 Yajing Gan
Yajing Gan Zihan Wei
Zihan Wei Chao Liu
Chao Liu Guoyan Li
Guoyan Li Yan Feng
Yan Feng Yanchun Deng
Yanchun Deng- 1Department of Neurology, Epilepsy Center of Xijing Hospital, Fourth Military Medical University, Xi'an, China
- 2Xijing Institute of Epilepsy and Encephalopathy, Xi'an, China
The International League Against Epilepsy officially revised its classification in 2017, which amended “epileptic encephalopathy” to “developmental and epileptic encephalopathy”. With the development of genetic testing technology, an increasing number of genes that cause developmental and epileptic encephalopathies are being identified. Among these, solute transporter dysfunction is part of the etiology of developmental and epileptic encephalopathies. Solute carrier transporters play an essential physiological function in the human body, and their dysfunction is associated with various human diseases. Therefore, in-depth studies of developmental and epileptic encephalopathies caused by solute carrier transporter dysfunction can help develop new therapeutic modalities to facilitate the treatment of refractory epilepsy and improve patient prognosis. In this article, the concept of transporter protein disorders is first proposed, and nine developmental and epileptic encephalopathies caused by solute carrier transporter dysfunction are described in detail in terms of pathogenesis, clinical manifestations, ancillary tests, and precise treatment to provide ideas for the precise treatment of epilepsy.
Introduction
The concept of epileptic encephalopathy was widely accepted and used in the early years, but later scholars have found that many genetic disorders can cause developmental disorders in addition to the direct effects of epileptic activity on development (1). In 2017, the International League Against Epilepsy (ILAE) introduced the concept of developmental and epileptic encephalopathy (DEE) (2). Most DEEs have a genetic cause. With the advances in genetic testing and neuroimaging in recent years, an increasing number of variants of new genes have been discovered, advancing the understanding of DEE etiology. Transporter dysfunction accounts for one etiology of DEE, with solute carrier (SLC) transporters predominating. SLC transporters not only play an essential role in maintaining human physiological functions but also are important targets for drug action in the human body, and in-depth analysis of SLC transporters can significantly contribute to the development of treatments for refractory epilepsy. To date, nine SLC genes related to DEE have been identified. The study of the pathogenesis of these nine types of DEEs will improve targeted treatment and the prognosis of patients.
Transporter proteins and transporter protein diseases
Transporter proteins are widely expressed on human cellular and organelle membranes. It is believed that 10% of human genes are associated with transport functions, which control the passage of substances across membranes and are essential for cell growth and reproduction (3). In total, four superfamilies of transporter proteins have been identified: the ATP-binding cassette (ABC) superfamily, ATPase superfamily, ion channel superfamily, and SLC superfamily (4). The ATP-binding cassette and SLC superfamilies are the main components of transporter proteins. SLCs are present in almost every organelle and cell membrane and play a significant role in maintaining environmental homeostasis in the human body. Broadly speaking, we refer to all diseases caused by variants of genes encoding transporter proteins as transporter protein diseases, and narrowly speaking, for those mainly caused by variants of the SLC transporter genes, we call them SLC transporter diseases.
Solute carrier transporter disease
There are 458 different transmembrane SLCs in humans, which are categorized into 65 families (5). SLC transporters, one of the major mammalian cell membrane proteins, are involved in a wide range of transport functions between various organelles. Since they rely on electrochemical gradients or ion gradients generated by proton pumps to transport substrates, SLC transporters are considered secondary active transporter proteins (6). In the brain, SLC transporters are involved in the transport of various solutes across membranes, such as across the blood–brain barrier, cell membranes, and mitochondrial membranes. This transport plays an essential role in maintaining normal brain function, and SLC transporter dysfunction can contribute to various neurological diseases, such as Huntington's disease, Parkinson's disease, Alzheimer's disease, and epilepsy (7, 8). Furthermore, an increasing number of monogenic diseases have been found to be associated with SLC, suggesting that transporter proteins have very high potential for exploitation. In this article, we only describe the association of SLC with DEE because an in-depth study of SLC may reveal some precise therapeutic targets that could promote the development of treatments for drug-refractory epilepsy. Based on the literature, we found that variants of SLC1A2, SLC2A1, SLC6A1, SLC12A5, SLC13A5, SLC25A22, SLC35A2, and SLC38A3 are associated with a series of DEEs, and the pathogenesis and precise therapeutic measures for each of these nine DEEs are described in the following text.
Pathogenesis, clinical manifestations, and treatment of solute carrier transporter diseases
SLC1A2 and developmental and epileptic encephalopathy type 41
Pathogenesis
The SLC1 family mediates the cellular uptake of glutamate, thus reducing the intersynaptic glutamate concentration and helping to terminate excitatory neurotransmission (9). SLC1A2 is a member of the SLC1 family located on chromosome 11p13 and encodes sodium-dependent glutamate transporter 2, which is also known as excitatory amino acid transporter 2 (EAAT2) (10). Pathogenic variants of SLC1A2 can lead to developmental and epileptic encephalopathy type 41 (DEE41, OMIM 600300) (11). EAAT2 is expressed on the surface of astrocytes and is responsible for transporting more than 80–90% of glutamate by the codirectional transfer of three Na+ and one H+ ions and receiving one K+ ion in exchange (9). EAAT2 reduces the extracellular concentration of glutamate, thus preventing excessive stimulation of cell membrane receptors by glutamate, which can lead to neuronal damage or death (Figure 1) (12, 13).
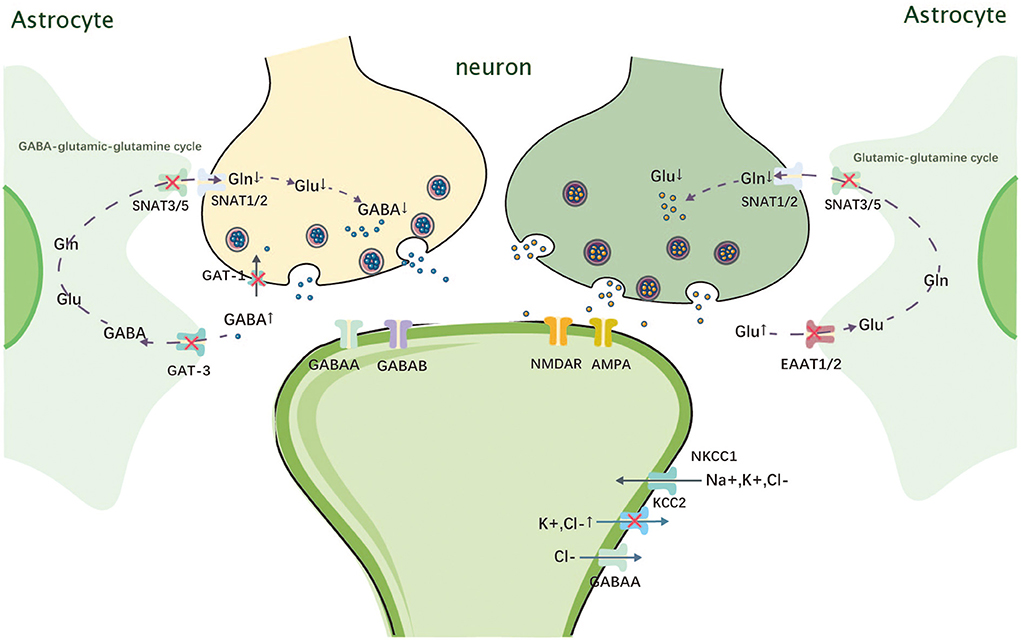
Figure 1. Mechanism of action of SLC1A2, SLC6A1, SLC12A5, and SLC38A3. Gln, glutamine; Glu, glutamate; GABA, gamma-amino butyric acid; SNAT, sodium-coupled neutral amino acid transporter; NMDAR, N-methyl-D-aspartate receptor; AMPA, alpha-amino-3-hydroxy-5-methyl-4 -isoxazolepropionic acid receptor; GAT-1, Na+-Cl–coupled γ-aminobutyric acid transporter 1; NKCC1, Na+-K+-2Cl- cotransporter 1; KCC2, K+-Cl-cotransporter 2; EAAT, excitatory amino acid transporter.
Clinical presentation
Variants of SLC1A2 are autosomal dominant, most of which are heterozygous de novo variants (14). Wagner et al. found that autosomal recessive inherited double-allele variants of SLC1A2 can also cause epilepsy (15). All patients with autosomal dominant inheritance of SLC1A2 developed seizures within the first 6 months of life (16). They typically begin as tonic or myoclonic seizures and then gradually evolve into multiple seizure types, such as atonic seizures, tonic–clonic seizures, and generalized tonic seizures, with global developmental delay, axial hypotony, optic nerve atrophy, and cortical visual dysplasia (16). The homozygous splicing variant reported by Wagner et al. had less severe symptoms, with the first seizure at the age of 2 years, starting with vomiting and nervous eye deviation, followed by generalized tonic–clonic and atonic seizures (15). This patient also had autism and attention-deficit/hyperactivity disorder (ADHD), and a single medication was sufficient to bring the developmental delay under control (15). The EEG of most patients showed multifocal or diffuse epileptiform discharges with abnormal background activity (16). EEG of patients with autosomal recessive inheritance showed diffuse hemispheric dysfunction (15).
Treatment options
DEE41 is a treatment-refractory epilepsy, while DEE41 in one patient was well controlled with valproic acid monotherapy (17). Translational activators of EAAT2 may provide a new therapeutic approach to protect neurons from excitatory neurotransmitter damage (12, 18). Dyomina et al. showed that a recombinant form of the interleukin 1 receptor antagonist (anakinra) reduced neuronal loss in the hippocampal region and enhanced EAAT2 expression levels, thereby suppressing recurrent seizures in the chronic phase (19). This may also prevent some behavioral disorders, such as motor hyperactivity and disturbances in social interactions (19). A pyrazine derivative named LDN/OSU-0212320, identified by Kong et al., can reduce excitotoxic effects and increase glutamate uptake by glial cells by upregulating EAAT2 translation (20). LDN/OSU-0212320 was found to significantly reduce neuronal death and seizure frequency in mice (20). Parawixin10 is an EAAT1 and EAAT2 allosteric modulator, which has neuroprotective effects and reduces the severity and duration of seizures (18). However, tamoxifen and growth factor α-selective agonists enhance EAAT1 and EAAT2 expression through the activation of the NF-κB pathway (21, 22). In addition, ceftriaxone, a β-lactam antibiotic, is one of the well-studied SLC1A2 modulators, and it is an EAAT2 protein enhancer that reduces neuronal death and seizure frequency (23, 24). An animal study also showed that low doses of ceftriaxone with valproate increased GABAergic activity and decreased glutamatergic activity, favoring seizure suppression and improved cognitive function (25). However, Stergachis et al. did not find significant improvements in SLC1A2-associated epilepsy in 20-month-old patients treated with ceftriaxone (16).
