 Hao Cheng
Hao Cheng He Lv
He Lv Yan Wang
Yan Wang Gui-Qiang Wang
Gui-Qiang Wang- 1Department of Infectious Diseases and the Center for Liver Diseases, Peking University First Hospital, Beijing, China
- 2Department of Neurology, Peking University First Hospital, Beijing, China
Peripheral nerve injury is one of the rare complications of adult-onset Still's disease (AOSD). We report a 20-year-old woman diagnosed with AOSD combined with severe sensory neuropathy. She presented with a sore throat, joint pain, rash, and lymphadenopathy. After receiving glucocorticoid therapy, her fever, rash, and inflammatory markers improved. Unexpectedly, 3 weeks after the onset, she experienced sudden paresthesia in her extremities, decreased muscle strength, and diminished tendon reflexes. The electrophysiological examination and peripheral nerve biopsy confirmed immune-mediated severe sensory neuropathy. For the first time, we report typical Wallerian degeneration in AOSD patients with sensory neuropathy by nerve biopsy. Compared with other common symptoms, the delayed aggravation of neurological symptoms may be an important characteristic of sensory neuropathy secondary to AOSD. We emphasize that intensive attention to neurological symptoms after general symptoms control, administration of adequate and appropriate prolonged immunosuppressive therapy, and long-term follow-up are essential for these patients.
Introduction
Peripheral nerve injury is a rare complication of adult-Onset Still's Disease (AOSD). Wallerian degeneration was observed in distal myelinated fibers, which may cause the delayed aggravation of neurological symptoms during glucocorticoid treatment in an AOSD patient.
The rapidly progressive clinical deterioration of the AOSD patient presented here posed a diagnostic challenge requiring an extensive diagnostic workup. In the literature, only a few case reports describe the main manifestations of AOSD with sensory neuropathy, while most were short of pathological evidence. For the first time, we report typical Wallerian degeneration in AOSD patients with severe sensory neuropathy by nerve biopsy. Careful distinction and prompt diagnosis are critical for adherence to immunosuppressive therapy.
Case description
A 20-year-old woman was presented to the emergency clinic with fever and sore throat for one week and joint pain, weakness in limbs, and slight numbness of fingertips and tongue. She denied any tobacco or alcohol (illicit drugs) use, and her family history was not notable. After admission, she continued to experience a high fever (Tmax 40°C). One week after her admission, scattered congestive pinpoint itching rashes appeared on her chest, abdomen, back, and limbs, without ulceration or scaling, accompanied by liver function abnormalities (alanine aminotransferase, ALTmax 227 IU/L, reference range of 7–40 IU/L; aspartate transaminase, ASTmax 193 IU/L, reference range of 13–35 IU/L). The rash was noticeable when the body temperature rose and faded when the body temperature dropped. Inflammatory markers such as serum ferritin (max to >7,500 ng/ml, reference range 11–306.8 ng/ml), erythrocyte sedimentation rate (ESR, max to 50 mm/h, reference range 0–20 mm/h), C-reactive protein (CRP, max to 104 mg/L, normal <5 mg/L), and lactate dehydrogenase (LDH, max to 900 IU/L, reference range 100–240 IU/L) were all significantly elevated. The lymph node ultrasound revealed extensive enlarged superficial lymph nodes (Figure 1).
