
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 01 February 2022
Sec. Pediatric Neurology
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.806516
This article is part of the Research Topic Genetically Determined Epilepsies: Perspectives in the Era of Precision Medicine View all 19 articles
 Lorenzo Perilli1*
Lorenzo Perilli1* Gioia Mastromoro2
Gioia Mastromoro2 Manuel Murciano1,3
Manuel Murciano1,3 Ilaria Amedeo1Federica Avenoso1
Ilaria Amedeo1Federica Avenoso1 Antonio Pizzuti2
Antonio Pizzuti2 Cristiana Alessia Guido1
Cristiana Alessia Guido1 Alberto Spalice1
Alberto Spalice1We report on the rare case of a male toddler presenting with myoclonic epilepsy characterized by daily episodes of upward movements of the eyebrows, and myoclonic jerks of both head and upper limbs. In addition, the child showed speech delay, tremors, and lack of motor coordination. Next Generation Sequencing analysis (NGS) performed in trio revealed in the proband the c.889C>T de novo missense variant in the KCNA2 gene in heterozygous state. This is the first case of myoclonic epilepsy in a toddler due to a c.889C>T KCNA2 missense variant. The patient was treated with valproic acid and ethosuximide with a good clinical response. At 6 years old, follow-up revealed that the proband was seizure-free with tremors and clumsiness in movements. According to the literature, this case supports the correlation between myoclonic epilepsy and KCNA2 alterations. This evidence suggests that performing genomic testing including the KCNA2 gene in preschool patients affected by myoclonic epilepsy, especially when associated with delayed neurodevelopment. Our goal is to expand the phenotypical spectrum of this rare condition and adding clinical features following a genotype-first approach.