SLC2A1 and glucose transporter 1 deficiency syndromes 1 and 2
Pathogenesis
The SLC2 family plays an important role in the control of systemic glucose homeostasis (26). The SLC2A1 gene belongs to the first identified member of the SLC2 family of glucose transporters and encodes glucose transporter 1 (GLUT1). GLUT1 is the main transporter protein that mediates glucose crossing the blood–brain barrier. GLUT1 deficiency not only decreases glucose transport in the brain but also prevents cerebral angiogenesis, resulting in a smaller than normal brain microvasculature (27). Both brain glucose deficiency and a smaller microvasculature deprive the brain of the energy resources it needs to function properly.
Clinical presentation
GLUT1 dysfunction can lead to glucose transporter 1 deficiency syndrome (GLUT1DS), including GLUT1DS1 (OMIM 606777) and GLUT1DS2 (OMIM 612126). Approximately 90% of GLUT1DS is caused by de novo heterozygous variants, whereas GLUT1DS homozygote variants can be morphologically abnormal during the embryonic stage and are incompatible with life (28).
GLUT1DS1 is mainly autosomal dominant and, to a lesser extent, autosomal recessive. Approximately 80% of patients develop seizures in infancy, which can present as generalized tonic–clonic, myoclonic, atonic, or atypical absence seizures (29, 30). Patients mostly present with microcephaly, hypotony, dystonia, ataxia, hyperreflexia, developmental delay, mental retardation, and motor disorders such as paroxysmal hemiparesis, paroxysmal limb stiffness and purple limb, paroxysmal oculomotor abnormalities, and chorea (29, 31).
GLUT1DS2 is autosomal dominant, and its main phenotype is paroxysmal exercise-induced dyskinesia (PED), which commonly develops concurrently with prolonged exercise as the main trigger but can also be triggered by anxiety and emotional stress in a few patients (32). The seizures may be preceded by autonomic symptoms such as hyperventilation and sweating (33). Some patients also have progressive spastic paraplegia, paroxysmal chorea, paroxysmal limb weakness, ataxia, developmental delay, dysarthria, and epilepsy (31, 34, 35). Their epilepsy mainly manifests as absence and, to a lesser extent, as atypical absence seizures, myoclonic seizures, and atonic seizures (33, 36).
It is worth noting that GLUT1DS has significantly lower epileptiform abnormalities on postprandial EEG than on preprandial EEG (37, 38). MRIs are mostly normal or mildly abnormal, including mild brain atrophy, myelin dysplasia, ventricular enlargement, and corpus callosum dysplasia (39). 18F-fluorodeoxyglucose positron emission tomography (18F-FDG-PET) showed reduced glucose metabolism in the bilateral thalamus, cerebellum, and neocortex, which may be associated with epilepsy (40). Cerebrospinal fluid lactate glucose measurement can be a rapid and effective method to diagnose GLUT1DS because almost all patients with GLUT1DS have reduced glucose levels in the cerebrospinal fluid and normal or reduced lactate levels (41).
Treatment options
The ketogenic diet is a very effective tool that increases blood ketone levels, and ketones cross the blood–brain barrier via monocarboxylate transporter 1 and supply the brain with energy (42–44). Thus, a ketogenic diet can improve seizure symptoms, motor deficits, and cognition of most patients (45–47). Triheptanoin is metabolized to acetyl coenzyme A and propionyl coenzyme A, which can stimulate the tricarboxylic acid cycle and effectively improve brain energy deficiency in patients with GLUT1DS (48). Joshi et al. reported a case of a boy with partial seizure control through the use of a medium-chain triglyceride oil-based ketogenic diet (35). The addition of carnitine and alpha-lipoic acid resulted in complete seizure control and significant improvements in motor and cognitive function (35). Phenobarbital and valproic acid inhibit GLUT1-DS transport and should be avoided (49). A study by Tang et al. showed that using adeno-associated virus serotype 9 (AAV9) as a vector for SLC2A1 in model mice increased GLUT1 expression in the mouse brain, increased the cerebrospinal fluid glucose concentration, increased glucose uptake in the brain, and restored the brain microvessel size (50). Directly combining GLUT1 with transduction domains or cell-penetrating peptides for delivery into cells is a possible new therapy (51).
SLC6A1 and myoclonic–atonic epilepsy
Pathogenesis
SLC6A1 belongs to the γ-aminobutyric acid (GABA) transporter subfamily, is the most abundantly expressed GABA transporter protein in the central nervous system (CNS), and is one of the most intensively studied members of the SLC6 family (52). It is located on chromosome 3p25.3, encoding Na+-Cl−-coupled γ-aminobutyric acid transporter 1 (GAT-1), which is mainly responsible for the reuptake of GABA from the synapse (Figure 1). GAT-1 dysfunction leads to GABA accumulation between synapses and overstimulation of GABAA and GABAB receptors on the postsynaptic membrane (42). GABAA overstimulation can cause continuous hyperpolarization and outbreak discharge of cell membranes, which cause seizures (43).
Clinical presentation
SLC6A1 pathogenic variants cause myoclonic–atonic epilepsy (OMIM 616421), but there is no interaction between seizures and cognitive impairment (44). The main clinical features also include cognitive developmental impairment, mild to moderate intellectual disability, autism spectrum disorders, and language disorders. However, a small number of patients carrying SLC6A1 variants do not have seizures and show only mild intellectual disability, suggesting that the expression of the gene is related to the environment and is subject to epigenetic modifications (44, 53). The EEG of patients with the SLC6A1 variant may have some similar features, such as the appearance of irregular spikes, multiple spikes, slow waves over a wide range of 2.5–3.5 Hz, and slowed background activity in the EEG (52). MRI does not show specific results and may show enlarged frontal lobe gaps and cerebellar vermis hypoplasia (44).
Treatment options
A ketogenic diet makes astrocytes metabolically active and increases the amount of glutamine in the brain, allowing for a more efficient conversion of glutamine to GABA, which exerts an antiepileptic effect (54, 55). Seizures can be partially or even completely controlled with valproate monotherapy or in combination with other antiepileptic drugs in patients with epilepsy (44, 56). This may be related to the fact that valproate increases the concentration of the neurotransmitter GABA in the brain and reduces the excitability of neurons (57). Tiagabine is a selective inhibitor of GAT-1 in the brain, and vigabatrin is a GABA amino transaminase, both of which act by increasing the concentration of GABA in the brain (20, 58). Since SLC6A1 can be packaged in an AAV9 vector, AAV-mediated gene therapy shows good therapeutic promise (59). Antisense oligonucleotide treatment also specifically increases GAT-1 protein (60). Nwosu et al. found that patients with SLC6A1 pathogenic variants are heterozygous and that all carry wild-type GAT-1 (61). 4-Phenylbutyrate alone can increase GAT-1 expression and promote positive transport of wild-type GAT-1, thereby increasing GABA reuptake by astrocytes with SLC6A1-mutant neurons (61). Thus, targeting GAT-1 could open new avenues to treat refractory epilepsy.
SLC12A5 and developmental and epileptic encephalopathy type 34
Pathogenesis
The SLC12 family encodes a neutral cation–chloride cotransporter protein involved in transepithelial ion transport, which is associated with the regulation of cell volume and intracellular chloride ion concentration (62). The SLC12A5 gene located on chromosome 20q13.12 encodes mammalian K+-Cl− cotransporter 2 (KCC2), which is expressed only in neurons (63). However, the Na+-K+-2Cl− cotransporter (NKCC1) encoded by the SLC12A2 gene is widely expressed in the brain (64). GABA is the most important inhibitory neurotransmitter in the CNS, whose function depends on KCC2 activity. KCC2 maintains a low intracellular chloride ion concentration, which helps improve GABA and glycine function, whereas NKCC1 has the exact opposite function (Figure 1) (65, 66). The effect of GABA depends on the intracellular chloride concentration: when the intracellular chloride concentration is too high, GABA switches from an inhibitory to an excitatory transmitter (67). According to the current literature, NKCC1 is gradually downregulated as development progresses, while KCC2 expression increases (64). Therefore, during the early stages of development, GABAergic neurotransmission can lead to chloride efflux and membrane depolarization due to high NKCC1 expression levels and high intracellular chloride ion concentrations (67). As the brain develops, the expression of KCC2 gradually increases, causing a shift from GABAergic excitation to inhibition (68). When KCC2 is dysfunctional, the excitatory–inhibitory balance of GABA is disrupted, which may lead to the development of epilepsy (69, 70).
Clinical presentation
Variants of SLC12A5 can lead to developmental epileptic encephalopathy type 34 (DEE34) (OMIM 616645) and idiopathic generalized epilepsy (OMIM 616685). Few cases of DEE34 have been reported, and the phenotype can be consistent with epilepsy of infancy with migrating focal seizures (EIMFS). The clinical features are mainly seizures within the first 6 months of life, developmental delay or regression, microcephaly, and low eye pressure (71, 72). It mainly starts with focal epilepsy and gradually changes to multifocal seizures that are unresponsive to antiseizure medications (71, 72). Patients with EIMFS have EEGs that are typical of migrating seizures, with regular background waves at the end of the episode, followed by slow-wave activity (71, 72). Some patients' MRI may present with myelin retardation, brain atrophy, and corpus callosum thinning (71).
Treatment options
DEE34 in general is treatment-refractory epilepsy, but treatment with a ketogenic diet and potassium bromide has been reported to reduce the seizure frequency in some patients (66, 72). Bumetanide is an NKCC1 inhibitor, and its specific mechanism of action remains unclear. Bumetanide is effective in refractory temporal lobe epilepsy, but some patients have experienced adverse effects, such as loss of appetite, nausea, vomiting, and increased seizure frequency (73). Studies by Kahle et al. have also shown that bumetanide significantly reduces the duration and frequency of seizures in neonates (74). However, in an open-label clinical trial, bumetanide as an add-on treatment to phenobarbital did not improve seizures in neonates with hypoxic ischemic encephalopathy and even increased the risk of hearing damage (75). Therefore, the effectiveness and safety of bumetanide need further study (76, 77). Stimulation of IGF-1 receptors accelerates activation of the KCC2 protein, so IGF-1 can increase KCC2 expression and decrease the NKCC1/KCC2 ratio (78). In neurons, the WNK-SPAK/OSR1 pathway regulates NKCC1 and KCC2, and inhibition of this pathway promotes intracellular chloride ion elimination and GABAergic inhibition, which exerts an antiepileptic effect (79, 80).
