolamide was prescribed but led to a reduction of the frequency or duration of the vertigo attacks in only one case. 4-Aminopyridine was administered in three children/adolescents (the youngest being 1 year and 6 months old) and led to a decrease in attack frequency and duration in two cases. Magnesium which was administered in three patients did not influence vertigo attacks. In the adolescent with EA 9, especially the behavioral recommendations (in combination with magnesium) led to a significant decrease in attack frequency and severity, but also to a reduced SARA score (see Table 1).
Table 4 shows the results of the comparison between EA, VM, and RCV in detail. A positive family history of genetically determined EA and delayed motor and/or mental development occurred significantly more often (p = 0.0012) in EA patients compared to patients with VM and RVC (40 vs. 0%). The attack characteristics (type of vertigo, attack frequency, attack duration) did not differ between the three diagnoses. Accompanying symptoms such as “speech disturbances” and “paleness” differed significantly (p = 0.0429 and p = 0.0212 respectively) between groups, with the highest frequency observed in EA patients (30 and 50% respectively), while “headache” (p <0.0001) and “photo-/phonophobia” (p < 0.0001) were most often observed in children/adolescents with VM (80% each). Furthermore, physical activity (“sport”) as a trigger factor occurred more often in EA patients (60%, p = < 0.0001). Provocation, gaze-holding, and down-beat nystagmus were more frequently observed in EA, but also occurred, in a milder form, in some patients with VM, but no patients with RVC. Similarly, an impaired smooth pursuit was observed in all EA patients, in 47% of VM patients, and in 7% of RVC patients, significantly differing between the groups (p < 0.0001). In all VM and RVC patients with an impaired smooth pursuit, the gain was only slightly reduced (gain between 0.65 and 0.85) in contrary to the EA patients (see above).
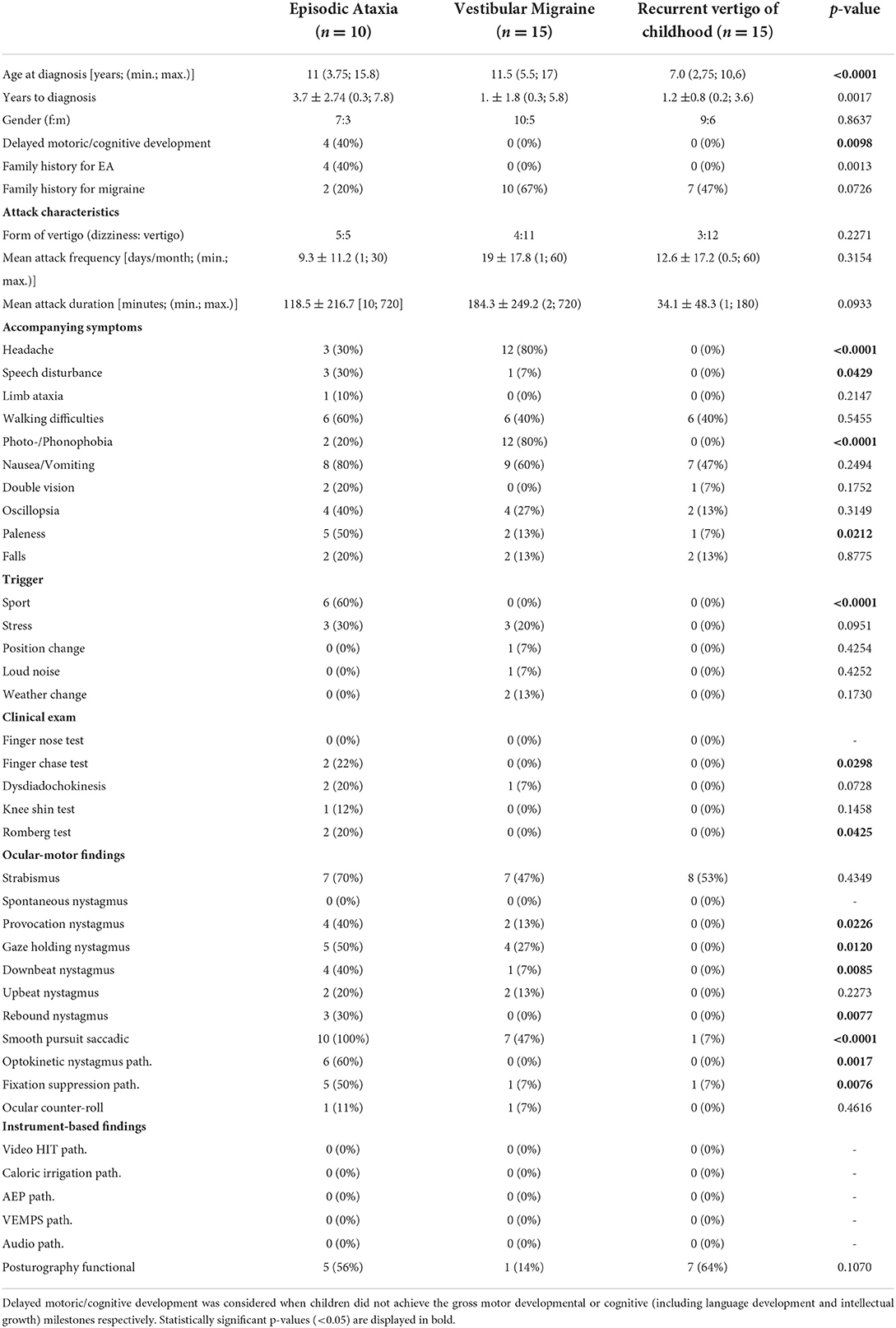
Table 4. Comparison of clinical and instrument-based findings between children and adolescents with episodic ataxia, vestibular migraine, and recurrent vertigo of childhood.