Among the genetically determined forms of epilepsy, many genes remain unknown. Cases of epilepsy caused by a KCNA2 mutation are known in literature (as shown in Table 1) different from the one reported in this manuscript. In 2017, Sachdev et al. (8) described a case of genetically determined epilepsy carrying the same mutation of our patient, but with different phenotype, described in Table 2. We report for the first time a pediatric patient affected by myoclonic epilepsy due to the heterozygous c.889C>T missense variant in the KCNA2 gene. According to the literature, this case supports the correlation between myoclonic epilepsy and KCNA2 alterations. This evidence suggests that performing genomic testing including the KCNA2 gene in preschool patients affected by myoclonic epilepsy, especially when associated with delayed neurodevelopment. Our goal is to expand the phenotypical spectrum of this rare condition and adding clinical features following a genotype-first approach.
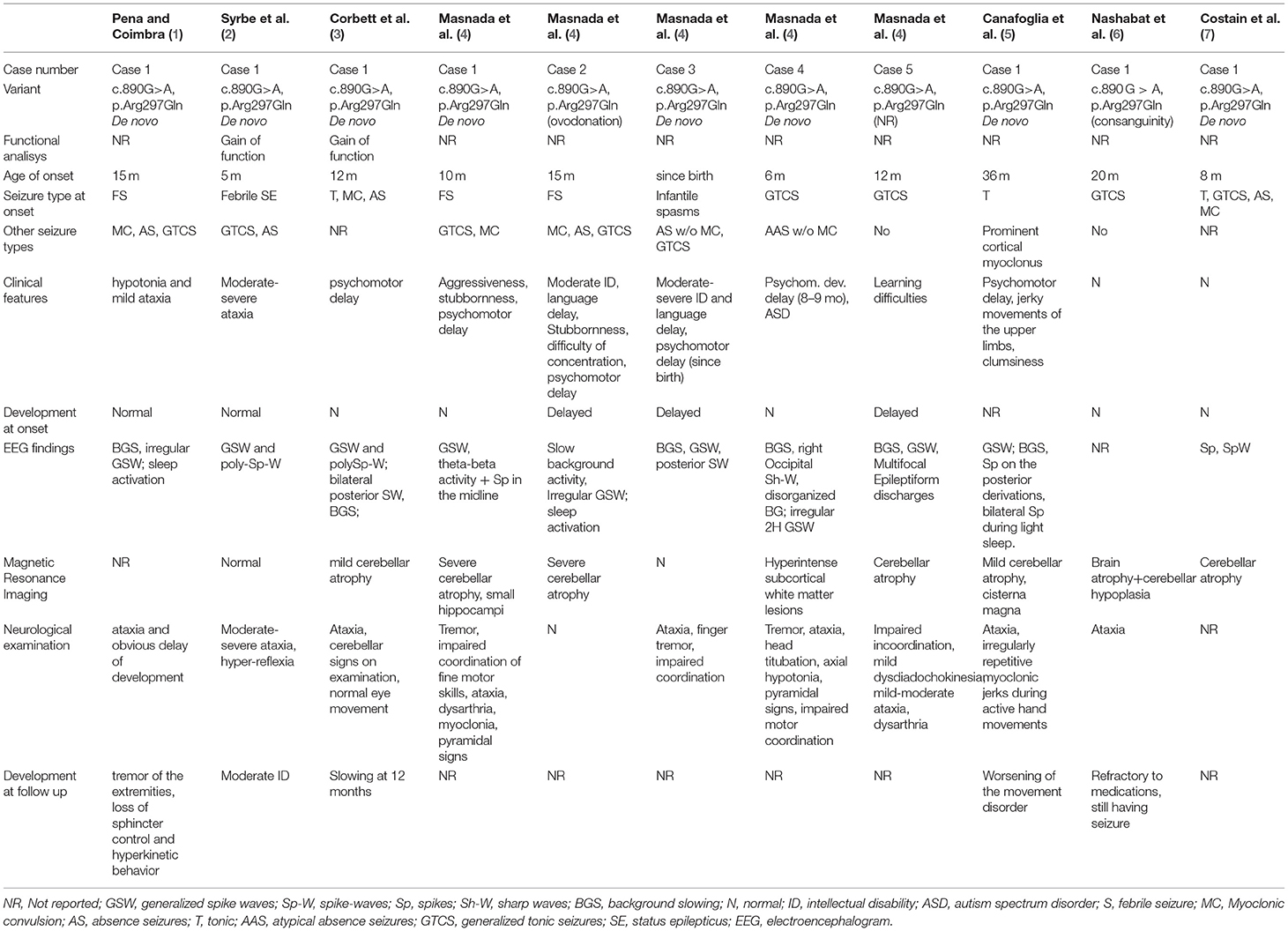
Table 1. Comparison between the characteristics of patients reported in the literature with mutation in the same triplet encoding an amino acid as the patient analyzed in this study.
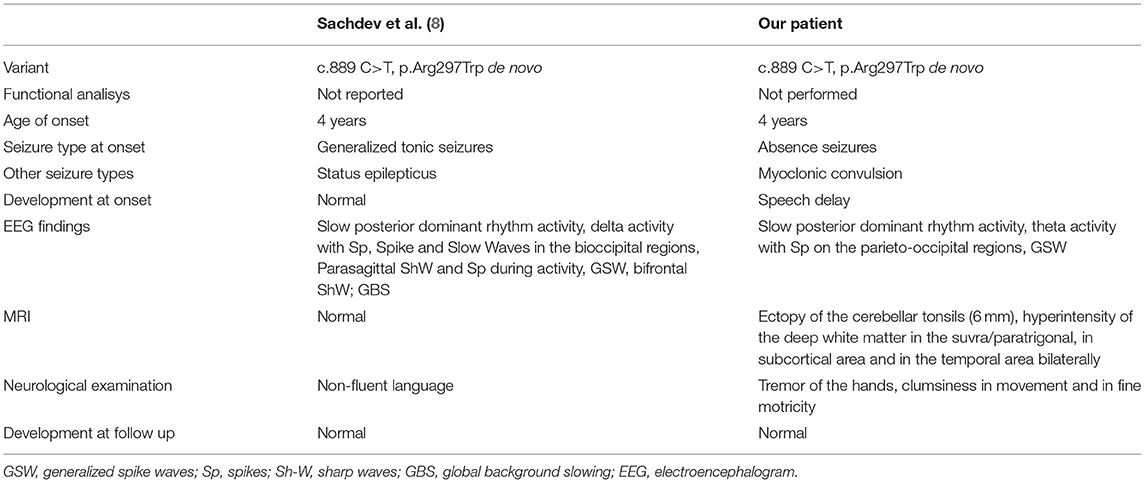
Table 2. Comparison between the characteristics of the patient studied with the only previous case reported in the literature with the same mutation.