SLC13A5 and developmental and epileptic encephalopathy type 25
Pathogenesis
In mammals, the SLC13 superfamily transports metabolic intermediates of the citric acid cycle in a 3Na+/1 substrate stoichiometry ratio (81, 82). SLC13A5 is located on chromosome 17p13.1 and encodes Na+/citric acid transporter (NACT) protein. NACT dysfunction can cause developmental epileptic encephalopathy type 25 (DEE25, OMIM 615905) with dental enamel hypoplasia (83). NACT is primarily expressed on the surface of neurons, where Na+ transport provides electrochemical dynamics, which can transport circulating citric acid into neurons, promote the utilization of circulating citric acid, and provide energy to neuronal cells. The exact mechanism by which variants of SLC13A5 cause epilepsy and neurodevelopmental disorders is still unclear, but researchers have proposed three hypotheses to explain this pathogenesis: (1) Cytoplasmic citric acid deficiency hypothesis: NACT gene variants cause a decrease in the cytoplasmic citric acid content and an insufficient supply of neuronal energy, resulting in epilepsy and delayed brain development (84); (2) interneuron energy hypothesis: in the brain, the metabolic energy consumption of inhibitory neurons is higher than that of excitatory neurons, and SLC13A5 pathogenic variants make neurons less able to restore ionic gradients, leading to an excitatory–inhibitory imbalance (84); and (3) zinc chelation hypothesis: variants of the SLC13A5 gene lead to a relative increase in extracellular citric acid concentration, causing partial zinc and NCTA chelation, which enhances NMDA receptor-mediated synaptic transmission and leads to an imbalance in excitatory inhibition (85).
Clinical presentation
Variants of SLC13A5 are autosomal recessive and characterized clinically by seizures in the first weeks of life; most patients present with focal myoclonic seizures and a few with secondary generalized seizures in the neonatal period (86, 87). Patients may also present with dental dysplasia, ataxia, axial hypotony, motor deficits, and developmental delays (86, 87). Dental hypoplasia is a characteristic presentation and can be used as a screening indicator for key populations. Speech impairment is particularly evident in this group of patients and can manifest as altered speech intonation (86, 87). The prognosis of DEE25 varies widely, with most patients having significantly less frequent seizures as they grow up, but some children with DEE25 die during the neonatal period due to persistent epilepsy and respiratory insufficiency (87, 88). Yang et al. studied the EEG characteristics of 23 patients with SLC13A5 variants and found that the patients in the neonatal period showed frequent epileptiform abnormalities with multifocal spikes and sharps, while the EEG background remained essentially normal after the neonatal period (88). MRI showed characteristic punctate white matter damage and periventricular white matter softening (89). By 6 months of age, the punctate white matter high signal disappeared, followed by glial scarring (89).
Treatment options
The efficacy of a ketogenic diet in these patients is uncertain. The three patients reported by Hardies et al. responded well to a ketogenic diet, but some patients who used a ketogenic diet experienced a deterioration of their condition (87, 90). High concentrations of lithium (Li) can enhance the transport function of NACT, and Li can occupy the four sodium-binding sites of NACT, producing a stimulatory effect, but whether it has an antiepileptic effect remains to be verified (84, 91). Acetazolamide controls seizures in some patients and may increase plasma acidity and decrease intracranial pressure by reducing urinary citrate excretion, but the specific antiepileptic effects of acetazolamide are still unclear (90, 92). Antiseizure medications that act through the GABA system, such as valproic acid and phenobarbital, are the most commonly used drugs to treat this disorder, and they significantly decrease the seizure frequency (90). A patient reported by Pellegrino et al. had a significant reduction in seizure frequency and significant improvements in motor skills and speech comprehension after treatment with a combination of valproic acid and acetazolamide (93).
SLC25A12 and developmental and epileptic encephalopathy type 39
Pathogenesis
SLC25A12 is located on chromosome 2q31.1 and encodes a mitochondrial aspartate/glutamate transporter (AGC) (94). SLC25A12 is expressed mainly in neurons and skeletal muscles and is a major component of the malate/aspartate shuttle (Figure 2) (94, 95). It allows aspartate to be exchanged for glutamate and protons in the mitochondria and transfers their cytosolic reducing equivalents into mitochondria (95). When AGC is dysfunctional, aspartate cannot be transported into the cytoplasm, resulting in reduced production of N-acetyl glutamate in the cytoplasm, which leads to poor myelin production (95, 96). However, the exact mechanism by which variants of SLC25A12 cause epilepsy remains unclear. It is speculated that it may be related to intracellular glutamate accumulation and cellular damage.
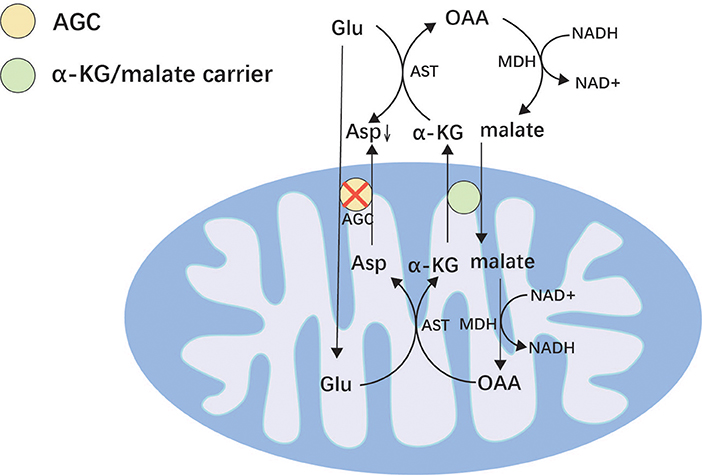
Figure 2. Malate/aspartate shuttle. Mitochondria are impermeable to NADH and transport NADH into the mitochondria via the malate/aspartate form to supply the respiratory chain to generate ATP. AGC can reverse the transport of glutamate and aspartate and plays an important role in the malate/aspartate shuttle. Asp, aspartic acid; OAA, oxaloacetic acid; AST, aspartate aminotransferase; MDH, malate dehydrogenase; α-KG, α-ketoglutaric acid; AGC, mitochondrial aspartate/glutamate transporter; Glu, glutamic.
Clinical presentation
SLC25A12 variants are associated with developmental epileptic encephalopathy type 39 (DEE39, OMIM 612949), which is autosomal recessive. In previous studies, four patients have been reported, and their main clinical features are early-onset epilepsy, psychomotor retardation, hypotonia, short stature, and microcephaly (96–98). Brain MRI shows a characteristic myelin developmental disorder and brain atrophy (97). Proton magnetic resonance wave analysis may show a reduced N-acetyl aspartate peak, which indicates neuronal damage (96).
Treatment options
A ketogenic diet significantly controls seizures in AGC1 deficiency, suggesting that a ketogenic diet may be linked to the pathogenesis of AGC1 deficiency. (1) A ketogenic diet provides acetyl coenzyme A directly to the mitochondrial tricarboxylic acid cycle, ameliorating neuronal energy deficiency (99). (2) A ketogenic diet reduces NADH production from cellular glycolysis, and malate dehydrogenase is an NADH-dependent dehydrogenase, so oxaloacetate is converted into aspartate, rather than malate, promoting neuronal use of aspartate and N-acetyl glutamate for myelin production (100). (3) β-Hydroxybutyrate has a protective effect on neurons and restores mitochondrial respiration, increasing cytoplasmic aspartate and N-acetyl glutamate, which can promote myelin formation (101). In previous studies, two patients with SLC25A12 pathogenic variants had a dramatic decrease in seizure frequency and some improvement in psychomotor development after adopting a ketogenic diet (99, 100).
SLC25A22 and developmental and epileptic encephalopathy type 3
Pathogenesis
The SLC25 gene family encodes mitochondrial carriers that transport various metabolites through the inner mitochondrial membrane and hence the name mitochondrial carrier family (102). SLC25A22 is one of the SLC25 gene isoforms located on chromosome 11p15.5, encoding the mitochondrial glutamate carrier 1(GC1) (103, 104). This protein is expressed only in the brain, mainly on astrocytes, and is an essential channel for glutamate entry into mitochondria (105, 106). With variants of SLC25A22, mitochondrial transport of glutamate is impaired. NADPH cannot be transferred to the respiratory chain, resulting in an inadequate supply of intracellular energy and a significant accumulation of glutamate in the cytoplasm, which is released into the synaptic gap, leading to neuronal excitation (105, 107). In addition, the SLC25A22 variant also leads to an impaired mitochondrial pyrroline-5-carboxylic acid (P5C) cycle, which increases P5C in mitochondria. P5C is eventually further metabolized to glutamate, ornithine, and arginine (108). Therefore, plasma metabolic screening may show elevated levels of these amino acids (107). The excess glutamate produced by the impaired P5C cycle in mitochondria leads to inactivation of the urea cycle, resulting in the production of large amounts of citric acid, and the excess NADPH and citric acid together promote lipid synthesis (Figure 3) (107). Thus, muscle biopsies from patients with SLC25A22 show a large number of fat vacuoles within the muscle fibers.
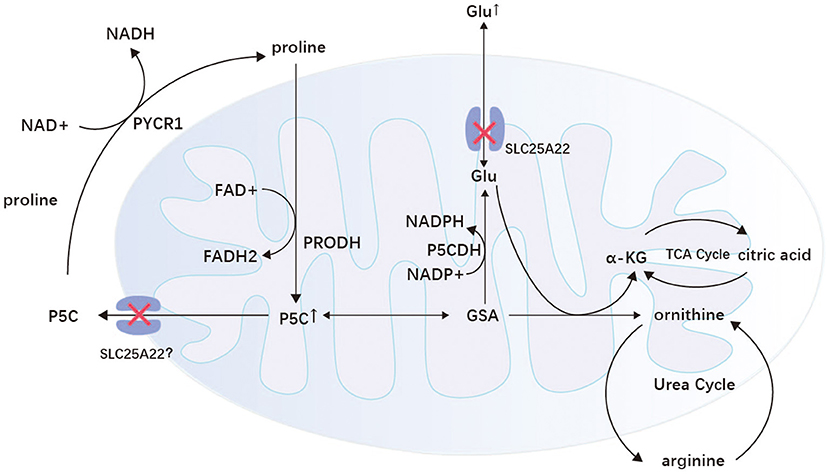
Figure 3. Proline/P5C cycle and P5C metabolism in mitochondria. GC1 is found on the mitochondria of astrocytes and transports glutamate in both directions. When the SLC25A22 gene is mutated, glutamate transport and the mitochondrial P5C cycle are impaired, leading to abnormal cell excitation and metabolism. P5C, delta (1)-pyrroline-5-carboxylate; GSA, glutamate semialdehyde; α-KG, α-ketoglutaric acid; Glu, glutamic acid; P5CDH, P5C dehydrogenase; PRODH, proline dehydrogenase/proline oxidase; PYCR1, pyrroline-5-carboxylate reductase 1; TCA cycle, tricarboxylic acid cycle.
