 Ersilia Mirabelli
Ersilia Mirabelli Stella Elkabes
Stella Elkabes
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 16 December 2021
Sec. Multiple Sclerosis and Neuroimmunology
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.793745
This article is part of the Research TopicPain in Multiple Sclerosis and Experimental Autoimmune EncephalomyelitisView all 5 articles
Multiple sclerosis (MS) is a multifaceted, complex and chronic neurological disease that leads to motor, sensory and cognitive deficits. MS symptoms are unpredictable and exceedingly variable. Pain is a frequent symptom of MS and manifests as nociceptive or neuropathic pain, even at early disease stages. Neuropathic pain is one of the most debilitating symptoms that reduces quality of life and interferes with daily activities, particularly because conventional pharmacotherapies do not adequately alleviate neuropathic pain. Despite advances, the mechanisms underlying neuropathic pain in MS remain elusive. The majority of the studies investigating the pathophysiology of MS-associated neuropathic pain have been performed in animal models that replicate some of the clinical and neuropathological features of MS. Experimental autoimmune encephalomyelitis (EAE) is one of the best-characterized and most commonly used animal models of MS. As in the case of individuals with MS, rodents affected by EAE manifest increased sensitivity to pain which can be assessed by well-established assays. Investigations on EAE provided valuable insights into the pathophysiology of neuropathic pain. Nevertheless, additional investigations are warranted to better understand the events that lead to the onset and maintenance of neuropathic pain in order to identify targets that can facilitate the development of more effective therapeutic interventions. The goal of the present review is to provide an overview of several mechanisms implicated in neuropathic pain in EAE by summarizing published reports. We discuss current knowledge gaps and future research directions, especially based on information obtained by use of other animal models of neuropathic pain such as nerve injury.
Multiple sclerosis (MS) is the most common chronic inflammatory and demyelinating disease of the central nervous system (CNS) (1, 2). MS is a multifaceted disease. Both genetic and environmental factors contribute to the risk of developing the disorder. It is estimated that close to a million people live with MS in the United States alone (3). Young adults are the most affected with the range of onset being 20–40 years of age. The prevalence of the disease is three-times higher in women than in men (4, 5). Even if the majority of the patients initially present with relapsing-remitting (R-R) MS, the disease eventually progresses into secondary-progressive form within 10–20 years from the onset (6). The major symptoms include limb weakness, fatigue, spasticity, sensory impairments, loss of coordination, cognitive decline, pain and paralysis. Because MS is not curable, the goal of the various therapeutic approaches is to slow disease progression and alleviate the symptoms to improve quality of life (7).
Among the MS population, pain is a frequent symptom that affects from 28 to 87% of individuals, with variations in time of onset and type of pain. Pain impacts both the physical and emotional well-being of the individual (8) and interferes with the majority of daily life activities such as sleep, work, and participation in recreational and social activities (9), reducing the quality of life and leading to depression and other comorbidities (10–12).
The pain associated with MS can be classified into four groups: musculoskeletal pain (painful tonic spasms, pain secondary to spasticity), intermittent central neuropathic pain (trigeminal neuralgia, Lhermitte's sign), continuous central neuropathic pain, and mixed neuropathic and non-neuropathic pain (headache). Regardless from the type of pain, there is a correlation between pain and the disease course, its duration, and age of the affected individual (13). Neuropathic pain is more common in women with higher disability and longer disease duration (14).
Painful spasms, especially in the lower limbs, are due to ectopic impulses, generated from the motor fibers, as a result of axonal damage and demyelination. These painful spasms are more frequent at night (15). Headaches and low back pain are very common among affected individuals throughout the course of the disease (16–18). Importantly, other manifestations of pain occur as the disease progresses. The spasticity and progressive weakness compromise the posture and motility of the individual, leading to osteoporosis and the dysfunctions of tendons, ligaments and/or joints, which evoke secondary pain (15). Even the pharmaceutical treatments commonly used for MS symptomatology exacerbate some of the most common pains (19). For example, Interferon-β exacerbates headaches and migraines (20).
Primary sensory neurons of the dorsal root ganglia (DRG) sense noxious stimuli via their peripheral projections, and convey the pain information to the dorsal horn (DH) of the spinal cord (SC) through their central projections. The central projections synapse with second-order sensory neurons and excitatory or inhibitory interneurons in the DH. The DH also receives projections from supraspinal locations which modulate pain transmission. These signals are integrated in the DH and then conveyed to various brain regions where pain perception and affective responses to pain develop (21). The cingulate and insular cortices, the amygdala and brainstem, are among brain regions implicated in pain states (22–25).
Neuropathic pain is caused by damage or disease that affects the central or peripheral somatosensory systems, and is referred to as central or peripheral neuropathic pain, respectively (26, 27). The pathophysiological changes associated with neuropathic pain include hyperexcitability of neurons in pain pathways. Neuronal hyperexcitability is an essential mechanism underlying the increased sensitivity to pain in various pathological conditions.
