 Antonio Bruno
Antonio Bruno Ettore Dolcetti
Ettore Dolcetti Diego Centonze
Diego Centonze
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Neurol. , 07 February 2022
Sec. Multiple Sclerosis and Neuroimmunology
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.802918
In patients with multiple sclerosis (MS), a typical pattern of muscle tone alteration, known as spasticity, is frequently observed in combination with other signs or symptoms such as spasms, cramps, pain, bladder dysfunction, sleep disturbances, fatigue, and tremor. Recently, the concept of spasticity-plus syndrome (SPS) has been proposed to take into account the frequent coexistence of all these complaints in patients with MS and a common pathophysiological basis for this putative new clinical entity has been proposed. Muscle tone, sleep, bladder function, and the pain pathway are controlled by cannabinoid CB1 (CB1R) and CB2 receptors (CB2R) that are particularly enriched in the brainstem. Axons with smaller diameters are particularly susceptible to conduction block and the irritative, ephaptic, consequences of demyelination and their involvement in the demyelination process caused by MS in the brainstem might underlie the various clinical manifestations of SPS. The adoption of SPS in clinical practice could be useful to improve symptomatic treatments in a significant proportion of patients with MS, possibly limiting the adverse events produced by polypharmacotherapy.
Clinical neurology generally refers to spasticity as a well-defined clinical sign characterized by a velocity-dependent increase of muscle tone (hypertonus) that can be objectivated during the neurological examination (1, 2). This hypertone is generally believed to follow the loss of inhibitory control that the corticospinal tract exerts over spinal tonic stretch reflexes, derived from its degeneration or interruption at different levels due to demyelinating lesions (3). The clinical practice of multiple sclerosis (MS) teaches us that spasticity is not only a sign to be objectified, but it can be better captured taking into account also the subjective complaints or sensations of altered muscle tone and pain occurring during daily activities. It is usually reported by patients and its severity can be described by subjective and objective items reported, respectively, in the Numeric Rating Scale (NRS, variable on a scale of 0–10, where 0 is no spasticity and 10 is the worst possible spasticity), and in the Ashworth Scale (AS, variable from 0—no spasticity to 4—affected parts rigid in flexion or extension) (2), among other scales. According to a classical model trying to provide a plausible explanation for all clinical manifestations typically accompanying spasticity in MS, muscle tone increase caused by corticospinal tract demyelination induces a cascade of downstream effects, giving rise to spasms, cramps, fatigue, tremor, pain, and sleep disturbances. Similarly, the overexcitability or reduced activity of bladder detrusor can cause incontinence or urinary retention (4, 5). This model implies a proportionality between the severity of spasticity and that of the other downstream symptoms, and the idea that the resolution of spasticity, as the primary upstream event, should improve the other signs and symptoms downstream. Unfortunately, this model is not able to cover all the possible eventualities that recur in clinical practice.
A syndrome is defined as a variable combination of signs and symptoms forming a distinctive clinical entity and indicative of a particular disease or disorder. An interesting and recent study proposes a new approach to spasticity in MS, introducing the concept of spasticity plus syndrome (SPS), that hypothesizes a new theoretical collocation for this condition (4). According to this proposition, the signs and symptoms accompanying the classically defined spasticity are not connected in a sequential manner after hypertone occurrences in patients with MS, but constitute, along with the so-defined spasticity, a cluster of clinical manifestations, all sharing one specific underlying pathophysiology (4). The novelty of this approach is that it allows putting spasticity and other symptoms (pain, allodynia, bladder dysfunction, fatigue, and sleep disturbances) on the same level (Figure 1).
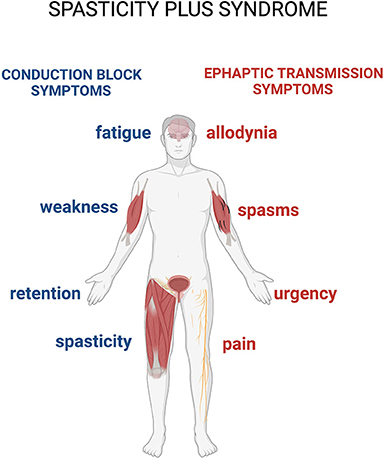
Figure 1. The spasticity-plus syndrome (SPS) model characteristics. The new spasticity-plus syndrome, differently from the classical model, supports the hypothesis that spasticity, triggered by central nervous system (CNS) damage, and other symptoms caused by axon demyelination in terms of conduction block (spasticity, fatigue, weakness, and retention) and ephaptic transmission (spasms, pain, allodynia, and urgency) (see further in the text) are placed on the same level. This model implies that spasticity could be even milder compared to the other associated symptoms.