Clinical presentation
Variants of SLC25A22 can cause developmental and epileptic encephalopathies type 3 (DEE3) (OMIM 609304), and their clinical manifestations may be consistent with early infantile myoclonic epilepsy, migratory focal epilepsy in infancy, and epileptic spasms (103, 107, 109). The main clinical features are neonatal seizures, developmental delay, low eye pressure, abnormal electroretinogram, microcephaly, hypotonia, and active tendon reflexes. The seizures mostly appear within the first few months of life and are mainly focal and take various forms, including myoclonic, tonic, and tonic–clonic seizures. These patients mostly have a severe epilepsy phenotype and a poor prognosis, but a case with a milder phenotype was reported in which the seizures appeared only at the age of 7 years (107). Molinari et al. (103) reported a case of early myoclonic epilepsy in an infant with autosomal recessive inheritance, characterized by an EEG with a burst suppression pattern and a general slowing of background waves (110, 111). However, in infants with migratory focal epilepsy, the characteristic EEG is epilepsy of multifocal origin (109). Brain MRI examinations of patients with DEE3 always show developmental or structural brain abnormalities, such as varying degrees of brain atrophy, delayed myelin formation, and thinning of the corpus callosum (106, 108).
Treatment options
The disease has a poor prognosis with high mortality. Epilepsy caused by the SLC25A22 pathogenic variant is in most cases unresponsive to antiseizure medications and adrenocorticotropic hormones (ACTHs). A 12-year-old girl with seizures starting on the 15th day of life was treated with valproic acid, vigabatrin, levetiracetam, phenobarbital, ethosuximide, clobazam, and ACTH with poor results (112). Giacominia et al. reported a case of a SLC25A22 heterozygous mutant whose seizures did not respond to levetiracetam and pyridoxine, but the addition of phenobarbital and topiramate resulted in good control of the seizures (110). In conclusion, there are few specific approaches to treat epilepsy caused by the SLC25A22 pathogenic variant, and new targeted therapies for this gene are urgently needed.
SLC35A2 and developmental and epileptic encephalopathy type 22
Pathogenesis
SLC35A2, located on chromosome Xp11.23, encodes UDP-galactose transporter (UGT), a nucleotide–sugar transporter protein (113, 114). Under physiological conditions, complex polysaccharides are attached to proteins by N-glycosylation to ensure protein functional and structural stability. UGT transports UDP-galactose into the Golgi lumen as a substrate for protein galactosylation. Variants of SLC35A2 lead to a decrease in galactose within the Golgi and a deficiency of galactose and sialic acid in the N-glycan branch, disrupting the glycosylation process and resulting in congenital disorders of glycosylation (SLC35A2-CDG) (115, 116).
Clinical presentation
Kodera et al. first demonstrated that variants of SLC35A2 are one of the genetic causes of DEE, which is now classified as developmental and epileptic encephalopathy type 22 (DEE22, OMIM 300896) (117). N-Glycosylation defects caused by SLC35A2 variants may alter the neurotransmission and excitability of neural circuits, leading to seizures (118). Both X-chromosome dominant inheritance and somatic mosaic mutations can lead to SLC35A2-CDG, which is an age-dependent seizure disorder. All patients present with seizures within the first 6 months of life, mainly infantile spasms (115, 117–120). Notably, over time, the seizure form of epilepsy can change. SLC35A2-CDG cases mostly have severe intellectual disability, hypotonia, and delayed language development, with some patients unable to say meaningful words even by age 12 years (117). These patients mostly have dysmorphic features such as coarse facial features, sunken nipples, shortened limbs, skeletal defects, hip subluxation, and scoliosis (119, 121, 122). Some patients also have tetralogy of Fallot, atrial septal defect, ventricular septal defect, and a small amount of pericardial effusion (119). More than half of the patients have ocular abnormalities such as nystagmus (which may stop with seizure control), severe hypotony, iris heterochromia sign, and retinitis pigmentosa (121–123). In addition, patients may also develop systemic manifestations such as coagulation disorders, immune dysfunction, cardiac dysfunction, and liver failure (124). The two cases of somatic mosaic variants reported thus far were both male patients and both showed mental retardation, developmental delay, shortened limbs, microcephaly, small cerebellum, and cerebral hypoplasia/atrophy compared with X-dominant patients (120). Patients with somatic mosaic variants can be seizure-free and have acute nephrotic syndrome, coagulation disorders, gastroesophageal reflux, esophagitis, and duodenal perforation (120).
A majority of patients with SLC35A2-CDG have an EEG with hypsarrhythmia, with a small percentage having a focal multispine wave, a diffuse spike or sharp and slow-wave complex, and a burst suppression (117, 119, 122). Common MRI manifestations in patients with SLC35A2-CDG include corpus callosum thinning, brain atrophy, delayed myelin formation, and a multifocal inhomogeneous abnormal patchy white matter high signal (121, 124). Abnormal glycosylation of serum transferrin is detectably altered in the presence of glycosylation dysfunction. However, the altered transferrin in patients may return to normal after 2–3 years of age (119, 120).
Treatment options
Patients with SLC35A2-CDG with standard treatment of epileptic spasms have temporary seizure control but are prone to recurrence, and most recurrences are unresponsive to antiepileptic drugs and ketogenic diet therapy (117, 119, 122). Only one patient with seizure recurrence had complete seizure control with the same dose of ACTH applied again (117, 122). A patient reported by Yates et al. had seizure control with a ketogenic diet combined with vigabatrin and nitrazepam (119). UGT dysfunction can lead to reduced galactose levels in the Golgi lumen and disrupt the glycosylation process, so galactose supplementation can increase the intracytoplasmic UDP-galactose concentration. A total of 11 patients with SLC35A2-CDG were treated with galactose, which was well tolerated and led to a continuous improvement in clinical symptoms and a significant reduction in seizure frequency (121, 123). Only one patient did not achieve any clinical improvement with galactose supplementation (123).
SLC38A3 and developmental and epileptic encephalopathy 102
Pathogenesis
The SLC38 family is mainly expressed in actively growing cells throughout the body and is responsible for mediating the transport of sodium-coupled neutral amino acids (125). SLC38A3 is located on chromosome 3p21.31 and encodes a sodium-coupled neutral amino acid transporter protein (SNAT3), which is mainly responsible for the transport of asparagine, glutamine, and histidine (126). SLC38A3 is most strongly expressed in the liver, skeletal muscle, and pancreas, followed by the kidney, brain, and heart (127). Among them, SLC38A3 is predominantly expressed in astrocytes and endothelial cells in the brain (128). In the brain, glutamate and GABA in the synaptic gap enter astrocytes via EAAT2 and GABA transporter 3 (GAT3), respectively, where they are converted into glutamine. After glutamine is released into the extracellular fluid via SNAT3 and SNAT5, it enters neurons via SNAT1 and SNAT2 to be metabolized into glutamate and GABA, which are used to replenish depleted neurotransmitters (Figure 1) (126). This metabolic process is known as the GABA–glutamate–glutamine cycle. SNAT3, which is expressed in blood–brain barrier endothelial cells, is also responsible for glutamine transport out of the brain (129). Therefore, variants of SLC38A3 can lead to increased glutamine levels in the brain, which may reduce the levels of the inhibitory neurotransmitter GABA and the excitatory neurotransmitter glutamate. However, the effect of SLC38A3 variants on neurons is not clear, and the mechanism leading to DEE needs to be further investigated.
Clinical presentation
The disease caused by SLC38A3 pathogenic variants is currently classified as developmental and epileptic encephalopathy type 102 (OMIM 619881) and was first reported by Marafi et al. (130). Currently, 10 patients were reported with DEE102 from six families, five of which had consanguineous marriages (130). All 10 patients had axial hypotonia, absent speech, and global developmental delay/mental retardation (130). Most patients also had epilepsy, peripheral hypertonia, constipation, and visual impairment (129). Visual impairment includes cortical blindness and cone-rod dystrophy (129). Epilepsy typically presents within the first 15 months of life and manifests primarily as generalized tonic–clonic, followed by tonic, gelastic, focal status epilepticus, and generalized myoclonic–tonic seizures (129). Patients in this group may also present with dysphagia, movement disorder, hyperreflexia, hepatomegaly, gastroesophageal reflex disease, and atrial septal defect. A total of six patients had background slowing of the EEG and epileptiform abnormalities, but no characteristic EEG manifestations have been found (129). MRI is mainly characterized by callosal abnormalities, dysmyelination, and brain atrophy (129). Rarely, thin brainstem, generous extra-axial CSF spaces, dolichocephaly, mild posterior plagiocephaly, and mild superior vermian hypoplasia are present (129). In one patient, cerebrospinal fluid N-acetyl glutamine and N-acetyl aspartate levels were elevated. His brother, who was also affected, had significantly lower plasma glutamine and N-acetylglutamine levels (129). This may be related to the fact that SNAT3 is expressed in the endothelial cells of the blood–brain barrier and is responsible for the transfer of glutamine.
Treatment options
SLC38A3 variant-associated epilepsy is typically unresponsive to multiple antiepileptic drugs. The ketogenic diet has been reported to be only partially effective in patients with DEE102 (130). Vigabatrin is the most effective treatment for reducing the frequency of seizures 3-fold (130). In addition, patients with DEE102 respond positively to benzodiazepines (130). In two patients, not only did the seizures improve with benzodiazepines but also their neurological function remained at baseline levels (130). This is probably because benzodiazepines act on GABAA receptors and promote GABAergic inhibition (131). Vigabatrin irreversibly inhibits GABA transaminase and increases GABA concentrations in the CNS (132). Thus, benzodiazepines and aminocaproic acid partially restored the reduction in GABA due to SNAT3 dysfunction.
Discussion
In this article, we first proposed the concept of transporter protein disease. We described the nine DEE phenotypes in detail in terms of pathogenesis, clinical features, ancillary tests, and precise treatment (Table 1). All nine DEEs have early-onset refractory epilepsy and global developmental delay, but they can be distinguished based on their characteristic presentation. Targeting the pathogenesis of each specific disease may lead to the development of new antiepileptic drugs to facilitate the treatment of refractory epilepsy. For example, enhanced EAAT2 expression increases glutamate uptake by glial cells and protects neurons from excitatory neurotransmitter damage (12, 18). Recombinant forms of interleukin 1 receptor antagonists, LDN/OSU-0212320, ceftriaxone, and Parawixin10 all act by enhancing EAAT2 expression (18–20). Among them, ceftriaxone has been applied to one clinical patient, but no significant efficacy was found (16, 24, 25). Therefore, more clinical studies are needed to validate the efficacy of ceftriaxone in SLC1A2-associated epilepsy.