Episodic ataxia syndromes are rare genetic disorders, but as demonstrated here, in a specialized vertigo/dizziness clinic they might be present in about 5% of patients under the age of 18 years (see Figure 1). The most common EA syndrome was EA type 2 followed by EA type 1. Of the seven patients with EA type 2 included in the study, five had mutations that have not been described before (patients 5–9). Additionally, the mutation of the EA type 1 patient (patient 1) also constitutes a novel description (details presented in Table 1). According to criteria suggested by Jen 2008 (8) these new mutations were considered disease-causing, as all newly described mutations were heterozygous and caused a premature stop or altered amino acid residues. The high number of spontaneous mutations in the present study supports the suggestion to consider EA also in children and adolescents with a negative family history.
The patient included here with a mutation in the FGF14 gene is to our knowledge the thirteenth case described with the clinical syndrome of episodic ataxia (7, 15, 16). FGF14 mutations are considered a rare cause of spinocerebellar ataxia type 27 (SCA 27) and show a broad phenotypic spectrum (17–20). The case presented here showed recurrent attacks of vertigo and dizziness lasting between a few minutes and 2 h, accompanied by severe walking difficulties partly leading to falls. Furthermore, the patient had a developmental delay (mental and motor) and psychiatric comorbidity. Further characteristics are presented in Tables 1–3. Overall, this case extends the phenotypic spectrum of EA related to a mutation in the FGF14 gene and further supports the previous suggestion to characterize such patients as EA type 9 (7).
A high genotype-phenotype variability has been shown especially in patients with EA type 2 and EA type 1 (6, 17, 21, 22). Furthermore, mutations in the most common genes associated with EA, namely in the CACNA1A gene (associated with EA type 2) and KCNA1 (associated with EA type 1) might lead to different clinical entities without episodes of vertigo/ataxia, such as epilepsy, paroxysmal dyskinesia, or hemiplegic migraine (4, 6, 21, 23). Also, in a considerable number of patients with a clinical syndrome of EA, no disease-causing mutation might be found (4, 24–26).
