 Océane Perdaens
Océane Perdaens Vincent van Pesch
Vincent van Pesch
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 25 February 2022
Sec. Multiple Sclerosis and Neuroimmunology
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.811518
This article is part of the Research Topic Immunosenescence and Multiple Sclerosis: Prognostic and Therapeutic Implications View all 13 articles
Aging is characterized, amongst other features, by a complex process of cellular senescence involving both innate and adaptive immunity, called immunosenescence and associated to inflammaging, a low-grade chronic inflammation. Both processes fuel each other and partially explain increasing incidence of cancers, infections, age-related autoimmunity, and vascular disease as well as a reduced response to vaccination. Multiple sclerosis (MS) is a lifelong disease, for which considerable progress in disease-modifying therapies (DMTs) and management has improved long-term survival. However, disability progression, increasing with age and disease duration, remains. Neurologists are now involved in caring for elderly MS patients, with increasing comorbidities. Aging of the immune system therefore has relevant implications for MS pathogenesis, response to DMTs and the risks mediated by these treatments. We propose to review current evidence regarding markers and molecular mechanisms of immunosenescence and their relevance to understanding MS pathogenesis. We will focus on age-related changes in the innate and adaptive immune system in MS and other auto-immune diseases, such as systemic lupus erythematosus and rheumatoid arthritis. The consequences of these immune changes on MS pathology, in interaction with the intrinsic aging process of central nervous system resident cells will be discussed. Finally, the impact of immunosenescence on disease evolution and on the safety and efficacy of current DMTs will be presented.
With the aging of the world population, seniors aged over 65 years, that account for 9.3% of the global population in 2020, are predicted to have doubled in absolute number by 2050, representing 15.9% (1). This increase in life expectancy inevitably has an impact on disease prevalence and incidence, especially of chronic diseases. Health care systems worldwide must face this demographic evolution within the next decades. Biological aging is the decline in homeostasis, with functional alterations of all organs and tissues, resulting in an increase of morbidity and mortality (2). On a cellular level, senescent cells, accumulating with age, are arrested in their cell cycle, but are still active, although functionally dysregulated and affecting their microenvironment by secreting soluble signaling factors (interleukins, chemokines, growth factors), proteases, or insoluble proteins/extracellular components. These constitute the so-called senescence-associated secretory phenotype (SASP) exerting a paracrine pro-inflammatory effect (3, 4). The immune system, which is continuously operating throughout life, is prone to these age-related changes, referred to as immunosenescence (5). Immunosenescence affects both the innate, and, to a greater extent, the adaptive immunity. It is postulated to explain increased prevalence of infections, cancers and auto-immune diseases and reduced response to vaccination in the elderly (6). On the contrary, the purpose of cell cycle arrest in senescent cells is to prevent cellular escape into tumoral processes (7). However, no single immune change associated with senescence explains health-related consequences of aging. Hence, a longitudinal study proposes an age-related ‘immune risk phenotype' associated with poorer survival, characterized by an inversion in the CD4+/CD8+ T cell ratio, the expansion of the terminally differentiated CD8+CD28− T cells, lower B cell numbers and seroconversion for cytomegalovirus (CMV) (8–10). Moreover, a chronic, sterile low-grade inflammation occurs concurrently, named inflammaging, mutually interacting with immunosenescence. Continuous antigenic load and stressors stimulate the innate immune system, mainly macrophages, to produce pro-inflammatory cytokines, such as interleukin (IL) 1, IL6, or tumor necrosis factor (TNF), also part of the SASP (11). However, centenarians aging healthily have an inverted immune risk phenotype and a heightened inflammaging profile properly counterbalanced by anti-inflammaging (12–14). Hence, Franceschi et al. argument that diseases arise when this equilibrium is broken (11, 15).
The multiple sclerosis (MS) population older than 65 years is increasing worldwide due to improving life expectancy with MS, although the latter remains 6–10 years shorter as compared to the general population (16, 17). There is growing awareness about the implications of aging with MS, due to immunosenescence, the high burden of comorbidities and the lack of knowledge on long-term effects of exposure to disease modifying therapies (DMTs). The safety, efficacy and benefit of DMTs in this population are unknown, since patients over 55-to-60-years-old are generally excluded from clinical trials (17). Furthermore, while remitting-relapsing MS (RRMS) is the prominent phenotype in younger patients, older patients more likely have primary or secondary progressive MS (PPMS/SPMS), in which chronic inflammation and neurodegeneration, due to failure in myelin repair and axonal loss, is considered to prevail (18). However, the pathophysiology underlying the progression of the disease with aging remains incompletely understood.
We aim to review and compare current knowledge on immunosenescence and inflammaging, relative to MS and other autoimmune diseases (AIDs), such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA), as these AIDs are amongst the most studied in this context (19–21). In the setting of MS, we will also focus on the concomitant senescence of central nervous system (CNS) resident cells in order to answer several questions. (a) What is the evidence or the lack thereof to consider MS a disease of premature immunosenescence? (b) What are the common or different immunosenescence features found in MS and other autoimmune diseases? (c) Are epigenetic changes involved in immunosenescence? (d) Is the age-related evolution of MS toward a progressive phenotype linked to immunosenescence? (e) Does immunosenescence expose aging MS patients to increased risks of infection and cancer, especially when taking DMTs?
Immunosenescence is defined as the physiological aging of the immune system (22). Immune cells are generated from hematopoietic stem cells (HSCs) throughout life and differentiate stepwise, undergoing selection and proliferation pressure upon antigenic contact. They are thus especially prone to senescent processes. Changes related to immunosenescence are more preeminent within the adaptive than the innate immune system [reviewed by (23, 24)].
Cell cycle arrest of aging cells is initially a protective phenomenon against increasing cellular damage and tumorigenesis (7). The pro-inflammatory SASP (IL6, IL8, matrix metalloproteinase (MMP) 1/3, monocyte chemotactic protein (MCP) 2/3, insulin growth factor binding proteins) of senescent cells constitutes a removal-signal directed toward immune cells (4). However, due to age-related dysfunctions in the immune system, this clearance partly fails (25). The pro-inflammatory and oxidative context occurring during the aging process, enhances the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) pathway, a key regulator of inflammation (26). This compensatory mechanism may become self-deleterious, since cumulative cell debris, self-antigens and the inflammatory SASP contribute together to inflammaging, altering cell, tissue, organ and organism homeostasis [reviewed by (23)].
Immunosenescence has been implicated in reduced defenses against infections and reduced response to vaccination (due to a reduced antigenic response by T and B cells), an increased risk of cancer (due to an imbalance between the function of regulatory cells and cytotoxic CD8+ T cells) and auto-immune diseases (due to reduced clearance of apoptotic cells and reduced antibody diversity, with however an increased susceptibility to molecular mimicry) (27–30). These risks might be counterbalanced by the subject's intrinsic (e.g., genetic polymorphisms, epigenetics) and extrinsic factors (e.g., the individual's history of past immune reactions, referred to as immunobiography, as well as environmental factors) (11, 15).
With aging, the pool of naïve T cells is reduced due to thymic involution and reduced bone marrow proliferative capacity. Both the thymus and the bone marrow lose their epithelial/stromal cell frame, which is replaced by adipocytes, resulting in a reduction in HSC proliferation (31, 32). Moreover, a general shift of HSCs from the lymphoid to the myeloid lineage is observed. Thymic T cell output, measurable by T cell receptor (TCR) excision circles (TRECs), is reduced with age. TRECs are stable extrachromosomal DNA byproducts resulting from thymic TCR rearrangements. TRECs are not replicated and are therefore diluted with cell division (33).
Homeostatic proliferation, driven by dendritic and B cells upon exposure to IL7 and IL15, occurs initially to compensate for the reduced peripheral input of naïve T cells, but results in the clonal expansion of memory T cells and a depleted TCR repertoire (34–37). The proportion of T helper (Th) cells decreases due to defective antigen presentation and an impaired TCR response, resulting in a reduction of TCR-mediated proliferation (38). The reduced expression of CD40 ligand (CD40L) on CD4+ T cells impairs their binding to B cells and thus their ability to function as T helper cells (39).
With aging, a shift from Th1 to Th2 cells is observed, due to decreased IL2 production, although this is disputed (40, 41), while the percentage of Th17 cells is increased in subjects aged over 65 as compared to younger subjects (42). Moreover, memory T cells are resistant to apoptosis, hence reenforcing their numerical increase (43).
Overall, during senescence, the number of CD4+ T cells decreases and CD8+ T cells increases resulting in an inverted CD4+/CD8+ ratio (<1) (10). Antigen-experienced T cells proliferate and differentiate into terminally differentiated memory cells with shortened telomeres that eventually lose CD28 expression, a costimulatory signal involved in T cell activation and survival (44). This loss, mainly observed in memory CD8+ T cells, has been linked to aging and immunosenescence, and is partly enhanced by chronic antigenic stimulation, especially by CMV, with a ten-fold factor for CD4+ and 2-fold for CD8+ T cells (45, 46). These CD28− cells express the natural killer (NK) receptor NKG2D which provides an antigen-independent activation signal (along with the NK adaptor molecule DAP12), bypassing the missing costimulatory signal CD28, and enhancing their autoreactivity (47, 48). Moreover, these cells express cytokines [interferon (IFN)g, TNFa] and cytotoxic molecules (granzyme A/B, perforin) upon expression of the eomesodermin factor, and are resistant to apoptosis [by expressing B cell lymphoma 2 (BCL2) and Fas-associated death domain-like IL-1-converting enzyme inhibitory protein (FLIP)] (49–51). Finally, they express chemokine receptors [e.g., C-X3-C Motif Chemokine Receptor 1 (CX3CR1)], which might favor their migration to inflammation sites (51, 52).
In summary, immunosenescence in T cells is characterized by a physiologically reduced pool of naïve T cells and an increase in memory, particularly CD8+, T cells, that have lost CD28 and express NKG2D, the first increasing T cell self-reactivity in secondary lymphoid organs, the second reducing their threshold for antigen-specific activation hence enabling an antigen-independent activation. The autoreactivity of senescent T cells is enhanced by the clonal expansion of memory T cells and the reduced TCR repertoire. These changes can also partly explain the reduced immune defenses against new pathogens observed during aging, as senescent cells are considered functionally deficient, contrary to exhausted T cells, which are considered dormant and can still respond to a previously encountered antigen (15).
B cell numbers, phenotypes and functions change with age [reviewed by (53, 54)]. Reduced B cell output is attributed to global changes in hematopoiesis, as described above. Moreover, peripheral B cell survival factor levels, such as B cell activating factor (BAFF) and A proliferation-inducing ligand (APRIL) are reduced in the elderly (55). In addition, stromal cell-derived IL7 production is reduced, whereas the increased pro-inflammatory cytokine levels [TNFa, IL1b, and transforming growth factor (TGF)b] withhold the B progenitor cells from the IL7-rich niches, hence impairing B lymphopoiesis and reducing the pro-B cell immunoglobulin (Ig) heavy chain V-DJ rearrangement and thus the pre-B cell receptor (BCR) repertoire (56–59). As a consequence, absolute and relative numbers of peripheral CD19+ B cells are reduced, while the proportions of B subsets remain stable with age in humans (53).
With aging, naïve mature (IgD+CD27−) B cells decrease while exhausted double negative memory (IgD−CD27−) B cells increase. IgM unswitched (IgD+CD27+) and switched (IgD−CD27+) memory B cells remain generally stable (53, 54). The immature transitional immunoregulatory CD24highCD38high B cell subset decreases with age, so does its IL10 production (60).
The humoral immune response is altered during senescence, since antibodies are reduced not in quantity but in their diversity and affinity and show cross-reactivity to self- and foreign antigens. This is due to a decrease in antibody class switch and affinity maturation in clonally expanding B cells, related to the downregulation of the E47 transcription factor and activation-induced deaminase (61, 62). This alters the ability to mount a rapid secondary antibody response. Furthermore, a progressive decline in germinal center formation during aging decreases somatic hypermutation, i.a. in IgD−CD27+ B cells and even more in double negative B cells (63). Moreover, immunosenescent B cells lack the support of the Th cells, since Th cells are reduced in number, express less CD40L, and are less exposed to antigen presentation by antigen presenting cells (APCs), due to a reduced expression of the major histocompatibility complex of class II (MHC-II) on the latter (39, 64).
Interestingly, double negative memory B cells express chemokine receptors, C-X-C Motif Chemokine Receptor 3 (CXCR3), although reduced with age, C-C Motif Chemokine Receptor (CCR)6 and CCR7, and are thus prone to migrate to the inflammation sites (65). Moreover, these cells are pre-activated and can produce pro-inflammatory cytokines, and granzyme (66, 67). They undergo an antigen-driven BCR hypermutation.
Finally, low but steadily expanding CD11b+CD11c+CD21− age-associated B cells (ABCs) have been identified in the elderly, in response to antigenic stimulation, and linked to autoreactivity. This functionally exhausted memory subset is driven by the T-box transcription factor (TBET) and is activated synergically upon stimulation of the BCR and Toll-like receptors (TLR)7 and 9. ABCs produce pro-inflammatory cytokines (e.g., TNFa), inhibit B lymphopoiesis and favor Th17 polarization (68–70). They possibly derive from follicular B cells and exhibit downregulation of Epstein Barr Virus (EBV) receptor CD21 due to chronic EBV stimulation.
In summary, the changes in B cell phenotypes, and recirculation, along with their altered humoral response contribute to immunosenescence and can explain the reduced response to vaccination and increased susceptibility to infections, while the clonal expansion of B cells cross-reactive to self-antigens can favor autoimmunity.
Natural, thymic-derived CD4+CD25+FOXP3+ regulatory T cells [(n)Tregs], mainly with an effector memory phenotype (CD45RO+/CD45RA−), increase with age in human in relative and absolute numbers, so does the expression of their transcription factor, forkhead box P3 (FOXP3), possibly due to their better survival in the periphery, since they reduce the expression of the pro-apoptic BCL2 interacting mediator of cell death (BIM) (71–74). Functionally, CD4+ Tregs of aged humans and mice can suppress CD4+ and CD8+ T cell proliferation and IFNg production, but in aged mice they could not suppress IL17 production (75, 76).
Likewise, natural CD8+FOXP3+ nTregs increase with age, while their peripheral inducible capacity is reduced (77, 78). Functionally CD8+ nTregs retain the same suppressive ability independently of aging. Interestingly, a CD8+CD28−FOXP3+ cell subset has been described, in agreement with the overall increase of CD8+CD28− T cells (79). Finally regulatory B cells and myeloid-derived suppressor cells also appear increased with age but have been less studied (80).
In summary, Tregs participate to the immunosenescent process by their increased number and their safeguarded suppressive activity, except against Th17 cells [reviewed by (79, 81)]. This correlates with increased cancer incidence, since Tregs suppress the CD8+ T cell anti-tumor response, and with an increased risk of infection and viral reactivation, since they suppress the anti-pathogen response (72, 82, 83). They have also been linked to neurodegeneration due to their differential interaction with microglia both in the presence and absence of effector T cells (84).
Although less affected by immunosenescence, partly because HSCs are redirected toward the myeloid lineage, innate immunity still displays mainly functional changes [reviewed by (85)].
With aging, dendritic cells show less migration abilities, less responsiveness to TLR stimulation, reduced pathogen processing (phagocytosis, endocytosis) and antigen presentation. This is attributed to mitochondrial dysfunction, resulting in the production of reactive oxygen species (ROS) (86, 87). These alterations affect T cell stimulation and consequently the CD8+ T cell cytotoxic response. Type I (IFN-I, i.e., IFNa/b) and III (IFN-lambda) IFN production is decreased, but they still produce IL6 and TNFa (87, 88).
Several important functions of neutrophils are reduced with aging: chemotaxis, phagocytosis, production of ROS and neutrophil extracellular traps (NET). Opsonization of antibody-bound pathogens is dwindled (89, 90).
Monocytes shift from the classical (CD14++CD16−) to the pro-inflammatory non-classical (CD14+CD16++) phenotype, however with some discrepancies on their expression of Human Leukocyte Antigen-DR isotype (HLA-DR) and CX3CR1 (91–93).
Macrophages also produce less ROS, IL6, and TNFa. They display impaired phagocytosis resulting in reduced antiviral response and impaired auto-/mitophagy, resulting in the accumulation of altered organelles and molecules (25, 94). Moreover, they express less TLRs and MHC-II on their surfaces, thus impairing their ability to present antigens to CD4+ T cells (95, 96).
The slight net increase in the total number of NK cells is in fact due to a decrease of the immunoregulatory CD56bright and an increase of the cytotoxic CD56dim NK cell subsets. These show however impaired degranulation and thus decreased cytotoxic abilities on a per cell basis. IL2/12-mediated secretion of immunomodulatory cytokines (e.g., IFNg) and chemokines is reduced, while production of IL1/4/6/8 and TNFa is increased (97, 98). Furthermore, the central maturation of NK cells is incomplete (99).
Insight into the function of microRNAs (miRNAs) has rapidly grown over the past two decades. miRNAs are small non-coding RNAs regulating gene expression post-transcriptionally by binding to their target messenger RNA (mRNA) and mostly inhibiting its translation (100). Overall, miRNA transcription decreases with age [e.g., miR-17/92a/181a in peripheral blood mononuclear cells (PBMCs)] (101, 102), and they have been linked to several mechanisms underlying cellular senescence [reviewed by (103)]. Oxidative stress can affect positively or negatively miRNA expression (104–107). On the opposite, the downregulation of miR-146a enhances NADPH oxidase (NOX) as it targets its subunit NOX4 (108). The downregulation of the miR-17-92 cluster and the upregulation of miR-210 enhance ROS production (109, 110). Furthermore, miR-210 induces senescence-associated heterochromatin loci and double-strand DNA breaks and is involved in mitochondrial dysfunction by targeting a subunit of the electron transport chain (110, 111). miR-34a and -101 inhibit autophagy (112, 113). By targeting the pro-proliferative cyclin A2, an antagonist of p21, miR-29a and -124, both induced by p53, enhance p21 expression, and thus senescence by cell cycle arrest (114). miR-20a is also an indirect p53-senescence inducer (115). Furthermore, the miR-17-92 cluster and miR-106b family target p21, while p53 inhibits the miR-17-92 cluster (107, 116–119). Finally, miR-9/96/145 were upregulated concomitantly to the downregulation of insulin growth factor 1 receptor (a miR-96/182-target) and forehead box protein O1 (FOXO1, a miR-145/132-target) in the PBMCs of elderly subjects, but miR-132 and -182 were not differentially expressed in this study (120).
Telomeres [reviewed by (121)] are repetitive hexameric sequences (TTAGGG) at the chromosome end of 10–15 kb at birth that shorten by 40–200 bp with each cell division, although length of shortening per mitosis might vary as it is higher in memory than in naïve cells. Critically short telomeres induce a signal for p53-dependent cell cycle arrest. Telomerase is a ribonucleoprotein complex comprising a catalytic subunit, telomerase reverse transcriptase (TERT), which can elongate the hexameric sequences. Telomere length depends on the balance between telomere shortening and telomerase activity, but overall decreases with age. Telomerase activity is increased in stem cells, but also in lymphocytes, where it is the highest in the germinal center (122, 123). However, this activity in the latter is not enough to slow down telomere shortening. Oxidative stress and an increased replication rate upon repetitive stimulation during inflammation, progressively reduce telomerase activity, paralleling the loss of CD28, and hastens telomere attrition and thus cellular senescence (121, 124, 125). Remarkably, centenarians have longer telomeres with lower levels of basal inflammation (14). Interestingly, miRNAs can induce telomere dysfunction and cellular senescence, as miR-138 and -512-5p inhibit TERT. miR-155 targets telomeric repeat-binding factor (TRF)1, miR-23a targets TRF2, which both ensure telomere maintenance (126–129).
Several miRNAs are considered as major immuno-microRNAs, playing a role in immune cell homeostasis and senescence, but also in inflammatory responses and inflammaging [reviewed by (130, 131)]. Age-dependent changes in miRNAs diverge between naïve, central and effector memory CD8+ T cells, but miR-181a is commonly downregulated in the aged cells of all 3 subsets. The most changes are uncovered in the naïve T cell subset, where they are correlated with the decline of FOXO1 signaling, evidenced by the downregulation of IL7 receptor and CCR7, and the alteration of TNFa and NF-kB signaling (132). The SASP in senescent cells induces the delayed expression of miR-146a/b to target IL1 receptor associated kinase (IRAK)1 and to compensate downstream NF-kB-dependent inflammation mediated by IL6/IL8 (133). miR-223 downregulates the NF-kB pathway and the inflammasome NLRP3 (134). Contrarily, NF-kB induces miR-155, which inhibits suppressor of cytokine signaling (SOCS)1, allowing T effector expansion and T memory formation and maintenance (135). miR-17/19b/20a/106a were downregulated in CD28− vs. CD28+ and in CD28+ T cells of old vs. young donors alongside the upregulation of p21 (109). The miR-17-92 cluster and miR-21 support the differentiation into T effector cells (136, 137). On the contrary, the T effector response upon viral infection is delayed in miR-155- or miR-181a-deficient (CD8+) T cells, and cells differentiate to central rather than effector memory cells (138, 139). Moreover, with age, the decrease of Yin-Yang 1 and T cell factor 1 results in the downregulation of miR-181a, which induces dual specific phosphatase (DUSP)6 expression. The latter impairs extracellular signal-regulated kinase (ERK)-dependent TCR sensitivity (140). Furthermore, miRNAs can impair B cell differentiation in the elderly. miR-155, that targets activation-induced deaminase, and miR-16, that targets E47, are increased in memory B cells and even more in double negative B cells (141, 142).
Epigenetics translate the effect of the environment on gene expression [reviewed by (143)]. While methylation, catalyzed by DNA methyltransferases (DNMT), can vary at a cell-base level, the global methylation rate is reduced with age, possibly by passive demethylation and reduced activity of DNMT1 (144). Hypomethylation allows gene expression. Naïve CD4+ T cells display age-associated hypomethylation sites in immune-related pathways [TCR signaling, Fc gamma receptor (FCgR)-mediated phagocytosis, mammalian target of rapamycin (mTOR) and insulin signaling, antigen processing and presentation], while hypermethylation was observed in cell proliferation pathways [Wnt and mitogen-activated protein kinase (MAPK) signaling] (145, 146). Interestingly, centenarians display a slower reduction of DNA methylation level (147). Sirtuins (SIRT) encode NAD+-dependent histone deacetylases (HDAC) and maintain the genome's integrity during cellular stress. Downregulation of SIRT1 and SIRT3 in PBMCs of healthy elderly subjects was accompanied by the upregulation of miR-9, which targets SIRT1, and miR-34a (148). Oxidative stress induces miR-195, that targets SIRT1, which is associated with reduced telomerase activity (149).
In summary, telomeres shorten with age due to cell replication and oxidative stress, despite sustained telomerase activity. Epigenetically, global hypomethylation occurs with aging. Interestingly, miRNAs are involved in cellular senescence through different mechanisms [oxidative stress, mitochondrial dysfunction, cell cycle arrest (p53 pathway), telomere attrition, and inflammation].
Immunosenescence is associated with an increase in the incidence of several AIDs (150). Some diseases show a bimodal age of onset (e.g., RRMS vs. PPMS, MOG-antibody diseases), others almost exclusively occur in the elderly (e.g., polymyalgia rheumatica, giant cell arteritis) (30, 151–153). A German study showed an age-decreasing female incidence of SLE peaking at the age of 20–25 and to a lesser extent at menopausal age, and an age-increasing male incidence peaking at the age of 65–70 (154). RA-incidence and prevalence increase with age to peak at age 50–54 and 60–64, respectively (155). Serum autoantibody titers are generally higher in elderly subjects, even without overt AID. Moreover, the binding of circulating antibodies to random peptides, especially with a di-serine motif, increases with age (150, 156).
Interestingly, AIDs show inflammaging and features of immunosenescence at an earlier age. Age-associated defects at the cellular level, classified under the nine common denominators of aging (2), and the resulting impaired immune function create an unstable state, that may predispose for tolerance failure and occurrence of autoimmunity (152, 157). Herein, we will focus on the effect of stem cell exhaustion, altered intercellular communication (e.g., by inflammaging), proteostasis (i.e., the maintenance of a functional cellular protein pool) loss, telomere attrition, genomic instability, mitochondrial dysfunction, and epigenetic alterations in MS, SLE, and RA.
Lymphopenia-induced homeostatic proliferation leads to clonal expansion and TCR repertoire contraction over time. Furthermore, priming by cytokines produced during inflammaging, can transiently reduce the TCR stimulation threshold (by ERK phosphorylation), consequently interfering with tolerance maintenance and promoting autoreactive T cells [reviewed by (152)]. TNFa engages cellular senescence by inducing interferon response genes, cytokine secretion, and ROS production (158). Moreover, the Th17/Treg imbalance contributes to trigger autoimmune diseases (42).