In the pharmacological management of the various clinical manifestations of MS, many different molecules are generally used as symptomatic agents, alone or in combination. These include anticonvulsants (for example, gabapentin, pregabalin, and lamotrigine), antispasmodics (for example, baclofen), and antidepressants (for example, vortioxetine or citalopram), all aiming at targeting one specific symptom. This approach inevitably implies a loss of the overall vision over the disease processes and at the same time increases the risk of therapy side effects, with a potential worsening of the symptoms complained by the patients. Recognition of SPS as a distinct clinical entity implies that a single drug interfering with its underlying pathophysiology may be able to act simultaneously on all the symptoms that are part of it. It also implies that a given treatment can differently affect the various manifestations of the syndrome in a variable manner, depending on the patient-specific characteristics and pharmacological responsiveness (4) (Figure 1). This approach may also help to shift the focus of clinicians from spasticity alone to the whole cluster of symptoms, encouraging comprehensive understanding and management of the SPS in patients with MS. In their article, Fernandez and colleagues suggest that cannabinoid drugs, whose receptors are diffused at all levels of the cortical spinal tract, could have pleiotropic effects on the various signs and symptoms composing the SPS (4).
In this article, we welcome the theoretical frame implied in the SPS hypothesis, provide a unifying model of SPS signs and symptoms generation in patients with MS and, based on recent literature, suggest that cannabinoid derivatives can act even more extensively on the corticospinal tract through unprecedentedly considered mechanisms.
In humans, the corticospinal tract originates from the primary motor area (BA 4, 65% fibers) and to a lesser extent from the supplementary motor area (BA 6, 25% fibers) and associative somatosensorial cortex (BA 5, 5%). It is divided into a crossed corticospinal pathway, composed of larger diameter fibers that decussate in bulbar pyramids and course along the lateral column in the spinal cord, and in a direct corticospinal pathway, which contains a lower quantity of fibers, that course along the anteromedial column in the spinal cord, decussating on anterior horns (6). From a pathophysiological point of view, a lesion of the upper motor neuron along the corticospinal tract results in both negative deficient manifestations (weakness) and positive irritative signs (spasticity). In patients with MS, neurologists frequently objectify severe spasticity even in the absence of significant weakness. This feature differentiates the clinical presentation of MS from other diseases also damaging the upper motor neuron, such as ischemic stroke. We propose that this phenomenon could be tentatively explained considering the differential sensitivity of large and small axons to the effects of demyelination.
As already known from neuroanatomy, the corticospinal tract course along with the anteromedial and lateral columns in the spinal cord (3). Axons reach Ia sensory fibers, spinal interneurons, and gamma-motor neurons, and are therefore responsible for the control of the stretch reflex among other phylogenetically preserved spinal reflexes, are predominantly part of the phylogenetically older lateral or cruciate corticospinal tract. On the other hand, fibers that monosynaptically excite alpha-motor neurons in the laminae IX and X of spinal cord anterior horns are predominantly responsible for the control of voluntary movements and are part of the phylogenetically newer anterior corticospinal tract (Figure 2). A similar difference is observed in sensitivity pathways, where axons that conduce pain and visceral sensations have smaller diameters and present a different localization from those that convey tactile and proprioceptive sensitivity (7).
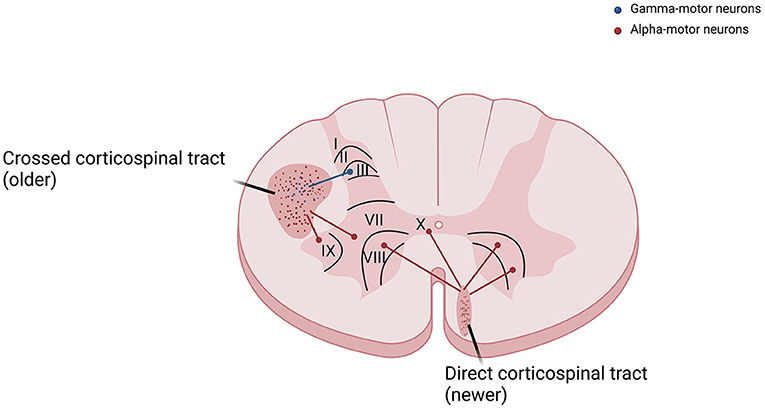
Figure 2. The corticospinal tract is divided into crossed corticospinal tract and direct corticospinal tract. Fibers that reach gamma-motor neurons, responsible for the control of the stretch reflex, are predominantly part of the phylogenetically older lateral or cruciate corticospinal tract. Fibers that reach alpha-motor neurons in the laminae IX and X of spinal cord anterior horns, responsible for the control of voluntary movements, are predominantly part of the newer anterior corticospinal tract.