The proband was born at 38 weeks of gestational age through Cesarean section, performed due to maternal-fetal disproportion. Psychomotor development was characterized by autonomous walking at 16 months (referred balance disorder) and mild language delay improved at the age of 30 months after schooling. The patient has been fed with homogenized food up to 24 months of age and showed selectivity in choosing new ones. The parents reported frequent episodes of vomiting (once a week) without nausea, mostly after physical activity. At the age of 2, the boy experienced head trauma after loss of consciousness. Subsequently, the child showed generalized hypertonus, cyanosis, and deviation of the buccal rhyme that lasted about 2 min with self-resolution. The CT scan highlighted a skull fracture without encephalic lesions and a Chiari type 1 malformation, confirmed during the MRI that showed caudal ectopia of cerebellar tonsils (6 mm from Foramen Magnum), hyperintensity of the deep white matter in the suvra/paratrigonal, in subcortical area, and in the temporal area bilaterally, compatible with terminal areas of myelination. Based on the evidence of cerebellar tonsils ectopia, the toddler underwent clinic evaluation for hypermobility, but did not meet Beighton Criteria for Ehlers Danlos Syndrome diagnosis.
At the age of 4 years (Figure 1A), the child started experiencing daily episodes of upward movements of the eyebrows associated with staring spells. The first neurological examination showed tremor of both hands at the end of the index-nose test. The toddler asks for support while climbing stairs and when walking. Electroencephalogram (EEG) recordings while awake showed a trace characterized by poorly organized brain electrical activity and differentiated by age, slow anomalies with interictal paroxysmal abnormalities on bilateral parieto-occipital regions, and abundant epileptiform anomalies with diffuse expression, the most prolonged associated with clinical correlates (Figures 1B,C). The physical examination does not underline any dysmorphism except for the presence of flaring of the lateral side of the eyebrows. After 3 months, frequency and duration of the seizures had increased (up to 20 episodes/day in clusters), subsequently associated with massive ictal myoclonus of the upper limbs or head of very short duration (80 episodes/day in clusters). A 48-h EEG recording showed multiple episodes of myoclonus and pathologic graphoelements.