Three cell types are unique to immunosenescence, i.e., CD28− T cells, linked to cytotoxicity, double negative B cells and ABCs, linked to autoantibody production, and might play a role in autoimmunity (48, 66, 68). The CD4+CD28− cell population is enlarged in subjects with autoimmune diseases compared to age-matched controls, with the highest percentage in RA, followed by RRMS and SPMS and finally SLE, and is positively correlated with age and CMV seropositivity. These cells are enriched in granzyme A and B, and perforin and their TCR repertoire is contracted as compared to CD28+ T cells. The latter appears even stronger in MS/RA than in healthy controls (159).
Cells recycle long-lived proteins, damaged organelles, and aggregates by autophagy via the lysosomes, for the synthesis of new proteins or for energy production, thus ensuring cellular homeostasis, especially under nutrient-/energy-poor conditions (160, 161). Autophagy declines with age, as seen by the downregulation of autophagy-related protein (ATG)5, ATG7 in the human aged brain (162, 163). Interestingly, autophagy and inflammation can reciprocally induce and suppress each other. Autophagy is induced by TLRs but inhibited by Th2 cytokines. Conversely, it blocks the inflammasome, and thus the IL1b response. It prevents ROS production by degrading dysfunctional mitochondria, but it also promotes the survival and differentiation of immune cells [reviewed by (164)].
In AIDs, metabolic reprogramming for energy production may fail leading to hyperreactive immune cells and an increase in oxidative stress. Oxidative stress and mitochondrial dysfunction contribute to (immuno-)senescence and inflammation through decreased redox capacity (glutathione depletion), activation/oxidation-induced cell apoptosis (with defective clearance and release of the cell content inducing TLR), mitochondrial DNA (mtDNA) damage, defective bioenergetics (ATP depletion) and production of neoantigens (165, 166). Moreover, oxidative stress, inflammation, and increased leukocyte renewal accelerate telomere shortening [reviewed by (166)].
In MS, activated peripheral autoreactive CD4+ T cells, migrate through a disrupted blood brain barrier (BBB) into the CNS. They are reactivated upon antigenic contact, and interact with other peripheral immune cells (CD8+ T and B lymphocytes, monocytes, and macrophages). They activate microglial cells and astrocytes to induce demyelination, oligodendrocyte apoptosis and axonal damage (Figure 1A) (18). MS patients present at a younger age with some features of immunosenescence seen in aged healthy controls, suggesting that it is possibly involved in MS pathogenesis [reviewed by (19, 167)].
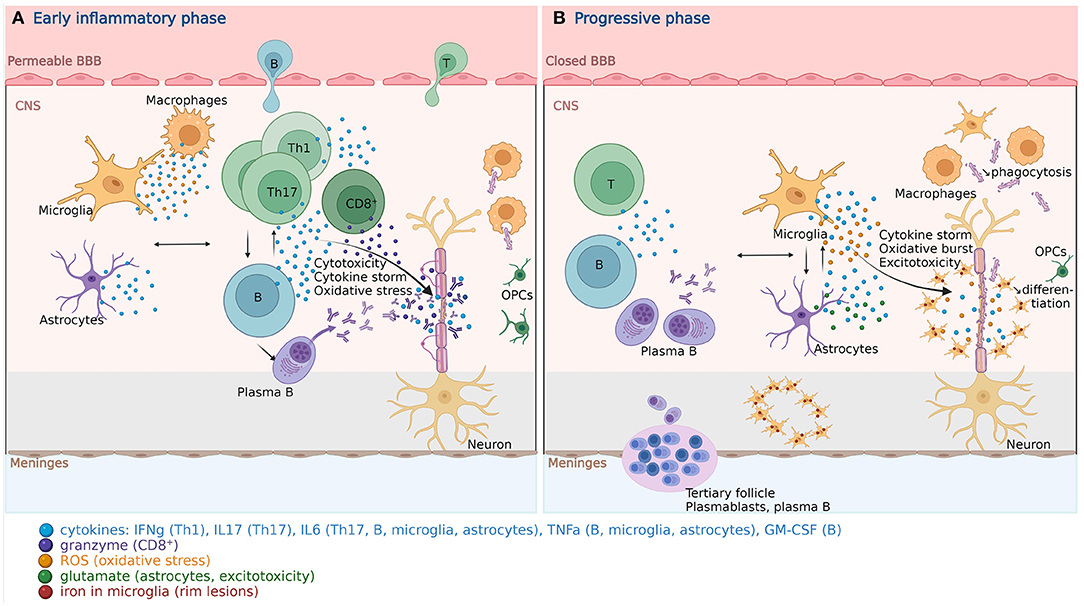
Figure 1. Pathophysiology of multiple sclerosis. (A) In the early inflammatory phase of MS, peripheral adaptive immune cells infiltrate the CNS through a disrupted BBB. These activated cells interact with each other and the resident cells of the CNS. They secrete cytokines (e.g., IFNg by Th1, IL6/17 by Th17, GM-CSF, IL6, TNFa by B cells) and cytotoxic molecules (e.g., granzyme B by CD8+ T cells). B cells can further evolve into autoantibody-producing plasma cells. As a consequence, T and B cells activate macrophages and microglia, which produce cytokines, nitric oxide, and ROS. This cytotoxic pro-inflammatory environment breaks down the myelin sheaths around axons and induces energy failure in the axon. Macrophages and microglia can still clear the myelin debris, allowing for the recruitment of OPCs that will partially remyelinate the lesion. (B) In the progressive phase of MS, T and B cell infiltrates are reduced. Remarkably, plasmablasts and plasma B cells form tertiary follicle-like structures in the meninges. The BBB is closed and the inflammation is sustained by innate CNS-resident cells, i.e., microglia and astrocytes. They produce cytokines (TNFa, IL6) and release ROS, causing myelin damage. Although microglia are primed into a pro-inflammatory phenotype, their phagocytic capacities are reduced. These features also characterize the senescence-associated phenotype of microglia and astrocytes. Myelin debris are improperly cleared, OPCs are less recruited and fail to differentiate. TNFa-mediated glutamate release from astrocytes results in excitotoxicity causing axonal damage. The ferrous iron released from the myelin, where it accumulates with age, is oxidized, which produces ROS, and incorporated by microglia, forming a phenotypical rim around the demyelinating lesions, both in the white and gray matter. These successive events are self-sustained and enhanced by senescent processes, resulting in a major oxidative burst, causing mitochondrial dysfunction, mitochondrial DNA damage, energy failure and axonal loss. BBB, blood brain barrier; B, B cell; CNS, central nervous system; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; MS, multiple sclerosis; OPC, oligodendrocyte progenitor cell; ROS, reactive oxygen species; T, T cell; Th, T helper cell; TNF, tumor necrosis factor. Created with BioRender.com.
Circulating neutrophils in RRMS patients produce more inflammatory markers and NETosis, and are resistant to apoptosis. The serum/plasma levels of neutrophil-activating chemokines and neutrophil-derived enzymes [e.g., C-X-C motif ligand (CXCL)1, CXCL8, elastase] are positively correlated with new inflammatory lesions. Neutrophils are also found in the cerebrospinal fluid (CSF) at onset and early in relapse, but decrease with disease duration (168–170). Regarding monocytes, discrepancies exist due to study population and staining strategy differences. Some describe an increase in classical and non-classical monocytes in inactive RRMS as compared to progressive MS, while others state an increase in non-classical monocytes in a mixed MS population (171, 172). The beneficial or detrimental role of NK cells is still debated [reviewed by (173)]. The peripheral blood of PP/SPMS counts more CD56dim NK cells, while the CD56bright NK cells are expanded in the CSF of untreated RRMS patients due to their higher migratory capacity, which might counterbalance the CNS inflammation (174, 175). However, their immunoregulatory and cytolytic functions appear to be impaired (176, 177). Moreover, NK cells in the CNS could delay remyelination, as they suppress the reparative properties of neural stem cells in experimental autoimmune encephalitis (EAE) (178).
The bone marrow cellularity is reduced and the in vitro proliferative capacity of mesenchymal stromal cells, supportive of hematopoiesis, declines with age, even more in PP/SPMS, while CD34+ HSC numbers remain stable in MS but the frequency of colony forming cells is low (179, 180). Moreover, thymic involution is accelerated in MS, given that TREC levels are, at all ages lower than in age-matched healthy controls and progressively decrease with age (159). Hence, the CD8+ naïve T cell pool is reduced, while data regarding the effect of age on naïve CD4+ T cells in MS are more discrepant (181–183). Interestingly, the TCR repertoire is more diversified in MS (184). Moreover, Th17 cells are largely involved in MS and increased in the periphery (185, 186). The inverted CD4+/CD8+ ratio, the hallmark of the immune risk phenotype, mostly does not apply in MS (187–189). This ratio was however decreased in the CSF of patients on natalizumab treatment (190, 191).
Both effector memory CD4+ and CD8+ T cells may enhance the chronic inflammatory responses to neuroantigens in MS and EAE (192, 193). Notably, inoculation with CD4+ memory rather than effector T cells in EAE preferentially induced marked CNS inflammation (194, 195). Herein, memory CD4+ T cells are increased in the blood and the CSF during active disease (196). Central and effector memory CD8+ T cells are increased, independently of disease activity, in the blood, and the latter also in the CSF, and the CNS tissue (192, 196–198).
The cytotoxic CD4+CD28− population is enriched with advancing age in RR/SP/PPMS, while it remains stable in healthy controls, and has been linked to disease severity in EAE and MS (159, 199–201). They are partly autoreactive to myelin basic protein (MBP) (199). Since these cells express CX3CR1, they might infiltrate the CNS where the CSF levels of its ligand fractalkine were found elevated in MS (52).
B cells play a central role in MS development and progression [reviewed by (202)]. Antigen-driven clonally expanded B cells produce pro-inflammatory cytokines [TNF, lymphotoxin (LT)a, IL6, granulocyte-macrophage colony-stimulating factor (GM-CSF)] and chemokines through the NF-kB pathway. Memory B cells act as APCs, and hereby prompt the proliferation and activation of T and myeloid cells. B cells, stimulated by Th follicular cells, differentiate into immunoglobulin-producing plasmablasts and plasma cells that accumulate to eventually form tertiary follicle-like structures in the leptomeninges during disease progression and are notably involved in inducing subpial demyelination.
Transitional B cells (CD24highCD38high) are reduced in the blood and are functionally defective in RRMS (produce less IL10). They have been found in the CSF while they were absent in CSF samples of other inflammatory neurological diseases (203, 204). The proportions of peripheral naïve B cells decrease and memory B cells increase with age in controls (54), but remain stable in MS, except during relapse. Interestingly, the proportion of CSF class-switched memory B cells is increased in adult MS whereas the relative numbers of unswitched memory B cells are increased in pediatric MS (205). In B cells from MS patients, a preferential naïve-to-memory transition possibly occurs as the production of LTa and TNFa by memory CD27+ B cells was high and comparable to that of healthy controls, whereas the production of IL10, normally expressed by naïve CD27− B cells was reduced (206).
Double negative (IgD−CD27−) B cells and ABCs (CD11c+CD21− or CD21low) are increased in a proportion of MS patients before the age of 60 years, whereas they are mainly found above 60 in healthy controls. This increase is positively correlated with age in healthy controls but not in MS patients and with CD4+CD28− T cell numbers in all subjects (63, 67). Remarkably, double negative B cells and ABCs are also increased in the CSF of MS patients (67). Double negative B cells from MS patients have a higher activation potential than those from controls. They are involved in antigen presentation as well as costimulation, and can produce proinflammatory cytokines (TNFa, LTa), and granzyme B after stimulation (67).
In RR/PPMS, Tregs (CD4+CD25+CD127−) levels were stable as compared to controls, although resting Tregs (CD45RA+CD25dim) were reduced, while activated Tregs (CD45RA−CD25bright) were increased in active MS (207, 208). There are some discrepancies whether Treg numbers and expression of surface markers are different between MS and control subjects (207–209). Overall, it is considered that the suppressive activity of Tregs is reduced in (RR)MS, but it seems to improve in SPMS (210–213). Several miRNAs have been found to target the TGFb pathway limiting the differentiation of CD4+ naïve T cells to Tregs. These miRNAs, however, did not affect their suppressive function (214).
CSF levels of TNFa, CXCL10 and IL8 increased with age in healthy controls, while IL10 level was the lowest in the middle age group (40-to-59-years-old). This inflection point of IL10 production possibly occurs 10–20 years earlier in MS, due to premature immunosenescence, which might correspond to disease onset. On the contrary, in MS, TNFa, CXCL10, IL8, and IL10 levels were higher than in controls, but only IL8 and CXCL10 increased with age. Moreover, there is a shift from Th1 to non-Th1 cytokine profiles in aging and MS, as the age-related increase of CXCL10 was relatively lower than for the other cytokines (41).
Circulating and CSF levels of a few markers are overall in favor of the presence of inflammaging in MS, although there are some discrepancies between studies. IL6 and TNFa are increased in the serum and the CSF in RRMS, mainly relapse, and SP/PPMS. IL6 correlates with disease duration, serum TNFa in PPMS correlates with disease progression. Serum IL10 levels were increased with remission, while CSF levels were high during relapse [reviewed by (215)].
Autophagy is increased in active RRMS, evidenced by the upregulation of ATG5 in peripheral T cells and in encephalitogenic T cells on brain autopsy samples (216). It exhibits both detrimental and protective effects dependent on the cell type. It enhances neuroinflammation by supporting autoantigen presentation by DCs and the survival of autoreactive B and T cells. Conversely it protects neuronal integrity, oligodendrocyte survival and the fragile pro/anti-inflammatory balance in astrocytes and microglia [reviewed by (217)]. However, sustained autophagy due to unresolved damage might lead to its detrimental dysregulation, paradoxical inflammasome activation and apoptosis (162, 218).
Telomeres in whole blood DNA (thus mainly PBMCs) were shorter in all MS subtypes (219) as compared to controls, and their length was negatively correlated with age. Telomere shortening was associated with a higher relapse rate, disability, and brain atrophy (220). It was predictive of transition to SPMS (220, 221).
Peripheral lymphocytes of MS patients exhibit an increased glucose demand with impaired oxidative phosphorylation, alongside mitochondrial dysfunction (marked by a reduced enzymatic activity and a decoupling of the respiratory chain) (222–224). Concurrently, oxidative stress can promote T cell activation and Th17 differentiation (225–227). Interestingly lymphocytic resistance to apoptosis might partly be due to an impaired mitochondria-mediated apoptotic deletion, as observed in CD4+CCR5+ T cells of PPMS (228).
Contrary to aging, methylation appears to be globally increased in MS [reviewed by (229)], with different methylation profiles between MS phenotypes, higher in PPMS compared to RRMS (230), but slightly higher in RRMS compared to SPMS (231). Lymphocyte signaling, T cell activation and migration were common pathways to RRMS and SPMS methylation profiles, while myeloid cell function and neuronal and neurodegenerative genes and pathways were SPMS-specific (231). Thirteen N6-methyladenosine (m6) regulatory genes were overexpressed in the CSF of MS patients as compared to healthy controls, of which 9 were negatively correlated with age. Remarkably, non-supervision consensus clustering separated RRMS and progressive MS patients in 2 distinct clusters, with higher levels of the m6 regulatory genes and m6 RNA methylation in RRMS patients (232).
miRNAs are upregulated in the CSF of mainly relapsing MS patients and associated to inflammatory (NF-kB, FOXO, TNFa, TGFb), cell cycle and p53 signaling pathways (233). miR-155-5p targets SOCS1 and hence promotes Th17 and Treg differentiation, and microglia-mediated immune response through expression of IL6, TNFa and induced nitric oxide synthase (iNOS) (234, 235). It also disrupts the BBB while miR-146a-5p protects it by modulating leukocyte adhesion to endothelial cells (236, 237). Moreover, miR-146a-5p inhibits Th17 differentiation by repressing TNF receptor-associated factor (TRAF)6 and IRAK1, transducers of NF-kB (238). Remarkably, miR-150 that targets CMYB, promotes terminally effector rather than precursor memory CD8+ T cells and is also expressed in mature B cells (239, 240).
MS-related inflammatory processes influenced by immunosenescence, potentially alter the function of CNS-resident cells by promoting senescence and a pro-inflammatory phenotype, which enhances the oxidative burden, resulting in alteration of mitochondrial function and DNA integrity. Moreover, cell cycle arrest and phenotypic changes in senescent cells might affect their functions and their regenerative capacity (241).
The adult brain encloses its remyelination potential into a pool of oligodendrocyte progenitor cells (OPCs). OPCs represent 5–10% of all CNS cells, can undergo asymmetric division and migrate to the site of demyelination to differentiate into mature oligodendrocytes thereby forming new myelin sheaths. This remyelination potential naturally decreases and slows down with age [reviewed by (241, 242)]. In addition, OPCs are improperly recruited to chronically demyelinated MS lesions and fail to differentiate with disease progression, due to intrinsic and extrinsic factors (243, 244).
Intrinsic factors include age-related decline of histone deacetylation and methylation in OPCs and oligodendrocytes (by reduced HDAC class I expression), enhancing the heterochronic expression of transcriptional inhibitors [e.g., inhibitor of DNA-binding (Id)4] as well as global hypomethylation by downregulation of Dnmt1 in OPCs of aged mice (245–247). Likewise, DNA methylation of ID2/ID4 allows OPC differentiation, but their methylation levels were lower in MS lesions on human brain samples than in controls (248). Extrinsic factors from the OPC environment can also affect their differentiation. Unlike induced pluripotent stem cell (iPS)-derived neural progenitor cells (NPCs) from age-matched healthy controls, NPCs from PPMS patients expressed senescence markers (p16INK4, p53, increased senescence-associated beta-galactosidase activity), and failed to induce OPC differentiation. This was reversed by treating the NPCs (and not the OPCs) with rapamycin or a blocking antibody against high-mobility group box (HMGB)1, a mediator of neuroinflammation in the SASP of NPCs (249, 250).
Moreover, immune reactive OPCs can contribute to neuroinflammation and to their own functional impairment in demyelinating conditions, as they express IL1b, MMP9, MHC-I/II, and immunoproteasome genes, facilitating the early disruption of the BBB, the recruitment of activated immune and glial cells and their production of cytokines (e.g., IL6 by astrocytes) (242, 251–254). They are also involved in neuronal cytotoxicity, by enhancing glutamatergic transmission through IL1b or dysregulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors through IFNg, directly or indirectly by inducing lymphocytic cytokines (255–257). Inversely, the SASP of the surrounding cells can interfere with OPC differentiation (258).
Microglia, maintained in a quiescent state by TGFb (259) and inhibitory ligand-receptor interactions with neurons, astrocytes, and oligodendrocytes, scan their environment through their ramifications, for danger signal and can sense extracellular ATP/UDP changes mirroring neuronal or astroglial injury/activity. Activated microglia will transiently change into pro-inflammatory subsets, particularly during myelin clearance, which sustains inflammation and hinders remyelination, while regulatory subsets support neuroprotection. However, their physiological age-related functional changes decrease their reparative ability toward CNS damage [reviewed by (260)].
Since microglia have a relatively long lifespan and a slow turnover rate, they are more prone to accumulate DNA damage and experience changes during aging [reviewed by (261)]. The motility and ramifications of microglia are reduced, and their sensome gene expression profile changes with age, delaying their recruitment to site and reducing their ability to sense their surroundings. Moreover, aged microglia are chronically activated and exhibit an elevated immunoreactivity and an exaggerated pro-inflammatory response, the so-called microglial priming. TLRs and advanced glycan-end products are upregulated while immune-suppressive factors (CD200R, CX3CR1) are downregulated, enhancing the expression of MHC-II, pro-inflammatory cytokines (IL1, IL6, TNFa), and the production of ROS/reactive nitrogen species (RNS) (by overexpressing NOS and NADPH oxidase) (262–265). Conversely, the age-related increase in TGFb levels, with senescence promoting roles [reviewed by (266)], induces changes in aging microglia that interfere with their ability to acquire a regulatory phenotype and to promote OPC differentiation (267). Moreover, activated microglia initiate a TNFa-mediated synaptic degeneration, and reciprocally influence astrocytes through TNFa and ATP to prompt the astroglial release of glutamate (257, 268).
With age, the phagocytic activity of microglia declines, impairing the clearance of myelin debris and delaying remyelination [reviewed by (269)]. Furthermore, as lysosomal degradation and cholesterol efflux are defective, lipofuscin granules (insoluble aggregates of myelin) accumulate, which increase inflammasome signaling and protein expression (270, 271).
Although microglia and macrophages are phenotypically related and complement one another in MS pathogenesis, their age-related changes partially differ. Aging macrophages are deficient in phagocytosis and chemotaxis, as microglia. Contrarily to microglia, they lose their pro-inflammatory and regulatory functionality (i.e., reduced activation of NF-kB, downregulation of TLR4, TNFa, IL6) [reviewed by (269)]. Interestingly, transferring young macrophages into an aging demyelinating brain enhanced remyelination (272).
Astrocytes are part of the CNS innate immune system and participate to demyelination by impairing the BBB, by controlling the passage of immune cells through the BBB (cellular adhesion molecules), by attracting peripheral immune cells and resident CNS cells to the lesion site (chemokines), by guiding T cell phenotypes, by inducing B cells (BAFF), by modulating microglial recruitment and function, and by acting as APCs. Although astrocytes can prevent excitotoxicity by glutamate uptake, they can worsen it by secreting several cytotoxic factors (ROS, RNS, glutamate, ATP) in response to IFNg and IL1b stimulation. Furthermore, TNFa downregulates glutamate receptors in astrocytes, thus elevating the extracellular levels of glutamate, which is directly toxic to oligodendrocytes, axons and neurons. Astrocytes further secrete fibroblast growth factor 2 (FGF2) and produce glycosaminoglycan hyaluronan, which promote OPC proliferation instead of their differentiation, and produce chondroitin sulfate proteoglycans, ephrins, and myelin-associated inhibitors, which inhibit axonal growth. The glial scars formed by reactive astrogliosis try to contain the demyelination by surrounding the damaged area, but their rigidity hinders remyelination and axonal regeneration [reviewed by (273)].
In aging astrocytes, the overexpression of the intermediate filaments, glial fibrillary acidic protein (GFAP) and vimentin, parallels increased p16INK4 expression and cell cycle arrest. Moreover, TGFb1 and HMGB1 induce pro-inflammatory cytokines (IL6, TNFa, IL1b, prostaglandins) and chemokines constituting the SASP of aging astrocytes (274, 275). Interestingly, EAE improves by blocking HMGB1 in the CNS (276). Furthermore, during EAE, oxidative stress sustains excitotoxicity (273, 277).
While 80–85% of patients present with RRMS at a younger age, the relapse rate is reduced with aging. Moreover, the post-relapse recovery potential decreases with age. The decline in white matter integrity and neuro-axonal reserve might precipitate the onset of progression, and increase the risk of accumulating disability (278–281). It is now established that subclinical neurodegeneration starts long before clinical progression becomes more evident, explaining the occurrence of progression independent of relapse activity (PIRA) in earlier phases of the disease (282). Therefore, according to natural history studies, up to 50% of RRMS patients might transition to SPMS, 15–20% present with disability progression from onset (PPMS) (18). Remarkably, both PPMS and SPMS onset occurs on average around the age of 45 years. Transition to SPMS happens independently of the duration of the prior relapsing course (283). Aging and underlying senescence might therefore, at least partially, be involved in the evolution and pathogenesis of the disease.
The CNS inflammatory infiltration and acute axonal injury are negatively correlated with age, while in inactive progressive MS, the CNS inflammation declines to the same level as in healthy controls (284). While RRMS is characterized by a disrupted BBB allowing the invasion of the CNS by peripheral immune cells, progressive MS is characterized by a compartmentalized CNS inflammation, behind a closed BBB [reviewed by (285, 286)]. Follicle-like structures, enriched in B and plasma cells, form in the meninges and these cells have a higher relative contribution within the infiltrates (287). Perivascular and parenchymal T/B cell infiltrates are limited. New active lesions are infrequent. Slowly expanding white matter lesions, also called smoldering lesions, with low-grade myelin destruction and axonal degeneration, are formed by a moderate lymphocytic infiltration and a dense network of reactive astrogliosis in their center (288, 289), surrounded by activated microglia and macrophages forming a narrow rim (290). Cortical lesions are frequent and are also mainly caused by activated microglia, resulting in synaptic and neuronal loss (285, 286). In the normal appearing white matter, the proinflammatory state induces microglial and astrocytic activation resulting in diffuse axonal injury (291).