From these considerations, it follows that a selective interruption of the corticospinal lateral tract can give rise to a clinical phenomenon where spasticity would appear even in the absence of weakness. In MS, demyelination of the motor pathway likely involves without distinction both the lateral and the anterior corticospinal tract, thus letting unexplained why spasticity often prevails over weakness in these patients. Our hypothesis is that fibers from the lateral corticospinal tract have a greater sensitivity to the demyelination process than the anterior tract, due to their distinct sizes.
The hypothesized greater sensitivity to demyelination effects of the smaller sized corticospinal later tract could be explained by basic electrophysiology: axons with smaller diameters are more sensitive to conduction block and develop more easily ephaptic transmission to surrounding nerve fibers in response to demyelination. This can be well highlighted by the space constant (λ) of a fiber with electrotonic features, which describes how easily a given perturbation of the resting membrane potential can propagate at a distance (Figure 3A). It can be hypothesized that conduction blocks could be at the basis of the negative, deficitary, and persisting phenomena of SPS such as spasticity, fatigue, weakness, and urinary retention (due to reactivity of the detrusor muscle). Moreover, many other positive, irritative, and recurrent symptoms, such as spasms, pain, allodynia, and urgency, can be better explained in terms of hyperexcitability of demyelinated fibers and secondary ephaptic propagation of excitation. This phenomenon is observed when the depolarizing current of an action potential is forced to pass through a damaged point of an otherwise myelinated tract, thus causing current shunt in the extracellular space and cross-stimulation of other nearby damaged fibers. This type of conduction, which is opposed to synaptic transmission, refers to the fact that a demyelinated axon shares with other demyelinated axons of the same district the same extracellular medium (8) (Figure 3B).
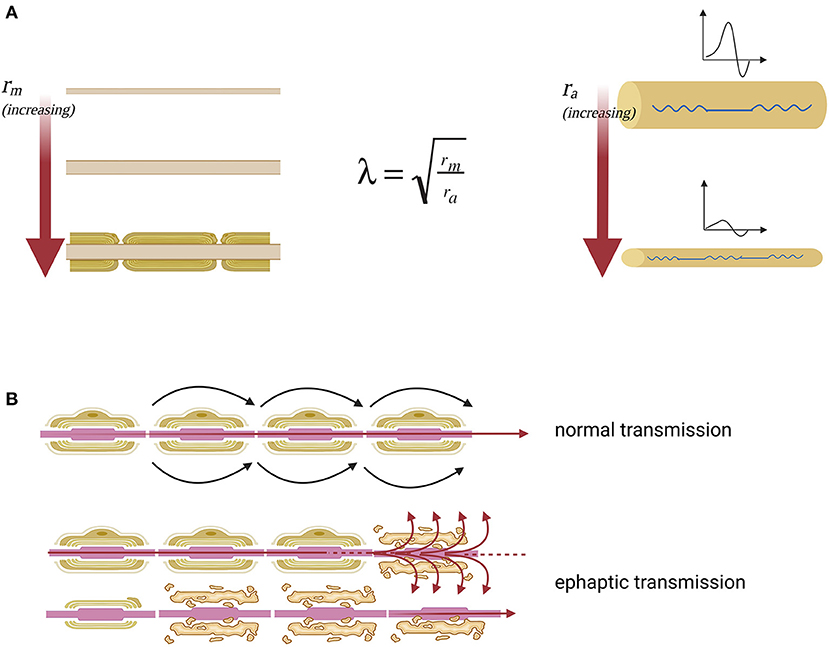
Figure 3. Nerve impulse propagation in axons with normal and reduced diameter. (A) Lambda space constant (λ) is an index of how the difference in membrane potential propagates along with the fiber at distance. Moreover, space constant is directly proportional to the membrane resistance (rm), which opposes the escape of ions that must proceed along the axon to transmit the signal and inversely proportional to axial resistance (ra), which opposes the propagation of the action potential along the axon. The membrane resistance increases enormously with the myelination of the fiber, which isolates the axon from current leakage. The axial resistance instead is a direct measure of axonal diameter, so the greater the diameter of the axon, the easier it will be the propagation of excitation at a distance, the smaller the diameter of the axon, the greater the probability that the fiber will suffer a conduction block. From this model, it is clear that the fibers of small diameter are more sensitive to the effects of demyelination because they have high axial resistance. (B) The nerve fiber function deficit in the case of SPS can be explained as an effect of ephaptic transmission. In this way when an action potential arises, perturbing the extracellular medium, the excitation is transferred to neighboring axons. Specifically, there is a transverse propagation of excitation from one axon to another, independently of the synaptic junction.