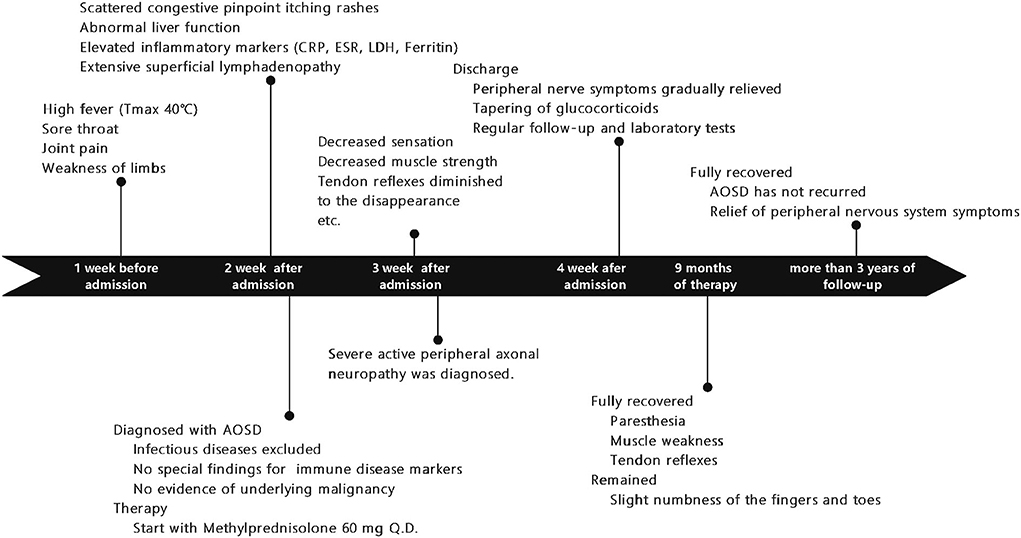
Figure 1. A timeline showing symptoms and exams/interventions performed on the patient. AOSD, Adult-onset Still's disease; CRP, C-reactive protein; LDH, lactate dehydrogenase, ESR, erythrocyte sedimentation rate.
The clinical diagnosis of AOSD is generally reached by exclusion while investigating a patient with a fever of unknown origin. We carried out detailed screenings for infectious diseases, autoimmune diseases, and tumors. Laboratory examinations for common pathogens (respiratory pathogens, such as influenza virus, mycoplasma, chlamydia, legionella, adenovirus, etc.) and specific disease-related pathogens (cytomegalovirus/Epstein-Barr virus, mycobacterium tuberculosis, rickettsia of typhus, Coxiella burnetii of Q fever, Salmonella typhi, Brucella, etc.) were all negative. The blood culture was repeatedly negative, and screening tests for AIDS, hepatitis B, hepatitis C, and syphilis were negative. Antibodies that indicate some autoimmune diseases, such as the anti-double-stranded DNA antibody, antineutrophil cytoplasmic antibodies (ANCA), anti-extractable nuclear antigen (ENA) antibodies, immunoglobulins, protein electrophoresis, immunofixation electrophoresis, complements, rheumatoid factor were all no meaningful found. No significant findings were found in tumor markers, bone marrow biopsy, and imaging examinations of underlying malignant tumors. According to the Yamaguchi criteria (1), the patient was diagnosed with AOSD.
Methylprednisolone (60 mg Q.D.) was administrated as starting dose with subsequent tapering. After glucocorticoid treatment, her main symptoms were relieved, and inflammatory markers rapidly decreased, but the paresthesia and decreased sensation in her extremities decreased muscle strength with MRC (Medical Research Council) grades 2–3, tendon reflexes diminished to the disappearance, and imbalance progressed remarkably so that she was not able to walk or write; even words started slurring in the third week. We ordered an MRI of the spine with no positive findings, excluding the possibility of the patient's lumbosacral ganglion compression. Heavy metal and toxicants blood tests were negative. Head MRI and cerebrospinal fluid (CSF) examination were normal. The anti-ganglioside GQ1b antibody was negative in both serum and cerebrospinal fluid. Polymyositis antibodies, and paraneoplastic syndrome-related antibodies (Hu, Yo, and Ri antibodies) showed no meaningful findings. We performed the electrophysiological examination, and nerve and muscle biopsy revealed immune-mediated extensive peripheral nerve injury (Supplementary Figures 1–3). Intriguingly, electron microscopy images of nerve biopsy demonstrate the typical manifestation of distal axonal degeneration due to nerve injury—Wallerian degeneration (Figure 2). Paresthesia, muscle weakness, and tendon reflexes were fully recovered with MRC grades five after 9 months of glucocorticoid therapy in the patient, and slight numbness of the fingers and toes remained. She got fully recovered after more than 3 years of regular follow-up.