Since a broad clinical spectrum has been described in patients with different types of EA, a precise clinical evaluation of patients suspected of suffering from EA is of the utmost importance in order to reach a correct diagnosis and initiate appropriate therapeutic measures including medical treatment option with acetazolamide or 4-aminopyridin.
In the present study, all the EA children/adolescents included suffered from vertigo or dizziness attacks lasting between 5 min and 12 h, in the mean 2 ± 3.6 h The lowest attack frequency was one attack per month, the highest was daily attacks. All children/adolescents had accompanying symptoms, such as paleness, speech disturbances, and walking difficulties. Walking difficulties were often described as the “inability to walk straight” or children/adolescents “lying down and not being able to stand up”. Many children/adolescents reported trigger factors, most frequently physical exercise (“sports”), but two children did not report any triggers. Physical exercise leading to an attack most commonly was described while participating in school sports, but also in lighter activities such as climbing a few stairs. These findings seem to constitute the core symptoms of EA children/adolescents. This is also in line with findings from previous studies and case descriptions of patients with EA (4, 6, 7, 22, 26–35).
In the interictal interval only three children/adolescents had clinical signs of ataxia, two with EA type 2 and the above-described adolescent with EA type 9. As expected, in the latter, ataxia was much more pronounced (see Table 1), since patients with an FGF14 mutation show an overlap between episodic and chronic progressive ataxia (7). No pathological findings were found in any child/adolescent in the instrument-based findings such as vHIT, caloric irrigation, AEP's, VEMPs, or the audiogram (see Table 3).
To our knowledge, this is the first prospective study to evaluate the ocular-motor function of children/adolescents with EA in detail. One previous study retrospectively reported on neuro-ophthalmological findings in chronic ataxia, which included 7 patients with EA and described the presence of nystagmus in these patients, however without further differentiation (36). A second study retrospectively analyzed eye movement disorders in children with CACNA1A mutations, most frequently describing paroxysmal tonic upgaze, saccade dysmetria, and strabismus (37). Strabismus was also present in most children/adolescents (7/10, see Table 3) of our cohort. The prevalence of strabismus among children/adolescents varies between different countries in the world between <1% and 8.6% (38), thus suggesting a much higher prevalence among children/adolescents with EA. Paroxysmal tonic upgaze and saccade dysmetria were not present in our cohort. The most common finding in all patients in the present study was a pathological smooth pursuit, either horizontally, vertically, or in both directions. Furthermore, most children/adolescents showed a nystagmus in the interictal interval, either a vertical spontaneous nystagmus (4/10 down-beat, 2/10 up-beat) and/or a gaze-holding nystagmus (5/10, see Table 3). The optokinetic nystagmus was disrupted in 6/10 children/adolescents and a pathological fixation suppression was found in half of the children/adolescents. These findings are in line with a study describing ocular-motor findings in nearly only adults with EA type 2 (39, 40), so it can be assumed that ocular-motor disturbances are also distinct clinical findings in children and adolescents with EA.
Treatment with acetazolamide or 4-aminopyridine improved attack frequency and duration in some, but not all patients, which is in line with previous findings (8, 24, 28). Both treatments are not approved for use in patients with EA, so it is an individual treatment that children/adolescents and parents must be made aware of. Additionally behavioral recommendations (e.g., reducing physical activity and stress) should not be underestimated, since they might reduce EA attacks.
The most common differential diagnoses of EA are RVC and more importantly VM. These diseases can show similar vertigo/dizziness attacks and accompanying symptoms such as headaches (see Table 4), so that a distinct differentiation in order to reach the correct diagnosis and subsequently initiate the correct treatment is merited.