In physiological conditions, the myelin sheath is essential to ensure high-speed transmission of action potentials along the axons. The ion current associated with action potentials is mediated predominantly by sodium ions flowing through voltage-gated sodium channels (NaV) expressed between two myelinated areas. The intermediate tract between two myelinated areas is called Ranvier's node and expresses the highest density of voltage-gated channel proteins along the entire axon. Since the action potential is renewed at each Ranvier node by “jumping” the myelinated areas, this type of conduction is called saltatory conduction (9).
As reported, demyelination mediated by autoreactive B and T cells causes an action potential propagation disorder along the axons in patients with MS (10). In the case of demyelination, in particular, the absence of voltage-gated channels in demyelinated tracts between Ranvier's nodes causes a progressive amplitude decline of the action potentials during its propagation, possibly resulting in conduction block when the residual amplitude is below the threshold of action potential generation at the next Ranvier's node. In this condition, neurons activate different compensation mechanisms aimed at trying to allow a correct propagation of the action potential at constant amplitude, including ectopic expression of voltage-dependent Na+ in demyelinated membrane tracts between Ranvier's nodes. In MS, there is a diffuse expression of both the Nav1.2 and Nav1.6 channels all over the axons (11). Regarding NaV1.6 channels, it is well known that their activation results in persistent Na+ influx, imbalance of the Na+/Ca2+ pump and, intra-axonal Ca++ accumulation. Unfortunately, in the long run, these compensatory mechanisms can cause axonal damage by altering the distribution of Na+, K+, and Ca2+ ions across the plasma membrane leading to activation of damaging injury cascades (11, 12) (Table 1). It follows that reducing NaV1.6-mediated hyperexcitability is vitally important to alleviate secondary symptoms in MS (16). Moreover, their wide distribution throughout the CNS makes them crucial targets for the control of SPS symptoms, due to the origin of spasticity from different areas of the brain and spinal cord. A possible class of compounds effective in controlling SPS symptoms could be cannabinoids.

Table 1. Multiple sodium channel isoforms are expressed in different tissues and initiate action potentials in neurons, skeletal muscles, and cardiac muscles.
In humans, the endocannabinoid system has a pleiotropic role in the CNS and acts on several elements of the synaptic transmissions. Endocannabinoids interact with different receptors placed at both presynaptic and postsynaptic levels. The main receptors are named CB1 and CB2, among others (17). CB1 is mainly localized at the presynaptic terminal and is involved in synaptic modulation by inhibiting the release of other neurotransmitters (17). The distribution of CB2 receptors in the CNS is concentrated mainly along the sensory pathway, making this receptor a target for neuropathic pain therapy (18). Moreover, CB2 is also localized on microglia and plays a role in immune modulation and neuronal-microglial interaction (17, 19). Converging pieces of evidence are now suggesting that cannabinoids may interact with axonal channels reducing their excitotoxic potential in several pathological conditions. To understand the relationship between the Na+/Ca2+ pump and cannabinoids, a whole-cell patch-clamp experimental study was conducted on a model of myocardial ischemic damage on rats (20). The authors demonstrated that exposing myocardiocytes to a simulated ischemic medium causes a Na+/Ca2+ exchanger reversal of function from its physiological role, with an increase of intracellular free Ca2+ concentration and a reduction of intracellular free Na+ (20). Exposition of myocardiocytes to N-arachidonylethanolamide (anandamide), one of the endocannabinoids, suppresses free Ca2+ concentration overload through suppression of the inverted Na+/Ca2+ exchanger, further demonstrating that this action is specifically mediated by CB2 receptor (20). Moreover, these authors discovered that this effect of anandamide is disease specific, observing that anandamide did not effect on current and intracellular free Ca2+ concentration in normal conditions (20). In a second whole-cell patch-clamp study it was studied the relationship between the NaV channels and the cannabinoids in a transgenetic mouse carrying a NaV1.6 gene mutation (16). The authors examined the electrophysiological effects of anandamide and cannabidiol (CBD) in striatal neuron cultures of mutant NaV1.6 compared to wild-type mice. Cannabinoids selectively reduced currents due to pathological expressed NaV1.6 channels on the neuron, influencing both peak current-density and peak resurgent current, thus reducing overall action potential firing of striatal neurons. In particular, cannabinoids did not interfere with the genesis of the action potentials, avoiding a conduction block. Moreover, cannabinoids block the terminal part of repetitive neuronal firing preventing the dispersion of charges through the cell membrane, responsible for ephaptic transmission (16).