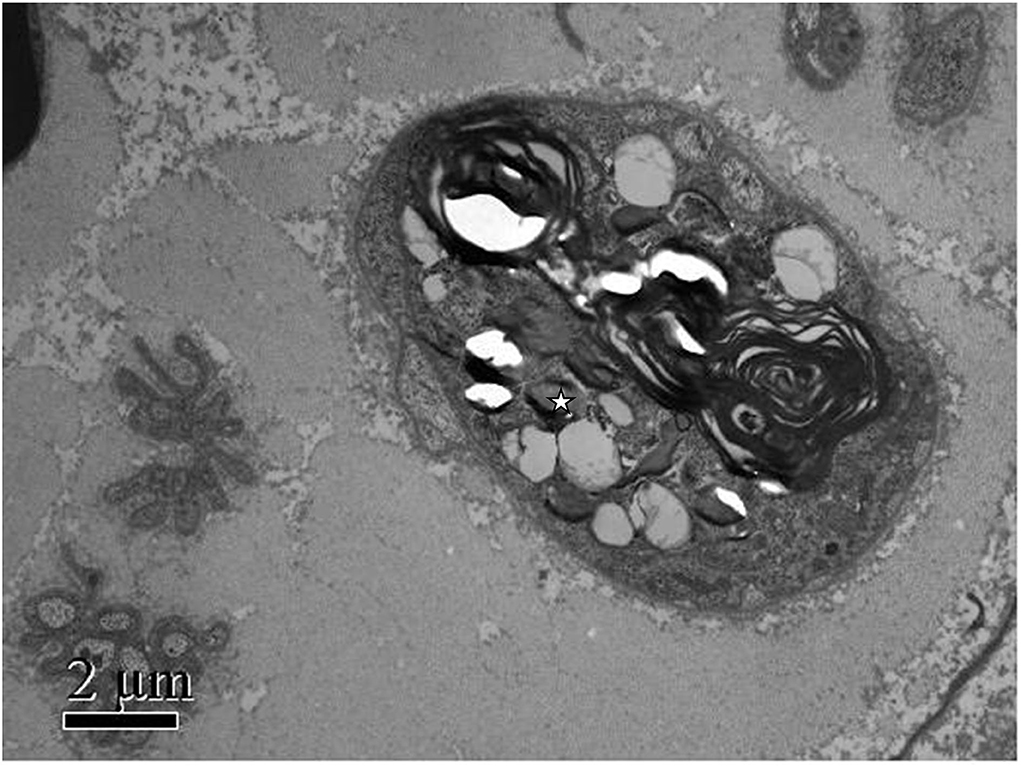
Figure 2. Patient's photograph of the sural nerve biopsy specimen. Electron microscopic findings: Myelinated nerve fibers were extensively destroyed. Wallerian degeneration could be observed (white star), and Schwann cells and macrophages were not observed. Scale bars: 2μm.
Discussion
Neurological complications of AOSD are rare (2), usually seizures, cranial nerve palsy, aseptic meningoencephalitis, and Miller-Fisher syndrome, of which immune-mediated peripheral nerve injury has been reported minimally. We summarized previous case reports of AOSD combined with peripheral nerve injuries in Table 1. The delayed onset of peripheral nerve injury involving medium and large myelinated sensory fibers occurred 2 weeks to 3 years after the onset of the early symptoms, which was a great challenge for early identification and accurate diagnosis.