Attack frequency was similar in all three patient groups, while attack duration was shorter in RVC with a maximum of 3 h compared to a maximum of 12 h in EA and VM patients. The most common accompanying symptoms of VM attacks were headache, photo-/phonophobia, and nausea/vomiting, which was expected in accordance with the diagnostic criteria (13). EA patients in our cohort showed typical clinical attack features (i.e. sport or stress induced ataxia or vertigo, accompanied by walking-difficulties, oscillopsia, and paleness) as previously described (9, 28). The two included EA type 1 patients had no evidence of myokymia which is often reported in EA type 1 (41, 42). As EA type 9 overlaps with SCA 27, this patient had a high SARA score, while the SARA score was slightly increased in only two EA type 2 patients and none of the EA type 1 patients. Furthermore, RVC patients more frequently showed a functional sway pattern in posturography, which may indicate a higher risk of secondary psychosomatic development, similarly to VM and migraine-related disorders (43, 44).
Ocular-motor findings in children with VM have to our knowledge not been reported previously. However, the most common finding of a slight saccadic smooth pursuit in the present VM cohort is similar to findings in adult VM populations (45, 46). In contrast, ocular-motor deficits were rarely noted in RVC (see Table 4). All EA patients showed considerable ocular-motor deficits compared to only slight deficits in VM. Especially EA type 1 is typically reported to not show any interictal nystagmus (41, 42); however, ocular-motor deficits have to our knowledge not been examined in detail before. In our study we find a clearly saccadic smooth pursuit in both EA type 1 patients, as well as a pathological fixation suppression in one of them (see Table 3). The present findings suggest, that distinct interictal ocular-motor findings are present in the most frequent EA types, nevertheless the number of EA type 1 patients in the present study is low and further evaluation of a larger EA type 1 cohort is needed.
In summary, the findings of the present study suggest that EA patients more often report speech disturbances and paleness as accompanying symptoms during the attacks, while VM patients more often report headaches and photo-/phonophobia. Regarding trigger factors, only patients with EA reported physical activity (sport) as an attack trigger. Pathological findings in the clinical evaluation were almost exclusively found in EA patients. The most striking differences between these groups though are the ocular-motor findings, especially various types of spontaneous nystagmus and/or a pathological smooth pursuit in patients with EA. Nevertheless, ocular-motor disturbances, such as gaze-holding nystagmus and a pathological smooth pursuit might also be found in some patients with VM and only rarely in RVC, but overall to a much lesser extent and scarcely with more than one ocular-motor abnormality.
Besides the recommendations by Jen 2004 (4), diagnostic criteria for EA have not been defined. Defining criteria for EA syndromes is especially important, since genetic findings in patients with EA might be negative or inconclusive (4, 24–26). In Figure 2, we propose diagnostic criteria for identifying children/adolescents with “probable EA” according to the findings of the present study, considering a broad literature review (4–12, 15, 16, 22, 24–30, 32–35, 39–42, 47) and our expert opinions. The diagnosis of a “definite EA” should additionally include a pathological genetic finding with a “disease-causing mutation” as characterized by Jen 2008 (8).

Figure 2. Proposed clinical and ocular-motor criteria for diagnosing children and adolescents with episodic ataxia. With the fulfillment of all criteria (A–E) the diagnosis of “probable episodic ataxia” can be made and genetic testing should be initiated. A positive genetic finding confirms the diagnosis of episodic ataxia.
The diagnostic criteria include data from medical history such as details on the duration of vertigo attacks and accompanying symptoms (see Figure 2 points A. and B.) as well as ocular-motor findings (see Figure 2, point C). Because ocular-motor findings in EA in some cases might be subtle and only distinguishable by experienced physicians (mostly early in the disease course), we suggest performing a standardized orthoptic examination conducted by a trained orthoptician to evaluate the ocular-motor system. Except gait analysis, which may show an atactic gait in the attack free interval of EA patients, all instrument-based examinations (VEMP's, AEP's, audiometry, posturography, MRI) were unremarkable in EA children, but are necessary to rule out other vestibular conditions (see Figure 2, point E.). We therefore suggest a basic vestibulo-cochlear work-up in all children suspected to suffer from EA comprising of a caloric irrigation, one examination of the auditory function (either AEP's or audiometry), and an MRI. Latter is suggested, since anatomical variants of the vestibular/cochlear organ and central pathologies are not uncommon in childhood (see Figure 1).