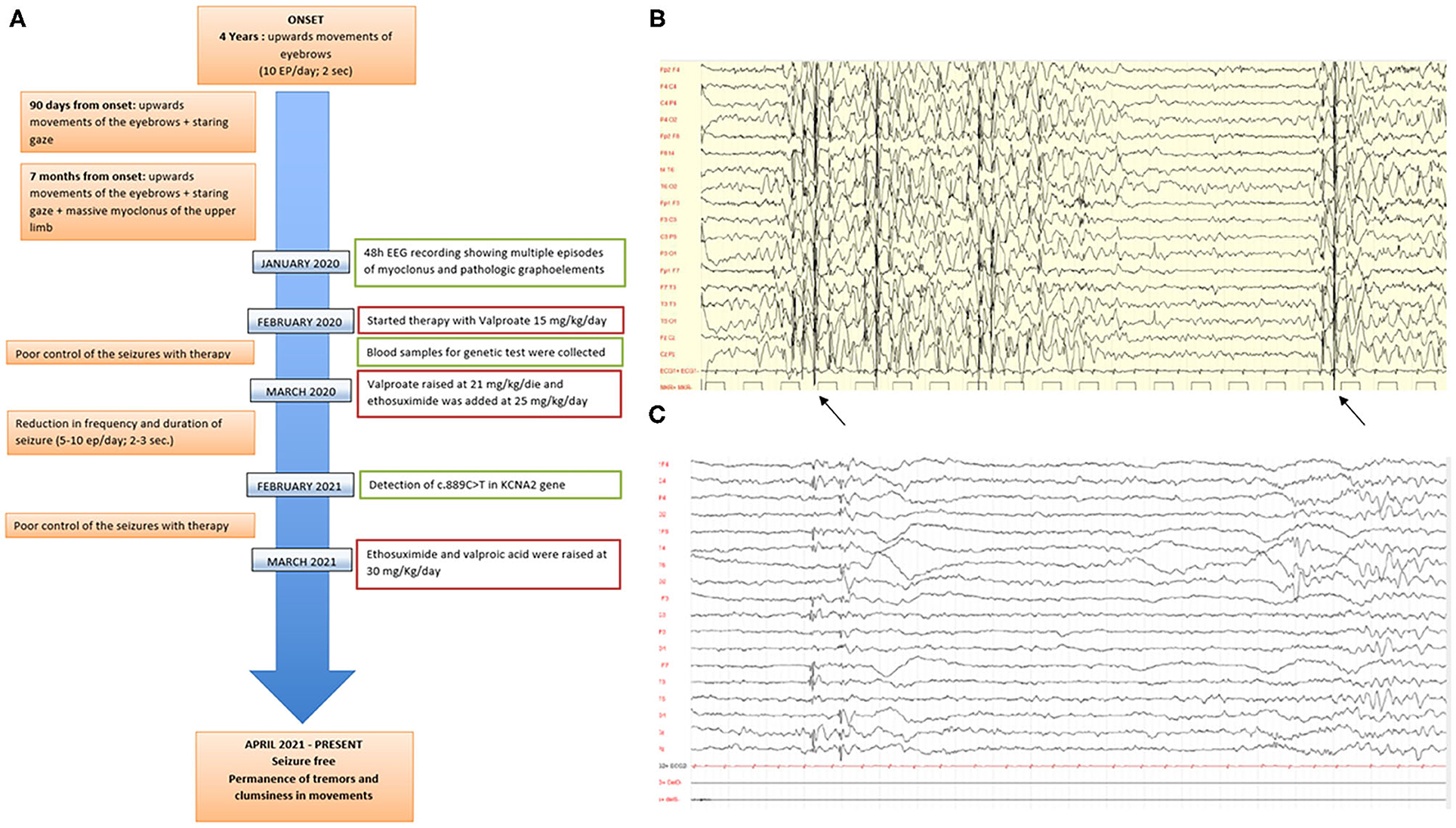
Figure 1. (A) Timeline of clinical features, instrumental characteristics, and therapy approaches in the proband since the toddler started showing epilepsy. (B,C) Electroencephalogram (EEG) performed when the child was 4 years old. The trace is characterized by poorly organized brain electrical activity, slow anomalies with interictal paroxysmal abnormalities on bilateral parieto-occipital sites, and abundant epileptiform anomalies with diffuse expression.
Next Generation Sequencing analysis (NGS) performed in trio on a panel of 77 genes related to epilepsy revealed the missense variant c.889C>T; p.Arg297Trp: NM_004974 in the third exon of the KCNA2 gene (potassium channel, voltage-gated, shaker-related subfamily, member 2, chr1:110,593,579-110,631,535;GRCh38, MIM *176262) in a heterozygous state. Sanger sequencing did not detect this variant in parental DNA, suggesting a de novo origin of the alteration.
So, the toddler started antiepileptic therapy with sodium valproate at a dosage of 15 mg/kg/day which required, given the poor control of events, a raise to 21 mg/kg/day. This therapy did not result in any change of the clinical picture and EEG pattern. Therefore, after about 2 months of treatment, ethosuximide was added at a dosage of 25 mg/day with a reduction in the seizures and EEG abnormalities. Approximately a year later, due to the poor control of the therapy, valproic acid was raised to 30 mg/Kg/day and ethosuximide to 30 mg/Kg/day with complete resolution of the pathological picture.
At the age of 5 years and 11 months, the toddler has been tested with WPPSI-III, CPM, CBCL, and ABAS II. Based on the evaluation carried out, a normal cognitive functioning emerges (IQ 91), with abilities adequate to the expected level in the verbal performance and general language scale.