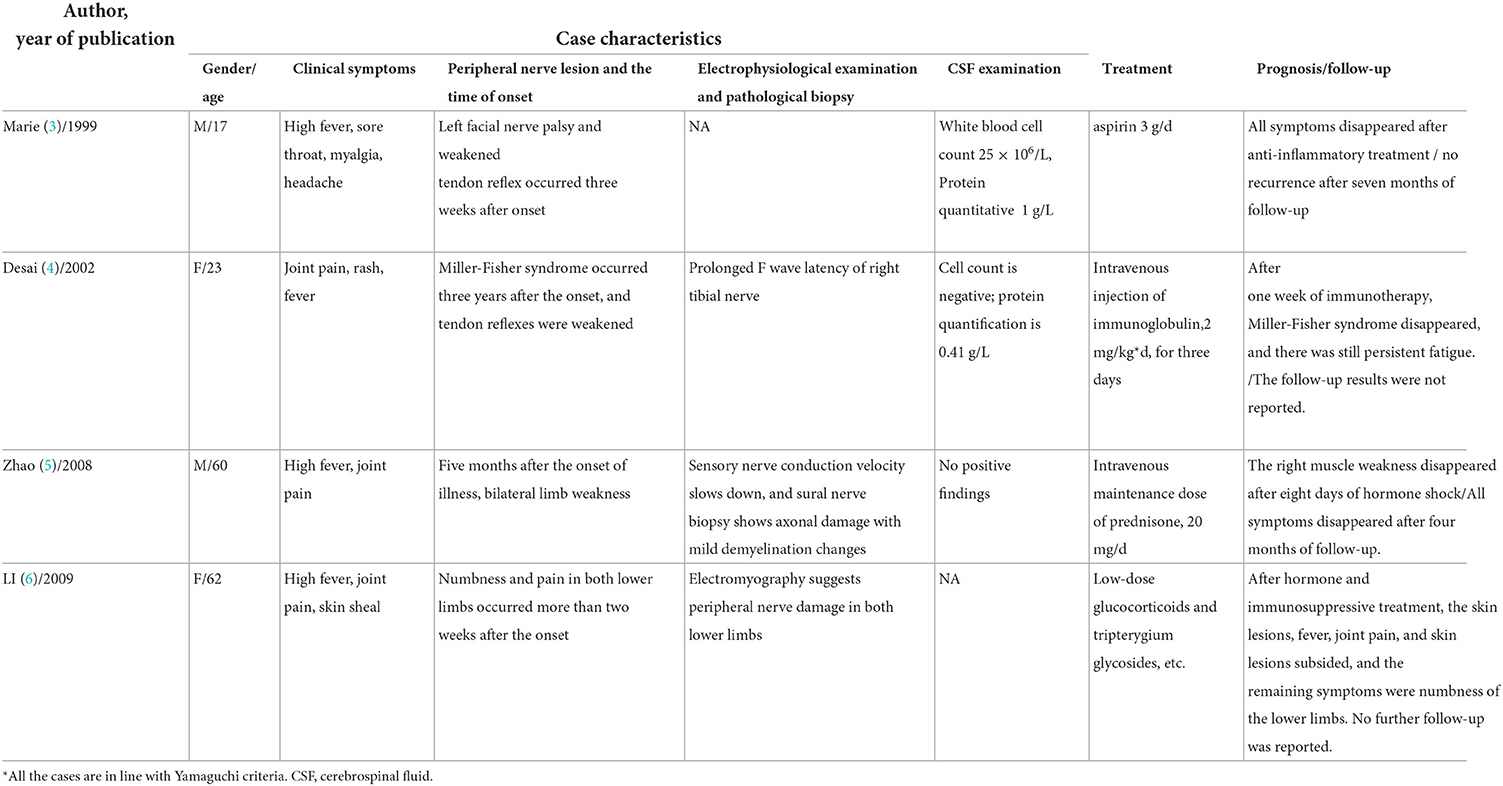
Table 1. Summary of literature on adult-onset Still's disease* with peripheral nerve injury.
We confirmed the patient's severe immune-mediated sensory neuropathy by electrophysiological examination and pathological findings from nerve and muscle biopsies. For etiologies of sensory nerve injury, sensory ganglionopathy should be ruled out. After detailed examinations and long-term follow-up observation, we excluded the common causes of sensory ganglionopathies, such as lumbosacral ganglion compression, paraneoplastic sensory ganglionopathy, and sensory ganglionopathy secondary to Sjögren disease.