When applying the proposed criteria to the present findings, all patients could be identified. Furthermore, the diagnostic criteria were applied on patient descriptions from previous studies of EA including types 1–9 (see Supplementary Table 1). Only studies including a clinical characterization of the attacks as well as interictal findings, at least regarding the presence of nystagmus and/or myokymia/myotonia, were used for analysis. Sensitivity was calculated as the number of correctly identified EA patients (173; including the children of the present study) divided by the number of all EA patients included (223) and reached 78% (Supplementary Table 1). The most frequent reason to not fulfill the proposed diagnostic criteria was a missing interictal clinical finding (see Figure 2, point C.). This might be due to the lack of reported broad ocular-motor findings such as smooth pursuit, optokinetic nystagmus, and fixation suppression, which were only reported in one study (33). Therefore, and according to the present findings, where all EA patients showed considerable ocular-motor deficits, the above reported sensitivity might be underestimated.
Specificity could only be calculated according to the control group defined in the present study (including children with RVC and VM) and reached 90% (3 children with VM fulfilled the here suggested diagnostic criteria). These three children suffered from headache attacks, which might segregate EA from VM. However, we decided not to use “headache attacks” as an exclusion criterium for the suggested diagnostic criteria, since headaches (irrespective of the fulfillment of the diagnostic criteria of VM or other ICHD diagnoses) are commonly present in EA children/adolescents (4, 22, 28). Overall, this suggests a good sensitivity and specificity for the suggested criteria for children with EA; nevertheless the applicability and accuracy must be further evaluated in larger cohorts of precisely characterized children with vertigo, dizziness, or ataxia attacks.
EA is a rare genetic disorder, but might be present in a considerable amount of children and adolescents presenting with episodic vertigo and/or dizziness. Six new genetic mutations are presented, five for EA type 2 and one for EA type 1, so that a broad genetic testing (e.g., next generation sequencing) in suspected EA patients is recommended. Clinical and instrument-based findings that help to differentiate EA from VM and RVC include attacks triggered by physical activity and accompanied by speech disturbances and paleness. Furthermore, a pathological smooth pursuit and the presence of a nystagmus in the interictal interval seem to be indicative for EA, especially type 1, 2, and 9. In accordance with the findings of the present study, we propose diagnostic criteria for EA, which should be further evaluated as to their sensitivity and specificity for detecting EA in future research.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Ethics Committee of the Medical Faculty of the LMU (414-15). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
FF: patient recruitment, data acquisition, statistical analysis, interpretation of data, and drafting the manuscript. LS and KD: patient recruitment and data acquisition. RS: statistical analysis and interpretation of data. DH: patient recruitment, data acquisition, interpretation of data, and drafting the manuscript. All authors approved the final version of the manuscript.