During the progressive phase of the disease, the oxidative burst by activated microglia is prominent (285). Iron accumulates with age in the brain and is stored with ferritin in oligodendrocytes. The oligodendrocytes, harmed by inflammation/oxidation, release ferrous iron. Ferrous iron (Fe2+) reacts with H2O2 to form ferric iron (Fe3+) and a hydroxyl radical, which increases the oxidative stress (285). Ferric iron is incorporated by microglia and macrophages at the active lesion margins, forming the magnetic rim lesions detectable by MRI in about 50% of the cases (290). This iron uptake causes dystrophy of macrophages and microglia, leading to the secondary release of iron and fueling the oxidative stress. Although autophagy is increased in progressive MS in an attempt to ensure cellular homeostasis, it is not enough to compensate the mechanisms at play in the periplaque environment causing cellular senescence (216, 285). Moreover, the oxidative stress results in and is subsequently amplified by mtDNA damage and mitochondrial dysfunction of the respiratory chain complexes. Furthermore, synaptopathy, which happens also in normal aging brain, is caused by reduced neurotrophic factors and excitotoxicity resulting from a glutamatergic/gamma-aminobutyric acid (GABA)-ergic imbalance, as well as by pro-inflammatory cytokines (IL1b, IL6, TNF) of activated microglia, astroglia, and infiltrating lymphocytes (257, 292, 293). IL1b can also alter synaptic plasticity. Both are exacerbated by neuroinflammation and accelerated with age during MS [reviewed by (293)]. These features contribute to neurodegeneration and translate at the macroscopic level into accelerated brain atrophy, which can be viewed as premature aging of the MS brain, at a rate of 0.7–1% per year, compared to 0.1–0.3% per year in healthy subjects (285, 294, 295).
Exosome-associated miR-15b-5p/23a-3p/30b-5p/223-3p/342-3p/374a-5pand miR-432-5p/433-3p/485-5p are, respectively, up- and downregulated in RRMS vs. PP/SPMS (296). Interestingly, miR-15b-5p and -23a-3p are predicted to target FGF2, a promoter of OPC migration present in active and in the periphery of chronic lesions and elevated in the CSF of RR/SPMS (297–299). miR-342-3p is required for NF-kB induction in TNFa-activated microglia (300). miRNAs dysregulated in cortical lesions as compared to myelinated gray matter, are involved in axonal guidance, TGFb, and FOXO signaling. Furthermore, miR-20a/25/29c/149* are associated to gray matter atrophy (301).
In summary (Table 1, Figures 1B, 2), with disease progression, the involvement of the peripheral immune system becomes secondary, while increasing oxidative stress, sustained by the pro-inflammatory phenotype of glial cells, is the major mechanism causing mitochondrial dysfunction in all CNS-resident cells, inducing their complete functional decline (impaired clearance of myelin debris, impaired remyelination, energy failure, loss of neurotrophic support, release of neurotoxic factors), resulting in irreversible neurodegeneration.
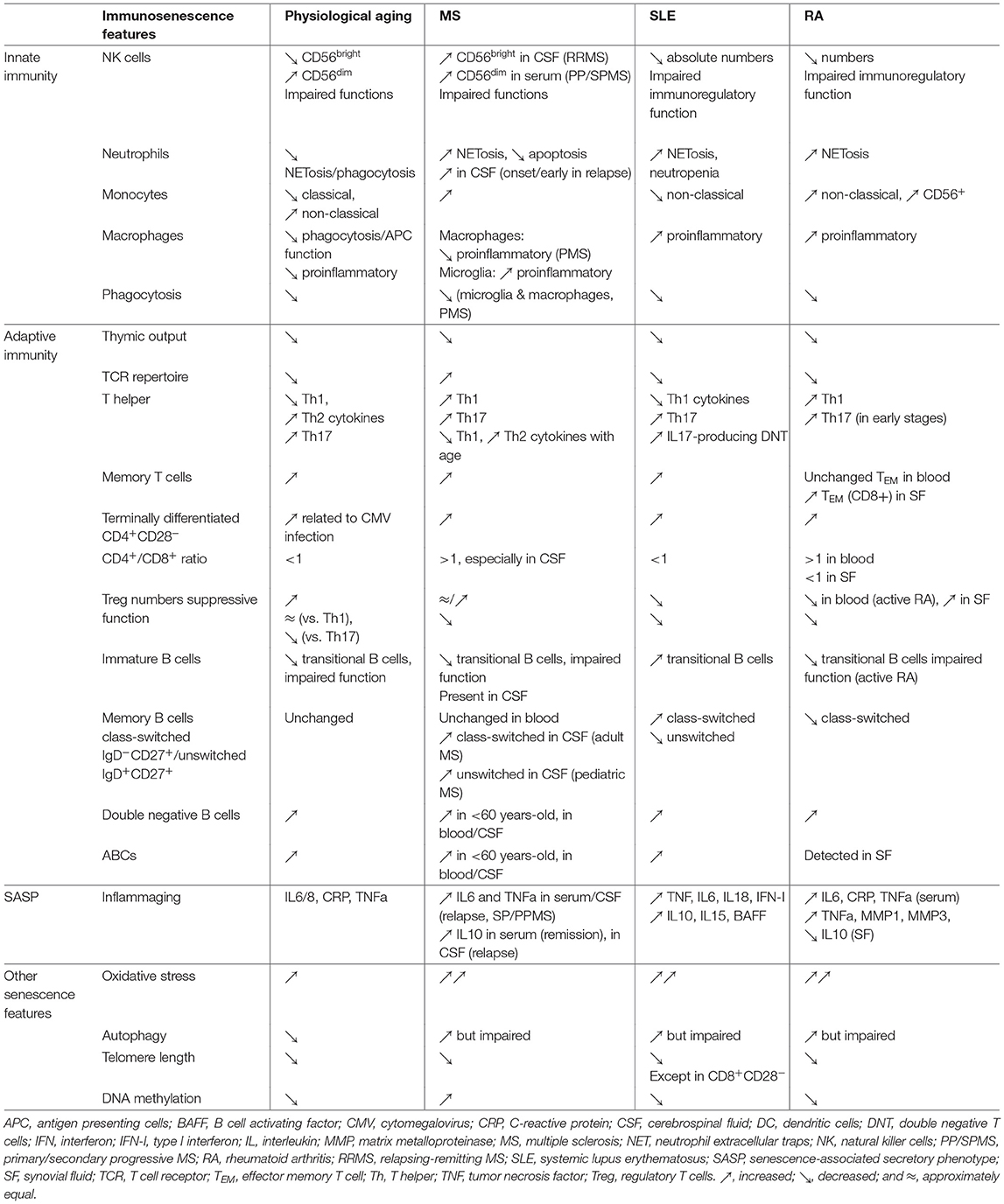
Table 1. Comparison between immunosenescence features in aging, SLE, RA, and MS.
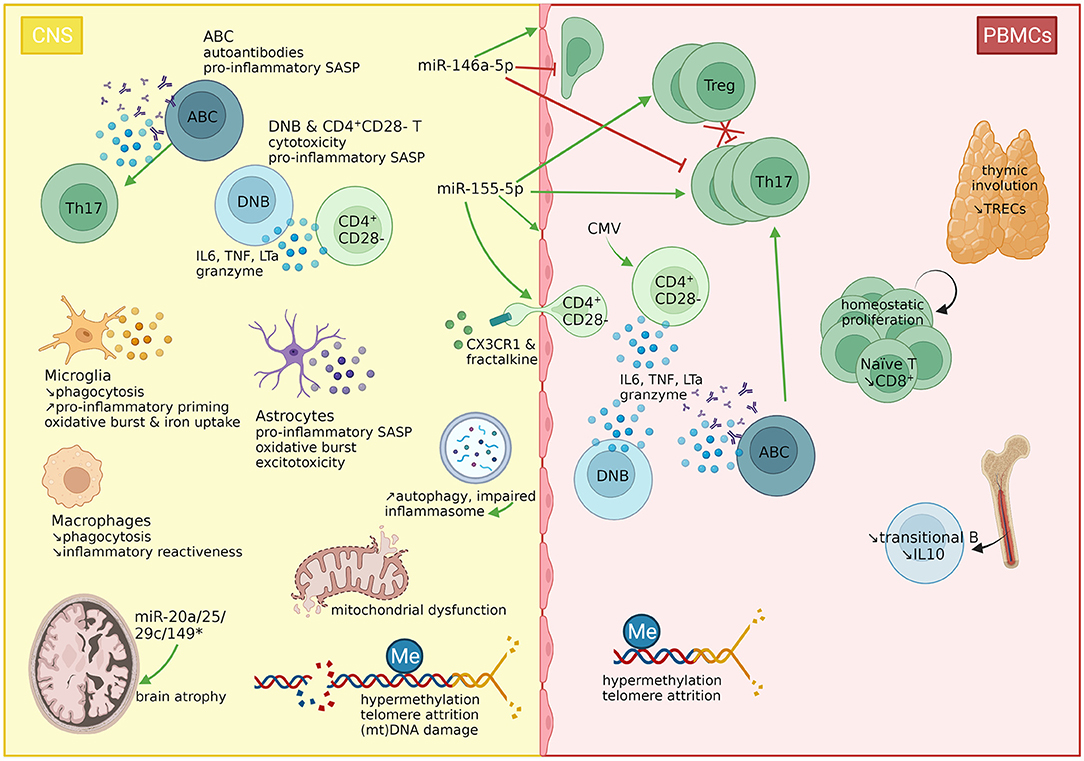
Figure 2. immunosenescence features in multiple sclerosis. Features of immunosenescence have been described in MS. The thymic involution (measurable by reduced TREC levels) induces the homeostatic proliferation of T cells. However, naïve T cells rapidly differentiate into memory T cells due to antigenic stimulation. While Th17 cells, and to a lesser extent, Tregs are expanded in the periphery, the latter fail to suppress Th17 cells. A subset of CD4+ T cells has lost the costimulatory signal CD28, marking the T cell exhaustion mainly through sustained CMV stimulation. These cells express the CX3CR1 receptor, favoring their migration through the BBB, as their ligand, fractalkine, has been found overexpressed in the cerebrospinal fluid (CSF). The B cell compartment is characterized by a reduction in immunoregulatory transitional B cells, but an increase in double negative B cells (DNBs) and ABCs. These three subsets (CD4+CD28− T cells, DNBs, ABCs) have been linked to immunosenescence and detected in the CNS of MS patients. CD4+CD28− T cells and double negative B cells produce TNFa, and granzyme B. CD4+CD28− T cells produce also IL6, and double negative B cells produce LTa, hence corresponding to the senescence-associated secretory phenotype of these cells. ABCs produce TNFa and autoantibodies and polarize Th17 cells. In the CNS, both microglia and macrophages have impaired phagocytic properties, but while microglia are primed, macrophages lose their inflammatory reactiveness. Microglia further produce ROS and incorporate iron. Astrocytes produce also proinflammatory cytokines and ROS and release glutamate, inducing excitotoxicity. The oxidative burst causes mitochondrial dysfunction, and (mitochondrial) DNA damage. Autophagy is increased but impaired, which might induce the inflammasome. Moreover, telomeres shorten with age and disease progression. Interestingly, hypermethylation is a common feature in PBMCs from MS patients, found in several subsets as well as in brain tissues, while hypomethylation has on the contrary been linked to aging. Finally, miR-146a-5p and miR-155-5p are two major immuno-microRNAs with opposite effects on the integrity of the BBB, T cell migration and the differentiation of Th17 cells. Herein, miR-155-5p displays pro-inflammatory characteristics, but also supports the differentiation of Tregs. Four miRNAs (miR-20a/25/29c/149*) have been linked to brain atrophy. CMV, cytomegalovirus; CNS, central nervous system; CX3CR1, C-X3-C Motif Chemokine Receptor 1; B, B cell; ABC, age-associated B cell; DNB, double negative B cell; IL, interleukin; LTa, lymphotoxin A; Me, methylation; mt, mitochondrial; MS, multiple sclerosis; PBMCs, peripheral blood mononuclear cells; SASP, senescence-associated secretory phenotype; T, T cell; Th, T helper cell; TRECs, T cell receptor excision circles; Treg, regulatory T cell; TNF, tumor necrosis factor. Created with BioRender.com.
Disease modifying therapies (DMTs) are efficient to reduce clinical relapses and radiological disease activity in active MS. However, due to the predominant CNS-restricted inflammation concurrent to neurodegeneration, treatments for progressive MS remain scarce, possibly more effective to patients with superimposed active inflammation [reviewed by (302)]. Moreover, since clinical trials classically exclude patients over the age of 55 years, the safety and efficacy of DMTs in older MS patients is still debated, while these patients represent a growing proportion of the MS population (17, 302). In patients younger than 40.5 years, high-efficacy drugs (ocrelizumab, mitoxantrone, alemtuzumab, and natalizumab) initiated without delay, were more powerful than lower-efficacy drugs (fingolimod, dimethyl fumarate, interferon-beta, teriflunomide, and glatiramer acetate), but may already lose their benefits on disability progression after that age. However, this model could not distinguish benign from active MS courses. The same meta-analysis by Weideman et al. found DMT efficacy to be negatively correlated with age and predicted no efficacy of DMTs after the age of 53 years (303). Moreover, the intrinsic effects of DMTs on immune cells in addition to the age-related changes in the immune system might become deleterious for remyelination and immunosurveillance with age, since DMTs deplete, sequestrate or functionally impair lymphocytes (191, 304–307). Discontinuing DMTs in the elderly might be reasonable for these reasons, however, studies are sparse. Herein, stable patients discontinuing DMTs experienced the same time to relapse as patients still on DMTs but a shorter time to disability progression. The latter was also correlated with age. However, this study included MS patients from 18 years and older, and did not focus specifically on the elderly MS patients (308). For patients older than 45 years, a 4-year relapse-free disease course under DMTs was predictive of absence of relapse following DMT discontinuation while longer disease duration and higher EDSS were predictive of disability progression (309).
Since DMTs directly act on the immune system, there has always been a major concern about risk for cancer and infections (191, 310). While some found initially a slightly increased risk for cancer (e.g., urogenital, breast, CNS cancers, lymphomas, melanomas) (311, 312), the overall risk considering all current DMTs is not increased [reviewed by (313)], although a higher incidence of neoplasm with depletive DMTs (alemtuzumab, cladribine, ocrelizumab) was found by a meta-regression analysis, especially after the age of 45 years (314). Switching from DMTs, especially more than twice, was also a risk factor for cancer (311). Furthermore, awareness is raised for the possible link between natalizumab, fingolimod, cladribine, or alemtuzumab and several types of immune malignancies, melanomas, carcinomas etc. [reviewed by (313)].
Natalizumab, dimethyl fumarate, and fingolimod increase the risk of progressive multifocal leukoencephalopathy (PML) caused by JC-polyomavirus. The risk related to natalizumab remains for several months after switching to another DMT, which explains carry-over cases as seen with ocrelizumab, fingolimod, or teriflunomide. Age, later age at DMT initiation (>50 years), prior immunosuppressive treatment, and lymphopenia, particularly inside the CNS, are important risk factors for PML, although cases with normal blood lymphocyte count have been reported (310, 315–317).
Other opportunistic infections increasing with age can be cryptococcal meningitis and herpes encephalitis (fingolimod, natalizumab) (318, 319), mucocutaneous herpes infection [sphingosine-1-phosphate receptor modulators, natalizumab, alemtuzumab], and varicella-zoster reactivation [sphingosine-1-phosphate receptor modulators, dimethyl fumarate (320), natalizumab, cladribine, alemtuzumab, ocrelizumab], human papilloma virus (fingolimod), and Listeria meningitis (alemtuzumab) [reviewed by (310, 321)].
Most DMTs act through different mechanisms on the subsets of the adaptive immune system that might undergo immunosenescence in parallel. For example, DMTs have been found to differentially affect the thymic and bone marrow output after treatment initiation but also in an age-related fashion. Both are, respectively, measured through TRECs, which decrease with age in healthy control, and K-deleting recombination excision circles (KRECs), which remain stable. While TREC levels did not change between DMT-naïve and DMT-discontinued patients, KREC levels were significantly enhanced in the latter. Interestingly, fingolimod that sequestrates lymphocytes in lymph nodes, showed a reduction in thymic and bone marrow output at 6 and 12 months after treatment initiation while the opposite was observed in natalizumab, that sequestrates lymphocytes in blood vessels, apart from the inflammation site. With the immunomodulatory IFNb both thymic and bone marrow output were stable within the first months, while KREC levels did not further decrease with age, contrary to what was observed in patients with fingolimod or natalizumab. With alemtuzumab, which temporarily depletes peripheral lymphocytes to induce their repopulation, only KREC levels increased following treatment and both parameters further remained stable with age (322).
In summary, the MS population is growing older, concurrently accumulating the comorbidities related to aging. The efficacy and safety of DMTs seem to decrease with age, although robust data are still missing. DMTs might increase the risk of opportunistic infections and cancers in the elderly by changing immune cell population distributions and by affecting the functions of the immune system, hence possibly promoting certain immunosenescence features, in combination with MS-related premature immunosenescence.
SLE pathogenesis is characterized by Th17 polarization, autoreactive B cells producing autoantibodies targeting nucleic acid-bound antigens and the innate immune system providing a strong IFN-I signature (323). The tissue damage in SLE, resulting in organ dysfunction (e.g., kidney, brain, lungs, cardiovascular system) corresponds to premature biological aging. The immune dysregulation in SLE presents some features resembling immunosenescence, mainly in the adaptive immune system, but underlying mechanisms might be different. In the innate immune system, the effects of SLE and aging appear to be more divergent [reviewed by (20)].
Unlike immunosenescence, SLE-derived neutrophils primed by IFNa and autoantibodies produce more ROS and are engaged in NETosis, which causes tissue damage and partly explains the neutropenia observed in this disease (324–326). Moreover, the non-classical monocytes (CD14+CD16++), present in a decreased proportion in SLE contrarily to aging, display a reduced phagocytosis capacity, but an increased expression of TLR, TNFa and IL10 (20, 327). Macrophages contribute to SLE by their defective phagocytosis of apoptotic cells, their polarization toward a proinflammatory phenotype, and an aberrant activation of their autophagy and inflammasome machinery (328). The imbalance between the decreased immunoregulatory (CD56bright) and the increased pro-inflammatory function (CD56dim) of NK cells is correlated with disease activity, although their relative frequencies are unchanged, while their absolute numbers are decreased (329, 330). Moreover, increased serum levels of IFNa in active SLE parallel the frequency of IFNg-producing NK cells (330) [as seen in a TNFa/IL12-mediated viral infection response (331, 332)].
Thymic output and TCR repertoire are reduced in SLE (333, 334). SLE patients often exhibit the key features of the immune risk phenotype, an inverted CD4+/CD8+ ratio, due to CD8+ T cell expansion and a higher CD4+ T cell turnover (335). Th17 and IL17-producing double negative T cells are involved in SLE pathogenesis [reviewed by (336)]. A dominant granzyme-producing CD8+ T cell population is found in patients with severe nephritis, leucopenia, and clinically active disease (337, 338). Expanding CD4+CD28− T cells produce IFNg in moderately active SLE and are positively correlated with the clinical disease score (339). Conversely, some autoreactive CD8+CD28− clones secrete less IFNg and comparatively, relatively more IL10 but with impaired suppressive capacities (340). Remarkably, TCR signaling is driven by the FCgR chain in SLE rather than the TCR-zeta chain, due to its altered composition, resulting in a lower activation threshold, higher calcium influx, increased excitability and baseline stimulation [reviewed by (341)].
Contrary to aging, there is a shift toward immature B cells in SLE, due to a two-fold increase in transitional B cells with a defective tolerance checkpoint resulting in autoreactive B cells producing autoantibodies (342, 343). Frequent cycles of B cell activation and differentiation shape peripheral B cells, marked by an expansion in switched (IgD−CD27+) memory B cells and double negative (IgD−CD27−) B cells, as well as a subset of activated memory B cells (IgD−CD27−CD95+CD21−), the latter being increased during disease flares (344–347). The CD11highTBET+ ABCs as well as two ABC-like subtypes, found in African American patients, have been linked to disease severity (348, 349). Double negative type 2 CXCR5− cells with a unique cytokine, cytokine receptor, transcription factor and signaling factor expression profile, are increased in young patients, and do not further expand with age (350).
Data on the number and function of Tregs in SLE are very disparate, mainly due to study population and staining strategy differences. However, Tregs appear to be largely outpaced by the T and B cell activation in SLE, possibly due to the decrease in IL2 production, that mediates Treg homeostasis, and the increase in IL6 production, that induces effector T cell activation. Furthermore, the Th17/Treg ratio increases in SLE alongside the decrease of TGFb. Finally, the effect of IL10 remains unclear as it has both anti-inflammatory effects (when produced by Tregs and type 1 T-regulatory cells) and it induces autoreactive B cell proliferation (when produced by monocytes and B cells) [reviewed by (351–353)].
The binding of immune complexes induces the production of pro-inflammatory inflammaging-associated cytokines, such as TNF, IL6, and IL18 by monocytes/macrophages, but also of IFN-I by plasmacytoid DCs, and immunoregulatory cytokines IL10, IL1, and BAFF. Interestingly, they have been linked to disease activity, while CRP for instance is not [reviewed by (354)].
Contrary to aging, autophagy is increased in SLE, as highlighted by the increased autophagy-associated markers in naïve CD4+ T cells (355). Moreover, the autophagosome density of B cells is positively correlated with disease activity (356). However, while autoantibodies from lupus patients can induce autophagy in T cells of healthy controls in vitro, T cells from SLE patients are resistant to it (355).
Overall, telomere length is shorter compared to aged-matched healthy controls in SLE patients, with the reduction being even more pronounced in younger subjects, and without the typically progressive age-related decline seen during physiological aging. On the contrary, telomerase activity is increased in T and B cells. However, this fails to compensate for the accelerated telomere attrition in T cells (357, 358). Telomerase activity, but not telomere length, is positively correlated with disease activity (357). Unlike what is observed during aging, CD8+CD28− T cells in SLE have longer telomeres, an increased telomerase activity and a preserved proliferative potential (359).
Activated SLE T cells produce an excess of ROS and RNS and deplete the glutathione reserve, which leads to mitochondrial dysfunction (166, 360, 361). Oxidated DNA and mtDNA damage are correlated with the high serum levels of cytokines (IL10, IL23, IFNa, IFNg) and chemokines (CXCL10 and MCP1) (362). Remarkably, only higher mtDNA damage levels are related to disease duration (363, 364). Moreover, peroxynitrite-modified histones, due to amino-acid nitration by RNS, induce high titers of anti-histone antibodies and UV-induced DNA damage potentially induces IFN-I (365, 366).
DNA methylation levels are globally decreased in T cells from SLE patients. Herein, the hypomethylation of interferon signature genes [i.a. interferon regulatory factor (IRF)5, IRF7] is a hallmark of SLE pathogenesis and immune response genes are associated with chromatin remodeling (e.g., trimethylation of H3K4) (367–369). Moreover, nitration of the protein kinase C (PKC/ERK pathway) inhibits its delta catalytic activity, resulting in decreased activity of DNMT1 and thus low methylation levels in CD4+ T cells allowing the transcription of CD70, possibly CD11a, and perforin (370–372). Interestingly, miR-21/29b/126/148, which are overexpressed in CD4+ T cells from SLE patients, downregulate DNMT1 (373–375), while miR-199a-5p increased splenic CD4+ T cell senescence by inhibiting SIRT1 and thus increasing the acetylation and consequently the activation of p53 (376). Upregulation of miR-7 and -30a in B cells of SLE ensures B cell proliferation, differentiation to plasma cells and antibody production (377, 378), while miR-15a activates the apoptosis of Bregs by targeting BCL2 (379, 380). Furthermore, miR-142 downregulation by histone and DNA methylation of its regulatory region, results in the activation of T cells, hyperstimulation of B cells and suppression of Treg function (381, 382). miR-146a inhibits and miR-155 enhances IFN-I (383, 384). Finally, miR-125a stabilizes Treg-mediated homeostasis but is downregulated in SLE, and mir-31 and -34a, induced by the NF-kB pathway, target FOXP3 (385–388).
RA is likely due to a systemic immune dysregulation, possibly driven by DCs and macrophages that present citrullinated antigens to autoreactive T cells, inducing the production of pro-inflammatory and joint damaging mediators and causing synovial inflammation, and articular and extra-articular tissue damage (389, 390). A premature senescent phenotype of immune cells has been evidenced in RA and partly linked to its pathogenesis [reviewed by (21)].