There is a significant decline in the domain that evaluates the processing speed. Non-verbal intelligence is adequate for the chronological age. Assessment of the adaptive profile, as reported by the mother of child, showed subnormal scores in the conceptual, social, and practical domains. Critical issues were observed in the categories related to preschool skills, communication, self-control, play, and use of the environment. Currently, the proband presents no epileptic events and improvement in the EEG picture, which still maintains theta-delta slow wave bursts in the occipital regions. Neurological examination appears to be normal, except for tremor of the hands and mild fine motor skills deficit. Some behavioral problems persist. In the EEG assessments carried out from 2017 to 2021, it was evident that the permanence of the slow theta-delta variable rhythm anomalies in the posterior regions, with isolated spike-and-wave complexes on the parieto-occipital sites bilaterally, sometimes fronto- or temporo-central that tend to present in diffuse paroxysms with diffuse expression of varying duration. The associated clinical correlation consists in a reduction in contact, ideomotor slowdown with spontaneous resolution, and asynchronous four-limb myoclonus. Overall, the EEG picture appeared disorganized for the age of child. After 7 months being seizure free, in the latest evaluation, the child no longer presents generalized epileptiform abnormalities.
The psychological tests carried out showed performance at the normal range in the ability to analyze and synthesize visual stimuli based on correct visual perception and visuomotor coordination. In addition, they highlighted results in the average range in logical reasoning and in the ability to abstract reason and categorize visual stimuli. The child showed good expressive and receptive language skills. The scale that assesses skills related to comprehension of verbal terms and instructions associated with long-term memory reached levels above normal. There was a decline in the ability to perform cognitive tasks smoothly and automatically especially under the pressure to maintain focused attention and concentration. Scores showed significantly below normal abilities related to the visual discrimination associated with visual short-term memory and visual-motor coordination and cognitive flexibility; even the ability to work quickly with unusual material achieved below normal performance. The toddler presented a cognitive profile that is adequate to the expected level for gender and age, although there is a significant drop in the domain that assesses the speed of processing. The adaptive framework presents criticalities that currently do not seem to affect the life of the child.
This is the first case of a toddler affected by myoclonic epilepsy due to a de novo missense pathogenic variant (c.889C>T) in the KCNA2 gene in the heterozygous state.
The KCNA2 gene encodes for 2 NCBI Refseq transcripts (NM_004974.4 and NM_001204269.2). Monoallelic KCNA2 pathogenic variants, encoding the voltage-gated K+ channel Kv1.2, have been reported as a cause of developmental delay and seizures (Developmental and epileptic encephalopathy 32, MIM #616366) (1–3, 9–13).
Mice models, carrying a KCNA2 mutation, show motor incoordination, myoclonic jerks, tremor, and small body size (14), while null animals present increased seizure susceptibility (15). Previous functional studies showed that KCNA2 mutations cause either a dominant-negative loss-of-function, or a drastic gain-of function (2), determining distinct phenotypes in patients (4, 16). Alterations, causing loss-of-function, are characterized by a better prognosis than gain-of-function and mixed forms. In dominant-negative loss-of-function, focal predominant seizures with greater sleep activation are reported in literature. In gain-of-function, critical events seem to be prevalently severe generalized seizures, most frequently are problems in neurodevelopment, such as ataxia. At a structural level, the cerebellar or whole brain atrophy has been frequently reported. Severe early onset epilepsies appear to be more frequent in mixed forms. Neonatal onset epilepsies with developmental impairments or generalized ictal events are rare (4, 16, 17). Recently the phenotypic spectrum has been expanded to include forms of progressive myoclonus epilepsy and myoclonic-atonic epilepsy (18) and functional studies of pathogenic variants had been performed (19).
The p.Arg297Trp variant, detected in the present case, is ranked as “Likely Pathogenic” according to the American College of Medical Genetics guidelines and “Pathogenic” according to ClinVar. Allele frequency is not available. The case we describe here represents the first in pediatric age and the second overall case of myoclonic epilepsy caused by this heterozygous pathogenic variant in KCNA2.
The toddler presented at the onset generalized epileptiform abnormalities that were often associated with upward movements of the eyebrows, fixation gaze, and ictal myoclonus of head and upper limbs. The toddler still presents difficulties in autonomous walking and while climbing stairs. The parents reported weekly vomiting episodes not preceded by nausea mostly associated with physical exercise, that could represent a clinical manifestation of seizure. After the therapeutical adjustments, given the considerable reduction in ictal events and the inability to perform a functional analysis of the gene, it has not been possible to attempt therapeutic strategies as recently suggested by the literature, such as 4-aminopyridine (20).