The authors thank Katie Göttlinger for Copy Editing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.1016856/full#supplementary-material
1. Jahn K, Langhagen T, Schroeder A, Heinen F. Vertigo and dizziness in childhood– update on diagnosis and treatment. Neuropediatrics. (2011) 42:129–34. doi: 10.1055/s-0031-1283158
2. Batu ED, Anlar B, Topçu M, Turanli G, Aysun S. Vertigo in childhood: a retrospective series of 100 children. Eur J Paediatr Neurol. (2015) 19:226–32. doi: 10.1016/j.ejpn.2014.12.009
3. Davitt M, Delvecchio MT, Aronoff SC. The differential diagnosis of vertigo in children: a systematic review of 2726 cases. Pediatr Emerg Care. (2020) 36:368–71. doi: 10.1097/PEC.0000000000001281
4. Jen J, Kim G, Baloh R. Clinical spectrum of episodic ataxia type 2. Neurology. (2004) 62:17–22. doi: 10.1212/01.WNL.0000101675.61074.50
5. Ophoff RA, Terwindt GM, Vergouwe MN, Van Eijk R, Oefner PJ, Hoffman SM, et al. Familial hemiplegic migraine and episodic Ataxia type-2 are caused by mutations in the Ca2+ channel gene Cacnl1a4. Cell. (1996) 87:543–52. doi: 10.1016/S0092-8674(00)81373-2
6. Paulhus K, Ammerman L, Glasscock E. Clinical spectrum of Kcna1 mutations: new insights into episodic ataxia and epilepsy comorbidity. Int J Mol Sci. (2020) 21:2802. doi: 10.3390/ijms21082802
7. Piarroux J, Riant F, Humbertclaude V, Remerand G, Hadjadj J, Rejou F, et al. Fgf14-related episodic ataxia: delineating the phenotype of episodic ataxia type 9. Annal Clin Transl Neurol. (2020) 7:565–72. doi: 10.1002/acn3.51005
8. Jen JC. Hereditary episodic ataxias. Ann N Y Acad Sci. (2008) 1142:250–3. doi: 10.1196/annals.1444.016
9. Jen JC, Wan J. Episodic ataxias. Handb Clin Neurol. (2018) 155:205–15. doi: 10.1016/B978-0-444-64189-2.00013-5
10. Spilioti M, Kiryttopoulos A, Moschou M, Kimiskidis VK. Episodic ataxia 1 & 2: clinical characteristics, diagnosis, and management. Arch Clin Neurol. (2022) 31:79–86.
11. Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, Kcna1. Nat Genet. (1994) 8:136–40. doi: 10.1038/ng1094-136
12. D'Adamo MC, Hasan S, Guglielmi L, Servettini I, Cenciarini M, Catacuzzeno L, et al. New insights into the pathogenesis and therapeutics of episodic ataxia type 1. Front Cell Neurosci. (2015) 9:317. doi: 10.3389/fncel.2015.00317
13. van de Berg R, Widdershoven J, Bisdorff A, Evers S, Wiener-Vacher S, Cushing SL, et al. Vestibular migraine and recurrent vertigo of childhood: diagnostic criteria consensus document of the classification committee of vestibular disorders of the BÁRÁNy society and the international headache society. J Vestib Res. (2021) 31:1–9. doi: 10.3233/VES-200003
14. R Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing (2021).
15. Choquet K, La Piana R, Brais B. A novel frameshift mutation in Fgf14 Causes an autosomal dominant episodic ataxia. Neurogenetics. (2015) 16:233–6. doi: 10.1007/s10048-014-0436-7
16. Schesny M, Joncourt F, Tarnutzer AA. Acetazolamide-responsive episodic ataxia linked to novel splice site variant in Fgf14 gene. Cerebellum. (2019) 18:649–53. doi: 10.1007/s12311-018-0997-3
17. Coebergh JA, van de Putte DEF, Snoeck IN, Ruivenkamp C, van Haeringen A, Smit LM, et al. New variable phenotype in spinocerebellar ataxia 27 (Sca 27) caused by a deletion in the Fgf14 gene. Eur J Paediatr Neurol. (2014) 18:413–5. doi: 10.1016/j.ejpn.2013.10.006