In another preclinical study, Ghovanloo and colleagues characterized the electrophysiological effects of CBD on culture cells engineered to expose human NaV (hNaV) channels (21). CBD inhibited hNaV1.1–1.7 currents of HEK293 cells when perfusion reaches therapeutic concentration relevant for humans, thus showing the direct interaction of CBD with NaV channels. They discovered that CBD preferentially stabilizes the inactivated NaV channel states also slowing their recovery from inactivation. Moreover, they also tested the effects of tetrahydrocannabinol (THC) on hNav1.2 channels suggesting that it may represent a different mechanism of inhibition of NaV (21). Recently, an in-depth multidisciplinary study confirmed the functional inhibitory effect of CBD highlighting, by the means of crystallography technique, direct interaction on NaV at the hydrophobic cavity of the channel (22). Finally, another recent electrophysiological study, where the authors demonstrated the ability of CBD to inhibit repetitive action potential firing binding to slow inactivated states of Nav1.8 channels in primary nociceptive neurons from mouse dorsal root ganglia, suggesting that CBD can exert analgesic effects directly inhibiting repetitive firing of primary nociceptors (23).
The finding of a direct effect of cannabinoids on NaV channels further supports their possible use to treat SPS symptoms. The potential efficacy of cannabinoids is also supported by both the widespread diffusion of NaV in the CNS and by their ability to reduce the hyperexcitability of NaV channels, also counteracting ephaptic transmission (20).
In the previous work, we showed how cannabinoids may have a direct role in axonal pathological conduction in MS and may counteract the symptoms of SPS. This new interpretation integrates what was already stated on SPS (4). In their paper, Fernandez and colleagues mainly focused on the effects of cannabinoids within the CNS, recognizing some points of crucial importance, due to their high concentration of CB1 and CB2 receptors (4). They suggested the brainstem as a critical brain area for the action of cannabinoid-derived drugs, where most of the motor and premotor cortex efferents and sensory afferents converge into relay nuclei and centers responsible for muscle tone and autonomic nervous system functions (4). Here we propose a new insight on the functioning mechanism of the cannabinoid by direct and indirect interaction with demyelinated axons. The importance of cannabinoids is progressively finding a counterpart in clinical practice. A new body of studies showed that THC and CBD are effective in relieving symptoms in patients with MS with moderate to severe spasticity and pain (24). In accord with the SPS model, cannabinoids demonstrate a positive trend in the improvement of bladder function, tremor, fatigue, and sleep disorders in several clinical studies (18, 24). Moreover, recent pieces of evidence suggest that the treatment with THC is associated with a clinically relevant improvement in the Patient Global Impression of Change Scale to quantify and track patient progress and treatment response over time (24). Despite the proven clinical efficacy of synthetic cannabinoids in the treatment of spasticity and pain, the effects of cannabinoids on the other secondary symptoms of MS have not yet been clearly demonstrated and further studies are needed. Moreover, several studies draw attention to the possible effects of synthetic cannabinoid drugs on cognitive functions. In this regard, studies conducted on small cohorts of patients have presented conflicting results. In some cases, showing a cognitive dysfunction associated with cortical thickness reduction in several cortical areas, in other cases showing no significant effect on the cognitive functions of patients with MS (25). In this perspective, a large-scale clinical trial would be needed considering not only spasticity but also all the other secondary symptoms that make up SPS with a particular focus on the potential harm on cognition from chronic use of synthetic cannabinoids in patients with MS.