During sleep EEG showed fast/slow wave and polypoint/slow wave of medium- or large-voltage, on the fronto-central regions bilaterally, electrical status epilepticus during sleep (ESES) was not present as described by previous authors (21).
A literature review was conducted to identify the characteristics of reported patients who presented a mutation at the same triplet encoding an amino acid (as shown in Table 1). A case with the same mutation was described (8): the patient, differently from our toddler, was diagnosed with unprovoked generalized tonic-clonic seizures. Age of onset was 4 years old in a normal psychomotor neurodevelopment. After carbamazepine treatment, the boy showed seizure remittance. The toddler was worsened from 19 years old, when started presenting episodes of refractory status epilepticus strongly drug resistant. Similarly to our patient, EEG at the age of 4 was characterized by continuous polymorphic delta and theta slowing of the posterior dominant rhythm, intermittent bursts of rhythmic 2–2.5 Hz delta activity intermixed with spikes, spike-and-slow wave discharges over the bi-occipital regions, and isolated parasagittal sharp wave and spike activity during sleep.
Sachdev et al. (8) compared the similar clinical picture of their patient carrying the c.889 C>T mutation, to the reported cases detected with the c.890 G>A mutation in the same amino acidic site, differently from our patient that presents a mild clinical history. In Table 2, we compare the characteristics of the patient analyzed in this study with the only previous case reported in the literature with the same mutation (8).
Several cases detected with the c.890G>A p.(Arg297Gln) variant in the KCNA2 gene had been reported, expanding the clinical spectrum of developmental and epileptic encephalopathy and highlighting the variable expressivity. Progressive myoclonic epilepsy was described in one of the patients described by Canafoglia (5). This case showed ataxia and psychomotor delay, whereas epilepsy was delayed and limited to relatively rare tonic seizures, associated with prominent cortical myoclonus, occurred at 3 years. Syrbe et al. (2) reported the case of a toddler carrying a c.890G>A p.(Arg297Gln) de novo mutation in the KCNA2 gene, whose functional analysis showed a gain-of-function alteration. The patient, differently from the reported case, experienced generalized tonic-clonic seizures, absences, status epilepticus often during febrile events. Moreover, the patient presented moderate-severe ataxia and moderate intellectual disability. The same nucleotidic variant was described in a patient with myoclonic and absence seizures. The age of onset, the clinical, and the neuropsychological deterioration was different from our case, resulting in a progressive worsening and drug resistance.
Masnada et al. (4) enrolled five patients carrying this mutation in the KCNA2 gene. Differently from the present case, all of them showed generalized tonic-clonic seizures, but they all presented tremors, ataxia, and a moderate intellectual disability. Two of them experienced myoclonic seizures associated with absences. Only one was seizure-free from 18 months of age. Corbett et al. (3) reported a patient that started having tonic seizures at 12 months, progressing in erratic myoclonic tremors. The proband showed different ictal phenotypes, while EEG and the neuropsychological evaluation showed similar results. Nashabat et al. (6) described the case of a patient that, differently from the present case, experienced earlier onset of generalized tonic-clonic seizures in a good global development, but with poor response to therapy. Costain et al. (7) reported a patient showing various ictal events, such as generalized tonic and tonic-clonic seizures, absences, and myoclonic seizures. This toddler presented an earlier age of onset, a good global development, and ataxia. Ngo et al. (22) described a case with apparently isolated ataxia as primary symptom, but no more clinical information was provided.
Interestingly, an unusual case of mosaicism of two novel missense variants in KCNA2 had been reported, expanding the phenotypic spectrum associated with this mutations of gene, but not presenting with myoclonic epilepsy (23).