Neutrophils are involved in generating citrullinated autoantigens that are afterwards externalized by NETosis, while anti-citrullinated protein antibodies promote NETosis. Neutrophils in joints also produce cytokines and ROS and release proteases by degranulation (391). Intermediate (non-classical) pro-inflammatory CD14+CD16+ monocytes were similarly increased in elderly subjects with atherosclerosis and middle-aged RA patients as compared to young healthy controls (392, 393). Young RA patients have also a higher frequency of CD56+ monocytes, producing more TNFa, IL10, IL23, and ROS, although this is normalized by TNFa blocking therapy. Interestingly, in RA patients, the age-dependency of circulating CD56+ monocytes is lost (394). Overall, macrophages drive joint inflammation in RA by secreting cytokines, chemokines and tissue degrading enzymes, activating fibroblast-like synoviocytes (FLSs) and promoting T cell infiltration and osteoclastogenesis. However, it is currently unknown how the heterogeneity between infiltrating monocyte-derived and tissue-resident macrophages might impact disease pathogenesis (395). Furthermore, in RA, the NK cells are reduced in number and functionally impaired, seen their increased production of ROS and proinflammatory cytokines, hence hindering their immunoregulatory properties (396, 397).
The alteration in T cell homeostasis occurs early in RA and is independent of disease duration (398, 399). The proliferative capacity of CD34+ HSCs in the bone marrow is reduced due to a decreased ERK signaling pathway. Both CD34+ HSCs and naïve CD4+ T cells from RA patients are more susceptible to apoptosis, hence reinforcing the burden on homeostatic proliferation in the periphery (398, 400–402). RA patients have a thymic output of healthy individuals aged 20–30 years older. TREC levels are already lower than normal in young RA patients (159, 399). The TCR repertoire is prematurely contracted in both naïve and memory T cells (403). The CD4+/CD8+ ratio is increased in the blood, and inverted in the synovial fluid (404). Th17 cells are possibly increased in the blood of RA patients and might be more important at early stages of the disease [reviewed by (405)]. Memory T cells are unchanged in the periphery as compared to controls. However, effector memory CD8+ T cells are increased in the synovial fluid (404).
Circulating CD4+CD28− T cells produce higher levels of TNFa and IFNg than the CD28+ T cells. Their rate is correlated with disease severity and the extent of extra-articular manifestations (406, 407). These cells easily react to neoantigens as they express de novo NK receptors (CD56, NKG2D) in RA patients (408, 409). Interestingly, in vitro generated CD56+CD28− T cells by repeated stimulation of CD56−CD28+ T cells of young healthy donors, expressed BCL2, p53, and p16INK4 that induce cell cycle arrest, a hallmark of cellular senescence, and activated the NF-kB pathway (410). Since they also express CX3CR1, they can migrate into the synovial fluid where FLSs express the ligand fractalkine, induced by TNFa and IFNg. This interaction activates the CD28− T cells, induces the expression of pro-inflammatory cytokines and facilitates the proliferation of FLSs and the secondary activation of T cells (411). The expansion of CD4+CD28− T cells in RA patients has been associated with the HLA-DR4 risk factor and a TNFa polymorphism, alongside increased TNFa and IFNg production (159, 412).
The effect of senescent B cells is not well-described in RA (21). However, transitional B cells are reduced and have impaired functions in PBMCs of active RA (413). Moreover, double negative B cells were increased in the periphery and ABCs have been detected in the synovial fluid of RA patients (414, 415).
Although discrepancies exist, Tregs appear to be reduced among the PBMCs in active RA, and normalized during remission as compared to controls (416, 417). In the synovial fluid they are overall increased but functionally impaired (417, 418). Interestingly, a novel subset of senescent CD28− Treg-like cells, characterized as CD4+FOXP3+CD28−, was discovered in the blood and synovial fluid of RA patients. They express markers such as CD25, cytotoxic T-lymphocyte-associated protein (CTLA)4 and FOXP3, and also exhibit premature senescence, as shown by reduced TREC levels and an accumulation of phosphorylated gamma-H2AX (upon DNA double-strand break). Moreover, their SASP consists of high levels of pro- (TNFa, IFNg, IL2, IL4, IL17) and anti-inflammatory (IL10) cytokines. However, CD28− Treg-like cells had also impaired suppressive capacities. They could be obtained in vitro by stimulating CD28+ Tregs with TNFa. Although CD28− Tregs numbers correlated with age, nor CD28− nor CD28+ Tregs correlated with disease duration and clinical features (419).
Similarly to inflammaging in the elderly, RA patients have increased systemic levels of pro-inflammatory cytokines (IL6, CRP, TNFa) (420). In the synovial fluid TNFa, MMP1, and MMP3 levels were increased in early and established RA, IL10 decreased during established RA only as compared to osteoarthritis (421). Moreover, RA-derived FLSs produce more IL6, IL8, vascular endothelial growth factor, and prostaglandin E2 in response to IL1b during in vitro induced senescence (422). Both TNFa and IL6 play a major role in activating effector cells, inducing cytokine/chemokine/autoantibody production and tissue damage [reviewed by (389)].
While autophagy decreases with age, it is increased in RA FLSs due to stress-induced endoplasmic reticulum hyperactivity and an elevated protein turnover, but the ubiquitin-proteasome system is impaired in RA (423, 424). This altered proteostasis may enhance inflammation.
The increased telomerase activity of HSCs is insufficient to compensate for telomere shortening (400, 401). Naïve T cells fail to induce telomerase activity following antigen priming. Telomere attrition was observed in granulocytes, naïve and memory T cells (398, 399, 425). Interestingly, telomerase activity of infiltrating cells correlated with synovial lining hyperplasia, but was independent of disease duration or severity (426). The HLA-DR4 risk factor in RA induces premature immunosenescence by accelerating telomere shortening (425).
Naïve and memory T cells from RA patients have high levels of DNA double-strand breaks due to impaired DNA repair mechanisms [e.g., reduced DNA repair kinase ataxia telangiectasia mutated (ATM)] (427). Moreover, T cells isolated from RA patients enhance the activity of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), a DNA repair enzyme. The DNA-PKcs-Janus kinase-axis causes chronically cellular stress and intensifies the inflammatory activity of T cells (428). Cell-free mtDNA, released by tissue damage, was found in plasma and synovial fluid of RA patients. Interestingly, intra-articular injection of oxidized mtDNA in mice caused arthritis (429). MtDNA damage in RA (induced by TNFa and ROS) is positively correlated with macroscopic synovitis, and synovial TNFa and IFNg levels, but does not depend on age (430). Furthermore, p16INK4 and p16 INK4-encoding genes along with IL6 could be induced in FLSs by H2O2 or TNFa (431). Likewise, p53 was upregulated in synovial tissues from early and late-stage RA as compared to normal synovial tissue (432). Interestingly p53 mutations, secondary to chronic oxidative stress, have been detected in RA synovial tissue and promoted clonal FLSs expansion and IL6 expression (433).
Epigenetic changes in RA promote the pro-inflammatory profile involved in disease pathogenesis. Global hypomethylation, along with a decrease in active DNMT1 in the FLSs was found in RA (434). Hypomethylation in PBMCs is correlated with the disease activity score (435). The promoter gene of IL6 and TNFa is hypomethylated in PBMCs and in peripheral naïve CD4+ T cells, respectively (436, 437). The IFNg locus is hypomethylated in CD4+CD28− T cells as compared to CD28+ counterparts resulting in increased expression of IFNg and TNF in the periphery and of IL17, CXCR3, CCR6 in the synovial fluid (406). Histones are globally hyperacetylated by decreased HDAC activity in RA, in particular H3 acetylation in the IL6 promoter was increased in the FLSs (438, 439). TNFa-mediated SIRT1 overexpression in FLSs induced IL6 and IL8 expression and protected cells from apoptosis (440). Moreover, miR-16 and -146a are elevated in synovial fluid, plasma and PBMCs of RA patients and are linked to disease activity (441, 442). Interestingly, HDAC downregulation restored the expression of miR-16 (443). miR-146a and -155 were upregulated in FLSs of RA, both induced by TNFa and IL1b. While miR-155 appeared to be compensatory to joint destruction by reducing MMP1/3, the role of miR-146a in FLSs is unknown, but in PBMCs it fails to properly repress IRAK1/TRAF6 and thus the NF-kB pathway (444–446).
We have compared key features of immunosenescence occurring in physiological aging with changes of the immune system evidenced in MS, SLE, and RA (Table 1). While some features of immunosenescence are found in the 3 AIDs, others differ from physiological aging but also between them. Hence, it seems still unclear whether these findings are inherent to the disease course or causative of its pathogenesis.
In MS (Figure 2), innate immunity does not seem affected by senescence, at least in the early stages of the disease. However, the role of NK cells remains debated, and neutrophilic NETosis is increased contrarily to aging. Macrophages that are strongly involved in the neuroinflammatory processes in the early stages of the disease, lose their function with aging, while microglia remain highly primed. T and B cells display some immunosenescence features: CD4+CD28− T cells, ABCs, and double negative B cells are expanded, and exhibit properties supporting autoreactivity. However, the inversion of the CD4+/CD8+ ratio is missing. Memory Tregs are increased, but their functionality is presumably reduced. In SLE, the innate immune function strikingly differs from what is observed during aging (increased NETosis, decreased non-classical monocytes, a proinflammatory macrophage shift). However, T and B cells display immunosenescence features (CD4+/CD8+ inversion, CD4+CD28− T cell, ABC, ABC-like and double negative B cell expansion), contrarily Tregs are possibly reduced in number and function. In RA, innate immunity shows features of immunosenescence in monocytes as well as a reduced immunosurveillance by NK cells. Macrophages have reduced phagocytic properties despite actively contributing to the inflammation as do neutrophils through increased NETosis. The T cell compartment is marked by an expansion of CD4+CD28− T cells, including functionally impaired CD28− Tregs, which are possibly involved in RA pathogenesis. The CD4+/CD8+ ratio is inversed in the synovial fluid but not in the blood. Double negative B cells and ABCs have been detected in RA, but are barely characterized.
The released inflammatory mediators (e.g., IL6, IL10, TNFa) contribute to the disease pathogenesis of all 3 AIDs and even mirror disease activity in MS. In all 3 AIDS, telomeres are shortened, except in CD4+CD28− T cells in SLE, which consequently have a preserved proliferative potential. Mitochondrial dysfunction is increased as is observed in physiological aging, and dysregulated miRNAs are largely involved in inflammatory pathways. However, contrary to aging, autophagy is increased; but impaired. Distinctively, MS appears to feature a global hypermethylation, with distinct clusters between the disease subtypes, rather than the hypomethylation observed during physiological aging as well as in SLE and RA.
Immunosenescence encompasses functional and phenotypic changes within the immune system occurring naturally during aging. Among other features the resulting loss of self-tolerance, alongside inflammaging might be involved in the pathogenesis of AIDs. In this review, we have discussed and compared the similarities and discrepancies between hallmarks of immunosenescence in MS, SLE, and RA. Notably, cell types that are characteristic of immunosenescence and prone to autoreactivity, i.e., CD4+CD28− T cells, ABCs and double negative B cells are expanded in MS. Although their functional features support a possible involvement in MS pathogenesis, it is currently not clear how and to which extent they contribute to the inflammatory processes in the different stages of MS. Hence, they might only reflect the consequences of chronic inflammation rather than the cause of disease. Moreover, the self-generated and self-sustained pro-inflammatory and oxidative environment within the CNS under ongoing recruitment of inflammatory and glial cells, possibly potentiates or causes premature immunosenescence. However, with disease progression the compartmentalized CNS inflammation is also governed by a distinct cellular senescence mechanism. Oxidative stress-induced mitochondrial dysfunction within CNS-resident cells progressively and irreversibly contributes to cellular and continued tissue damage, reduced remyelination capacity, impaired brain plasticity and finally loss of neuro-axonal reserves. Further research is needed to unravel the clinical relevance of these mechanisms, in relationship to immunosenescence, to improve treatments for MS at all ages and disease stages, with an acceptable risk-benefit profile.
OP and VvP contributed to the conceptualization of this work and to the research of the current literature on the subject. OP extensively compiled current knowledge on the subject to write and correct the first draft. VvP commented, corrected, and validated the first draft. All authors contributed to the article and approved the submitted version.
OP is currently supported by the Fonds de la Recherche Scientifique (F.R.S.-FNRS, Belgium) as a research fellow (Grant No. 40005159).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ABC, age-associated B; AID, autoimmune disease; BBB, blood brain barrier; BCR, B cell receptor; CMV, cytomegalovirus; CNS, central nervous system; CSF, cerebrospinal fluid; DC, dendritic cell; DMT, disease-modifying therapy; EAE, experimental autoimmune encephalomyelitis; EBV, Epstein Barr Virus; FLS, fibroblast-like synoviocyte; HLA-DR, Human Leukocyte Antigen-DR isotype; HSC, hematopoietic stem cell; Ig, immunoglobulin; IFN-I, type I interferon; KREC, K-deleting recombination excision circles; MHC-I/II, major histocompatibility complex of class I or II; miRNA, microRNA; MS, multiple sclerosis; mtDNA, mitochondrial DNA; NET, neutrophil extracellular trap; NK, natural killer; NPC, neural progenitor cell; OPC, oligocendrocyte progenitor cell; PBMC, peripheral blood mononuclear cell; PML, progressive multifocal leukoencephalopathy; PPMS, primary progressive MS; RA, rheumatoid arthritis; RNS, reactive nitrogen species; ROS, reactive oxygen species; RRMS, remitting-relapsing MS; SASP, senescence-associated secretory phenotype; SLE, systemic lupus erythematosus; SPMS, secondary progressive MS; TCR, T cell receptor; Th, T helper cell; TREC, T cell receptor excision circles; Treg, regulatory T cell.
1. United Nations Department Department of Economic and Social Affairs Population Division. World Population Prospects 2019, Volume II: Demographic Profiles. New York, NY (2019).
2. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
3. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. (2014) 15:482–96. doi: 10.1038/nrm3823
4. Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
5. Pawelec G. Age and immunity: what is “immunosenescence”? Exp Gerontol. (2018) 105:4–9. doi: 10.1016/j.exger.2017.10.024
6. Feehan J, Tripodi N, Apostolopoulos V. The twilight of the immune system: the impact of immunosenescence in aging. Maturitas. (2021) 147:7–13. doi: 10.1016/j.maturitas.2021.02.006
7. Collado M, Serrano M. The senescent side of tumor suppression. Cell Cycle Georget Tex. (2005) 4:1722–4. doi: 10.4161/cc.4.12.2260
8. Ferguson FG, Wikby A, Maxson P, Olsson J, Johansson B. Immune parameters in a longitudinal study of a very old population of swedish people: a comparison between survivors and nonsurvivors. J Gerontol Ser A. (1995) 50A:B378–82. doi: 10.1093/gerona/50A.6.B378
9. Wikby A, Maxson P, Olsson J, Johansson B, Ferguson FG. Changes in CD8 and CD4 lymphocyte subsets, T cell proliferation responses and non-survival in the very old: the Swedish longitudinal OCTO-immune study. Mech Ageing Dev. (1998) 102:187–98. doi: 10.1016/S0047-6374(97)00151-6
10. Olsson J, Wikby A, Johansson B, Löfgren S, Nilsson B-O, Ferguson FG. Age-related change in peripheral blood T-lymphocyte subpopulations and cytomegalovirus infection in the very old: the Swedish longitudinal OCTO immune study. Mech Ageing Dev. (2001) 121:187–201. doi: 10.1016/S0047-6374(00)00210-4
11. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging: an evolutionary perspective on immunosenescence. Ann N Y Acad Sci. (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
12. Strindhall J, Nilsson B-O, Löfgren S, Ernerudh J, Pawelec G, Johansson B, et al. No immune risk profile among individuals who reach 100 years of age: findings from the Swedish NONA immune longitudinal study. Exp Gerontol. (2007) 42:753–61. doi: 10.1016/j.exger.2007.05.001
13. Franceschi C, Bonafè M. Centenarians as a model for healthy aging. Biochem Soc Trans. (2003) 31:457–61. doi: 10.1042/bst0310457
14. Arai Y, Martin-Ruiz CM, Takayama M, Abe Y, Takebayashi T, Koyasu S, et al. Inflammation, but not telomere length, predicts successful ageing at extreme old age: a longitudinal study of semi-supercentenarians. EBioMedicine. (2015) 2:1549–58. doi: 10.1016/j.ebiom.2015.07.029
15. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol. (2018) 8:1960. doi: 10.3389/fimmu.2017.01960
16. Sanai SA, Saini V, Benedict RH, Zivadinov R, Teter BE, Ramanathan M, et al. Aging and multiple sclerosis. Mult Scler J. (2016) 22:717–25. doi: 10.1177/1352458516634871
17. Vaughn CB, Jakimovski D, Kavak KS, Ramanathan M, Benedict RHB, Zivadinov R, et al. Epidemiology and treatment of multiple sclerosis in elderly populations. Nat Rev Neurol. (2019) 15:329–42. doi: 10.1038/s41582-019-0183-3
18. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. (2015) 15:545–58. doi: 10.1038/nri3871
19. Dema M, Eixarch H, Villar LM, Montalban X, Espejo C. Immunosenescence in multiple sclerosis: the identification of new therapeutic targets. Autoimmun Rev. (2021) 20:102893. doi: 10.1016/j.autrev.2021.102893
20. van den Hoogen LL, Sims GP, van Roon JAG, Fritsch-Stork RDE. Aging and systemic lupus erythematosus - immunosenescence and beyond. Curr Aging Sci. (2015) 8:158–77. doi: 10.2174/1874609808666150727111904
21. Chalan P, van den Berg A, Kroesen B-J, Brouwer L, Boots A. Rheumatoid arthritis, immunosenescence and the hallmarks of aging. Curr Aging Sci. (2015) 8:131–46. doi: 10.2174/1874609808666150727110744
23. Freund A, Orjalo AV, Desprez P-Y, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. (2010) 16:238–46. doi: 10.1016/j.molmed.2010.03.003
24. Nikolich-Žugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. (2018) 19:10–9. doi: 10.1038/s41590-017-0006-x
25. Aprahamian T, Takemura Y, Goukassian D, Walsh K. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin Exp Immunol. (2008) 152:448–55. doi: 10.1111/j.1365-2249.2008.03658.x
26. Adler AS, Sinha S, Kawahara TLA, Zhang JY, Segal E, Chang HY. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. (2007) 21:3244–57. doi: 10.1101/gad.1588507
27. Lian J, Yue Y, Yu W, Zhang Y. Immunosenescence: a key player in cancer development. J Hematol OncolJ Hematol Oncol. (2020) 13:151. doi: 10.1186/s13045-020-00986-z
28. Castle SC. Clinical relevance of age-related immune dysfunction. Clin Infect Dis. (2000) 31:578–85. doi: 10.1086/313947
29. Frasca D, Diaz A, Romero M, Blomberg BB. The generation of memory B cells is maintained, but the antibody response is not, in the elderly after repeated influenza immunizations. Vaccine. (2016) 34:2834–40. doi: 10.1016/j.vaccine.2016.04.023
30. Ramos-Casals M, García-Carrasco M, Brito MP, López-Soto A, Font J. Autoimmunity and geriatrics: clinical significance of autoimmune manifestations in the elderly. Lupus. (2003) 12:341–55. doi: 10.1191/0961203303lu383ed
31. Dixit VD. Thymic fatness and approaches to enhance thymopoietic fitness in aging. Curr Opin Immunol. (2010) 22:521–8. doi: 10.1016/j.coi.2010.06.010
32. Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci USA. (2011) 108:20012–7. doi: 10.1073/pnas.1116110108
33. Hazenberg MD, Borghans JAM, de Boer RJ, Miedema F. Thymic output: a bad TREC record. Nat Immunol. (2003) 4:97–9. doi: 10.1038/ni0203-97
34. Naylor K, Li G, Vallejo AN, Lee W-W, Koetz K, Bryl E, et al. The influence of age on T cell generation and TCR Diversity. J Immunol. (2005) 174:7446–52. doi: 10.4049/jimmunol.174.11.7446
35. Goldrath AW. Maintaining the status quo: T-cell homeostasis. Microbes Infect. (2002) 4:539–45. doi: 10.1016/S1286-4579(02)01570-8
36. Pfister G, Weiskopf D, Lazuardi L, Kovaiou RD, Cioca DP, Keller M, et al. Naive T cells in the elderly: are they still there? Ann N Y Acad Sci. (2006) 1067:152–7. doi: 10.1196/annals.1354.018
37. Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee J-Y, et al. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci USA. (2014) 111:13139–44. doi: 10.1073/pnas.1409155111
38. Haynes L, Eaton SM. The effect of age on the cognate function of CD4+ T cells. Immunol Rev. (2005) 205:220–8. doi: 10.1111/j.0105-2896.2005.00255.x
39. Eaton SM, Burns EM, Kusser K, Randall TD, Haynes L. Age-related defects in CD4 T cell cognate helper function lead to reductions in humoral responses. J Exp Med. (2004) 200:1613–22. doi: 10.1084/jem.20041395
40. Desai A, Grolleau-Julius A, Yung R. Leukocyte function in the aging immune system. J Leukoc Biol. (2010) 87:1001–9. doi: 10.1189/jlb.0809542
41. Hu WT, Howell JC, Ozturk T, Gangishetti U, Kollhoff AL, Hatcher-Martin JM, et al. CSF cytokines in aging, multiple sclerosis, and dementia. Front Immunol. (2019) 10:480. doi: 10.3389/fimmu.2019.00480
42. Lee JS, Lee W-W, Kim SH, Kang Y, Lee N, Shin MS, et al. Age-associated alteration in naive and memory Th17 cell response in humans. Clin Immunol. (2011) 140:84–91. doi: 10.1016/j.clim.2011.03.018
43. Rudd BD, Venturi V, Li G, Samadder P, Ertelt JM, Way SS, et al. Nonrandom attrition of the naive CD8+ T-cell pool with aging governed by T-cell receptor:pMHC interactions. Proc Natl Acad Sci USA. (2011) 108:13694–9. doi: 10.1073/pnas.1107594108
44. Weng N, Akbar AN, Goronzy J. CD28– T cells: their role in the age-associated decline of immune function. Trends Immunol. (2009) 30:306–12. doi: 10.1016/j.it.2009.03.013
45. Weinberger B, Lazuardi L, Weiskirchner I, Keller M, Neuner C, Fischer K-H, et al. Healthy aging and latent infection with CMV lead to distinct changes in CD8+ and CD4+ T-cell subsets in the elderly. Hum Immunol. (2007) 68:86–90. doi: 10.1016/j.humimm.2006.10.019
46. Fletcher JM, Vukmanovic-Stejic M, Dunne PJ, Birch KE, Cook JE, Jackson SE, et al. Cytomegalovirus-Specific CD4+ T cells in healthy carriers are continuously driven to replicative exhaustion. J Immunol. (2005) 175:8218–25. doi: 10.4049/jimmunol.175.12.8218
47. Pereira BI, De Maeyer RPH, Covre LP, Nehar-Belaid D, Lanna A, Ward S, et al. Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat Immunol. (2020) 21:684–94. doi: 10.1038/s41590-020-0643-3