Different from the classical approach to spasticity, defined as a neurological sign objectivated by neurologists and influencing other clinical manifestations of MS, there are growing pieces of evidence that lead to a new integrated view of MS symptoms. According to the SPS, the symptoms that commonly accompany the patient with MS (fatigue, insomnia, weakness, spasticity, and urinary disturbances) would constitute a single complex syndrome where all symptoms are placed at the same level. In this context, the use of cannabinoid drugs, thanks to their pleiotropic effects both at the axonal and synaptic level, could play a key role in controlling all clinical manifestations composing the SPS. Cannabinoids usefulness lies both in their proven efficacy in improving all symptoms of SPS and in their safety profile. Furthermore, using a single class of drugs could help to simplify the treatment of these patients who require complex management with polypharmacy by avoiding interactions between multiple drugs. In this regard, a large-scale randomized controlled clinical trial could provide an effective contribution to the involvement of cannabinoids in SPS.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
AB, ED, and DC wrote the manuscript. All authors reviewed the manuscript before submission. All authors contributed to the article and approved the submitted version.
This study received funding from Almirall. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
DC is an Advisory Board member of Almirall, Bayer Schering, Biogen, GW Pharmaceuticals, Merck Serono, Novartis, Roche, Sanofi-Genzyme, and Teva and received honoraria for speaking or consultation fees from Almirall, Bayer Schering, Biogen, GW Pharmaceuticals, Merck Serono, Novartis, Roche, Sanofi-Genzyme, and Teva. He is also the principal investigator in clinical trials for Bayer Schering, Biogen, Merck Serono, Mitsubishi, Novartis, Roche, Sanofi-Genzyme, and Teva. His preclinical and clinical research was supported by grants from Bayer Schering, Biogen Idec, Celgene, Merck Serono, Novartis, Roche, Sanofi-Genzyme, and Teva.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge Irene Scagnol, Biologist with Ph.D. in Clinical Pharmacology—the University of Milan for the collaboration in the drafting of this article.
1. Banky M, Clark RA, Mentiplay BF, Olver JH, Williams G. Clinical spasticity assessment using the Modified Tardieu Scale does not reflect joint angular velocity or range of motion during walking: assessment tool implications. J Rehabil Med. (2021) 53:jrm00137. doi: 10.2340/16501977-2777
2. Zorowitz RD, Wein TH, Dunning K, Deltombe T, Olver JH, Davé SJ, et al. A screening tool to identify spasticity in need of treatment. Am J Phys Med Rehabil. (2017) 96:315–20. doi: 10.1097/PHM.0000000000000605
4. Fernández Ó, Costa-Frossard L, Martínez-Ginés M, Montero P, Prieto JM, Ramió L. The broad concept of “Spasticity-Plus Syndrome” in multiple sclerosis: a possible new concept in the management of multiple sclerosis symptoms. Front Neurol. (2020) 11:152. doi: 10.3389/fneur.2020.00152
5. Rizzo MA, Hadjimichael OC, Preiningerova J, Vollmer J. Prevalence and treatment of spasticity reported by multiple sclerosis patients. Mult Scler. (2004) 10:589–95. doi: 10.1191/1352458504ms1085oa
6. Welniarz Q, Dusart I, Roze E. The corticospinal tract: evolution, development, and human disorders. Devel Neurobiol. (2017) 77:810–29. doi: 10.1002/dneu.22455