The present case aims to expand the clinical spectrum associated with heterozygous KCNA2 pathogenic variants. This evidence suggests a genotype-first approach, performing genomic tests that include the KCNA2 gene, in patients with myoclonic epilepsy diagnosis. According to the literature, we highlight the variability in expression that can be clinically observed in patients with heterogeneous ictal events (15), even in those who show mutations in the same triplet encoding an amino acid. Indeed, the patient we describe presented a good prognosis characterized by a good response to therapy, and an improved neurodevelopment, in contrast with the previously reported cases, presenting with a more severe clinical picture (1, 2, 11).
We suggest that an accurate clinical examination improve the detection of genotype-phenotype correlations and expand the possibility of predicting the functional impact of KCNA2 variants (18). Even if it is a rare condition, a KCNA2 gene mutation test should be performed in patients over 3 years of age.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
LP, GM, MM, IA, FA, AP, CG, and AS: conceptualization, data curation, resources, investigation, writing—original draft, methodology, visualization, supervision, and writing—review and editing. All authors contributed to the article and approved the submitted version.
All phases of this study were supported by the Department of Pediatrics of Sapienza University of Rome, Italy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pena SD, Coimbra RL. Ataxia and myoclonic epilepsy due to a heterozygous new mutation in KCNA2: proposal for a new channelopathy. Clin Genet. (2015) 87:e1–3. doi: 10.1111/cge.12542
2. Syrbe S, Hedrich UBS, Riesch E, Djémié T, Müller S, Møller RS, et al. De novo loss- or gain-of function mutations in KCNA2 cause epileptic encephalopathy. Nat Genet. (2015) 47:393–9. doi: 10.1038/ng.3239
3. Corbett MA, Bellows ST, Li M, Carroll R, Micallef S, Carvill GL, et al. Dominant KCNA2 mutation causes episodic ataxia and pharmacoresponsive epilepsy. Neurology. (2016) 87:1975–84. doi: 10.1212/WNL.0000000000003309
4. Masnada S, Hedrich UBS, Gardella E, Schubert J, Kaiwar C, Klee EW, et al. Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain. (2017) 140:2337–54. doi: 10.1093/brain/awx184
5. Canafoglia L, Castellotti B, Ragona F, Freri E, Granata T, Chiapparini L, et al. Progressive myoclonus epilepsy caused by a gain-of-function KCNA2 mutation. Seizure. (2019) 65:106–8. doi: 10.1016/j.seizure.2019.01.005
6. Nashabat M, Al Qahtani XS, Almakdob S, Altwaijri W, Ba-Armah DM, Hundallah K, et al. The landscape of early infantile epileptic encephalopathy in a consanguineous population. Seizure. (2019) 69:154–72. doi: 10.1016/j.seizure.2019.04.018
7. Costain G, Cordeiro D, Matviychuk D, Mercimek-Andrews S. Clinical application of targeted next-generation sequencing panels and whole exome sequencing in childhood epilepsy. Neuroscience. (2019) 418:291–310. doi: 10.1016/j.neuroscience.2019.08.016
8. Sachdev M, Gaínza-Lein M, Tchapyjnikov D, Jiang YH, Loddenkemper T, Mikati MA. Novel clinical manifestations in patients with KCNA2 mutations. Seizure. (2017) 51:74–6. doi: 10.1016/j.seizure.2017.07.018
9. Tang S, Addis L, Smith A, Topp SD, Pendziwiat M, Mei D, et al. Phenotypic and genetic spectrum of epilepsy with myoclonic atonic seizures. Epilepsia. (2020) 61:995–1007. doi: 10.1111/epi.16508
10. Allen NM, Conroy J, Shahwan A, Lynch B, Correa RG, Pena SD, et al. Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia. (2016) 57:e12–7. doi: 10.1111/epi.13250