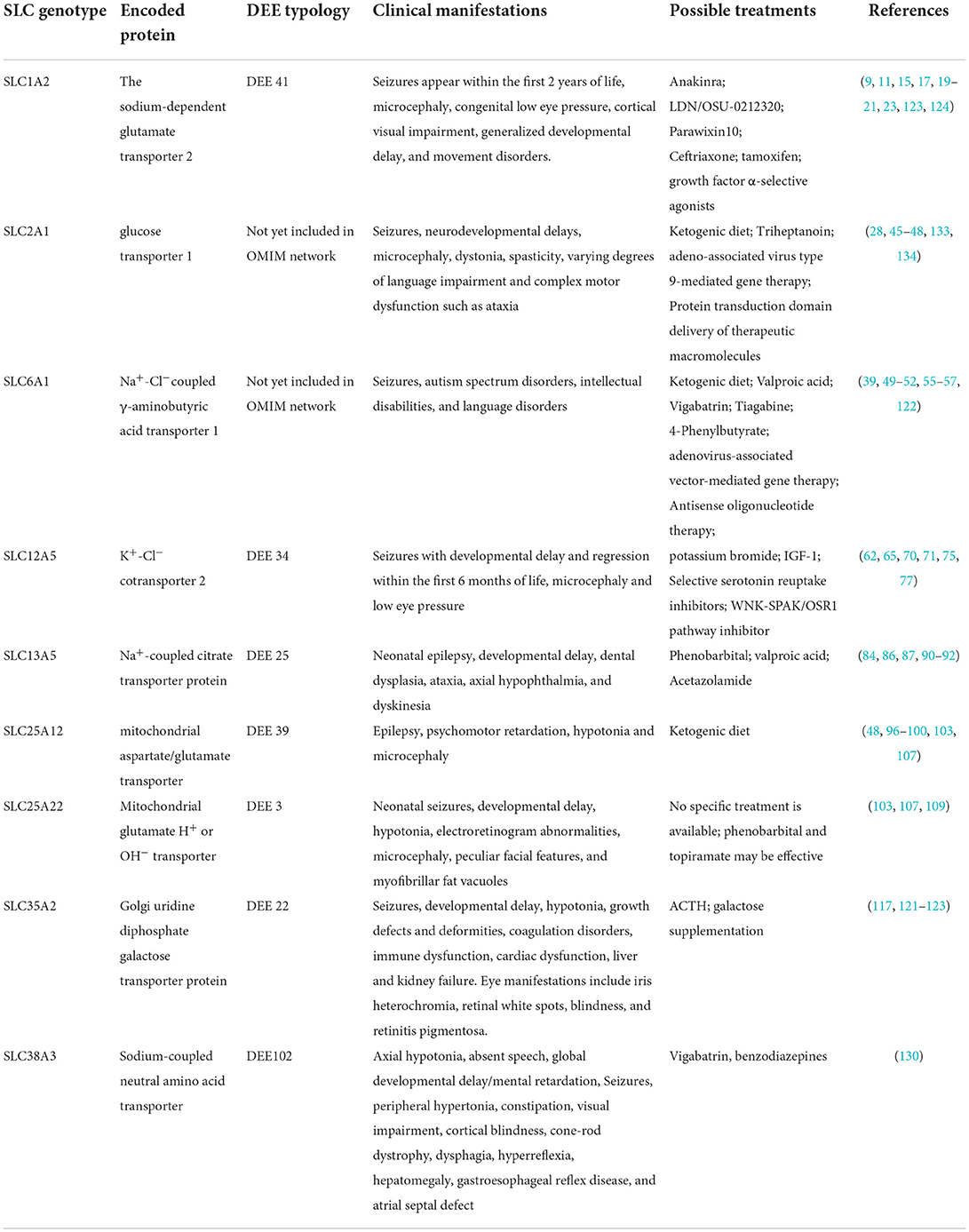
Table 1. Summary of SLC genotypes associated with DEE and their clinical features.
GLUT1DS1 is typical of DEE, and the degree of clinical symptoms and EEG abnormalities fluctuates with fasting (32). Low cerebrospinal fluid glucose is present in almost all patients with GLUT1DS. The use of alternative energy sources, such as a ketogenic diet, medium-chain triglyceride oil, and triheptanoin, can be effective in improving energy deficiency in the brain (35, 45–47). Further research is still needed to determine the optimal age to start a ketogenic diet. In addition, AAV9-mediated gene replacement therapy is a current research hot spot that has shown positive results in animal models, with the expectation of translating it into clinical treatment for GLUT1DS (50).
SLC6A1-associated epilepsy better responds to drugs that mainly act on increasing GABA concentrations in the brain, such as a ketogenic diet, valproate, tiagabine, and vigabatrin (57, 58, 135). A recent study revealed that 4-phenylbutyrate promotes wild-type GAT-1 transport and increases GABA reuptake by astrocytes and neurons, suggesting the opportunity to promote wild protein transport for therapeutic purposes, and this concept is being tested in a pilot clinical trial (61). Antisense oligonucleotide therapy and AAV9-mediated gene therapy are also potential treatments (59, 60).
DEE34 is predominantly a malignant migratory focal epilepsy in infants (71, 72). The current drugs for DEE34 mainly target NKCC1 and KCC2. Animal experiments have shown that inhibition of both the WNK-SPAK/OSR1 pathway and stimulation of IGF-2 reduce the intracellular chloride concentration and promote the inhibitory function of GABA (78–80). The efficacy of bumetanide as an antiepileptic agent needs to be further validated; it is effective in temporal lobe epilepsy and neonatal epilepsy but does not improve seizures due to neonatal ischemic-hypoxic encephalopathy (73–75).
Patients with SLC13A5 commonly present with seizures in the first week of life and have characteristic dental hypoplasia and speech changes (86, 87). MRI has characteristic punctate white matter damage (89). There is near normalization of the EEG background after the neonatal period in patients with SLC13A5 mutations (88). However, further studies are needed to compare the differences between the EEG of patients with well-controlled epilepsy and the normal EEG background of patients with persistent neurocognitive dysfunction (88). Acetazolamide is only effective in some patients, and more studies are expected to discover the specific antiepileptic mechanism of acetazolamide for early application (90, 92).
SLC25A12 is a severe epileptic encephalopathy with characteristic myelin developmental disorders and a reduced brain N-acetylaspartate content (96, 97). Patients with SLC25A12 variants mostly have a normal respiratory chain and can use a ketogenic diet to bypass their abnormal metabolism, protect their neurons, and promote myelin formation (99–101).
Due to metabolic impairment, patients with SLC25A22 can exhibit a large number of fat vacuoles within the muscle fibers and increased plasma ornithine, arginine, and glutamate levels (107). The use of multiple antiepileptic drugs was ineffective in 69% of patients. Only one patient responded well to phenobarbital and topiramate (110). There is a lack of effective treatment for patients with the SLC25A22 variant, so further research into its pathogenesis is necessary to discover targeted treatment options.
Patients with SLC35A2-CDG have seizure spasms and arrhythmias as their main manifestations, along with severe intellectual disability and some systemic manifestations (119). It is unclear whether congenital heart disease is part of this syndrome. Almost all patients with SLC35A2-CDG return to normal transferrin glycosylation after 3 years of age; therefore, genetic testing should be performed for early diagnosis in patients with a phenotype consistent with SLC35A2-CDG, even if ferritin glycosylation is normal (119, 120). Their seizures can be temporarily controlled with standard therapy for epileptic spasms but are prone to recurrence (117, 119, 122). However, researchers have proposed supplemental galactose therapy for the target of N-glycosylation deficiency, showing good therapeutic effects in current patients (121, 123). A long-term follow-up is still needed to observe its long-term effects.
Patients with SLC38A3 variants mainly present with axial hypotonia, absent speech, global developmental delay/mental retardation, epilepsy, and visual impairment (130). Decreased plasma glutamine and N-acetylglutamine levels and increased cerebrospinal fluid N-acetylglutamine levels may serve as biomarkers in patients with DEE102 (130). However, not all patients exhibit this metabolic abnormality, so more plasma and cerebrospinal fluid metabolic profiles of patients with DEE102 need to be examined to determine their metabolic changes and to help clarify the mechanism of the disease onset (130). Drugs do not completely control their seizures. Therefore, the underlying mechanism of how SLC38A3 variants cause DEE needs to be clarified so that targeted treatments can be developed (130).
In conclusion, mutations in nine SLC genes are known to cause DEE, and numerous SLC gene variants are thought to be associated with epilepsy but have yet to be fully validated. SLC is involved in the transmembrane transport of various substances and is a therapeutic target for many diseases. As a result, a thorough investigation of SLC and its pathogenesis could lead to the identification of new precise therapeutic targets, which are critical for developing new approaches to treating refractory epilepsy.