48. Alonso-Arias R, Moro-García MA, López-Vázquez A, Rodrigo L, Baltar J, García FMS, et al. NKG2D expression in CD4+ T lymphocytes as a marker of senescence in the aged immune system. Age Dordr Neth. (2011) 33:591–605. doi: 10.1007/s11357-010-9200-6
49. Vallejo AN, Schirmer M, Weyand CM, Goronzy JJ. Clonality and longevity of CD4+CD28null T cells are associated with defects in apoptotic pathways. J Immunol. (2000) 165:6301–7. doi: 10.4049/jimmunol.165.11.6301
50. Thewissen M, Somers V, Hellings N, Fraussen J, Damoiseaux J, Stinissen P. CD4+CD28null T cells in autoimmune disease: pathogenic features and decreased susceptibility to immunoregulation. J Immunol. (2007) 179:6514–23. doi: 10.4049/jimmunol.179.10.6514
51. Fann M, Chiu WK, Wood WH, Levine BL, Becker KG, Weng N-P. Gene expression characteristics of CD28null memory phenotype CD8+ T cells and its implication in T-cell aging. Immunol Rev. (2005) 205:190–206. doi: 10.1111/j.0105-2896.2005.00262.x
52. Broux B, Pannemans K, Zhang X, Markovic-Plese S, Broekmans T, Eijnde BO, et al. CX3CR1 drives cytotoxic CD4+CD28– T cells into the brain of multiple sclerosis patients. J Autoimmun. (2012) 38:10–9. doi: 10.1016/j.jaut.2011.11.006
53. Scholz JL, Diaz A, Riley RL, Cancro MP, Frasca D. A comparative review of aging and B cell function in mice and humans. Curr Opin Immunol. (2013) 25:504–10. doi: 10.1016/j.coi.2013.07.006
54. Bulati M, Caruso C, Colonna-Romano G. From lymphopoiesis to plasma cells differentiation, the age-related modifications of B cell compartment are influenced by “inflamm-ageing”. Ageing Res Rev. (2017) 36:125–36. doi: 10.1016/j.arr.2017.04.001
55. Jin R, Kaneko H, Suzuki H, Arai T, Teramoto T, Fukao T, et al. Age-related changes in BAFF and APRIL profiles and upregulation of BAFF and APRIL expression in patients with primary antibody deficiency. Int J Mol Med. (2008) 21:233–8. doi: 10.3892/ijmm.21.2.233
56. Ueda Y, Yang K, Foster SJ, Kondo M, Kelsoe G. Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. J Exp Med. (2004) 199:47–58. doi: 10.1084/jem.20031104
57. Riley RL. Impaired B lymphopoiesis in old age: a role for inflammatory B cells? Immunol Res. (2013) 57:361–9. doi: 10.1007/s12026-013-8444-5
58. Szabo P, Shen S, Telford W, Weksler ME. Impaired rearrangement of IgH V to DJ segments in bone marrow Pro-B cells from old mice. Cell Immunol. (2003) 222:78–87. doi: 10.1016/S0008-8749(03)00084-4
59. Gibson KL, Wu Y-C, Barnett Y, Duggan O, Vaughan R, Kondeatis E, et al. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell. (2009) 8:18–25. doi: 10.1111/j.1474-9726.2008.00443.x
60. Duggal NA, Upton J, Phillips AC, Sapey E, Lord JM. An age-related numerical and functional deficit in CD19(+) CD24(hi) CD38(hi) B cells is associated with an increase in systemic autoimmunity. Aging Cell. (2013) 12:873–81. doi: 10.1111/acel.12114
61. Listì F, Candore G, Modica MA, Russo M, Lorenzo GD, Esposito-Pellitteri M, et al. A study of serum immunoglobulin levels in elderly persons that provides new insights into B cell immunosenescence. Ann N Y Acad Sci. (2006) 1089:487–95. doi: 10.1196/annals.1386.013
62. Frasca D, Landin AM, Lechner SC, Ryan JG, Schwartz R, Riley RL, et al. Aging down-regulates the transcription factor E2A, activation-induced cytidine deaminase, and Ig class switch in human B cells. J Immunol. (2008) 180:5283–90. doi: 10.4049/jimmunol.180.8.5283
63. Fraussen J, Marquez S, Takata K, Beckers L, Diaz GM, Zografou C, et al. Phenotypic and immunoglobulin repertoire analysis indicate a common origin of IgD–CD27– double negative (DN) B cells in healthy individuals and multiple sclerosis patients. J Immunol. (2019) 203:1650–64. doi: 10.4049/jimmunol.1801236
64. Herrero C, Sebastián C, Marqués L, Comalada M, Xaus J, Valledor AF, et al. Immunosenescence of macrophages: reduced MHC class II gene expression. Exp Gerontol. (2002) 37:389–94. doi: 10.1016/S0531-5565(01)00205-4
65. Bulati M, Buffa S, Martorana A, Candore G, Lio D, Caruso C, et al. Trafficking phenotype and production of granzyme B by double negative B cells (IgG+IgD–CD27–) in the elderly. Exp Gerontol. (2014) 54:123–9. doi: 10.1016/j.exger.2013.12.011
66. Colonna-Romano G, Bulati M, Aquino A, Pellicanò M, Vitello S, Lio D, et al. A double-negative (IgD–CD27–) B cell population is increased in the peripheral blood of elderly people. Mech Ageing Dev. (2009) 130:681–90. doi: 10.1016/j.mad.2009.08.003
67. Claes N, Fraussen J, Vanheusden M, Hellings N, Stinissen P, Wijmeersch BV, et al. Age-Associated B cells with proinflammatory characteristics are expanded in a proportion of multiple sclerosis patients. J Immunol. (2016) 197:4576–83. doi: 10.4049/jimmunol.1502448
68. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)–driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood. (2011) 118:1305–15. doi: 10.1182/blood-2011-01-331462
69. Hao Y, O'Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. (2011) 118:1294–304. doi: 10.1182/blood-2011-01-330530
70. Ratliff M, Alter S, Frasca D, Blomberg BB, Riley RL. In senescence, age-associated B cells secrete TNFα and inhibit survival of B-cell precursors. Aging Cell. (2013) 12:303–11. doi: 10.1111/acel.12055
71. Gregg R, Smith CM, Clark FJ, Dunnion D, Khan N, Chakraverty R, et al. The number of human peripheral blood CD4+ CD25high regulatory T cells increases with age. Clin Exp Immunol. (2005) 140:540–6. doi: 10.1111/j.1365-2249.2005.02798.x
72. Lages CS, Suffia I, Velilla PA, Huang B, Warshaw G, Hildeman DA, et al. Functional regulatory T cells accumulate in aged hosts and promote chronic infectious disease reactivation. J Immunol. (2008) 181:1835–48. doi: 10.4049/jimmunol.181.3.1835
73. Chougnet CA, Tripathi P, Lages CS, Raynor J, Sholl A, Fink P, et al. A major role for bim in regulatory T cell homeostasis. J Immunol. (2011) 186:156–63. doi: 10.4049/jimmunol.1001505
74. Santner-Nanan B, Seddiki N, Zhu E, Quent V, Kelleher A, de St Groth BF, et al. Accelerated age-dependent transition of human regulatory T cells to effector memory phenotype. Int Immunol. (2008) 20:375–83. doi: 10.1093/intimm/dxm151
75. Williams-Bey Y, Jiang J, Murasko DM. Expansion of regulatory T cells in aged mice following influenza infection. Mech Ageing Dev. (2011) 132:163–70. doi: 10.1016/j.mad.2011.03.001
76. Sun L, Hurez VJ, Thibodeaux SR, Kious MJ, Liu A, Lin P, et al. Aged regulatory T cells protect from autoimmune inflammation despite reduced STAT3 activation and decreased constraint of IL-17 producing T cells. Aging Cell. (2012) 11:509–19. doi: 10.1111/j.1474-9726.2012.00812.x
77. Simone R, Zicca A, Saverino D. The frequency of regulatory CD3+CD8+CD28- CD25+ T lymphocytes in human peripheral blood increases with age. J Leukoc Biol. (2008) 84:1454–61. doi: 10.1189/jlb.0907627
78. Suzuki M, Jagger AL, Konya C, Shimojima Y, Pryshchep S, Goronzy JJ, et al. CD8+CD45RA+CCR7+FOXP3+ T cells with immunosuppressive properties: a novel subset of inducible human regulatory T cells. J Immunol. (2012) 189:2118–30. doi: 10.4049/jimmunol.1200122
79. Jagger A, Shimojima Y, Goronzy JJ, Weyand CM. Regulatory T cells and the immune aging process: a mini-review. Gerontology. (2014) 60:130–7. doi: 10.1159/000355303
80. Salminen A. Activation of immunosuppressive network in the aging process. Ageing Res Rev. (2020) 57:100998. doi: 10.1016/j.arr.2019.100998
81. Raynor J, Lages CS, Shehata H, Hildeman DA, Chougnet CA. Homeostasis and function of regulatory T cells in aging. Curr Opin Immunol. (2012) 24:482–7. doi: 10.1016/j.coi.2012.04.005
82. Sharma S, Dominguez AL, Lustgarten J. High accumulation of T regulatory cells prevents the activation of immune responses in aged animals. J Immunol. (2006) 177:8348–55. doi: 10.4049/jimmunol.177.12.8348
83. Tanchot C, Terme M, Pere H, Tran T, Benhamouda N, Strioga M, et al. Tumor-Infiltrating regulatory T cells: phenotype, role, mechanism of expansion in situ and clinical significance. Cancer Microenviron. (2012) 6:147–57. doi: 10.1007/s12307-012-0122-y
84. Kipnis J, Avidan H, Caspi RR, Schwartz M. Dual effect of CD4+CD25+ regulatory T cells in neurodegeneration: a dialogue with microglia. Proc Natl Acad Sci USA. (2004) 101 (Suppl. 2):14663–9. doi: 10.1073/pnas.0404842101
85. Solana R, Tarazona R, Gayoso I, Lesur O, Dupuis G, Fulop T. Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin Immunol. (2012) 24:331–41. doi: 10.1016/j.smim.2012.04.008
86. Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, et al. Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J Immunol. (2015) 195:2624–32. doi: 10.4049/jimmunol.1501006
87. Splunter M van, Perdijk O, Fick-Brinkhof H, Floris-Vollenbroek EG, Meijer B, Brugman S, et al. Plasmacytoid dendritic cell and myeloid dendritic cell function in ageing: a comparison between elderly and young adult women. PLOS ONE. (2019) 14:e0225825. doi: 10.1371/journal.pone.0225825
88. Sridharan A, Esposo M, Kaushal K, Tay J, Osann K, Agrawal S, et al. Age-associated impaired plasmacytoid dendritic cell functions lead to decreased CD4 and CD8 T cell immunity. Age Dordr Neth. (2011) 33:363–76. doi: 10.1007/s11357-010-9191-3
89. Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A, et al. Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell. (2014) 13:690–8. doi: 10.1111/acel.12222
90. Butcher SK, Chahal H, Nayak L, Sinclair A, Henriquez NV, Sapey E, et al. Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J Leukoc Biol. (2001) 70:881–6. doi: 10.1189/jlb.70.6.881
91. Metcalf TU, Wilkinson PA, Cameron MJ, Ghneim K, Chiang C, Wertheimer AM, et al. Human monocyte subsets are transcriptionally and functionally altered in aging in response to pattern recognition receptor agonists. J Immunol. (2017) 199:1405–17. doi: 10.4049/jimmunol.1700148
92. Seidler S, Zimmermann HW, Bartneck M, Trautwein C, Tacke F. Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol. (2010) 11:30. doi: 10.1186/1471-2172-11-30
93. Verschoor CP, Johnstone J, Millar J, Parsons R, Lelic A, Loeb M, et al. Alterations to the frequency and function of peripheral blood monocytes and associations with chronic disease in the advanced-age, frail elderly. PLoS ONE. (2014) 9:e104522. doi: 10.1371/journal.pone.0104522
94. Stranks AJ, Hansen AL, Panse I, Mortensen M, Ferguson DJP, Puleston DJ, et al. Autophagy controls acquisition of aging features in macrophages. J Innate Immun. (2015) 7:375–91. doi: 10.1159/000370112
95. Herrero C, Marqués L, Lloberas J, Celada A. IFN-γ-dependent transcription of MHC class II IA is impaired in macrophages from aged mice. J Clin Invest. (2001) 107:485–93. doi: 10.1172/JCI11696
96. Renshaw M, Rockwell J, Engleman C, Gewirtz A, Katz J, Sambhara S. Cutting edge: impaired toll-like receptor expression and function in aging. J Immunol. (2002) 169:4697–701. doi: 10.4049/jimmunol.169.9.4697
97. Borrego F, Alonso MC, Galiani MD, Carracedo J, Ramirez R, Ostos B, et al. NK phenotypic markers and IL2 response in NK cells from elderly people. Exp Gerontol. (1999) 34:253–65. doi: 10.1016/S0531-5565(98)00076-X
98. Le Garff-Tavernier M, Béziat V, Decocq J, Siguret V, Gandjbakhch F, Pautas E, et al. Human NK cells display major phenotypic and functional changes over the life span. Aging Cell. (2010) 9:527–35. doi: 10.1111/j.1474-9726.2010.00584.x
99. Beli E, Duriancik DM, Clinthorne JF, Lee T, Kim S, Gardner EM. natural Killer cell development and maturation in aged mice. Mech Ageing Dev. (2014) 135:33–40. doi: 10.1016/j.mad.2013.11.007
100. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. (2004) 116:281–97. doi: 10.1016/S0092-8674(04)00045-5
101. Noren Hooten N, Abdelmohsen K, Gorospe M, Ejiogu N, Zonderman AB, Evans MK. microRNA expression patterns reveal differential expression of target genes with age. PLoS ONE. (2010) 5:e10724. doi: 10.1371/journal.pone.0010724
102. Xu Y, Xu L, Chen C, Zhang Y, Zeng C, Jin Z, et al. Age-Related immune profile of the t cell receptor repertoire, thymic recent output function, and miRNAs. BioMed Res Int. (2020) 2020:e5910823. doi: 10.1155/2020/5910823
103. Williams J, Smith F, Kumar S, Vijayan M, Reddy PH. Are microRNAs true sensors of ageing and cellular senescence? Ageing Res Rev. (2017) 35:350–63. doi: 10.1016/j.arr.2016.11.008
104. Nidadavolu LS, Niedernhofer LJ, Khan SA. Identification of microRNAs dysregulated in cellular senescence driven by endogenous genotoxic stress. Aging. (2013) 5:460–73. doi: 10.18632/aging.100571
105. Zhang R, Zhang Q, Niu J, Lu K, Xie B, Cui D, et al. Screening of microRNAs associated with Alzheimer's disease using oxidative stress cell model and different strains of senescence accelerated mice. J Neurol Sci. (2014) 338:57–64. doi: 10.1016/j.jns.2013.12.017
106. Magenta A, Cencioni C, Fasanaro P, Zaccagnini G, Greco S, Sarra-Ferraris G, et al. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. (2011) 18:1628–39. doi: 10.1038/cdd.2011.42
107. Li G, Luna C, Qiu J, Epstein DL, Gonzalez P. Alterations in microRNA expression in stress-induced cellular senescence. Mech Ageing Dev. (2009) 130:731–41. doi: 10.1016/j.mad.2009.09.002
108. Vasa-Nicotera M, Chen H, Tucci P, Yang AL, Saintigny G, Menghini R, et al. miR-146a is modulated in human endothelial cell with aging. Atherosclerosis. (2011) 217:326–30. doi: 10.1016/j.atherosclerosis.2011.03.034
109. Hackl M, Brunner S, Fortschegger K, Schreiner C, Micutkova L, Mück C, et al. miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human aging. Aging Cell. (2010) 9:291–6. doi: 10.1111/j.1474-9726.2010.00549.x
110. Faraonio R, Salerno P, Passaro F, Sedia C, Iaccio A, Bellelli R, et al. A set of miRNAs participates in the cellular senescence program in human diploid fibroblasts. Cell Death Differ. (2012) 19:713–21. doi: 10.1038/cdd.2011.143
111. Puisségur M-P, Mazure NM, Bertero T, Pradelli L, Grosso S, Robbe-Sermesant K, et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. (2011) 18:465–78. doi: 10.1038/cdd.2010.119
112. Frankel LB, Wen J, Lees M, Høyer-Hansen M, Farkas T, Krogh A, et al. microRNA-101 is a potent inhibitor of autophagy. EMBO J. (2011) 30:4628–41. doi: 10.1038/emboj.2011.331
113. Liu L, Ren W, Chen K. MiR-34a promotes apoptosis and inhibits autophagy by targeting HMGB1 in acute myeloid leukemia cells. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. (2017) 41:1981–92. doi: 10.1159/000475277
114. Xu S, Wu W, Huang H, Huang R, Xie L, Su A, et al. The p53/miRNAs/Ccna2 pathway serves as a novel regulator of cellular senescence: complement of the canonical p53/p21 pathway. Aging Cell. (2019) 18:e12918. doi: 10.1111/acel.12918
115. Poliseno L, Pitto L, Simili M, Mariani L, Riccardi L, Ciucci A, et al. The proto-oncogene lrf is under post-transcriptional control of MiR-20a: implications for senescence. PLoS ONE. (2008) 3:e2542. doi: 10.1371/journal.pone.0002542
116. Borgdorff V, Lleonart ME, Bishop CL, Fessart D, Bergin AH, Overhoff MG, et al. Multiple microRNAs rescue from Ras-induced senescence by inhibiting p21Waf1/Cip1. Oncogene. (2010) 29:2262–71. doi: 10.1038/onc.2009.497
117. Yan H, Xue G, Mei Q, Wang Y, Ding F, Liu M-F, et al. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. (2009) 28:2719–32. doi: 10.1038/emboj.2009.214
118. Ivanovska I, Ball AS, Diaz RL, Magnus JF, Kibukawa M, Schelter JM, et al. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol Cell Biol. (2008) 28:2167–74. doi: 10.1128/MCB.01977-07
119. Inomata M, Tagawa H, Guo Y-M, Kameoka Y, Takahashi N, Sawada K. MicroRNA-17-92 down-regulates expression of distinct targets in different B-cell lymphoma subtypes. Blood. (2009) 113:396–402. doi: 10.1182/blood-2008-07-163907
120. Budzinska M, Owczarz M, Pawlik-Pachucka E, Roszkowska-Gancarz M, Slusarczyk P, Puzianowska-Kuznicka M. miR-96, miR-145 and miR-9 expression increases, and IGF-1R and FOXO1 expression decreases in peripheral blood mononuclear cells of aging humans. BMC Geriatr. (2016) 16:200. doi: 10.1186/s12877-016-0379-y
121. Zhang J, Rane G, Dai X, Shanmugam MK, Arfuso F, Samy RP, et al. Ageing and the telomere connection: an intimate relationship with inflammation. Ageing Res Rev. (2016) 25:55–69. doi: 10.1016/j.arr.2015.11.006
122. Hiyama K, Hirai Y, Kyoizumi S, Akiyama M, Hiyama E, Piatyszek MA, et al. Activation of telomerase in human lymphocytes and hematopoietic progenitor cells. J Immunol. (1995) 155:3711–5.
123. Hu BT, Lee SC, Marin E, Ryan DH, Insel RA. Telomerase is up-regulated in human germinal center B cells in vivo and can be re-expressed in memory B cells activated in vitro. J Immunol. (1997) 159:1068–71.
124. von Zglinicki T. Role of oxidative stress in telomere length regulation and replicative senescence. Ann N Y Acad Sci. (2000) 908:99–110. doi: 10.1111/j.1749-6632.2000.tb06639.x
125. Valenzuela HF, Effros RB. Divergent telomerase and CD28 expression patterns in human CD4 and CD8 T cells following repeated encounters with the same antigenic stimulus. Clin Immunol. (2002) 105:117–25. doi: 10.1006/clim.2002.5271
126. Luo Z, Feng X, Wang H, Xu W, Zhao Y, Ma W, et al. Mir-23a induces telomere dysfunction and cellular senescence by inhibiting TRF2 expression. Aging Cell. (2015) 14:391–9. doi: 10.1111/acel.12304
127. Dinami R, Ercolani C, Petti E, Piazza S, Ciani Y, Sestito R, et al. miR-155 drives telomere fragility in human breast cancer by targeting TRF1. Cancer Res. (2014) 74:4145–56. doi: 10.1158/0008-5472.CAN-13-2038
128. Mitomo S, Maesawa C, Ogasawara S, Iwaya T, Shibazaki M, Yashima-Abo A, et al. Downregulation of miR-138 is associated with overexpression of human telomerase reverse transcriptase protein in human anaplastic thyroid carcinoma cell lines. Cancer Sci. (2008) 99:280–6. doi: 10.1111/j.1349-7006.2007.00666.x
129. Li J, Lei H, Xu Y, Tao Z. miR-512-5p suppresses tumor growth by targeting hTERT in telomerase positive head and neck squamous cell carcinoma in vitro and in vivo. PLoS ONE. (2015) 10:e0135265. doi: 10.1371/journal.pone.0135265
130. Kroesen B-J, Teteloshvili N, Smigielska-Czepiel K, Brouwer E, Boots AMH, van den Berg A, et al. Immuno-miRs: critical regulators of T-cell development, function and ageing. Immunology. (2015) 144:1–10. doi: 10.1111/imm.12367
131. Olivieri F, Procopio AD, Montgomery RR. Effect of aging on microRNAs and regulation of pathogen recognition receptors. Curr Opin Immunol. (2014) 29:29–37. doi: 10.1016/j.coi.2014.03.006
132. Gustafson CE, Cavanagh MM, Jin J, Weyand CM, Goronzy JJ. Functional pathways regulated by microRNA networks in CD8 T-cell aging. Aging Cell. (2019) 18:e12879. doi: 10.1111/acel.12879
133. Bhaumik D, Scott GK, Schokrpur S, Patil CK, Orjalo AV, Rodier F, et al. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging. (2009) 1:402–11. doi: 10.18632/aging.100042
134. Yan Y, Lu K, Ye T, Zhang Z. MicroRNA-223 attenuates LPS-induced inflammation in an acute lung injury model via the NLRP3 inflammasome and TLR4/NF-κB signaling pathway via RHOB. Int J Mol Med. (2019) 43:1467–77. doi: 10.3892/ijmm.2019.4075
135. Yang J, Zhang P, Krishna S, Wang J, Lin X, Huang H, et al. Unexpected positive control of NFκB and miR-155 by DGKα and ζ ensures effector and memory CD8 + T cell differentiation. Oncotarget. (2016) 7:33744–64. doi: 10.18632/oncotarget.8164
136. Wu T, Wieland A, Araki K, Davis CW, Ye L, Hale JS, et al. Temporal expression of microRNA cluster miR-17-92 regulates effector and memory CD8+ T-cell differentiation. Proc Natl Acad Sci USA. (2012) 109:9965–70. doi: 10.1073/pnas.1207327109
137. Kim C, Hu B, Jadhav RR, Jin J, Zhang H, Cavanagh MM, et al. Activation of miR-21-Regulated pathways in immune aging selects against signatures characteristic of memory T cells. Cell Rep. (2018) 25:2148–62.e5. doi: 10.1016/j.celrep.2018.10.074
138. Kim C, Jadhav RR, Gustafson CE, Smithey MJ, Hirsch AJ, Uhrlaub JL, et al. Defects in antiviral T Cell responses inflicted by aging-associated miR-181a deficiency. Cell Rep. (2019) 29:2202–16.e5. doi: 10.1016/j.celrep.2019.10.044
139. Tsai C-Y, Allie SR, Zhang W, Usherwood EJ. MicroRNA miR-155 affects antiviral effector and effector memory CD8 T cell differentiation. J Virol. (2013) 87:2348–51. doi: 10.1128/JVI.01742-12
140. Li G, Yu M, Lee W-W, Tsang M, Krishnan E, Weyand CM, et al. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat Med. (2012) 18:1518–24. doi: 10.1038/nm.2963
141. Frasca D, Diaz A, Romero M, Ferracci F, Blomberg BB. Micro-RNAs miR-155 and miR-16 decrease AID and E47 in B cells from elderly individuals. J Immunol. (2015) 195:2134–40. doi: 10.4049/jimmunol.1500520
142. Frasca D, Diaz A, Romero M, Blomberg BB. Human peripheral late/exhausted memory B cells express a senescent-associated secretory phenotype and preferentially utilize metabolic signaling pathways. Exp Gerontol. (2017) 87:113–20. doi: 10.1016/j.exger.2016.12.001
143. Barik RR, Bhatt LK. Emerging epigenetic targets in rheumatoid arthritis. Rheumatol Int. (2021) 41:2047–67. doi: 10.1007/s00296-021-04951-y
144. Lopatina N, Haskell JF, Andrews LG, Poole JC, Saldanha S, Tollefsbol T. Differential maintenance and de novo methylating activity by three DNA methyltransferases in aging and immortalized fibroblasts. J Cell Biochem. (2002) 84:324–34. doi: 10.1002/jcb.10015
145. Dozmorov MG, Coit P, Maksimowicz-McKinnon K, Sawalha AH. Age-associated DNA methylation changes in naive CD4+ T cells suggest an evolving autoimmune epigenotype in aging T cells. Epigenomics. (2017) 9:429–45. doi: 10.2217/epi-2016-0143
146. Reynolds LM, Taylor JR, Ding J, Lohman K, Johnson C, Siscovick D, et al. Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat Commun. (2014) 5:5366. doi: 10.1038/ncomms6366
147. Horvath S, Pirazzini C, Bacalini MG, Gentilini D, Di Blasio AM, Delledonne M, et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging. (2015) 7:1159–70. doi: 10.18632/aging.100861
148. Owczarz M, Budzinska M, Domaszewska-Szostek A, Borkowska J, Polosak J, Gewartowska M, et al. miR-34a and miR-9 are overexpressed and SIRT genes are downregulated in peripheral blood mononuclear cells of aging humans. Exp Biol Med. (2017) 242:1453–61. doi: 10.1177/1535370217720884
149. Kondo H, Kim HW, Wang L, Okada M, Paul C, Millard RW, et al. Blockade of senescence-associated microRNA-195 in aged skeletal muscle cells facilitates reprogramming to produce induced pluripotent stem cells. Aging Cell. (2016) 15:56–66. doi: 10.1111/acel.12411
150. Arvey A, Rowe M, Legutki JB, An G, Gollapudi A, Lei A, et al. Age-associated changes in the circulating human antibody repertoire are upregulated in autoimmunity. Immun Ageing A. (2020) 17:28. doi: 10.1186/s12979-020-00193-x
151. Cooper GS, Stroehla BC. The epidemiology of autoimmune diseases. Autoimmun Rev. (2003) 2:119–25. doi: 10.1016/S1568-9972(03)00006-5
152. Goronzy JJ, Li G, Yang Z, Weyand CM. The janus head of T cell aging - autoimmunity and immunodeficiency. Front Immunol. (2013) 4:131. doi: 10.3389/fimmu.2013.00131
153. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain J Neurol. (2017) 140:3128–38. doi: 10.1093/brain/awx276
154. Brinks R, Hoyer A, Weber S, Fischer-Betz R, Sander O, Richter JG, et al. Age-specific and sex-specific incidence of systemic lupus erythematosus: an estimate from cross-sectional claims data of 2.3 million people in the German statutory health insurance 2002. Lupus Sci Med. (2016) 3:e000181. doi: 10.1136/lupus-2016-000181
155. Safiri S, Kolahi AA, Hoy D, Smith E, Bettampadi D, Mansournia MA, et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: a systematic analysis of the global burden of disease study 2017. Ann Rheum Dis. (2019) 78:1463–71. doi: 10.1136/annrheumdis-2019-215920
156. Manoussakis MN, Tzioufas AG, Silis MP, Pange PJ, Goudevenos J, Moutsopoulos HM. High prevalence of anti-cardiolipin and other autoantibodies in a healthy elderly population. Clin Exp Immunol. (1987) 69:557–65.