Central sensitization manifests as increased sensitivity to innocuous (allodynia) or noxious (hyperalgesia) stimuli, or spontaneous pain. It occurs in many chronic pain conditions (28–30) including MS or its animal models (31) and is independent of peripheral damage or disease. Central sensitization is the consequence of maladaptive changes in pain circuits of the CNS and heightened excitability of neurons which is partly attributed to increased synaptic efficacy and reduced inhibition (32). The major mechanisms underlying central sensitization have been discussed in earlier comprehensive reports (33, 34) and include glutamate and glutamate receptors. The efficacy of excitatory synapses is enhanced, partly as a consequence of increased glutamate release, impaired glutamate uptake and overactivation of glutamate receptors in DH neurons involved in pain processing. Increased activity, expression and trafficking of ionotropic glutamate receptors, such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptors are observed in second order sensory neurons of the DH (34–36). Metabotropic glutamate receptors (mGluRs) also participate in the modulation of pain transmission by inducing Ca2+ release from intracellular stores, which, in turn, activates kinases, including phosphatidyl inositol 3 kinase (PI3K) and mitogen activated protein kinase (MAPK). Activated kinases phosphorylate ion channels and receptors implicated in pain mechanisms, altering their activity and leading to increased synaptic efficacy (33). These events, together with decreased inhibitory activity of gamma aminobutyric acid (GABA)ergic interneurons (34) and the additional effects of non-neuronal cells, enhance neuronal excitability. In particular, activated glia and infiltrating immune cells secrete pro-nociceptive cytokines, including Tumor Necrosis Factor α (TNFα) and Interleukin 1β (IL-1β), which increase the excitatory and reduce the inhibitory currents in DH neurons, supporting central sensitization (37). Reactive astrocytes have been associated with hyperalgesia under pathological conditions (38). Furthermore, descending noradrenergic and serotoninergic projections to the DH inhibit or facilitate pain transmission (23). Therefore, injury- or disease- induced aberrance in descending pain pathways modulate chronic pain (39). For example, in individuals with MS, lesions in the periaqueductal gray (PAG) are often reported (40). The PAG is an important control center for the modulation and propagation of pain along the descending pathways with analgesic effect upon stimulation (40). In contrast, the rostral ventromedial medulla (RVM) GABAergic cells project to the DH where they facilitate mechanical nociception by inhibiting the spinal GABAergic interneurons (41). Finally it is important to mention that supraspinal glia also contribute to the modulation of chronic pain along the descending pathways particularly through the release of soluble mediators (42).
Peripheral sensitization manifests as an intensification of the responsiveness of primary sensory neurons in the DRG and lowered pain thresholds when there is damage or pathology in tissues that they innervate. Stimulus-independent spontaneous pain is also one of the outcomes of peripheral sensitization. Multiple effectors, including chemokines, prostaglandins, calcitonin gene-related peptide (CGRP), adenosine triphosphate (ATP), substance P and nerve growth factor (NGF) are released at the affected site (43–46). These effectors initiate a molecular cascade partly mediated by tyrosine kinase- and G-protein-coupled receptors. Protein kinases are activated and phosphorylate ion channels and receptors at the peripheral terminals of nociceptors, and by doing so, alter their activity (47–49). In addition, the same modulators modify the expression, localization and stability of nociceptive ion channels and receptors (50). The overall outcomes of these alterations are increased activity of nociceptors and decreased pain thresholds (51).
Neuropathic pain is widely experienced among individuals with MS and can take several forms. Dysaesthetic (lower extremity) pain is the most common form of neuropathic pain described as a constant burning, tingling and throbbing, painful sensation in the legs and feet (10). Even though the specific mechanisms underlying the onset of dysaesthetic pain have not been elucidated, it has been suggested that lesions in the SC could lead to the disruption of pain transmission along the spinothalamic tract, or dysfunction of GABAergic interneurons (52). In accordance, magnetic resonance imaging (MRI) of individuals with dysaethetic extremity pain shows plaques in the cervical and thoracic spinal cord (53, 54).
Trigeminal neuralgia is described as a brief electric shock sensation resulting from the irritation of the trigeminal nerve. It primarily affects different facial regions. Pain can be induced even by mild stimulation of the face. Although the attacks can be brief (up to 2 min), they occur frequently during the day (up to 50/day) (55). Demyelination of the pons, which affects the trigeminal root entry zone, and neurovascular compression of the trigeminal root are observed in MS, and could potentially be a cause of trigeminal neuralgia (55). Similar to trigeminal neuralgia, Lhermitte's sign is described as an electric-shock sensation associated with neck movement, and running along the back and down the limbs (56). Lhermitte's sign is not specific to MS and manifests in other pathological conditions that include the compression or lesion of the cervical SC (57). In accordance with this idea, the MRI of individuals with MS shows demyelinated plaques in the dorsal columns at the cervical level (58).