7. Kandel E, Koester JD, Mack SH. Principles of Neural Sciences. New York, NY: McGraw Hill (2021). Chapters 8–11.
8. Anastassiou CA, Perin R, Markram H, Koch C. Ephaptic coupling of cortical neurons. Nat Neurosci. (2011) 14:217–23. doi: 10.1038/nn.2727
9. Lubetzki C, Sol-Foulon N, Desmazières A. Nodes of Ranvier during development and repair in the CNS. Nat Rev Neurol. (2020) 16:426–39. doi: 10.1038/s41582-020-0375-x
10. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nature. (2015) 15:545–58. doi: 10.1038/nri3871
11. Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav12 and Nav16 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A. (2004) 101:8168–73. doi: 10.1073/pnas.0402765101
12. Coggan JS, Bittner S, Stiefel KM, Meuth SG, Prescott SA. Physiological dynamics in demyelinating diseases: unraveling complex relationships through computer modeling. Int J Mol Sci. (2015) 16:21215–36. doi: 10.3390/ijms160921215
13. Osteen JD, Sampson K, Iyer V, Julius D. Bosmans F. Pharmacology of the Nav11 domain IV voltage sensor reveals coupling between inactivation gating processes. Proc Natl Acad Sci U S A. (2017) 114:6836–41. doi: 10.1073/pnas.1621263114
14. Bouafia A, Golmard JL, Thuries V, Sazdovitch V, Hauw JJ, Fontaine B, et al. Axonal expression of sodium channels and neuropathology of the plaques in multiple sclerosis. Neuropathol Appl Neurobiol. (2014) 40:579–90. doi: 10.1111/nan.12059
15. Schattling B, Fazeli W, Engeland B, Liu Y, Lerche H, Isbrandt D, et al. Activity of Na V 12 promotes neurodegeneration in an animal model of multiple sclerosis. JCI Insight. (2016) 1:e89810. doi: 10.1172/jci.insight.89810
16. Patel RR, Barbosa C, Brustovetsky T, Brustovetsky N, Cummins TR. Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain. (2016) 139(Pt 8):2164–81. doi: 10.1093/brain/aww129
17. Senn L, Cannazza G, Biagini G. Receptors and channels possibly mediating the effects of phytocannabinoids on seizures and epilepsy. Pharmaceuticals (Basel). (2020) 13:174. doi: 10.3390/ph13080174
18. Nielsen S, Germanos R, Weier M, Pollard J, Degenhardt L, Hall W, et al. The use of cannabis and cannabinoids in treating symptoms of multiple sclerosis: a systematic review of reviews. Curr Neurol Neurosci Rep. (2018) 18:8. doi: 10.1007/s11910-018-0814-x
19. Rojo-Bustamante E, Íñigo-Marco I, Abellanas MA, Vinueza-Gavilanes R, Baltanás A, Luquin E, et al. CB2 receptors and Neuron–Glia interactions modulate neurotoxicity generated by MAGL inhibition. Biomolecules. (2020) 10:1198. doi: 10.3390/biom10081198
20. Li Q, Cui N, Du Y, Ma H, Zhang Y. Anandamide reduces intracellular Ca2+ concentration through suppression of Na+/Ca2+ exchanger current in rat cardiac myocytes. PLoS ONE. (2013) 8:e63386. doi: 10.1371/journal.pone.0063386
21. Ghovanloo M, Shuart NG, Mezeyova J, Dean RA, Ruben PC, Goodchild SJ. Inhibitory effects of cannabidiol on voltage-dependent sodium currents. J Biol Chem. (2018) 293:16546–58. doi: 10.1074/jbc.RA118.004929
22. Sait GL, Sula A, Ghovanloo M, Hollingworth D, Ruben PC, Wallace BA. Cannabidiol interactions with voltage-gated sodium channels. Elife. (2020) 9:e58593. doi: 10.7554/eLife.58593
23. Zhang HB, Bean BP. Cannabidiol inhibition of murine primary nociceptors: tight binding to slow inactivated states of Nav18 channels. J Neurosci. (2021) 41:6371–87. doi: 10.1523/JNEUROSCI.3216-20.2021
24. Sativex. Summary of Product Characteristics. Last version July 2021. Available online at: https://www.medicines.org.uk/emc/product/602/smpc
Keywords: multiple sclerosis, spasticity, cannabinoid, symptoms therapy, axonal transmission
Citation: Bruno A, Dolcetti E and Centonze D (2022) Theoretical and Therapeutic Implications of the Spasticity-Plus Syndrome Model in Multiple Sclerosis. Front. Neurol. 12:802918. doi: 10.3389/fneur.2021.802918
Received: 27 October 2021; Accepted: 27 December 2021;
Published: 07 February 2022.
Edited by:
Samar S. Ayache, Hôpitaux Universitaires Henri Mondor, FranceReviewed by:
Clementina Manera, University of Pisa, ItalyCopyright © 2022 Bruno, Dolcetti and Centonze. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diego Centonze, Y2VudG9uemVAdW5pcm9tYTIuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.