11. Hundallah K, Alenizi A, AlHashem A, Tabarki B. Severe early-onset epileptic encephalopathy due to mutations in the KCNA2 gene: expansion of the genotypic and phenotypic spectrum. Eur J Paediatr Neurol. (2016) 20:657–60. doi: 10.1016/j.ejpn.2016.03.011
12. Drögemöller BI. Maintaining the balance: both gain- and loss-of-function KCNA2 mutants cause epileptic encephalopathy. Clin Genet. (2015) 88:137–9. doi: 10.1111/cge.12615
13. Allou L, Julia S, Amsallem D, El Chehadeh S, Lambert L, Thevenon J, et al. Rett-like phenotypes: expanding the genetic heterogeneity to the KCNA2 gene and first familial case of CDKL5-related disease. Clin Genet. (2017) 91:431–40. doi: 10.1111/cge.12784
14. Xie G, Harrison J, Clapcote SJ, Huang Y, Zhang JY, Wang LY, et al. A new Kv1.2 channelopathy underlying cerebellar ataxia. J Biol Chem. (2010) 285:32160–73. doi: 10.1074/jbc.M110.153676
15. Brew HM, Gittelman JX, Silverstein RS, Hanks TD, Demas VP, Robinson LC, et al. Seizures and reduced life span in mice lacking the potassium channel subunit Kv1.2, but hypoexcitability and enlarged Kv1 currents in auditory neurons. J Neurophysiol. (2007) 98:1501–25. doi: 10.1152/jn.00640.2006
16. Morrison-Levy N, Borlot F, Jain P, Whitney R. Early-onset developmental and epileptic encephalopathies of infancy: an overview of the genetic basis and clinical features. Pediatr Neurol. (2021) 116:85–94. doi: 10.1016/j.pediatrneurol.2020.12.001
17. Steel D, Symonds JD, Zuberi SM, Brunklaus A. Dravet syndrome and its mimics: beyond SCN1A. Epilepsia. (2017) 58:1807–16. doi: 10.1111/epi.13889
18. Döring JH, Schröter J, Jüngling J, Biskup S, Klotz KA, Bast T, et al. Refining genotypes and phenotypes in KCNA2-related neurological disorders. Int J Mol Sci. (2021) 22:2824. doi: 10.3390/ijms22062824ijms22062824
19. Arbini A, Gilmore J, King MD, Gorman KM, Krawczyk J, McInerney V, et al. Generation of three induced pluripotent stem cell (iPSC) lines from a patient with developmental epileptic encephalopathy due to the pathogenic KCNA2 variant c.869T>G; p.Leu290Arg (NUIGi052-A, NUIGi052-B, NUIGi052-C). Stem Cell Res. (2020) 46:101853. doi: 10.1016/j.scr.2020.101853
20. Imbrici P, Conte E, Blunck R, Stregapede F, Liantonio A, Tosi M, et al. A novel KCNA2 variant in a patient with non-progressive congenital ataxia and epilepsy: functional characterization and sensitivity to 4-aminopyridine. Int J Mol Sci. (2021) 22:9913. doi: 10.3390/ijms22189913
21. Gong P, Xue J, Jiao X, Zhang Y, Yang Z. Genetic etiologies in developmental and/or epileptic encephalopathy with electrical status epilepticus during sleep: cohort study. Front Genet. (2021) 12:607965. doi: 10.3389/fgene.2021.607965
22. Ngo KJ, Rexach JE, Lee H, Petty LE, Perlman S, Valera JM, et al. A diagnostic ceiling for exome sequencing in cerebellar ataxia and related neurological disorders. Hum Mutat. (2020) 41:487–501. doi: 10.1002/humu.23946
Keywords: epilepsy, KCNA2, epileptic encephalopathies, genetic variants, genotype-first approach, epilepsy—abnormalities, classification, drug therapy
Citation: Perilli L, Mastromoro G, Murciano M, Amedeo I, Avenoso F, Pizzuti A, Guido CA and Spalice A (2022) Myoclonic Epilepsy: Case Report of a Mild Phenotype in a Pediatric Patient Expanding Clinical Spectrum of KCNA2 Pathogenic Variants. Front. Neurol. 12:806516. doi: 10.3389/fneur.2021.806516
Received: 31 October 2021; Accepted: 31 December 2021;
Published: 01 February 2022.
Edited by:
Joseph Sullivan, UCSF Benioff Children's Hospital, United StatesReviewed by:
Maurizio Elia, IRCCS Oasi Maria SS, ItalyCopyright © 2022 Perilli, Mastromoro, Murciano, Amedeo, Avenoso, Pizzuti, Guido and Spalice. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorenzo Perilli, ZG90dG9ycGVyaWxsaUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.