Guillain-Barré syndrome was the disease we focused on differential diagnosis in the diagnosis of sensory neuropathy in this patient. Although we found that this patient had a number of inconsistencies with the common Guillain-Barré syndrome and its common variants, for example, we found no obvious antecedent events such as infection, vaccination, and other predisposing factors such as drugs, surgery, radiation, chemotherapy, etc. that contributed to the Guillain-Barré syndrome. The patient had normal CSF cell count and protein levels, negative for anti-ganglioside GQ1b antibody, and the patient's response to glucocorticoid therapy was good. However, based on the results of the nerve conduction study and nerve and muscle pathology, theoretically, we cannot in an absolute sense rule out the possibility of the Guillain-Barre syndrome, since both Guillain-Barre syndrome and sensory neuropathy secondary to AOSD are peripheral nervous system damage due to autoimmune abnormalities. From the perspective of disease “monism,” combined with the patient's symptoms, auxiliary examination and treatment response, and long-term regular follow-up monitoring, we currently tend to believe that the patient's severe sensory neuropathy is secondary to AOSD.
Although the pathogenesis of sensory neuropathy and AOSD is unclear, the possible mechanism of its peripheral nerve damage may be related to both inflammatory response and autoimmune response. The enhanced inflammatory factors may directly attack the dorsal root ganglion, anterior horn motor neurons, interstitial structure, myelin, and axons, or infringement of nerve nourishing blood vessels cause peripheral nerve lesions (7). The autoimmune response can directly cause disease or facilitate potential pathogenic conditions, resulting in peripheral nerve lesions similar to the Guillain-Barré syndrome. Although AOSD diagnosis requires exclusion of infection, it has also been suggested that one of the possible pathogenesis of peripheral nerve injury is triggered by multiple infectious factors in an autoimmune background. They may become potentially invasive antigens to trigger peripheral neuroimmune damage, which causes the immune recognition system to misjudge and trigger autoimmune attacks. The delayed aggravation of peripheral nerve injury symptoms is consistent with this kind of autoimmune injury characterized by a prodromal period (8).
Most previous case reports lack pathological evidence; only one of the case reports showed axonal damage with mild demyelination changes by nerve biopsy. For the first time, we report typical Wallerian degeneration in AOSD patients with sensory neuropathy by neuromuscular biopsy. Wallerian degeneration is a common pathological feature of neurological disorders. It has been said that Wallerian degeneration is an active and dynamic cellular process that provides a chance for axons to reconnect, rather than rebuilding the entire nerve projection by axon regeneration, which was regulated at molecular and cellular levels (9). In response to immune-mediated injury, Wallerian degeneration showed as an orchestrated interplay between innate-immune cells and molecules produced by immune and non-immune cells.
Previous studies reported that activation of innate immunity plays a major role in the pathogenesis of AOSD, and innate immunity is also proven to contribute to the mechanism of Wallerian degeneration. All the symptoms, including that of peripheral nerve injury, were recovered by long-term glucocorticoid therapy. Delayed aggravation of the patient's peripheral nervous system symptoms may be due to Wallerian degeneration and dynamic self-destruction progress, which is a response to active systemic immunity in AOSD. Effective innate immune response to Wallerian degeneration after the injury is key to neurological recovery.
Since reports of sensory neuropathy secondary to AOSD are rare, there is currently no unified treatment recommendation. The patient started immunosuppressive therapy for AOSD. After a diagnosis of severe sensory neuropathy, we recommended glucocorticoids in combination with cyclophosphamide (CTX), but the patient refused because of concerns about the side effects of CTX. Due to economic reasons, the patient refused to use intravenous immunoglobulin (IVIG) and plasma exchange therapy and maintained glucocorticoids alone, followed by long-term tapering and eventually got remission. Combined with the above immunopathological mechanisms of AOSD and Wallerian degeneration, we believe that immunotherapy, in addition to glucocorticoids, such as glucocorticoids combined with other immunosuppressive agents, IVIG, or plasma exchange, may improve patient outcomes. Therefore, attention to neurological symptoms, early diagnosis, and initiation of adequate immunosuppressive therapy is critical to improving prognosis in patients with such severe sensory neuropathy.