Author contributions
YG is the main author of the review and completed the collection and analysis of the relevant literature, and the writing of the first draft of the manuscript. ZW, CL, GL, and YF participated in the analysis and organization of the literature. YD was the conceptualizer and the person in charge of the project and directed the writing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the National Key R&D Program of China, Precision medicine program—Cohort study on nervous system disease [Grant number: 2017YFC0907700 (2017–2021)], and National Natural Science Foundation of China (Grant number: 81871007).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia. (2010) 51:676–85. doi: 10.1111/j.1528-1167.2010.02522.x
2. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
3. Perland E, Fredriksson R. Classification systems of secondary active transporters. Trends Pharmacol Sci. (2017) 38:305–15. doi: 10.1016/j.tips.2016.11.008
4. Pizzagalli MD, Bensimon A, Superti-Furga G. A guide to plasma membrane solute carrier proteins. FEBS J. (2021) 288:2784–835. doi: 10.1111/febs.15531
5. Povey S, Lovering R, Bruford E, Wright M, Lush M, Wain H. The HUGO gene nomenclature committee (HGNC). Hum Genet. (2001) 109:678–80. doi: 10.1007/s00439-001-0615-0
6. Liu X, SLC. Family Transporters. Adv Exp Med Biol. (2019) 1141:101–202. doi: 10.1007/978-981-13-7647-4_3
7. Ayka A, Sehirli AÖ. The role of the SLC transporters protein in the neurodegenerative disorders. Clin Psychopharmacol Neurosci. (2020) 18:174–87. doi: 10.9758/cpn.2020.18.2.174
8. Hu C, Tao L, Cao X, Chen L. The solute carrier transporters and the brain: Physiological and pharmacological implications. Asian J Pharmac Sci. (2020) 15:131–44. doi: 10.1016/j.ajps.2019.09.002
9. Kanai Y, Clémençon Clémençon B, Simonin A, Leuenberger M, Lochner M, Weisstanner M, et al. The SLC1 high-affinity glutamate and neutral amino acid transporter family. Mol Aspects Med. (2013) 34:108–20. doi: 10.1016/j.mam.2013.01.001
10. Li X, Francke U. Assignment of the gene SLC1A2 coding for the human glutamate transporter EAAT2 to human chromosome 11 bands p13-p12. Cytogenet Cell Genet. (1995) 71:212–3. doi: 10.1159/000134111
11. Hediger MA, Clémençon Clémençon B, Burrier RE, Bruford EA. The ABCs of membrane transporters in health and disease (SLC series): Introduction. Mol Aspects Med. (2013) 34:95–107. doi: 10.1016/j.mam.2012.12.009
12. Colton CK, Kong Q, Lai L, Zhu MX, Seyb KI, Cuny GD, et al. Identification of translational activators of glial glutamate transporter EAAT2 through cell-based high-throughput screening: an approach to prevent excitotoxicity. J Biomol Screen. (2010) 15:653–62. doi: 10.1177/1087057110370998
13. Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. (1997) 276:1699–702. doi: 10.1126/science.276.5319.1699
14. Epi4K Consortium. De Novo Mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am J Hum Genet. (2016) 99:287–98. doi: 10.1016/j.ajhg.2016.06.003
15. Wagner M, Gusic M, Günthner R, Alhaddad B, Kovacs-Nagy R, Makowski C, et al. Biallelic mutations in SLC1A2; an additional mode of inheritance for SLC1A2-related epilepsy. Neuropediatrics. (2018) 49:059–62. doi: 10.1055/s-0037-1606370
16. Stergachis AB, Pujol-Giménez J, Gyimesi G, Fuster D, Albano G, Troxler M, et al. Recurrent SLC1A2 variants cause epilepsy via a dominant negative mechanism. Ann Neurol. (2019) 85:921–6. doi: 10.1002/ana.25477
17. Mir A, Almudhry M, Alghamdi F, Albaradie R, Ibrahim M, Aldurayhim F, et al. SLC gene mutations and pediatric neurological disorders: diverse clinical phenotypes in a Saudi Arabian population. Hum Genet. (2022) 141:81–99. doi: 10.1007/s00439-021-02404-x
18. Green JL, dos Santos WF, Fontana ACK. Role of glutamate excitotoxicity and glutamate transporter EAAT2 in epilepsy: Opportunities for novel therapeutics development. Biochem Pharmacol. (2021) 193:114786. doi: 10.1016/j.bcp.2021.114786
19. Dyomina AV, Zubareva OE, Smolensky IV, Vasilev DS, Zakharova MV, Kovalenko AA, et al. Anakinra reduces epileptogenesis, provides neuroprotection, and attenuates behavioral impairments in rats in the lithium–pilocarpine model of epilepsy. Pharmaceuticals. (2020) 13:340. doi: 10.3390/ph13110340
20. Kong Q, Chang L-C, Takahashi K, Liu Q, Schulte DA, Lai L, et al. Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J Clin Invest. (2014) 124:1255–67. doi: 10.1172/JCI66163
21. Lee E-SY, Sidoryk M, Jiang H, Yin Z, Aschner M. Estrogen and tamoxifen reverse manganese-induced glutamate transporter impairment in astrocytes. J Neurochem. (2009) 110:530–44. doi: 10.1111/j.1471-4159.2009.06105.x
22. Figiel M, Maucher T, Rozyczka J, Bayatti N, Engele J. Regulation of glial glutamate transporter expression by growth factors. Exp Neurol. (2003) 183:124–35. doi: 10.1016/S0014-4886(03)00134-1
23. Zeng L-H, Bero AW, Zhang B, Holtzman DM, Wong M. Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of Tuberous Sclerosis Complex. Neurobiol Dis. (2010) 37:764–71. doi: 10.1016/j.nbd.2009.12.020
24. Ramandi D, Elahdadi Salmani M, Moghimi A, Lashkarbolouki T, Fereidoni M. Pharmacological upregulation of GLT-1 alleviates the cognitive impairments in the animal model of temporal lobe epilepsy. PLoS ONE. (2021) 16:e0246068. doi: 10.1371/journal.pone.0246068
25. Li H-H, Lin P-J, Wang W-H, Tseng L-H, Tung H, Liu W-Y, et al. Treatment effects of the combination of ceftriaxone and valproic acid on neuronal and behavioural functions in a rat model of epilepsy. Exp Physiol. (2021) 106:1814–28. doi: 10.1113/EP089624
26. Uldry M, Thorens B. The SLC2 family of facilitated hexose and polyol transporters. Pflugers Archiv Eur J Physiol. (2004) 447:480–9. doi: 10.1007/s00424-003-1085-0
27. Tang M, Gao G, Rueda CB Yu H, Thibodeaux DN, Awano T, et al. Brain microvasculature defects and Glut1 deficiency syndrome averted by early repletion of the glucose transporter-1 protein. Nat Commun. (2017) 8:14152. doi: 10.1038/ncomms14152
28. Wang D, Pascual JM, De Vivo D. Glucose transporter type 1 deficiency syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Gripp KW, et al, editors. GeneReviews®. Seattle, WA: University of Washington. (1993).
29. Rotstein M, Engelstad K, Yang H, Wang D, Levy B, Chung WK, et al. Glut1 deficiency: Inheritance pattern determined by haploinsufficiency. Ann Neurol. (2010) 68:955–8. doi: 10.1002/ana.22088
30. Klepper J, Scheffer H, Elsaid MF, Kamsteeg E-J, Leferink M, Ben-Omran T. Autosomal recessive inheritance of GLUT1 deficiency syndrome. Neuropediatrics. (2009) 40:207–10. doi: 10.1055/s-0030-1248264
31. Pérez-Dueñas B, Prior C, Ma Q, Fernández-Álvarez E, Setoain X, Artuch R, et al. Childhood chorea with cerebral hypotrophy: a treatable GLUT1 energy failure syndrome. Arch Neurol. (2009) 66:1410–4. doi: 10.1001/archneurol.2009.236
32. Weber YG, Kamm C, Suls A, Kempfle J, Kotschet K, Schule R, et al. Paroxysmal choreoathetosis/spasticity (DYT9) is caused by a GLUT1 defect. Neurology. (2011) 77:959–64. doi: 10.1212/WNL.0b013e31822e0479
33. Suls A, Mullen SA, Weber YG, Verhaert K, Ceulemans B, Guerrini R, et al. Early-onset absence epilepsy caused by mutations in the glucose transporter GLUT1. Ann Neurol. (2009) 66:415–9. doi: 10.1002/ana.21724
34. Suls A, Dedeken P, Goffin K, Van Esch H, Dupont P, Cassiman D, et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain. (2008) 131:1831–44. doi: 10.1093/brain/awn113
35. Joshi C, Greenberg CR, De Vivo D. Dong Wang, Chan-Lui W, Booth FA. GLUT1 deficiency without epilepsy: yet another case. J Child Neurol. (2008) 23:832–4. doi: 10.1177/0883073808314896
36. Schneider SA, Paisan-Ruiz C, Garcia-Gorostiaga I, Quinn NP, Weber YG, Lerche H, et al. GLUT1 gene mutations cause sporadic paroxysmal exercise-induced dyskinesias. Mov Disord. (2009) 24:1684–8. doi: 10.1002/mds.22507
37. Ito Y, Gertsen E, Oguni H, Nakayama T, Matsuo M, Funatsuka M, et al. Clinical presentation, EEG studies, and novel mutations in two cases of GLUT1 deficiency syndrome in Japan. Brain Develop. (2005) 27:311–7. doi: 10.1016/j.braindev.2004.09.010
38. Von Moers A, Brockmann K, Wang D, Korenke CG, Huppke P, De Vivo DC, et al. EEG features of glut-1 deficiency syndrome. Epilepsia. (2002) 43:941–5. doi: 10.1046/j.1528-1157.2002.50401.x
39. Levy B, Wang D, Ullner PM, Engelstad K, Yang H, Nahum O, et al. Uncovering microdeletions in patients with severe Glut-1 deficiency syndrome using SNP oligonucleotide microarray analysis. Mol Genet Metab. (2010) 100:129–35. doi: 10.1016/j.ymgme.2010.03.007
40. Akman CI, Provenzano F, Wang D, Engelstad K, Hinton V, Yu J, et al. Topography of brain glucose hypometabolism and epileptic network in glucose transporter 1 deficiency. Epilepsy Res. (2015) 110:206–15. doi: 10.1016/j.eplepsyres.2014.11.007
41. Leen WG, Wevers RA, Kamsteeg E-J, Scheffer H, Verbeek MM, Willemsen MA. Cerebrospinal fluid analysis in the workup of GLUT1 deficiency syndrome: a systematic review. JAMA Neurol. (2013) 70:1440. doi: 10.1001/jamaneurol.2013.3090
42. Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lorincz ML, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. (2009) 15:1392–8. doi: 10.1038/nm.2058
43. Walker MC, Kullmann DM. Tonic GABAA receptor-mediated signaling in epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies 4th ed. Bethesda, MD: National Center for Biotechnology Information. (2012).
44. Johannesen KM, Gardella E, Linnankivi T, Courage C, de Saint Martin A, Lehesjoki A-E, et al. Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia. (2018) 59:389–402. doi: 10.1111/epi.13986
45. Alter AS, Engelstad K, Hinton VJ, Montes J, Pearson TS, Akman CI, et al. Long-Term Clinical Course of Glut1 Deficiency Syndrome. J Child Neurol. (2015) 30:160–9. doi: 10.1177/0883073814531822
46. Schwantje M, Verhagen LM, Hasselt PM, Fuchs SA. Glucose transporter type 1 deficiency syndrome and the ketogenic diet. J Inher Metab Dis. (2020) 43:216–22. doi: 10.1002/jimd.12175
47. Wei Z, Wang L, Deng Y. Treatment of myoclonic-atonic epilepsy caused by SLC2A1 de novo mutation with ketogenic diet: A case report. Medicine (Baltimore). (2019) 98:e15428. doi: 10.1097/MD.0000000000015428
48. Pascual JM, Liu P, Mao D, Kelly DI, Hernandez A, Sheng M, et al. Triheptanoin for glucose transporter type I deficiency (G1D): modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol. (2014) 71:1255. doi: 10.1001/jamaneurol.2014.1584
49. Akman CI, De Vivo DC. Glucose transporter type 1 deficiency syndrome. In: Pearl PL, editor Inherited Metabolic Epilepsies. New York, NY: Springer Publishing Company. (2017).