157. Boren E, Gershwin ME. Inflamm-aging: autoimmunity, and the immune-risk phenotype. Autoimmun Rev. (2004) 3:401–6. doi: 10.1016/j.autrev.2004.03.004
158. Kandhaya-Pillai R, Miro-Mur F, Alijotas-Reig J, Tchkonia T, Kirkland JL, Schwartz S. TNFα-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging. (2017) 9:2411–35. doi: 10.18632/aging.101328
159. Thewissen M, Somers V, Venken K, Linsen L, van Paassen P, Geusens P, et al. Analyses of immunosenescent markers in patients with autoimmune disease. Clin Immunol. (2007) 123:209–18. doi: 10.1016/j.clim.2007.01.005
160. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. (2009) 43:67–93. doi: 10.1146/annurev-genet-102808-114910
161. Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol Biol Cell. (2009) 20:1981–91. doi: 10.1091/mbc.e08-12-1248
162. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. (2011) 146:682–95. doi: 10.1016/j.cell.2011.07.030
163. Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci USA. (2010) 107:14164–9. doi: 10.1073/pnas.1009485107
164. Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. (2017) 6:e24. doi: 10.1186/s40169-017-0154-5
165. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. (2016) 23:303–14. doi: 10.1016/j.cmet.2015.11.011
166. Tsai CY, Shen CY, Liao HT, Li KJ, Lee HT, Lu CS, et al. Molecular and cellular bases of immunosenescence, inflammation, and cardiovascular complications mimicking ‘inflammaging' in patients with systemic lupus erythematosus. Int J Mol Sci. (2019) 20:E3878. doi: 10.3390/ijms20163878
167. Bolton C, Smith PA. The influence and impact of ageing and immunosenescence (ISC) on adaptive immunity during multiple sclerosis (MS) and the animal counterpart experimental autoimmune encephalomyelitis (EAE). Ageing Res Rev. (2018) 41:64–81. doi: 10.1016/j.arr.2017.10.005
168. Naegele M, Tillack K, Reinhardt S, Schippling S, Martin R, Sospedra M. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J Neuroimmunol. (2012) 242:60–71. doi: 10.1016/j.jneuroim.2011.11.009
169. Rumble JM, Huber AK, Krishnamoorthy G, Srinivasan A, Giles DA, Zhang X, et al. Neutrophil-related factors as biomarkers in EAE and MS. J Exp Med. (2015) 212:23–35. doi: 10.1084/jem.20141015
170. Kostic M, Dzopalic T, Zivanovic S, Zivkovic N, Cvetanovic A, Stojanovic I, et al. IL-17 and glutamate excitotoxicity in the pathogenesis of multiple sclerosis. Scand J Immunol. (2014) 79:181–6. doi: 10.1111/sji.12147
171. Haschka D, Tymoszuk P, Bsteh G, Petzer V, Berek K, Theurl I, et al. Expansion of neutrophils and classical and nonclassical monocytes as a hallmark in relapsing-remitting multiple sclerosis. Front Immunol. (2020) 11:594. doi: 10.3389/fimmu.2020.00594
172. Gjelstrup MC, Stilund M, Petersen T, Møller HJ, Petersen EL, Christensen T. Subsets of activated monocytes and markers of inflammation in incipient and progressed multiple sclerosis. Immunol Cell Biol. (2018) 96:160–74. doi: 10.1111/imcb.1025
173. Gross CC, Schulte-Mecklenbeck A, Wiendl H, Marcenaro E, Kerlero de Rosbo N, Uccelli A, et al. Regulatory functions of natural killer cells in multiple sclerosis. Front Immunol. (2016) 7:606. doi: 10.3389/fimmu.2016.00606
174. Rodríguez-Martín E, Picón C, Costa-Frossard L, Alenda R, Sainz de la Maza S, Roldán E, et al. Natural killer cell subsets in cerebrospinal fluid of patients with multiple sclerosis. Clin Exp Immunol. (2015) 180:243–9. doi: 10.1111/cei.12580
175. Plantone D, Marti A, Frisullo G, Iorio R, Damato V, Nociti V, et al. Circulating CD56dim NK cells expressing perforin are increased in progressive multiple sclerosis. J Neuroimmunol. (2013) 265:124–7. doi: 10.1016/j.jneuroim.2013.10.004
176. Lünemann A, Tackenberg B, DeAngelis T, da Silva RB, Messmer B, Vanoaica LD, et al. Impaired IFN-γ production and proliferation of NK cells in multiple sclerosis. Int Immunol. (2011) 23:139–48. doi: 10.1093/intimm/dxq463
177. Gross CC, Schulte-Mecklenbeck A, Rünzi A, Kuhlmann T, Posevitz-Fejfár A, Schwab N, et al. Impaired NK-mediated regulation of T-cell activity in multiple sclerosis is reconstituted by IL-2 receptor modulation. Proc Natl Acad Sci USA. (2016) 113:E2973–82. doi: 10.1073/pnas.1524924113
178. Liu Q, Sanai N, Jin W-N, La Cava A, Van Kaer L, Shi F-D. Neural stem cells sustain natural killer cells that dictate recovery from brain inflammation. Nat Neurosci. (2016) 19:243–52. doi: 10.1038/nn.4211
179. Papadaki HA, Tsagournisakis M, Mastorodemos V, Pontikoglou C, Damianaki A, Pyrovolaki K, et al. Normal bone marrow hematopoietic stem cell reserves and normal stromal cell function support the use of autologous stem cell transplantation in patients with multiple sclerosis. Bone Marrow Transplant. (2005) 36:1053–63. doi: 10.1038/sj.bmt.1705179
180. Redondo J, Sarkar P, Kemp K, Virgo PF, Pawade J, Norton A, et al. Reduced cellularity of bone marrow in multiple sclerosis with decreased MSC expansion potential and premature ageing in vitro. Mult Scler. (2018) 24:919–31. doi: 10.1177/1352458517711276
181. Balint B, Haas J, Schwarz A, Jarius S, Fürwentsches A, Engelhardt K, et al. T-cell homeostasis in pediatric multiple sclerosis: old cells in young patients. Neurology. (2013) 81:784–92. doi: 10.1212/WNL.0b013e3182a2ce0e
182. Haegert DG, Hackenbroch JD, Duszczyszyn D, Fitz-Gerald L, Zastepa E, Mason H, et al. Reduced thymic output and peripheral naïve CD4 T-cell alterations in primary progressive multiple sclerosis (PPMS). J Neuroimmunol. (2011) 233:233–9. doi: 10.1016/j.jneuroim.2010.12.007
183. Nielsen BR, Ratzer R, Börnsen L, von Essen MR, Christensen JR, Sellebjerg F. Characterization of naïve, memory and effector T cells in progressive multiple sclerosis. J Neuroimmunol. (2017) 310:17–25. doi: 10.1016/j.jneuroim.2017.06.001
184. Alves Sousa AP, Johnson KR, Ohayon J, Zhu J, Muraro PA, Jacobson S. Comprehensive analysis of TCR-β repertoire in patients with neurological immune-mediated disorders. Sci Rep. (2019) 9:344. doi: 10.1038/s41598-018-36274-7
185. Cao Y, Goods BA, Raddassi K, Nepom GT, Kwok WW, Love JC, et al. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci Transl Med. (2015) 7:287ra74. doi: 10.1126/scitranslmed.aaa8038
186. Li YF, Zhang SX, Ma XW, Xue YL, Gao C, Li XY. Levels of peripheral Th17 cells and serum Th17-related cytokines in patients with multiple sclerosis: a meta-analysis. Mult Scler Relat Disord. (2017) 18:20–5. doi: 10.1016/j.msard.2017.09.003
187. Vrethem M, Dahle C, Ekerfelt C, Forsberg P, Danielsson O, Ernerudh J. CD4 and CD8 lymphocyte subsets in cerebrospinal fluid and peripheral blood from patients with multiple sclerosis, meningitis and normal controls. Acta Neurol Scand. (1998) 97:215–20. doi: 10.1111/j.1600-0404.1998.tb00640.x
188. Pender MP, Csurhes PA, Pfluger CMM, Burrows SR. CD8 T cell deficiency impairs control of Epstein-Barr virus and worsens with age in multiple sclerosis. J Neurol Neurosurg Psychiatry. (2012) 83:353–4. doi: 10.1136/jnnp-2011-300213
189. Mix E, Olsson T, Correale J, Kostulas V, Link H. CD4+, CD8+, and CD4- CD8- T cells in CSF and blood of patients with multiple sclerosis and tension headache. Scand J Immunol. (1990) 31:493–501. doi: 10.1111/j.1365-3083.1990.tb02797.x
190. Harrer A, Pilz G, Wipfler P, Oppermann K, Sellner J, Hitzl W, et al. High interindividual variability in the CD4/CD8 T cell ratio and natalizumab concentration levels in the cerebrospinal fluid of patients with multiple sclerosis. Clin Exp Immunol. (2015) 180:383–92. doi: 10.1111/cei.12590
191. Warnke C, Mausberg AK, Stettner M, Dehmel T, Nekrich L, Meyer zu Horste G, et al. Natalizumab affects the T-cell receptor repertoire in patients with multiple sclerosis. Neurology. (2013) 81:1400–8. doi: 10.1212/WNL.0b013e3182a84101
192. Ifergan I, Kebir H, Alvarez JI, Marceau G, Bernard M, Bourbonnière L, et al. Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on α4 integrin. Brain J Neurol. (2011) 134 (Pt. 12):3560–77. doi: 10.1093/brain/awr268
193. Zhou F, Zhang G-X, Rostami A. Apoptotic cell-treated dendritic cells induce immune tolerance by specifically inhibiting development of CD4+ effector memory T cells. Immunol Res. (2016) 64:73–81. doi: 10.1007/s12026-015-8676-7
194. Elyaman W, Kivisäkk P, Reddy J, Chitnis T, Raddassi K, Imitola J, et al. Distinct functions of autoreactive memory and effector CD4+ T cells in experimental autoimmune encephalomyelitis. Am J Pathol. (2008) 173:411–22. doi: 10.2353/ajpath.2008.080142
195. Williams JL, Kithcart AP, Smith KM, Shawler T, Cox GM, Whitacre CC. Memory cells specific for myelin oligodendrocyte glycoprotein (MOG) govern the transfer of experimental autoimmune encephalomyelitis. J Neuroimmunol. (2011) 234:84–92. doi: 10.1016/j.jneuroim.2011.02.008
196. Okuda Y, Okuda M, Apatoff BR, Posnett DN. The activation of memory CD4+ T cells and CD8+ T cells in patients with multiple sclerosis. J Neurol Sci. (2005) 235:11–7. doi: 10.1016/j.jns.2005.02.013
197. Haegele KF, Stueckle CA, Malin J-P, Sindern E. Increase of CD8+ T-effector memory cells in peripheral blood of patients with relapsing–remitting multiple sclerosis compared to healthy controls. J Neuroimmunol. (2007) 183:168–74. doi: 10.1016/j.jneuroim.2006.09.008
198. Liu G-Z, Fang L-B, Hjelmström P, Gao X-G. Increased CD8+ central memory T cells in patients with multiple sclerosis. Mult Scler J. (2007) 13:149–55. doi: 10.1177/1352458506069246
199. Markovic-Plese S, Cortese I, Wandinger KP, McFarland HF, Martin R. CD4+CD28– costimulation-independent T cells in multiple sclerosis. J Clin Invest. (2001) 108:1185–94. doi: 10.1172/JCI200112516
200. Vanheusden M, Broux B, Welten SPM, Peeters LM, Panagioti E, Van Wijmeersch B, et al. Cytomegalovirus infection exacerbates autoimmune mediated neuroinflammation. Sci Rep. (2017) 7:663. doi: 10.1038/s41598-017-00645-3
201. Peeters LM, Vanheusden M, Somers V, Van Wijmeersch B, Stinissen P, Broux B, et al. Cytotoxic CD4+ T cells drive multiple sclerosis progression. Front Immunol. (2017) 8:1160. doi: 10.3389/fimmu.2017.01160
202. Cencioni MT, Mattoscio M, Magliozzi R, Bar-Or A, Muraro PA. B cells in multiple sclerosis - from targeted depletion to immune reconstitution therapies. Nat Rev Neurol. (2021) 17:399–414. doi: 10.1038/s41582-021-00498-5
203. Cencioni MT, Ali R, Nicholas R, Muraro PA. Defective CD19+CD24hiCD38hi transitional B-cell function in patients with relapsing-remitting MS. Mult Scler. (2021) 27:1187–97. doi: 10.1177/1352458520951536
204. Lee-Chang C, Top I, Zéphir H, Dubucquoi S, Trauet J, Dussart P, et al. Primed status of transitional B cells associated with their presence in the cerebrospinal fluid in early phases of multiple sclerosis. Clin Immunol. (2011) 139:12–20. doi: 10.1016/j.clim.2010.11.004
205. Schwarz A, Balint B, Korporal-Kuhnke M, Jarius S, von Engelhardt K, Fürwentsches A, et al. B-cell populations discriminate between pediatric- and adult-onset multiple sclerosis. Neurol Neuroimmunol Neuroinflammation. (2017) 4:e309. doi: 10.1212/NXI.0000000000000309
206. Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H, et al. Distinct effector cytokine profiles of memory and naive human b cell subsets and implication in multiple sclerosis. J Immunol. (2007) 178:6092–9. doi: 10.4049/jimmunol.178.10.6092
207. Ciccocioppo F, Lanuti P, Pierdomenico L, Simeone P, Bologna G, Ercolino E, et al. The characterization of regulatory t-cell profiles in alzheimer's disease and multiple sclerosis. Sci Rep. (2019) 9:8788. doi: 10.1038/s41598-019-45433-3
208. McKay FC, Swain LI, Schibeci SD, Rubio JP, Kilpatrick TJ, Heard RN, et al. CD127 immunophenotyping suggests altered CD4+ T cell regulation in primary progressive multiple sclerosis. J Autoimmun. (2008) 31:52–8. doi: 10.1016/j.jaut.2008.02.003
209. Muls NG, Dang HA, Sindic CJ, van Pesch V. Regulation of Treg-associated CD39 in multiple sclerosis and effects of corticotherapy during relapse: Mult Scler J. (2015). Available online at: https://journals.sagepub.com/doi/10.1177/1352458514567215?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub++0pubmed
210. Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O'Farrelly C, et al. CD39 + Foxp3 + regulatory T cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol. (2009) 183:7602–10. doi: 10.4049/jimmunol.0901881
211. Dhaeze T, Peelen E, Hombrouck A, Peeters L, Van Wijmeersch B, Lemkens N, et al. Circulating follicular regulatory T cells are defective in multiple sclerosis. J Immunol. (2015) 195:832–40. doi: 10.4049/jimmunol.1500759
212. Haas J, Hug A, Viehöver A, Fritzsching B, Falk CS, Filser A, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol. (2005) 35:3343–52. doi: 10.1002/eji.200526065
213. Venken K, Hellings N, Thewissen M, Somers V, Hensen K, Rummens J-L, et al. Compromised CD4+ CD25high regulatory T-cell function in patients with relapsing-remitting multiple sclerosis is correlated with a reduced frequency of FOXP3-positive cells and reduced FOXP3 expression at the single-cell level. Immunology. (2008) 123:79–89. doi: 10.1111/j.1365-2567.2007.02690.x
214. Severin ME, Lee PW, Liu Y, Selhorst AJ, Gormley MG, Pei W, et al. MicroRNAs targeting TGFβ signalling underlie the regulatory T cell defect in multiple sclerosis. Brain. (2016) 139:1747–61. doi: 10.1093/brain/aww084
215. Bolton C. An evaluation of the recognised systemic inflammatory biomarkers of chronic sub-optimal inflammation provides evidence for inflammageing (IFA) during multiple sclerosis (MS). Immun Ageing A. (2021) 18:18. doi: 10.1186/s12979-021-00225-0
216. Alirezaei M, Fox HS, Flynn CT, Moore CS, Hebb ALO, Frausto RF, et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy. (2009) 5:152–8. doi: 10.4161/auto.5.2.7348
217. Misrielal C, Mauthe M, Reggiori F, Eggen BJL. Autophagy in multiple sclerosis: two sides of the same coin. Front Cell Neurosci. (2020) 14:603710. doi: 10.3389/fncel.2020.603710
218. Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. (2014) 15:81–94. doi: 10.1038/nrm3735
219. Habib R, Ocklenburg S, Hoffjan S, Haghikia A, Epplen JT, Arning L. Association between shorter leukocyte telomeres and multiple sclerosis. J Neuroimmunol. (2020) 341:577187. doi: 10.1016/j.jneuroim.2020.577187
220. Krysko KM, Henry RG, Cree BAC, Lin J, University of California San Francisco MS-EPIC Team, et al. Telomere length is associated with disability progression in multiple sclerosis. Ann Neurol. (2019) 86:671–82. doi: 10.1002/ana.25592
221. Hecker M, Fitzner B, Jäger K, Bühring J, Schwartz M, Hartmann A, et al. Leukocyte telomere length in patients with multiple sclerosis and its association with clinical phenotypes. Mol Neurobiol. (2021) 58:2886–96. doi: 10.1007/s12035-021-02315-y
222. De Riccardis L, Rizzello A, Ferramosca A, Urso E, De Robertis F, Danieli A, et al. Bioenergetics profile of CD4+ T cells in relapsing remitting multiple sclerosis subjects. J Biotechnol. (2015) 202:31–9. doi: 10.1016/j.jbiotec.2015.02.015
223. Gonzalo H, Nogueras L, Gil-Sánchez A, Hervás JV, Valcheva P, González-Mingot C, et al. Impairment of mitochondrial redox status in peripheral lymphocytes of multiple sclerosis patients. Front Neurosci. (2019) 13:938. doi: 10.3389/fnins.2019.00938
224. Armon-Omer A, Neuman H, Sharabi-Nov A, Shahien R. Mitochondrial activity is impaired in lymphocytes of MS patients in correlation with disease severity. Mult Scler Relat Disord. (2020) 41:102025. doi: 10.1016/j.msard.2020.102025
225. Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, et al. Glutathione primes T cell metabolism for inflammation. Immunity. (2017) 46:675–89. doi: 10.1016/j.immuni.2017.03.019
226. Previte DM, O'Connor EC, Novak EA, Martins CP, Mollen KP, Piganelli JD. Reactive oxygen species are required for driving efficient and sustained aerobic glycolysis during CD4+ T cell activation. PLoS ONE. (2017) 12:e0175549. doi: 10.1371/journal.pone.0175549
227. Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, et al. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol. (2010) 185:5247–58. doi: 10.4049/jimmunol.1001472
228. Julià E, Edo MC, Horga A, Montalban X, Comabella M. Differential susceptibility to apoptosis of CD4+T cells expressing CCR5 and CXCR3 in patients with MS. Clin Immunol. (2009) 133:364–74. doi: 10.1016/j.clim.2009.08.012
229. Kiselev IS, Kulakova OG, Boyko AN, Favorova OO. DNA methylation as an epigenetic mechanism in the development of multiple sclerosis. Acta Naturae. (2021) 13:45–57. doi: 10.32607/actanaturae.11043
230. Kulakova OG, Kabilov MR, Danilova LV, Popova EV, Baturina OA, Tsareva EY, et al. Whole-Genome DNA methylation analysis of peripheral blood mononuclear cells in multiple sclerosis patients with different disease courses. Acta Naturae. (2016) 8:103–10. doi: 10.32607/20758251-2016-8-3-103-110
231. Ewing E, Kular L, Fernandes SJ, Karathanasis N, Lagani V, Ruhrmann S, et al. Combining evidence from four immune cell types identifies DNA methylation patterns that implicate functionally distinct pathways during multiple sclerosis progression. EBioMedicine. (2019) 43:411–23. doi: 10.1016/j.ebiom.2019.04.042
232. Ye F, Wang T, Wu X, Liang J, Li J, Sheng W. N6-Methyladenosine RNA modification in cerebrospinal fluid as a novel potential diagnostic biomarker for progressive multiple sclerosis. J Transl Med. (2021) 19:316. doi: 10.1186/s12967-021-02981-5
233. Perdaens O, Dang HA, D'Auria L, van Pesch V. CSF microRNAs discriminate MS activity and share similarity to other neuroinflammatory disorders. Neurol Neuroimmunol Neuroinflammation. (2020) 7:e673. doi: 10.1212/NXI.0000000000000673
234. Yao R, Ma YL, Liang W, Li HH, Ma ZJ, Yu X, et al. MicroRNA-155 modulates Treg and Th17 cells differentiation and th17 cell function by targeting SOCS1. PLoS ONE. (2012) 7:e46082. doi: 10.1371/journal.pone.0046082
235. Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology. (2012) 135:73–88. doi: 10.1111/j.1365-2567.2011.03514.x
236. Lopez-Ramirez MA, Wu D, Pryce G, Simpson JE, Reijerkerk A, King-Robson J, et al. MicroRNA-155 negatively affects blood-brain barrier function during neuroinflammation. FASEB J. (2014) 28:2551–65. doi: 10.1096/fj.13-248880
237. Wu D, Cerutti C, Lopez-Ramirez MA, Pryce G, King-Robson J, Simpson JE, et al. Brain endothelial miR-146a negatively modulates T-cell adhesion through repressing multiple targets to inhibit NF-κB activation. J Cereb Blood Flow Metab. (2015) 35:412–23. doi: 10.1038/jcbfm.2014.207
238. Li B, Wang X, Choi IY, Wang YC, Liu S, Pham AT, et al. miR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J Clin Invest. (2017) 127:3702–16. doi: 10.1172/JCI94012
239. Chen Z, Stelekati E, Kurachi M, Yu S, Cai Z, Manne S, et al. MiR-150 regulates memory CD8 T cell differentiation via c-Myb. Cell Rep. (2017) 20:2584–97. doi: 10.1016/j.celrep.2017.08.060
240. Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc Natl Acad Sci USA. (2007) 104:7080–5. doi: 10.1073/pnas.0702409104
241. Koutsoudaki PN, Papadopoulos D, Passias P-G, Koutsoudaki P, Gorgoulis VG. Cellular senescence and failure of myelin repair in multiple sclerosis. Mech Ageing Dev. (2020) 192:111366. doi: 10.1016/j.mad.2020.111366
242. Psenicka MW, Smith BC, Tinkey RA, Williams JL. Connecting neuroinflammation and neurodegeneration in multiple sclerosis: are oligodendrocyte precursor cells a nexus of disease? Front Cell Neurosci. (2021) 15:654284. doi: 10.3389/fncel.2021.654284
243. Kuhlmann T, Miron V, Cuo Q, Wegner C, Antel J, Brück W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. (2008) 131:1749–58. doi: 10.1093/brain/awn096
244. Sim FJ, Zhao C, Penderis J, Franklin RJM. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci. (2002) 22:2451–9. doi: 10.1523/JNEUROSCI.22-07-02451.2002
245. Shen S, Liu A, Li J, Wolubah C, Casaccia-Bonnefil P. Epigenetic memory loss in aging oligodendrocytes in the corpus callosum. Neurobiol Aging. (2008) 29:452–63. doi: 10.1016/j.neurobiolaging.2006.10.026
246. Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJM, et al. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci. (2008) 11:1024–34. doi: 10.1038/nn.2172
247. Zhou J, Wu Y-C, Xiao B-J, Guo X-D, Zheng Q-X, Wu B. Age-related changes in the global DNA methylation profile of oligodendrocyte progenitor cells derived from rat spinal cords. Curr Med Sci. (2019) 39:67–74. doi: 10.1007/s11596-019-2001-y
248. Tiane A, Schepers M, Riemens R, Rombaut B, Vandormael P, Somers V, et al. DNA methylation regulates the expression of the negative transcriptional regulators ID2 and ID4 during OPC differentiation. Cell Mol Life Sci CMLS. (2021) 78:6631–44. doi: 10.1007/s00018-021-03927-2
249. Nicaise AM, Banda E, Guzzo RM, Russomanno K, Castro-Borrero W, Willis CM, et al. iPS-derived neural progenitor cells from PPMS patients reveal defect in myelin injury response. Exp Neurol. (2017) 288:114–21. doi: 10.1016/j.expneurol.2016.11.012
250. Nicaise AM, Wagstaff LJ, Willis CM, Paisie C, Chandok H, Robson P, et al. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc Natl Acad Sci USA. (2019) 116:9030–9. doi: 10.1073/pnas.1818348116
251. Seo JH, Miyamoto N, Hayakawa K, Pham L-DD, Maki T, Ayata C, et al. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J Clin Invest. (2013) 123:782–6. doi: 10.1172/JCI65863
252. Schönrock LM, Gawlowski G, Brück W. Interleukin-6 expression in human multiple sclerosis lesions. Neurosci Lett. (2000) 294:45–8. doi: 10.1016/S0304-3940(00)01543-3
253. Kirby L, Jin J, Cardona JG, Smith MD, Martin KA, Wang J, et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat Commun. (2019) 10:3887. doi: 10.1038/s41467-019-11638-3
254. Moyon S, Dubessy AL, Aigrot MS, Trotter M, Huang JK, Dauphinot L, et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J Neurosci. (2015) 35:4–20. doi: 10.1523/JNEUROSCI.0849-14.2015
255. Rossi S, Motta C, Studer V, Macchiarulo G, Volpe E, Barbieri F, et al. Interleukin-1β causes excitotoxic neurodegeneration and multiple sclerosis disease progression by activating the apoptotic protein p53. Mol Neurodegener. (2014) 9:56. doi: 10.1186/1750-1326-9-56