18. Groth CL, Berman BD. Spinocerebellar Ataxia 27: A Review and Characterization of an Evolving Phenotype. Tremor Other Hyperkinet Mov (NY). (2018) 8:534. doi: 10.7916/D80S0ZJQ
19. Miura S, Kosaka K, Fujioka R, Uchiyama Y, Shimojo T, Morikawa T, et al. Spinocerebellar ataxia 27 with a novel nonsense variant (Lys177x) in Fgf14. Eur J Med Genet. (2019) 62:172–6. doi: 10.1016/j.ejmg.2018.07.005
20. Paucar M, Lundin J, Alshammari T, Bergendal Å, Lindefeldt M, Alshammari M, et al. Broader phenotypic traits and widespread brain hypometabolism in spinocerebellar ataxia 27. J Intern Med. (2020) 288:103–15. doi: 10.1111/joim.13052
21. Gur-Hartman T, Berkowitz O, Yosovich K, Roubertie A, Zanni G, Macaya A, et al. Clinical phenotypes of infantile onset cacna1a-related disorder. Eur J Paediatr Neurol. (2021) 30:144–54. doi: 10.1016/j.ejpn.2020.10.004
22. Nachbauer W, Nocker M, Karner E, Stankovic I, Unterberger I, Eigentler A, et al. Episodic ataxia type 2: phenotype characteristics of a novel cacna1a mutation and review of the literature. J Neurol. (2014) 261:983–91. doi: 10.1007/s00415-014-7310-2
23. Zhang L, Wen Y, Zhang Q, Chen Y, Wang J, Shi K, et al. Cacna1a gene variants in eight chinese patients with a wide range of phenotypes. Front Pediatrics. (2020) 8:577544. doi: 10.3389/fped.2020.577544
24. Strupp M, Zwergal A, Brandt T. Episodic ataxia type 2. Neurotherapeutics. (2007) 4:267–73. doi: 10.1016/j.nurt.2007.01.014
25. Hirose H, Arayama T, Takita J, Igarashi T, Hayashi Y, Nagao Y, et al. Family of episodic ataxia type 2: no evidence of genetic linkage to the Cacna1a gene. Int J Mol Med. (2003) 11:187–9. doi: 10.3892/ijmm.11.2.187
26. Eunson LH, Graves TD, Hanna MG. New calcium channel mutations predict aberrant RNA splicing in episodic ataxia. Neurology. (2005) 65:308–10. doi: 10.1212/01.wnl.0000169020.82223.dd
27. Spacey SD, Materek LA, Szczygielski BI, Bird TD. Two novel Cacna1a gene mutations associated with episodic ataxia type 2 and interictal dystonia. Arch Neurol. (2005) 62:314–6. doi: 10.1001/archneur.62.2.314
28. Choi K-D, Choi J-H. Episodic ataxias: clinical and genetic features. J Mov Dis. (2016) 9:129. doi: 10.14802/jmd.16028
29. Kerber KA, Jen JC, Lee H, Nelson SF, Baloh RW. A new episodic ataxia syndrome with linkage to chromosome 19q13. Arch Neurol. (2007) 64:749–52. doi: 10.1001/archneur.64.5.749
30. Steckley J, Ebers G, Cader M, McLachlan R. An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus. Neurology. (2001) 57:1499–502. doi: 10.1212/WNL.57.8.1499
31. Damji KF, Allingham RR, Pollock SC, Small K, Lewis KE, Stajich JM, et al. Periodic vestibulocerebellar ataxia, an autosomal dominant ataxia with defective smooth pursuit, is genetically distinct from other autosomal dominant ataxias. Arch Neurol. (1996) 53:338–44. doi: 10.1001/archneur.1996.00550040074016
32. Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, Mayer T, et al. Coding and noncoding variation of the human calcium-channel B4-subunit gene Cacnb4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. (2000) 66:1531–9. doi: 10.1086/302909
33. Jen J, Wan J, Palos T, Howard B, Baloh R. Mutation in the glutamate transporter eaat1 causes episodic ataxia, hemiplegia, and seizures. Neurology. (2005) 65:529–34. doi: 10.1212/01.WNL.0000172638.58172.5a