50. Tang M, Park SH, De Vivo DC, Monani UR. Therapeutic strategies for glucose transporter 1 deficiency syndrome. Ann Clin Transl Neurol. (2019) 6:1923–32. doi: 10.1002/acn3.50881
51. van den Berg A, Dowdy SF. Protein transduction domain delivery of therapeutic macromolecules. Curr Opin Biotechnol. (2011) 22:888–93. doi: 10.1016/j.copbio.2011.03.008
52. Bröer S, Gether U. The solute carrier 6 family of transporters: The solute carrier family 6. Br J Pharmacol. (2012) 167:256–78. doi: 10.1111/j.1476-5381.2012.01975.x
53. Carvill GL, McMahon JM, Schneider A, Zemel M, Myers CT, Saykally J, et al. Mutations in the GABA transporter SLC6A1 cause epilepsy with myoclonic-atonic seizures. The American J Human Genetics. (2015) 96:808–15. doi: 10.1016/j.ajhg.2015.02.016
54. Yudkoff M, Daikhin Y, Horyn O, Nissim I, Nissim I. Ketosis and brain handling of glutamate, glutamine, and GABA. Epilepsia. (2008) 49:73–5. doi: 10.1111/j.1528-1167.2008.01841.x
55. Palmer S, Towne MC, Pearl PL, Pelletier RC, Genetti CA, Shi J, et al. SLC6A1 mutation and ketogenic diet in epilepsy with myoclonic-atonic seizures. Pediatr Neurol. (2016) 64:77–9. doi: 10.1016/j.pediatrneurol.2016.07.012
56. Devries S, Mulder M, Charron JG, Prokop JW, Mark PR. SLC6A1 G443D associated with developmental delay and epilepsy. Cold Spring Harb Mol Case Stud. (2020) 6:a005371. doi: 10.1101/mcs.a005371
57. Chateauvieux S, Morceau F, Dicato M, Diederich M. Molecular and therapeutic potential and toxicity of valproic acid. J Biomed Biotechnol. (2010) 2010:479364. doi: 10.1155/2010/479364
58. Meldrum BS, Chapman AG. Basic mechanisms of gabitril (Tiagabine) and future potential developments. Epilepsia. (1999) 40:S2–6. doi: 10.1111/j.1528-1157.1999.tb02087.x
59. Ozlu C, Bailey RM, Sinnett S, Goodspeed KD. Gene transfer therapy for neurodevelopmental disorders. Dev Neurosci. (2021) 43:230–40. doi: 10.1159/000515434
60. Lim KH, Han Z, Jeon HY, Kach J, Jing E, Weyn-Vanhentenryck S, et al. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat Commun. (2020) 11:3501. doi: 10.1038/s41467-020-17093-9
61. Nwosu G, Mermer F, Flamm C, Poliquin S, Shen W, Rigsby K, et al. 4-Phenylbutyrate restored γ-aminobutyric acid uptake and reduced seizures in SLC6A1 patient variant-bearing cell and mouse models. Brain Commun. (2022) 4:fcac144. doi: 10.1093/braincomms/fcac144
62. Gagnon KB, Delpire E. Physiology of SLC12 transporters: lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am J Physiol. (2013) 304:C693–714. doi: 10.1152/ajpcell.00350.2012
63. Sallinen R, Tornberg J, Putkiranta M, Horelli-Kuitunen N, Airaksinen MS, Wessman M. Chromosomal localization of SLC12A5/Slc12a5, the human and mouse genes for the neuron- specific K+-Cl– cotransporter (KCC2) defines a new region of conserved homology. Cytogenet Cell Genet. (2001) 94:67–70. doi: 10.1159/000048785
64. Löscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. (2013) 69:62–74. doi: 10.1016/j.neuropharm.2012.05.045
65. Chamma I, Chevy Q, Poncer JC, Lévi S. Role of the neuronal K-Cl co-transporter KCC2 in inhibitory and excitatory neurotransmission. Front Cell Neurosci. (2012) 6:5. doi: 10.3389/fncel.2012.00005
66. Liu R, Wang J, Liang S, Zhang G, Yang X. Role of NKCC1 and KCC2 in Epilepsy: From Expression to Function. Front Neurol. (2019) 10:1407. doi: 10.3389/fneur.2019.01407
67. Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. (2002) 3:728–39. doi: 10.1038/nrn920
68. Pozzi D, Rasile M, Corradini I, Matteoli M. Environmental regulation of the chloride transporter KCC2: switching inflammation off to switch the GABA on? Transl Psychiatry. (2020) 10:349. doi: 10.1038/s41398-020-01027-6
69. Barmashenko G, Hefft S, Aertsen A, Kirschstein T, Köhling R. Positive shifts of the GABAA receptor reversal potential due to altered chloride homeostasis is widespread after status epilepticus: Positive Shifts of the GABAA Receptor Reversal Potential. Epilepsia. (2011) 52:1570–8. doi: 10.1111/j.1528-1167.2011.03247.x
70. Moore YE, Deeb TZ, Chadchankar H, Brandon NJ, Moss SJ. Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc Natl Acad Sci USA. (2018) 115:10166–71. doi: 10.1073/pnas.1810134115
71. Saitsu H, Watanabe M, Akita T, Ohba C, Sugai K, Ong WP, et al. Impaired neuronal KCC2 function by biallelic SLC12A5 mutations in migrating focal seizures and severe developmental delay. Sci Rep. (2016) 6:30072. doi: 10.1038/srep30072
72. Saito T, Ishii A, Sugai K, Sasaki M, Hirose S. A de novo missense mutation in SLC12A5 found in a compound heterozygote patient with epilepsy of infancy with migrating focal seizures. Clin Genet. (2017) 92:654–8. doi: 10.1111/cge.13049
73. Gharaylou Z, Tafakhori A, Agah E, Aghamollaii V, Kebriaeezadeh A, Hadjighassem M, et al. Preliminary study evaluating the safety and efficacy of bumetanide, an NKCC1 inhibitor, in patients with drug-resistant epilepsy. CNS Drugs. (2019) 33:283–91. doi: 10.1007/s40263-019-00607-5
74. Kahle KT, Barnett SM, Sassower KC, Staley KJ. Decreased seizure activity in a human neonate treated with bumetanide, an inhibitor of the Na(+)-K(+)-2Cl(-) cotransporter NKCC1. J Child Neurol. (2009) 24:572–6. doi: 10.1177/0883073809333526
75. Pressler RM, Boylan GB, Marlow N, Blennow M, Chiron C, Cross JH, et al. Bumetanide for the treatment of seizures in newborn babies with hypoxic ischaemic encephalopathy (NEMO): an open-label, dose finding, and feasibility phase 1/2 trial. Lancet Neurol. (2015) 14:469–77. doi: 10.1016/S1474-4422(14)70303-5
76. Lykke K, Töllner K, Feit PW, Erker T, MacAulay N, Löscher W. The search for NKCC1-selective drugs for the treatment of epilepsy: Structure-function relationship of bumetanide and various bumetanide derivatives in inhibiting the human cation-chloride cotransporter NKCC1A. Epilepsy Behav. (2016) 59:42–9. doi: 10.1016/j.yebeh.2016.03.021
77. Ben-Ari Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. (2017) 40:536–54. doi: 10.1016/j.tins.2017.07.001
78. Kelsch W, Hormuzdi S, Straube E, Lewen A, Monyer H, Misgeld U. Insulin-like growth factor 1 and a cytosolic tyrosine kinase activate chloride outward transport during maturation of hippocampal neurons. J Neurosci. (2001) 21:8339–47. doi: 10.1523/JNEUROSCI.21-21-08339.2001
79. de Los Heros P, Alessi DR, Gourlay R, Campbell DG, Deak M, Macartney TJ, et al. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl- co-transporters. Biochem J. (2014) 458:559–73. doi: 10.1042/BJ20131478
80. Alessi DR, Zhang J, Khanna A, Hochdörfer T, Shang Y, Kahle KT. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal. (2014) 7:re3. doi: 10.1126/scisignal.2005365
81. Pajor AM. Sodium-coupled dicarboxylate and citrate transporters from the SLC13 family. Pflugers Arch. (2014) 466:119–30. doi: 10.1007/s00424-013-1369-y
82. Bergeron MJ, Clémençon Clémençon B, Hediger MA, Markovich D. SLC13 family of Na+-coupled di- and tri-carboxylate/sulfate transporters. Mol Aspects Med. (2013) 34:299–312. doi: 10.1016/j.mam.2012.12.001
83. Inoue K, Zhuang L, Ganapathy V. Human Na+ -coupled citrate transporter: primary structure, genomic organization, and transport function. Biochem Biophys Res Commun. (2002) 299:465–71. doi: 10.1016/S0006-291X(02)02669-4
84. Bhutia YD, Kopel JJ, Lawrence JJ, Neugebauer V, Ganapathy V. Plasma Membrane Na+-Coupled Citrate Transporter (SLC13A5) and Neonatal Epileptic Encephalopathy. Molecules. (2017) 22:E378. doi: 10.3390/molecules22030378
85. Vergnano AM, Rebola N, Savtchenko LP, Pinheiro PS, Casado M, Kieffer BL, et al. Zinc Dynamics and Action at Excitatory Synapses. Neuron. (2014) 82:1101–14. doi: 10.1016/j.neuron.2014.04.034
86. Thevenon J, Milh M, Feillet F, St-Onge J, Duffourd Y, Jugé C, et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am J Hum Genet. (2014) 95:113–20. doi: 10.1016/j.ajhg.2014.06.006
87. Hardies K, de Kovel CGF, Weckhuysen S, Asselbergh B, Geuens T, Deconinck T, et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain. (2015) 138:3238–50. doi: 10.1093/brain/awv263
88. Yang Q-Z, Spelbrink EM, Nye KL, Hsu ER, Porter BE. Epilepsy and EEG Phenotype of SLC13A5 Citrate Transporter Disorder. Child Neurol Open. (2020) 7:2329048X20931361. doi: 10.1177/2329048X20931361
89. Weeke LC, Brilstra E, Braun KP, Zonneveld-Huijssoon E, Salomons GS, Koeleman BP, et al. Punctate white matter lesions in full-term infants with neonatal seizures associated with SLC13A5 mutations. Eur J Paediatric Neurol. (2017) 21:396–403. doi: 10.1016/j.ejpn.2016.11.002
90. Klotz J, Porter BE, Colas C, Schlessinger A, Pajor AM. Mutations in the Na+/Citrate Cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol Med. (2016) 22:310–21. doi: 10.2119/molmed.2016.00077
91. Inoue K, Zhuang L, Maddox DM, Smith SB, Ganapathy V. Human sodium-coupled citrate transporter, the orthologue of Drosophila Indy, as a novel target for lithium action. Biochem J. (2003) 374:21–6. doi: 10.1042/bj20030827
92. Unwin RJ, Capasso G, Shirley DG. An overview of divalent cation and citrate handling by the kidney. Nephron Physiol. (2004) 98:p15–20. doi: 10.1159/000080259
93. Pellegrino F, Tardivo I. SLC13A5-related epileptic encephalopathy successfully treated with valproate and acetazolamide. Seizure. (2021) 91:244–5. doi: 10.1016/j.seizure.2021.06.029
94. Sanz R, del Arco A, Ayuso C, Ramos C, Satrústegui J. Assignment1 of the calcium-binding mitochondrial carrier Aralar1 gene (SLC25A12) to human chromosome band 2q31 by in situ hybridization. Cytogenet Genome Res. (2000) 89:143–4. doi: 10.1159/000015595