256. Mizuno T, Zhang G, Takeuchi H, Kawanokuchi J, Wang J, Sonobe Y, et al. Interferon-gamma directly induces neurotoxicity through a neuron specific, calcium-permeable complex of IFN-gamma receptor and AMPA GluR1 receptor. FASEB J. (2008) 22:1797–806. doi: 10.1096/fj.07-099499
257. Centonze D, Muzio L, Rossi S, Cavasinni F, De Chiara V, Bergami A, et al. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J Neurosci Off J Soc Neurosci. (2009) 29:3442–52. doi: 10.1523/JNEUROSCI.5804-08.2009
258. Moore CS, Cui Q-L, Warsi NM, Durafourt BA, Zorko N, Owen DR, et al. Direct and indirect effects of immune and central nervous system-resident cells on human oligodendrocyte progenitor cell differentiation. J Immunol. (2015) 194:761–72. doi: 10.4049/jimmunol.1401156
259. Abutbul S, Shapiro J, Szaingurten-Solodkin I, Levy N, Carmy Y, Baron R, et al. TGF-β signaling through SMAD2/3 induces the quiescent microglial phenotype within the CNS environment. Glia. (2012) 60:1160–71. doi: 10.1002/glia.22343
260. Correale J. The role of microglial activation in disease progression. Mult Scler. (2014) 20:1288–95. doi: 10.1177/1352458514533230
261. Costa J, Martins S, Ferreira PA, Cardoso AMS, Guedes JR, Peça J, et al. The old guard: Age-related changes in microglia and their consequences. Mech Ageing Dev. (2021) 197:111512. doi: 10.1016/j.mad.2021.111512
262. Raj DDA, Jaarsma D, Holtman IR, Olah M, Ferreira FM, Schaafsma W, et al. Priming of microglia in a DNA-repair deficient model of accelerated aging. Neurobiol Aging. (2014) 35:2147–60. doi: 10.1016/j.neurobiolaging.2014.03.025
263. O'Neil SM, Witcher KG, McKim DB, Godbout JP. Forced turnover of aged microglia induces an intermediate phenotype but does not rebalance CNS environmental cues driving priming to immune challenge. Acta Neuropathol Commun. (2018) 6:129. doi: 10.1186/s40478-018-0636-8
264. Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, et al. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain. (2012) 135:886–99. doi: 10.1093/brain/aws012
265. Chamera K, Trojan E, Szuster-Głuszczak M, Basta-Kaim A. The potential role of dysfunctions in neuron-microglia communication in the pathogenesis of brain disorders. Curr Neuropharmacol. (2020) 18:408–30. doi: 10.2174/1570159X17666191113101629
266. Tominaga K, Suzuki HI. TGF-β signaling in cellular senescence and aging-related pathology. Int J Mol Sci. (2019) 20:5002. doi: 10.3390/ijms20205002
267. Baror R, Neumann B, Segel M, Chalut KJ, Fancy SPJ, Schafer DP, et al. Transforming growth factor-beta renders ageing microglia inhibitory to oligodendrocyte generation by CNS progenitors. Glia. (2019) 67:1374–84. doi: 10.1002/glia.23612
268. Centonze D, Muzio L, Rossi S, Furlan R, Bernardi G, Martino G. The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death Differ. (2010) 17:1083–91. doi: 10.1038/cdd.2009.179
269. Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW. Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain. (2016) 139:653–61. doi: 10.1093/brain/awv395
270. Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S, et al. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat Neurosci. (2016) 19:995–8. doi: 10.1038/nn.4325
271. Cantuti-Castelvetri L, Fitzner D, Bosch-Queralt M, Weil M-T, Su M, Sen P, et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science. (2018) 359:684–8. doi: 10.1126/science.aan4183
272. Ruckh JM, Zhao J-W, Shadrach JL, van Wijngaarden P, Rao TN, Wagers AJ, et al. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. (2012) 10:96–103. doi: 10.1016/j.stem.2011.11.019
273. Correale J, Farez MF. The role of astrocytes in multiple sclerosis progression. Front Neurol. (2015) 6:180. doi: 10.3389/fneur.2015.00180
274. Salminen A, Ojala J, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur J Neurosci. (2011) 34:3–11. doi: 10.1111/j.1460-9568.2011.07738.x
275. Pedrazzi M, Patrone M, Passalacqua M, Ranzato E, Colamassaro D, Sparatore B, et al. Selective proinflammatory activation of astrocytes by high-mobility group box 1 protein signaling. J Immunol. (2007) 179:8525–32. doi: 10.4049/jimmunol.179.12.8525
276. Sun Y, Chen H, Dai J, Zou H, Gao M, Wu H, et al. HMGB1 expression patterns during the progression of experimental autoimmune encephalomyelitis. J Neuroimmunol. (2015) 280:29–35. doi: 10.1016/j.jneuroim.2015.02.005
277. Castegna A, Palmieri L, Spera I, Porcelli V, Palmieri F, Fabis-Pedrini MJ, et al. Oxidative stress and reduced glutamine synthetase activity in the absence of inflammation in the cortex of mice with experimental allergic encephalomyelitis. Neuroscience. (2011) 185:97–105. doi: 10.1016/j.neuroscience.2011.04.041
278. Novotna M, Paz Soldán MM, Abou Zeid N, Kale N, Tutuncu M, Crusan DJ, et al. Poor early relapse recovery affects onset of progressive disease course in multiple sclerosis. Neurology. (2015) 85:722–9. doi: 10.1212/WNL.0000000000001856
279. Conway BL, Zeydan B, Uygunoglu U, Novotna M, Siva A, Pittock SJ, et al. Age is a critical determinant in recovery from multiple sclerosis relapses. Mult Scler. (2019) 25:1754–63. doi: 10.1177/1352458518800815
280. Schwehr NA, Kuntz KM, Butler M, Enns EA, Shippee ND, Kingwell E, et al. Age-related decreases in relapses among adults with relapsing-onset multiple sclerosis. Mult Scler. (2020) 26:1510–8. doi: 10.1177/1352458519866613
281. Hasan KM, Kamali A, Abid H, Kramer LA, Fletcher JM, Ewing-Cobbs L. Quantification of the spatiotemporal microstructural organization of the human brain association, projection and commissural pathways across the lifespan using diffusion tensor tractography. Brain Struct Funct. (2010) 214:361–73. doi: 10.1007/s00429-009-0238-0
282. Kappos L, Butzkueven H, Wiendl H, Spelman T, Pellegrini F, Chen Y, et al. Greater sensitivity to multiple sclerosis disability worsening and progression events using a roving versus a fixed reference value in a prospective cohort study. Mult Scler. (2018) 24:963–73. doi: 10.1177/1352458517709619
283. Tutuncu M, Tang J, Zeid NA, Kale N, Crusan DJ, Atkinson EJ, et al. Onset of progressive phase is an age dependent clinical milestone in multiple sclerosis. Mult Scler. (2013) 19:188–98. doi: 10.1177/1352458512451510
284. Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain J Neurol. (2009) 132 (Pt. 5):1175–89. doi: 10.1093/brain/awp070
285. Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. (2015) 14:183–93. doi: 10.1016/S1474-4422(14)70256-X
286. Lassmann H. Pathology and disease mechanisms in different stages of multiple sclerosis. J Neurol Sci. (2013) 333:1–4. doi: 10.1016/j.jns.2013.05.010
287. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol Zurich Switz. (2004) 14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x
288. Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. (2009) 32:638–47. doi: 10.1016/j.tins.2009.08.002
289. Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol. (2015) 78:710–21. doi: 10.1002/ana.24497
290. Maggi P, Sati P, Nair G, Cortese ICM, Jacobson S, Smith BR, et al. Paramagnetic rim lesions are specific to multiple sclerosis: an international multicenter 3T MRI study. Ann Neurol. (2020) 88:1034–42. doi: 10.1002/ana.25877
291. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. (2005) 128:2705–12. doi: 10.1093/brain/awh641
292. Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation. (2000) 7:153–9. doi: 10.1159/000026433
293. Musella A, Gentile A, Rizzo FR, De Vito F, Fresegna D, Bullitta S, et al. Interplay between age and neuroinflammation in multiple sclerosis: effects on motor and cognitive functions. Front Aging Neurosci. (2018) 10:238. doi: 10.3389/fnagi.2018.00238
294. McMahon JM, McQuaid S, Reynolds R, FitzGerald UF. Increased expression of ER stress- and hypoxia-associated molecules in grey matter lesions in multiple sclerosis. Mult Scler. (2012) 18:1437–47. doi: 10.1177/1352458512438455
295. Vollmer T, Signorovitch J, Huynh L, Galebach P, Kelley C, DiBernardo A, et al. The natural history of brain volume loss among patients with multiple sclerosis: a systematic literature review and meta-analysis. J Neurol Sci. (2015) 357:8–18. doi: 10.1016/j.jns.2015.07.014
296. Ebrahimkhani S, Vafaee F, Young PE, Hur SSJ, Hawke S, Devenney E, et al. Exosomal microRNA signatures in multiple sclerosis reflect disease status. Sci Rep. (2017) 7:14293. doi: 10.1038/s41598-017-14301-3
297. Fenoglio C, Ridolfi E, Galimberti D, Scarpini E. MicroRNAs as active players in the pathogenesis of multiple sclerosis. Int J Mol Sci. (2012) 13:13227–39. doi: 10.3390/ijms131013227
298. Clemente D, Ortega MC, Arenzana FJ, de Castro F. FGF-2 and anosmin-1 are selectively expressed in different types of multiple sclerosis lesions. J Neurosci. (2011) 31:14899–909. doi: 10.1523/JNEUROSCI.1158-11.2011
299. Sarchielli P, Di Filippo M, Ercolani MV, Chiasserini D, Mattioni A, Bonucci M, et al. Fibroblast growth factor-2 levels are elevated in the cerebrospinal fluid of multiple sclerosis patients. Neurosci Lett. (2008) 435:223–8. doi: 10.1016/j.neulet.2008.02.040
300. Brás JP, Bravo J, Freitas J, Barbosa MA, Santos SG, Summavielle T, et al. TNF-alpha-induced microglia activation requires miR-342: impact on NF-kB signaling and neurotoxicity. Cell Death Dis. (2020) 11:415. doi: 10.1038/s41419-020-2626-6
301. Tripathi A, Pandit I, Perles A, Zhou Y, Cheng F, Dutta R. Identifying miRNAs in multiple sclerosis gray matter lesions that correlate with atrophy measures. Ann Clin Transl Neurol. (2021) 8:1279–91. doi: 10.1002/acn3.51365
302. Tur C, Montalban X. Progressive MS trials: lessons learned. Mult Scler. (2017) 23:1583–92. doi: 10.1177/1352458517729460
303. Weideman AM, Tapia-Maltos MA, Johnson K, Greenwood M, Bielekova B. Meta-analysis of the Age-dependent efficacy of multiple sclerosis treatments. Front Neurol. (2017) 8:577. doi: 10.3389/fneur.2017.00577
304. Loeffler C, Dietz K, Schleich A, Schlaszus H, Stoll M, Meyermann R, et al. Immune surveillance of the normal human CNS takes place in dependence of the locoregional blood-brain barrier configuration and is mainly performed by CD3(+)/CD8(+) lymphocytes. Neuropathol Off J Jpn Soc Neuropathol. (2011) 31:230–8. doi: 10.1111/j.1440-1789.2010.01167.x
305. Bieber AJ, Kerr S, Rodriguez M. Efficient central nervous system remyelination requires T cells. Ann Neurol. (2003) 53:680–4. doi: 10.1002/ana.10578
306. Zoehner G, Miclea A, Salmen A, Kamber N, Diem L, Friedli C, et al. Reduced serum immunoglobulin G concentrations in multiple sclerosis: prevalence and association with disease-modifying therapy and disease course. Ther Adv Neurol Disord. (2019) 12:1756286419878340. doi: 10.1177/1756286419878340
307. Szepanowski F, Warnke C, Meyer Zu Hörste G, Mausberg AK, Hartung H-P, Kleinschnitz C, et al. Secondary immunodeficiency and risk of infection following immune therapies in neurology. CNS Drugs. (2021) 35:1173–88. doi: 10.1007/s40263-021-00863-4
308. Kister I, Spelman T, Alroughani R, Lechner-Scott J, Duquette P, Grand'Maison F, et al. Discontinuing disease-modifying therapy in MS after a prolonged relapse-free period: a propensity score-matched study. J Neurol Neurosurg Psychiatry. (2016) 87:1133–7. doi: 10.1136/jnnp-2016-313760
309. Bsteh G, Feige J, Ehling R, Auer M, Hegen H, Di Pauli F, et al. Discontinuation of disease-modifying therapies in multiple sclerosis - clinical outcome and prognostic factors. Mult Scler. (2017) 23:1241–8. doi: 10.1177/1352458516675751
310. Winkelmann A, Loebermann M, Reisinger EC, Hartung H-P, Zettl UK. Disease-modifying therapies and infectious risks in multiple sclerosis. Nat Rev Neurol. (2016) 12:217–33. doi: 10.1038/nrneurol.2016.21
311. D'Amico E, Chisari CG, Arena S, Zanghì A, Toscano S, Lo Fermo S, et al. Cancer risk and multiple sclerosis: evidence from a large Italian cohort. Front Neurol. (2019) 10:337. doi: 10.3389/fneur.2019.00337
312. Horton DB, Reder AT. Medications for multiple sclerosis and risk of malignancy: what next? Neurother J Am Soc Exp Neurother. (2021) 18:1650–3. doi: 10.1007/s13311-021-01107-5
313. Melamed E, Lee MW. Multiple sclerosis and cancer: the ying-yang effect of disease modifying therapies. Front Immunol. (2019) 10:2954. doi: 10.3389/fimmu.2019.02954
314. Prosperini L, Haggiag S, Tortorella C, Galgani S, Gasperini C. Age-related adverse events of disease-modifying treatments for multiple sclerosis: a meta-regression. Mult Scler. (2021) 27:1391–402. doi: 10.1177/1352458520964778
315. Prosperini L, Scarpazza C, Imberti L, Cordioli C, De Rossi N, Capra R. Age as a risk factor for early onset of natalizumab-related progressive multifocal leukoencephalopathy. J Neurovirol. (2017) 23:742–9. doi: 10.1007/s13365-017-0561-9
316. Garcia J, Hendel-Chavez H, De-Goer M-G, L'Honneur AS, Dubessy AL, Taoufik Y, et al. Progressive multifocal leukoencephalopathy on dimethyl fumarate with preserved lymphocyte count but deep T-cells exhaustion. Mult Scler. (2021) 27:640–4. doi: 10.1177/1352458520942201
317. Gheuens S, Pierone G, Peeters P, Koralnik IJ. Progressive multifocal leukoencephalopathy in individuals with minimal or occult immunosuppression. J Neurol Neurosurg Psychiatry. (2010) 81:247–54. doi: 10.1136/jnnp.2009.187666
318. Grebenciucova E, Reder AT, Bernard JT. Immunologic mechanisms of fingolimod and the role of immunosenescence in the risk of cryptococcal infection: a case report and review of literature. Mult Scler Relat Disord. (2016) 9:158–62. doi: 10.1016/j.msard.2016.07.015
319. Scotto R, Reia A, Buonomo AR, Moccia M, Viceconte G, Pisano E, et al. Risk of invasive fungal infections among patients treated with disease modifying treatments for multiple sclerosis: a comprehensive review. Expert Opin Drug Saf. (2021) 20:925–36. doi: 10.1080/14740338.2021.1918673
320. Longbrake EE, Naismith RT, Parks BJ, Wu GF, Cross AH. Dimethyl fumarate-associated lymphopenia: risk factors and clinical significance. Mult Scler J - Exp Transl Clin. (2015) 1:2055217315596994. doi: 10.1177/2055217315596994
321. Schweitzer F, Laurent S, Fink GR, Barnett MH, Reddel S, Hartung H-P, et al. Age and the risks of high-efficacy disease modifying drugs in multiple sclerosis. Curr Opin Neurol. (2019) 32:305–12. doi: 10.1097/WCO.0000000000000701
322. Paghera S, Sottini A, Previcini V, Capra R, Imberti L. Age-Related lymphocyte output during disease-modifying therapies for multiple sclerosis. Drugs Aging. (2020) 37:739–46. doi: 10.1007/s40266-020-00789-4
323. Tsokos GC. Systemic lupus erythematosus. N Engl J Med. (2011) 365:2110–21. doi: 10.1056/NEJMra1100359
324. Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol. (2011) 7:691–9. doi: 10.1038/nrrheum.2011.132
325. Perazzio SF, Salomão R, Silva NP, Andrade LEC. Increased neutrophil oxidative burst metabolism in systemic lupus erythematosus. Lupus. (2012) 21:1543–51. doi: 10.1177/0961203312461060
326. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
327. Burbano C, Vasquez G, Rojas M. Modulatory effects of CD14+CD16++ monocytes on CD14++CD16- monocytes: a possible explanation of monocyte alterations in systemic lupus erythematosus. Arthritis Rheumatol Hoboken NJ. (2014) 66:3371–81. doi: 10.1002/art.38860
328. Ma C, Xia Y, Yang Q, Zhao Y. The contribution of macrophages to systemic lupus erythematosus. Clin Immunol. (2019) 207:1–9. doi: 10.1016/j.clim.2019.06.009
329. Henriques A, Teixeira L, Inês L, Carvalheiro T, Gonçalves A, Martinho A, et al. NK cells dysfunction in systemic lupus erythematosus: relation to disease activity. Clin Rheumatol. (2013) 32:805–13. doi: 10.1007/s10067-013-2176-8
330. Hervier B, Beziat V, Haroche J, Mathian A, Lebon P, Ghillani-Dalbin P, et al. Phenotype and function of natural killer cells in systemic lupus erythematosus: excess interferon-γ production in patients with active disease. Arthritis Rheum. (2011) 63:1698–706. doi: 10.1002/art.30313
331. Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. (1999) 17:189–220. doi: 10.1146/annurev.immunol.17.1.189
332. Marshall JD, Heeke DS, Abbate C, Yee P, Van Nest G. Induction of interferon-γ from natural killer cells by immunostimulatory CpG DNA is mediated through plasmacytoid-dendritic-cell-produced interferon-α and tumour necrosis factor-α. Immunology. (2006) 117:38–46. doi: 10.1111/j.1365-2567.2005.02261.x
333. Kurosaka D, Yasuda J, Ikeshima-Kataoka H, Ozawa Y, Yoshida K, Yasuda C, et al. Decreased numbers of signal-joint T cell receptor excision circle-containing CD4+ and CD8+ cells in systemic lupus erythematosus patients. Mod Rheumatol. (2007) 17:296–300. doi: 10.3109/s10165-007-0583-x
334. Liu X, Zhang W, Zhao M, Fu L, Liu L, Wu J, et al. T cell receptor β repertoires as novel diagnostic markers for systemic lupus erythematosus and rheumatoid arthritis. Ann Rheum Dis. (2019) 78:1070–8. doi: 10.1136/annrheumdis-2019-215442
335. Maeda N, Sekigawa I, Iida N, Matsumoto M, Hashimoto H, Hirose S. Relationship between CD4+/CD8+ T cell ratio and T cell activation in systemic lupus erythematosus. Scand J Rheumatol. (1999) 28:166–70. doi: 10.1080/03009749950154248
336. Suárez-Fueyo A, Bradley SJ, Tsokos GC. T cells in systemic lupus erythematosus. Curr Opin Immunol. (2016) 43:32–8. doi: 10.1016/j.coi.2016.09.001
337. Blanco P, Pitard V, Viallard J-F, Taupin J-L, Pellegrin J-L, Moreau J-F. Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. (2005) 52:201–11. doi: 10.1002/art.20745
338. Couzi L, Merville P, Deminière C, Moreau J-F, Combe C, Pellegrin J-L, et al. Predominance of CD8+ T lymphocytes among periglomerular infiltrating cells and link to the prognosis of class III and class IV lupus nephritis. Arthritis Rheum. (2007) 56:2362–70. doi: 10.1002/art.22654
339. Kosmaczewska A, Ciszak L, Stosio M, Szteblich A, Madej M, Frydecka I, et al. CD4+CD28null T cells are expanded in moderately active systemic lupus erythematosus and secrete pro-inflammatory interferon gamma, depending on the disease activity index. Lupus. (2020) 29:705–14. doi: 10.1177/0961203320917749
340. Fritsch-Stork R, Müllegger D, Skriner K, Jahn-Schmid B, Smolen JS, Steiner G. The spliceosomal autoantigen heterogeneous nuclear ribonucleoprotein A2 (hnRNP-A2) is a major T cell autoantigen in patients with systemic lupus erythematosus. Arthritis Res Ther. (2006) 8:R118. doi: 10.1186/ar2007
341. Konya C, Paz Z, Tsokos GC. The role of T cells in systemic lupus erythematosus: an update. Curr Opin Rheumatol. (2014) 26:493–501. doi: 10.1097/BOR.0000000000000082
342. Sims GP, Ettinger R, Shirota Y, Yarboro CH, Illei GG, Lipsky PE. Identification and characterization of circulating human transitional B cells. Blood. (2005) 105:4390–8. doi: 10.1182/blood-2004-11-4284
343. Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. (2005) 201:703–11. doi: 10.1084/jem.20042251
344. Odendahl M, Jacobi A, Hansen A, Feist E, Hiepe F, Burmester GR, et al. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol. (2000) 165:5970–9. doi: 10.4049/jimmunol.165.10.5970
345. Zhu L, Yin Z, Ju B, Zhang J, Wang Y, Lv X, et al. Altered frequencies of memory B cells in new-onset systemic lupus erythematosus patients. Clin Rheumatol. (2018) 37:205–12. doi: 10.1007/s10067-017-3877-1
346. Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A new population of cells lacking expression of cd27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol. (2007) 178:6624–33. doi: 10.4049/jimmunol.178.10.6624
347. Jacobi AM, Reiter K, Mackay M, Aranow C, Hiepe F, Radbruch A, et al. Activated memory B cell subsets correlate with disease activity in systemic lupus erythematosus: delineation by expression of CD27, IgD, and CD95. Arthritis Rheum. (2008) 58:1762–73. doi: 10.1002/art.23498
348. Wang S, Wang J, Kumar V, Karnell JL, Naiman B, Gross PS, et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chiT-bet+ B cells in SLE. Nat Commun. (2018) 9:1758. doi: 10.1038/s41467-018-03750-7
349. Sachinidis A, Xanthopoulos K, Garyfallos A. Age-Associated B cells (ABCs) in the prognosis, diagnosis and therapy of systemic lupus erythematosus (SLE). Mediterr J Rheumatol. (2020) 31:311–8. doi: 10.31138/mjr.31.3.311
350. Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, et al. Distinct effector B-cells induced by unregulated Toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity. (2018) 49:725–39.e6. doi: 10.1016/j.immuni.2018.08.015
351. Ohl K, Tenbrock K. Regulatory T cells in systemic lupus erythematosus. Eur J Immunol. (2015) 45:344–55. doi: 10.1002/eji.201344280
352. Alunno A, Bartoloni E, Bistoni O, Nocentini G, Ronchetti S, Caterbi S, et al. Balance between regulatory T and Th17 cells in systemic lupus erythematosus: the old and the new. Clin Dev Immunol. (2012) 2012:823085. doi: 10.1155/2012/823085
353. Geginat J, Vasco M, Gerosa M, Tas SW, Pagani M, Grassi F, et al. IL-10 producing regulatory and helper T-cells in systemic lupus erythematosus. Semin Immunol. (2019) 44:101330. doi: 10.1016/j.smim.2019.101330
354. Aringer M. Inflammatory markers in systemic lupus erythematosus. J Autoimmun. (2020) 110:102374. doi: 10.1016/j.jaut.2019.102374
355. Alessandri C, Barbati C, Vacirca D, Piscopo P, Confaloni A, Sanchez M, et al. T lymphocytes from patients with systemic lupus erythematosus are resistant to induction of autophagy. FASEB J. (2012) 26:4722–32. doi: 10.1096/fj.12-206060
356. Clarke AJ, Ellinghaus U, Cortini A, Stranks A, Simon AK, Botto M, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. (2015) 74:912–20. doi: 10.1136/annrheumdis-2013-204343
357. Kurosaka D, Yasuda J, Yoshida K, Yokoyama T, Ozawa Y, Obayashi Y, et al. Telomerase activity and telomere length of peripheral blood mononuclear cells in SLE patients. Lupus. (2003) 12:591–9. doi: 10.1191/0961203303lu426oa
358. Kurosaka D, Yasuda J, Yoshida K, Yoneda A, Yasuda C, Kingetsu I, et al. Abnormal telomerase activity and telomere length in T and B cells from patients with systemic lupus erythematosus. J Rheumatol. (2006) 33:1102–7.