34. De Vries B, Mamsa H, Stam AH, Wan J, Bakker SL, Vanmolkot KR, et al. Episodic ataxia associated with eaat1 mutation C186s affecting glutamate reuptake. Arch Neurol. (2009) 66:97–101. doi: 10.1001/archneurol.2008.535
35. Conroy J, McGettigan P, Murphy R, Webb D, Murphy SM, McCoy B, et al. A novel locus for episodic ataxia: Ubr4 the likely candidate. Eur J Hum Genet. (2014) 22:505–10. doi: 10.1038/ejhg.2013.173
36. Salman MS, Chodirker BN. Neuro-ophthalmological findings in children and adolescents with chronic ataxia. Neuroophthalmology. (2015) 39:125–31. doi: 10.3109/01658107.2015.1016579
37. Tantsis EM, Gill D, Griffiths L, Gupta S, Lawson J, Maksemous N, et al. Eye movement disorders are an early manifestation of Cacna 1a mutations in children. Dev Med Child Neurol. (2016) 58:639–44. doi: 10.1111/dmcn.13033
38. Hashemi H, Pakzad R, Heydarian S, Yekta A, Aghamirsalim M, Shokrollahzadeh F, et al. Global and regional prevalence of strabismus: a comprehensive systematic review and meta-analysis. Strabismus. (2019) 27:54–65. doi: 10.1080/09273972.2019.1604773
39. Baloh RW, Yue Q, Furman JM, Nelson SF. Familial episodic ataxia: clinical heterogeneity in four families linked to chromosome 19p. Ann Neurol. (1997) 41:8–16. doi: 10.1002/ana.410410105
40. Bertholon P, Chabrier S, Riant F, Tournier-Lasserve E, Peyron R. Episodic Ataxia Type 2: Unusual Aspects in Clinical and Genetic Presentation. Special Emphasis in Childhood. J Neurol Neurosurg Psychiatry. (2009) 80:1289–92. doi: 10.1136/jnnp.2008.159103
41. Graves TD, Cha Y-H, Hahn AF, Barohn R, Salajegheh MK, Griggs RC, et al. Episodic ataxia type 1: clinical characterization, quality of life and genotype–phenotype correlation. Brain. (2014) 137:1009–18. doi: 10.1093/brain/awu012
43. Langhagen T, Lehrer N, Borggraefe I, Heinen F, Jahn K. Vestibular migraine in children and adolescents: clinical findings and laboratory tests. Front Neurol. (2015) 5:292. doi: 10.3389/fneur.2014.00292
44. Langhagen T, Schroeder AS, Rettinger N, Borggraefe I, Jahn K. Migraine-related vertigo and somatoform vertigo frequently occur in children and are often associated. Neuropediatrics. (2013) 44:055–8. doi: 10.1055/s-0032-1333433
45. Dieterich M, Brandt T. Episodic vertigo related to migraine (90 cases): vestibular migraine? J Neurol. (1999) 246:883–92. doi: 10.1007/s004150050478
46. Neugebauer H, Adrion C, Glaser M, Strupp M. Long-term changes of central ocular motor signs in patients with vestibular migraine. Eur Neurol. (2013) 69:102–7. doi: 10.1159/000343814
Keywords: episodic ataxia, spinocerebellar ataxia 27, ocular motor disturbances, children, adolescents, vertigo, dizziness
Citation: Filippopulos FM, Schnabel L, Dunker K, Strobl R and Huppert D (2022) Episodic ataxias in children and adolescents: Clinical findings and suggested diagnostic criteria. Front. Neurol. 13:1016856. doi: 10.3389/fneur.2022.1016856
Received: 11 August 2022; Accepted: 06 October 2022;
Published: 24 October 2022.
Edited by:
Jun Yang, Shanghai Jiaotong University School of Medicine, ChinaReviewed by:
Nahid Olfati, University of California, San Diego, United StatesCopyright © 2022 Filippopulos, Schnabel, Dunker, Strobl and Huppert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Filipp Maximilian Filippopulos, ZmlsaXBwLmZpbGlwcG9wdWxvc0BtZWQudW5pLW11ZW5jaGVuLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.