Conclusion
In conclusion, we present the rare case of a 20-year-old woman who was diagnosed with AOSD with severe sensory neuropathy. For the first time, we report typical Wallerian degeneration found in the patient by neuromuscular biopsy. By elucidating possible autoimmune mechanisms and reviewing previous reports, we conclude that Wallerian degeneration is the innate immune response to AOSD peripheral nerve injury, which may cause the delayed aggravation of neurological symptoms during glucocorticoid treatment in an AOSD patient. We emphasize that intensive attention to neurological symptoms, administration of adequate and appropriate prolonged immunosuppressive therapy, and long-term follow-up are particularly important for these patients.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
HC wrote this case report, including the concept of this article, the definition of intellectual content, and data acquisition. HL guided the microphotograph identification of the patient's sural nerve biopsy specimens and the discussion on sensory neuropathy. YW and G-QW designed and reviewed the manuscript for its intellectual content. YW is the principal investigator. HC, HL, and G-QW are the main participants in the project. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (#82070605) and the Science and Technology Department of Xinjiang Uygur Autonomous Region (#2020A03004-3).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.1016393/full#supplementary-material
References
1. Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, Kashiwagi H, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. (1992) 19:424–30.
2. Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still's disease. J Autoimmun. (2018) 93:24–36. doi: 10.1016/j.jaut.2018.07.018
3. Marie I, Levesque H, Perraudin N, Cailleux N, Lecomte F, Courtois H. Aseptic meningitis and cranial nerve palsy revealing adult-onset Still's disease. Clin Infect Dis. (1999) 29:220–1. doi: 10.1086/520170
4. Desai SS, Allen E, Deodhar A. Miller Fisher syndrome in adult onset Still's disease: case report and review of the literature of other neurological manifestations. Rheumatology. (2002) 41:216–22. doi: 10.1093/rheumatology/41.2.216
5. Zhao H, Yuan Y, Li Y, Si C-W, Tian G-S, Wang G-Q, et al. Encephalic large arteries narrowness and peripheral neuropathy in a patient with adult-onset Still's disease. Rheumatol Int. (2008) 28:1261. doi: 10.1007/s00296-008-0599-3
6. Yanxi Li, Zhu Xi, Yang Shi, Diao Qingchun. Adult Still's disease complicated by peripheral neuropathy: a case report. J Clin Dermatol. (2009) 38:111–2. doi: 10.3969/j.issn.1000-4963.2009.02.023
7. Callaghan BC, Price RS, Chen KS, Feldman EL. The importance of rare subtypes in diagnosis and treatment of peripheral neuropathy. JAMA Neurol. (2015) 72:1510. doi: 10.1001/jamaneurol.2015.2347
8. Hughes RA, Cornblath DR. Guillain-Barré syndrome. Lancet. (2005) 366:1653–66. doi: 10.1016/S0140-6736(05)67665-9
Keywords: adult-onset Still's disease, sensory neuropathy, sensory ganglionopathy, Wallerian degeneration, delayed aggravation, innate-immune response
Citation: Cheng H, Lv H, Wang Y and Wang G-Q (2022) Case report: Wallerian degeneration: The innate-immune response to adult-onset Still's disease peripheral nerve injury. Front. Neurol. 13:1016393. doi: 10.3389/fneur.2022.1016393
Received: 10 August 2022; Accepted: 23 September 2022;
Published: 19 October 2022.
Edited by:
Giovanni Meola, University of Milan, ItalyReviewed by:
Mohamed Kazamel, University of Alabama at Birmingham, United StatesAngelo Torrente, University of Palermo, Italy
Copyright © 2022 Cheng, Lv, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Wang, d2FuZ3lhbndhbmdAYmptdS5lZHUuY24=; Gui-Qiang Wang, am9objEzMTIxMkAxMjYuY29t