359. Honda M, Mengesha E, Albano S, Nichols WS, Wallace DJ, Metzger A, et al. Telomere shortening and decreased replicative potential, contrasted by continued proliferation of telomerase-positive CD8+CD28lo T cells in patients with systemic lupus erythematosus. Clin Immunol. (2001) 99:211–21. doi: 10.1006/clim.2001.5023
360. Gergely P, Grossman C, Niland B, Puskas F, Neupane H, Allam F, et al. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. (2002) 46:175–90. doi: 10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H
361. Shah D, Aggarwal A, Bhatnagar A, Kiran R, Wanchu A. Association between T lymphocyte sub-sets apoptosis and peripheral blood mononuclear cells oxidative stress in systemic lupus erythematosus. Free Radic Res. (2011) 45:559–67. doi: 10.3109/10715762.2011.555765
362. Lee H-T, Wu T-H, Lin C-S, Lee C-S, Pan S-C, Chang D-M, et al. Oxidative DNA and mitochondrial DNA change in patients with SLE. Front Biosci. (2017) 22:493–503. doi: 10.2741/4497
363. López-López L, Nieves-Plaza M, Castro M del R, Font YM, Torres-Ramos CA, Vilá LM, et al. Mitochondrial DNA damage is associated with damage accrual and disease duration in patients with systemic lupus erythematosus. Lupus. (2014) 23:1133–41. doi: 10.1177/0961203314537697
364. Montalvão TM, Miranda-Vilela AL, Roll MM, Grisolia CK, Santos-Neto L. DNA damage levels in systemic lupus erythematosus patients with low disease activity: an evaluation by comet assay. Adv Biosci Biotechnol. (2012) 3:983–8. doi: 10.4236/abb.2012.327121
365. Khan MdA, Alam K, Zafaryab Md, Rizvi MMA. Peroxynitrite-modified histone as a pathophysiological biomarker in autoimmune diseases. Biochimie. (2017) 140:1–9. doi: 10.1016/j.biochi.2017.06.006
366. Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity. (2013) 39:482–95. doi: 10.1016/j.immuni.2013.08.004
367. Absher DM, Li X, Waite LL, Gibson A, Roberts K, Edberg J, et al. Genome-Wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLOS Genet. (2013) 9:e1003678. doi: 10.1371/journal.pgen.1003678
368. Imgenberg-Kreuz J, Almlöf JC, Leonard D, Alexsson A, Nordmark G, Eloranta M-L, et al. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann Rheum Dis. (2018) 77:736–43. doi: 10.1136/annrheumdis-2017-212379
369. Zhang Z, Shi L, Dawany N, Kelsen J, Petri MA, Sullivan KE. H3K4 tri-methylation breadth at transcription start sites impacts the transcriptome of systemic lupus erythematosus. Clin Epigenetics. (2016) 8:14. doi: 10.1186/s13148-016-0179-4
370. Gorelik G, Fang JY, Wu A, Sawalha AH, Richardson B. Impaired T cell protein kinase Cδ activation decreases ERK pathway signaling in idiopathic and hydralazine-induced lupus. J Immunol. (2007) 179:5553–63. doi: 10.4049/jimmunol.179.8.5553
371. Lu Q, Kaplan M, Ray D, Ray D, Zacharek S, Gutsch D, et al. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum. (2002) 46:1282–91. doi: 10.1002/art.10234
372. Kaplan MJ, Lu Q, Wu A, Attwood J, Richardson B. Demethylation of promoter regulatory elements contributes to perforin overexpression in CD4+ lupus T cells. J Immunol. (2004) 172:3652–61. doi: 10.4049/jimmunol.172.6.3652
373. Pan W, Zhu S, Yuan M, Cui H, Wang L, Luo X, et al. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. (2010) 184:6773–81. doi: 10.4049/jimmunol.0904060
374. Qin H, Zhu X, Liang J, Wu J, Yang Y, Wang S, et al. MicroRNA-29b contributes to DNA hypomethylation of CD4+ T cells in systemic lupus erythematosus by indirectly targeting DNA methyltransferase 1. J Dermatol Sci. (2013) 69:61–7. doi: 10.1016/j.jdermsci.2012.10.011
375. Zhao S, Wang Y, Liang Y, Zhao M, Long H, Ding S, et al. MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum. (2011) 63:1376–86. doi: 10.1002/art.30196
376. Cheng T, Ding S, Liu S, Li Y, Sun L. Human umbilical cord-derived mesenchymal stem cell therapy ameliorates lupus through increasing CD4+ T cell senescence via MiR-199a-5p/Sirt1/p53 axis. Theranostics. (2021) 11:893–905. doi: 10.7150/thno.48080
377. Wu X, Ye Y, Niu J, Li Y, Li X, You X, et al. Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Sci Transl Med. (2014) 6:246ra99. doi: 10.1126/scitranslmed.3009131
378. Liu Y, Dong J, Mu R, Gao Y, Tan X, Li Y, et al. MicroRNA-30a promotes B cell hyperactivity in patients with systemic lupus erythematosus by direct interaction with Lyn. Arthritis Rheum. (2013) 65:1603–11. doi: 10.1002/art.37912
379. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. (2005) 102:13944–9. doi: 10.1073/pnas.0506654102
380. Yuan Y, Kasar S, Underbayev C, Vollenweider D, Salerno E, Kotenko SV, et al. Role of microRNA-15a in autoantibody production in interferon-augmented murine model of lupus. Mol Immunol. (2012) 52:61–70. doi: 10.1016/j.molimm.2012.04.007
381. Ding S, Liang Y, Zhao M, Liang G, Long H, Zhao S, et al. Decreased microRNA-142-3p/5p expression causes CD4+ T cell activation and B cell hyperstimulation in systemic lupus erythematosus. Arthritis Rheum. (2012) 64:2953–63. doi: 10.1002/art.34505
382. Anandagoda N, Willis JC, Hertweck A, Roberts LB, Jackson I, Gökmen MR, et al. microRNA-142-mediated repression of phosphodiesterase 3B critically regulates peripheral immune tolerance. J Clin Invest. (2019) 129:1257–71. doi: 10.1172/JCI124725
383. Hou J, Wang P, Lin L, Liu X, Ma F, An H, et al. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol. (1950) (2009) 183:2150–8. doi: 10.4049/jimmunol.0900707
384. Wang P, Hou J, Lin L, Wang C, Liu X, Li D, et al. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol. (2010) 185:6226–33. doi: 10.4049/jimmunol.1000491
385. Zhang J, Chen C, Fu H, Yu J, Sun Y, Huang H, et al. MicroRNA-125a-loaded polymeric nanoparticles alleviate systemic lupus erythematosus by restoring effector/regulatory T cells balance. ACS Nano. (2020) 14:4414–29. doi: 10.1021/acsnano.9b09998
386. Rouas R, Fayyad-Kazan H, El Zein N, Lewalle P, Rothé F, Simion A, et al. Human natural Treg microRNA signature: role of microRNA-31 and microRNA-21 in FOXP3 expression. Eur J Immunol. (2009) 39:1608–18. doi: 10.1002/eji.200838509
387. Zhang L, Ke F, Liu Z, Bai J, Liu J, Yan S, et al. MicroRNA-31 negatively regulates peripherally derived regulatory T-cell generation by repressing retinoic acid-inducible protein 3. Nat Commun. (2015) 6:7639. doi: 10.1038/ncomms8639
388. Xie M, Wang J, Gong W, Xu H, Pan X, Chen Y, et al. NF-κB-driven miR-34a impairs Treg/Th17 balance via targeting Foxp3. J Autoimmun. (2019) 102:96–113. doi: 10.1016/j.jaut.2019.04.018
389. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
390. Lundberg K, Nijenhuis S, Vossenaar ER, Palmblad K, van Venrooij WJ, Klareskog L, et al. Citrullinated proteins have increased immunogenicity and arthritogenicity and their presence in arthritic joints correlates with disease severity. Arthritis Res Ther. (2005) 7:R458–67. doi: 10.1186/ar1697
391. O'Neil LJ, Kaplan MJ. Neutrophils in rheumatoid arthritis: breaking immune tolerance and fueling disease. Trends Mol Med. (2019) 25:215–27. doi: 10.1016/j.molmed.2018.12.008
392. Merino A, Buendia P, Martin-Malo A, Aljama P, Ramirez R, Carracedo J. Senescent CD14+CD16+ monocytes exhibit proinflammatory and proatherosclerotic activity. J Immunol. (2011) 186:1809–15. doi: 10.4049/jimmunol.1001866
393. Kawanaka N, Yamamura M, Aita T, Morita Y, Okamoto A, Kawashima M, et al. CD14+, CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. (2002) 46:2578–86. doi: 10.1002/art.10545
394. Krasselt M, Baerwald C, Wagner U, Rossol M. CD56+ monocytes have a dysregulated cytokine response to lipopolysaccharide and accumulate in rheumatoid arthritis and immunosenescence. Arthritis Res Ther. (2013) 15:R139. doi: 10.1186/ar4321
395. Siouti E, Andreakos E. The many facets of macrophages in rheumatoid arthritis. Biochem Pharmacol. (2019) 165:152–69. doi: 10.1016/j.bcp.2019.03.029
396. Aggarwal A, Sharma A, Bhatnagar A. Role of cytolytic impairment of natural killer and natural killer T-cell populations in rheumatoid arthritis. Clin Rheumatol. (2014) 33:1067–78. doi: 10.1007/s10067-014-2641-z
397. Thanapati S, Ganu M, Giri P, Kulkarni S, Sharma M, Babar P, et al. Impaired NK cell functionality and increased TNF-α production as biomarkers of chronic chikungunya arthritis and rheumatoid arthritis. Hum Immunol. (2017) 78:370–4. doi: 10.1016/j.humimm.2017.02.006
398. Fujii H, Shao L, Colmegna I, Goronzy JJ, Weyand CM. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci USA. (2009) 106:4360–5. doi: 10.1073/pnas.0811332106
399. Koetz K, Bryl E, Spickschen K, O'Fallon WM, Goronzy JJ, Weyand CM. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci USA. (2000) 97:9203–8. doi: 10.1073/pnas.97.16.9203
400. Colmegna I, Diaz-Borjon A, Fujii H, Schaefer L, Goronzy JJ, Weyand CM. Defective proliferative capacity and accelerated telomeric loss of hematopoietic progenitor cells in rheumatoid arthritis. Arthritis Rheum. (2008) 58:990–1000. doi: 10.1002/art.23287
401. Colmegna I, Weyand CM. Haematopoietic stem and progenitor cells in rheumatoid arthritis. Rheumatol Oxf Engl. (2011) 50:252–60. doi: 10.1093/rheumatology/keq298
402. Colmegna I, Pryshchep S, Oishi H, Goronzy JJ, Weyand CM. Dampened ERK signaling in hematopoietic progenitor cells in rheumatoid arthritis. Clin Immunol. (2012) 143:73–82. doi: 10.1016/j.clim.2012.01.007
403. Wagner UG, Koetz K, Weyand CM, Goronzy JJ. Perturbation of the T cell repertoire in rheumatoid arthritis. Proc Natl Acad Sci USA. (1998) 95:14447–52. doi: 10.1073/pnas.95.24.14447
404. Cho B-A, Sim JH, Park JA, Kim HW, Yoo W-H, Lee S-H, et al. Characterization of effector memory CD8+ T cells in the synovial fluid of rheumatoid arthritis. J Clin Immunol. (2012) 32:709–20. doi: 10.1007/s10875-012-9674-3
405. Chemin K, Gerstner C, Malmström V. Effector Functions of CD4+ T cells at the site of local autoimmune inflammation—lessons from rheumatoid arthritis. Front Immunol. (2019) 10:353. doi: 10.3389/fimmu.2019.00353
406. Pieper J, Johansson S, Snir O, Linton L, Rieck M, Buckner JH, et al. Peripheral and site-specific CD4(+) CD28(null) T cells from rheumatoid arthritis patients show distinct characteristics. Scand J Immunol. (2014) 79:149–55. doi: 10.1111/sji.12139
407. Pawlik A, Ostanek L, Brzosko I, Brzosko M, Masiuk M, Machalinski B, et al. The expansion of CD4+CD28- T cells in patients with rheumatoid arthritis. Arthritis Res Ther. (2003) 5:R210–3. doi: 10.1186/ar766
408. Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci USA. (2003) 100:9452–7. doi: 10.1073/pnas.1632807100
409. Michel JJ, Turesson C, Lemster B, Atkins SR, Iclozan C, Bongartz T, et al. CD56-expressing T cells that have features of senescence are expanded in rheumatoid arthritis. Arthritis Rheum. (2007) 56:43–57. doi: 10.1002/art.22310
410. Lemster BH, Michel JJ, Montag DT, Paat JJ, Studenski SA, Newman AB, et al. Induction of CD56 and TCR-independent activation of T cells with aging. J Immunol. (2008) 180:1979–90. doi: 10.4049/jimmunol.180.3.1979
411. Sawai H, Park YW, He X, Goronzy JJ, Weyand CM. Fractalkine mediates T cell-dependent proliferation of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheum. (2007) 56:3215–25. doi: 10.1002/art.22919
412. Rodríguez-Carrio J, Alperi-López M, López P, Alonso-Castro S, Ballina-García FJ, Suárez A. TNFα polymorphism as marker of immunosenescence for rheumatoid arthritis patients. Exp Gerontol. (2015) 61:123–9. doi: 10.1016/j.exger.2014.12.009
413. Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, et al. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med. (2013) 5:173ra23. doi: 10.1126/scitranslmed.3005407
414. Wang Y, Lloyd KA, Melas I, Zhou D, Thyagarajan R, Lindqvist J, et al. Rheumatoid arthritis patients display B-cell dysregulation already in the naïve repertoire consistent with defects in B-cell tolerance. Sci Rep. (2019) 9:19995. doi: 10.1038/s41598-019-56279-0
415. Nakayama T, Yoshimura M, Higashioka K, Miyawaki K, Ota Y, Ayano M, et al. Type 1 helper T cells generate CXCL9/10-producing T-bet+ effector B cells potentially involved in the pathogenesis of rheumatoid arthritis. Cell Immunol. (2021) 360:104263. doi: 10.1016/j.cellimm.2020.104263
416. Huang Y, Wang H, Ba X, Chen Z, Wang Y, Qin K, et al. Decipher manifestations and Treg /Th17 imbalance in multi-staging rheumatoid arthritis and correlation with TSDR/RORC methylation. Mol Immunol. (2020) 127:1–11. doi: 10.1016/j.molimm.2020.08.002
417. Boissier M-C, Assier E, Biton J, Denys A, Falgarone G, Bessis N. Regulatory T cells (Treg) in rheumatoid arthritis. Joint Bone Spine. (2009) 76:10–4. doi: 10.1016/j.jbspin.2008.08.002
418. Möttönen M, Heikkinen J, Mustonen L, Isomäki P, Luukkainen R, Lassila O. CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol. (2005) 140:360–7. doi: 10.1111/j.1365-2249.2005.02754.x
419. Fessler J, Raicht A, Husic R, Ficjan A, Schwarz C, Duftner C, et al. Novel senescent regulatory T-cell subset with impaired suppressive function in rheumatoid arthritis. Front Immunol. (2017) 8:300. doi: 10.3389/fimmu.2017.00300
420. Masi AT, Rehman AA, Elmore KB, Aldag JC. Serum acute phase protein and inflammatory cytokine network correlations: comparison of a pre-rheumatoid arthritis and non-rheumatoid arthritis community cohort. J Innate Immun. (2013) 5:100–13. doi: 10.1159/000345700
421. Fraser A, Fearon U, Billinghurst RC, Ionescu M, Reece R, Barwick T, et al. Turnover of type II collagen and aggrecan in cartilage matrix at the onset of inflammatory arthritis in humans: relationship to mediators of systemic and local inflammation. Arthritis Rheum. (2003) 48:3085–95. doi: 10.1002/art.11331
422. Park J, Choi HM, Yang H-I, Yoo MC, Kim KS. Increased expression of IL-1 receptors in response to IL-1β may produce more IL-6, IL-8, VEGF, and PGE2 in senescent synovial cells induced in vitro than in presenescent cells. Rheumatol Int. (2012) 32:2005–10. doi: 10.1007/s00296-011-1891-1
423. Connor AM, Mahomed N, Gandhi R, Keystone EC, Berger SA. TNFα modulates protein degradation pathways in rheumatoid arthritis synovial fibroblasts. Arthritis Res Ther. (2012) 14:R62. doi: 10.1186/ar3778
424. Kato M, Ospelt C, Gay RE, Gay S, Klein K. Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol. (2014) 66:40–8. doi: 10.1002/art.38190
425. Schönland SO, Lopez C, Widmann T, Zimmer J, Bryl E, Goronzy JJ, et al. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc Natl Acad Sci USA. (2003) 100:13471–6. doi: 10.1073/pnas.2233561100
426. Yudoh K, Matsuno H, Nezuka T, Kimura T. Different mechanisms of synovial hyperplasia in rheumatoid arthritis and pigmented villonodular synovitis: the role of telomerase activity in synovial proliferation. Arthritis Rheum. (1999) 42:669–77. doi: 10.1002/1529-0131(199904)42:4<669::AID-ANR9>3.0.CO;2-V
427. Shao L, Fujii H, Colmegna I, Oishi H, Goronzy JJ, Weyand CM. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J Exp Med. (2009) 206:1435–49. doi: 10.1084/jem.20082251
428. Shao L, Goronzy JJ, Weyand CM. DNA-dependent protein kinase catalytic subunit mediates T-cell loss in rheumatoid arthritis. EMBO Mol Med. (2010) 2:415–27. doi: 10.1002/emmm.201000096
429. Collins LV, Hajizadeh S, Holme E, Jonsson I-M, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. (2004) 75:995–1000. doi: 10.1189/jlb.0703328
430. Harty LC, Biniecka M, O'Sullivan J, Fox E, Mulhall K, Veale DJ, et al. Mitochondrial mutagenesis correlates with the local inflammatory environment in arthritis. Ann Rheum Dis. (2012) 71:582–8. doi: 10.1136/annrheumdis-2011-200245
431. Del Rey MJ, Valín Á, Usategui A, Ergueta S, Martín E, Municio C, et al. Senescent synovial fibroblasts accumulate prematurely in rheumatoid arthritis tissues and display an enhanced inflammatory phenotype. Immun Ageing. (2019) 16:29. doi: 10.1186/s12979-019-0169-4
432. Tak PP, Smeets TJ, Boyle DL, Kraan MC, Shi Y, Zhuang S, et al. p53 overexpression in synovial tissue from patients with early and longstanding rheumatoid arthritis compared with patients with reactive arthritis and osteoarthritis. Arthritis Rheum. (1999) 42:948–53. doi: 10.1002/1529-0131(199905)42:5<948::AID-ANR13>3.0.CO;2-L
433. Yamanishi Y, Boyle DL, Rosengren S, Green DR, Zvaifler NJ, Firestein GS. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. (2002) 99:10025–30. doi: 10.1073/pnas.152333199
434. Karouzakis E, Gay RE, Gay S, Neidhart M. Increased recycling of polyamines is associated with global DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. (2012) 64:1809–17. doi: 10.1002/art.34340
435. Liebold I, Grützkau A, Göckeritz A, Gerl V, Lindquist R, Feist E, et al. Peripheral blood mononuclear cells are hypomethylated in active rheumatoid arthritis and methylation correlates with disease activity. Rheumatol Oxf Engl. (2021) 60:1984–95. doi: 10.1093/rheumatology/keaa649
436. Ishida K, Kobayashi T, Ito S, Komatsu Y, Yokoyama T, Okada M, et al. Interleukin-6 gene promoter methylation in rheumatoid arthritis and chronic periodontitis. J Periodontol. (2012) 83:917–25. doi: 10.1902/jop.2011.110356
437. Pitaksalee R, Burska AN, Ajaib S, Rogers J, Parmar R, Mydlova K, et al. Differential CpG DNA methylation in peripheral naïve CD4+ T-cells in early rheumatoid arthritis patients. Clin Epigenetics. (2020) 12:54. doi: 10.1186/s13148-020-00837-1
438. Huber LC, Brock M, Hemmatazad H, Giger OT, Moritz F, Trenkmann M, et al. Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum. (2007) 56:1087–93. doi: 10.1002/art.22512
439. Wada TT, Araki Y, Sato K, Aizaki Y, Yokota K, Kim YT, et al. Aberrant histone acetylation contributes to elevated interleukin-6 production in rheumatoid arthritis synovial fibroblasts. Biochem Biophys Res Commun. (2014) 444:682–6. doi: 10.1016/j.bbrc.2014.01.195
440. Niederer F, Ospelt C, Brentano F, Hottiger MO, Gay RE, Gay S, et al. SIRT1 overexpression in the rheumatoid arthritis synovium contributes to proinflammatory cytokine production and apoptosis resistance. Ann Rheum Dis. (2011) 70:1866–73. doi: 10.1136/ard.2010.148957
441. Feng Z, Li J, Ren J, Lv Z. [Expression of miR-146a and miR-16 in peripheral blood mononuclear cells of patients with rheumatoid arthritis and their correlation to the disease activity]. Nan Fang Yi Ke Da Xue Xue Bao. (2011) 31:320–3.
442. Murata K, Yoshitomi H, Tanida S, Ishikawa M, Nishitani K, Ito H, et al. Plasma and synovial fluid microRNAs as potential biomarkers of rheumatoid arthritis and osteoarthritis. Arthritis Res Ther. (2010) 12:R86. doi: 10.1186/ar3013
443. Sampath D, Liu C, Vasan K, Sulda M, Puduvalli VK, Wierda WG, et al. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood. (2012) 119:1162–72. doi: 10.1182/blood-2011-05-351510
444. Nakasa T, Miyaki S, Okubo A, Hashimoto M, Nishida K, Ochi M, et al. Expression of microRNA-146 in rheumatoid arthritis synovial tissue. Arthritis Rheum. (2008) 58:1284–92. doi: 10.1002/art.23429
445. Stanczyk J, Pedrioli DML, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, et al. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. (2008) 58:1001–9. doi: 10.1002/art.23386
Keywords: multiple sclerosis, immunosenescence, inflammaging, T/B cells, oligodendrocytes, microglia, astrocytes, disease modifying therapies
Citation: Perdaens O and van Pesch V (2022) Molecular Mechanisms of Immunosenescene and Inflammaging: Relevance to the Immunopathogenesis and Treatment of Multiple Sclerosis. Front. Neurol. 12:811518. doi: 10.3389/fneur.2021.811518
Received: 08 November 2021; Accepted: 27 December 2021;
Published: 25 February 2022.
Edited by:
Aurora Zanghì, Sant'Elia Hospital, ItalyReviewed by:
Niels Hellings, University of Hasselt, BelgiumCopyright © 2022 Perdaens and van Pesch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincent van Pesch, dmluY2VudC52YW5wZXNjaEBzYWludGx1Yy51Y2xvdXZhaW4uYmU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.