95. Jalil MdA, Begum L, Contreras L, Pardo B, Iijima M, Li MX, et al. Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J Biol Chem. (2005) 280:31333–9. doi: 10.1074/jbc.M505286200
96. Wibom R, Barbaro M, Naess K, Palmieri F. AGC1 deficiency associated with global cerebral hypomyelination. The New England J Medicine. (2009) 361:489–95. doi: 10.1056/NEJMoa0900591
97. Falk MJ, Li D, Gai X, McCormick E, Place E, Lasorsa FM, et al. AGC1 deficiency causes infantile epilepsy, abnormal myelination, and reduced n-acetylaspartate. In: Zschocke J, Gibson KM, Brown G, Morava E, Peters V, editors. JIMD Reports. Berlin, Heidelberg: Springer Berlin Heidelberg (2014). p. 77–85. doi: 10.1007/8904_2013_287
98. Kavanaugh BC, Warren EB, Baytas O, Schmidt M, Merck D, Buch K, et al. Longitudinal MRI findings in patient with SLC25A12 pathogenic variants inform disease progression and classification. Am J Med Genet. (2019) 179:2284–91. doi: 10.1002/ajmg.a.61322
99. Pfeiffer B, Sen K, Kaur S, Pappas K. Expanding Phenotypic Spectrum of Cerebral Aspartate–Glutamate Carrier Isoform 1 (AGC1) Deficiency. Neuropediatrics. (2020) 51:160–3. doi: 10.1055/s-0039-3400976
100. Dahlin M, Martin DA, Hedlund Z, Jonsson M, Döbeln U, Wedell A. The ketogenic diet compensates for AGC 1 deficiency and improves myelination. Epilepsia. (2015) 56:e176–81. doi: 10.1111/epi.13193
101. Pérez-Liébana I, Casarejos MJ, Alcaide A, Herrada-Soler E, Llorente-Folch I, Contreras L, et al. βOHB protective pathways in Aralar-KO neurons and brain: an alternative to ketogenic diet. J Neurosci. (2020) 40:9293–305. doi: 10.1523/JNEUROSCI.0711-20.2020
102. Kunji ERS, King MS, Ruprecht JJ, Thangaratnarajah C. The SLC25 carrier family: important transport proteins in mitochondrial physiology and pathology. Physiology (Bethesda). (2020) 35:302–27. doi: 10.1152/physiol.00009.2020
103. Molinari F, Raas-Rothschild A, Rio M, Fiermonte G, Encha-Razavi F, Palmieri L, et al. Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am J Human Genet. (2005) 76:334–9. doi: 10.1086/427564
104. Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Eur J Physiol. (2004) 447:689–709. doi: 10.1007/s00424-003-1099-7
105. Goubert E, Mircheva Y, Lasorsa FM, Melon C, Profilo E, Sutera J, et al. Inhibition of the mitochondrial glutamate carrier SLC25A22 in astrocytes leads to intracellular glutamate accumulation. Front Cell Neurosci. (2017) 11:149. doi: 10.3389/fncel.2017.00149
106. Fiermonte G, Palmieri L, Todisco S, Agrimi G, Palmieri F, Walker JE. Identification of the Mitochondrial Glutamate Transporter. J Biol Chem. (2002) 277:19289–94. doi: 10.1074/jbc.M201572200
107. Reid ES, Williams H, Anderson G, Benatti M, Chong K, James C, et al. Mutations in SLC25A22: hyperprolinaemia, vacuolated fibroblasts and presentation with developmental delay. J Inherit Metab Dis. (2017) 40:385–94. doi: 10.1007/s10545-017-0025-7
108. Baumgartner MR, Rabier D, Nassogne M-C, Dufier J-L, Padovani J-P, Kamoun P, et al. Δ1-pyrroline-5-carboxylate synthase deficiency: neurodegeneration, cataracts and connective tissue manifestations combined with hyperammonaemia and reduced ornithine, citrulline, arginine and proline. Eur J Pediatr. (2005) 164:31–6. doi: 10.1007/s00431-004-1545-3
109. Poduri A, Heinzen EL, Chitsazzadeh V, Lasorsa FM, Elhosary PC, LaCoursiere CM, et al. SLC25A22 is a novel gene for migrating partial seizures in infancy: SLC25A22 Mutation in MPSI. Ann Neurol. (2013) 74:873–82. doi: 10.1002/ana.23998
110. Giacomini T, Pisciotta L, Prato G, Meola I, Zara F, Fiorillo C, et al. Severe early-onset developmental and epileptic encephalopathy (DEE) associated with novel compound heterozygous pathogenic variants in SLC25A22: Case report and literature review. Seizure. (2019) 70:56–8. doi: 10.1016/j.seizure.2019.06.029
111. Milh M, Becq H, Villeneuve N, Ben-Ari Y, Aniksztejn L. Inhibition of glutamate transporters results in a “suppression-burst” pattern and partial seizures in the newborn rat. Epilepsia. (2007) 48:169–74. doi: 10.1111/j.1528-1167.2006.00839.x
112. Nicotera AG, Dicanio D, Pironti E, Bonsignore M, Cafeo A, Efthymiou S, et al. De novo mutation in SLC25A22 gene: expansion of the clinical and electroencephalographic phenotype. J Neurogenet. (2021) 35:67–73. doi: 10.1080/01677063.2021.1892094
113. Hara T, Yamauchi M, Takahashi E, Hoshino M, Aoki K, Ayusawa D, et al. The UDP-galactose translocator gene is mapped to band Xp11.23-p1122 containing the Wiskott-Aldrich syndrome locus. Somat Cell Mol Genet. (1993) 19:571–5. doi: 10.1007/BF01233383
114. Hadley B, Maggioni A, Ashikov A, Day CJ, Haselhorst T, Tiralongo J. Structure and function of nucleotide sugar transporters: Current progress. Comput Struct Biotechnol J. (2014) 10:23–32. doi: 10.1016/j.csbj.2014.05.003
115. Song Z. Roles of the nucleotide sugar transporters (SLC35 family) in health and disease. Mol Aspects Med. (2013) 34:590–600. doi: 10.1016/j.mam.2012.12.004
116. Amado M, Almeida R, Schwientek T, Clausen H. Identification and characterization of large galactosyltransferase gene families: galactosyltransferases for all functions. Biochim Biophys Acta. (1999) 1473:35–53. doi: 10.1016/S0304-4165(99)00168-3
117. Kodera H, Nakamura K, Osaka H, Maegaki Y, Haginoya K, Mizumoto S, et al. De novo mutations in SLC35A2 encoding a UDP-galactose transporter cause early-onset epileptic encephalopathy. Hum Mutat. (2013) 34:1708–14. doi: 10.1002/humu.22446
118. Sim NS, Seo Y, Lim JS, Kim WK, Son H, Kim HD, et al. Brain somatic mutations in SLC35A2 cause intractable epilepsy with aberrant N-glycosylation. Neurol Genet. (2018) 4:e294. doi: 10.1212/NXG.0000000000000294
119. Yates TM, Suri M, Desurkar A, Lesca G, Wallgren-Pettersson C, Hammer TB, et al. SLC35A2-related congenital disorder of glycosylation: Defining the phenotype. Eur J Paediatr Neurol. (2018) 22:1095–102. doi: 10.1016/j.ejpn.2018.08.002
120. Ng BG, Buckingham KJ, Raymond K, Kircher M, Turner EH, He M, et al. Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. Am J Hum Genet. (2013) 92:632–6. doi: 10.1016/j.ajhg.2013.03.012
121. Dörre K, Olczak M, Wada Y, Sosicka P, Grüneberg M, Reunert J, et al. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach. J Inherit Metab Dis. (2015) 38:931–40. doi: 10.1007/s10545-015-9828-6
122. Kimizu T, Takahashi Y, Oboshi T, Horino A, Koike T, Yoshitomi S, et al. A case of early onset epileptic encephalopathy with de novo mutation in SLC35A2: Clinical features and treatment for epilepsy. Brain Dev. (2017) 39:256–60. doi: 10.1016/j.braindev.2016.09.009
123. Witters P, Tahata S, Barone R, Õunap K, Salvarinova R, Grønborg S, et al. Clinical and biochemical improvement with galactose supplementation in SLC35A2-CDG. Genet Med. (2020) 22:1102–7. doi: 10.1038/s41436-020-0767-8
124. Freeze HH, Eklund EA, Ng BG, Patterson MC. Neurology of inherited glycosylation disorders. Lancet Neurol. (2012) 11:453–66. doi: 10.1016/S1474-4422(12)70040-6
125. Bröer S. The SLC38 family of sodium-amino acid co-transporters. Pflugers Arch. (2014) 466:155–72. doi: 10.1007/s00424-013-1393-y
126. Rubio-Aliaga I, Wagner CA. Regulation and function of the SLC38A3/SNAT3 glutamine transporter. Channels (Austin). (2016) 10:440–52. doi: 10.1080/19336950.2016.1207024
127. Lerman MI, Minna JD. The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: identification and evaluation of the resident candidate tumor suppressor genes The International Lung Cancer Chromosome 3p213 Tumor Suppressor Gene Consortium. Cancer Res. (2000) 60:6116–33.
128. Ruderisch N, Virgintino D, Makrides V, Verrey F. Differential axial localization along the mouse brain vascular tree of luminal sodium-dependent glutamine transporters Snat1 and Snat3. J Cereb Blood Flow Metab. (2011) 31:1637–47. doi: 10.1038/jcbfm.2011.21
129. Chan K, Busque SM, Sailer M, Stoeger C, Bröer S, Daniel H, et al. Loss of function mutation of the Slc38a3 glutamine transporter reveals its critical role for amino acid metabolism in the liver, brain, and kidney. Pflugers Arch. (2016) 468:213–27. doi: 10.1007/s00424-015-1742-0
130. Marafi D, Fatih JM, Kaiyrzhanov R, Ferla MP, Gijavanekar C, Al-Maraghi A, et al. Biallelic variants in SLC38A3 encoding a glutamine transporter cause epileptic encephalopathy. Brain. (2022) 145:909–24. doi: 10.1093/brain/awab369
131. Ochoa JG, Kilgo WA. The role of benzodiazepines in the treatment of epilepsy. Curr Treat Options Neurol. (2016) 18:18. doi: 10.1007/s11940-016-0401-x
132. French JA. Vigabatrin. Epilepsia. (1999) 40 Suppl 5:S11–16. doi: 10.1111/j.1528-1157.1999.tb00914.x
133. Konrad D, Somwar R, Sweeney G, Yaworsky K, Hayashi M, Ramlal T, et al. The antihyperglycemic drug α-lipoic acid stimulates glucose uptake via both GLUT4 translocation and GLUT4 activation. Diabetes. (2001) 50:1464–71. doi: 10.2337/diabetes.50.6.1464
134. Anheim M, Maillart E, Vuillaumier-Barrot S, Flamand-Rouvière C, Pineau F, Ewenczyk C, et al. Excellent response to acetazolamide in a case of paroxysmal dyskinesias due to GLUT1-deficiency. J Neurol. (2011) 258:316–7. doi: 10.1007/s00415-010-5702-5
Keywords: SLC, DEE, epilepsy, treatment, pathogenesis
Citation: Gan Y, Wei Z, Liu C, Li G, Feng Y and Deng Y (2022) Solute carrier transporter disease and developmental and epileptic encephalopathy. Front. Neurol. 13:1013903. doi: 10.3389/fneur.2022.1013903
Received: 08 August 2022; Accepted: 07 September 2022;
Published: 07 November 2022.
Edited by:
Tianfu Li, Sanbo Brain Hospital (CMV), ChinaCopyright © 2022 Gan, Wei, Liu, Li, Feng and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanchun Deng, eWFuY2h1bmRAZm1tdS5lZHUuY24=