The treatment of neuropathic pain is challenging, mostly due to the low efficacy and side effects of pharmacological agents (59–61). Tricyclic antidepressants, and serotonin/norepinephrine reuptake inhibitors have been used as first-line pharmacological treatments. Anticonvulsants, including gabapentine and pregabalin, are also considered first-line of treatment (62). Although cannabinoids and opioids have been utilized for the alleviation of neuropathic pain in MS, they are not considered as a first-line treatment option (63, 64). Neuromodulation therapies are becoming more common in clinical practice (65). Both brain and SC chemical and/or electrical stimulation and inhibition have been used for the management of different chronic pain conditions (66). Transcranial Direct Current Stimulation (67), peripheral nerve field stimulation (68), transcutaneous spinal direct current stimulation (69), are among treatments to manage neuropathic pain in MS. Alternative strategies such as water exercise and yoga have also been utilized (70).
The goal of the present review is to highlight select mechanisms implicated in the development and persistence of chronic neuropathic pain in MS and its animal models. We first focus on the cellular mechanisms, and then discuss potential molecular mechanisms with particular emphasis on ion channels, pumps and exchangers.
Experimental Autoimmune Encephalomyelitis (EAE) is one of the best characterized animal models utilized for the study of MS (71–73). For over 30 years, the use of EAE in different susceptible animal strains has proven essential for investigations on various aspects of MS pathophysiology, and the discovery of drugs that are widely used to treat MS such as Interferon-β or Glatiramer acetate (74–76). With all the limitations that an animal model could present, conventional EAE models and the spontaneous EAE model observed in T-cell receptor transgenic mice (77), have shed light on many aspects of MS etiology, pathophysiology and its pharmacological treatments (72, 78–80).
EAE can be induced by immunization of susceptible animal strains with myelin components and encephalitogenic peptides, or by adaptive transfer of encephalitogenic T cells (81). There are various EAE models in different species and susceptible strains, and they mimic distinct hallmarks of MS (82). In particular, a clear distinction can be made between two forms of EAE: chronic-progressive and R-R EAE. To induce chronic-progressive EAE, mice are inoculated with myelin oligodendrocyte glycoprotein (MOG). The MOG antigen in CNS tissue homogenates was long recognized to be crucial for provoking demyelinating lesions characteristic of MS (83). Currently, the use of recombinant MOG or synthetic encephalitogenic fragments such as MOG35−55 for immunization provides a reproducible animal model for the study of MS pathology (73). Alongside with MOG, myelin basic protein (MBP) has been used to induce chronic-progressive EAE (84). Proteolipid protein (PLP) and its encephalitogenic PLP fragment (PLP139−151) have been used as a standard approach to induce R-R EAE in susceptible rodent strains. The recombinant protein or synthetic peptide of choice is dissolved in a mineral oil-based adjuvant, Complete Freund's adjuvant (CFA), containing heat-inactivated mycobacteria tuberculosis (MT) that activates the innate immune system through actions on pattern recognition receptors. Injections of pertussis toxin enhance the immune response and perturb blood brain barrier integrity. The onset, course and severity of EAE differ depending on the antigen and adjuvant utilized, and the species, strain, sex and age of the animals used. Within the same species and strain, susceptibility to EAE can be different based on intrinsic and environmental factors such as colonization of the gut and the type of commensal flora, elements that are challenging to control (85).
Alongside with the classical EAE forms, induced by sensitization to a specific myelin protein, there are spontaneous and humanized EAE models which have been discussed in a comprehensive review (86). Among those models are two that have been developed by use of transgenic mouse lines: opticospinal EAE and spontaneous R-R EAE. These models have been useful to unravel the role of B cells and B and T cell interactions in disease pathogenesis (87). Mice manifesting opticospinal EAE develop a chronic progressive EAE-like disease that affects primarily the optic nerve and the SC, but not the brain. The neurological deficits that spontaneously develop are predominantly reminiscent of neuromyelitis optica (88). However, transcriptome profiling indicated that human MS risk genes are among differentially regulated transcripts, supporting the applicability of this model to the study of MS etiology (89). Mice affected by spontaneous R-R EAE develop the disease at high frequency and manifest unique clinical features with distinct symptoms at onset (e.g., ataxia) and during relapse (e.g., hindlimb paralysis). This is especially observed in female mice. The clinical symptoms are paralleled by formation of lesions in relevant CNS regions (e.g., lesions in the cerebellum and brainstem in mice manifesting ataxia, and SC lesions in mice with hindlimb paralysis) (90). Therefore, the spontaneous R-R EAE is a useful animal model for the study of specific disease aspects and treatments because it mimics the most frequent form of MS (88, 90).
Neuroinflammation, demyelinating lesions, axonal damage, and oligodendrocyte and neuronal death are observed in classical EAE, with the SC being the CNS region most affected. These histopathological features are similar to MS neuropathology (91, 92).
Ascending paralysis is the most pronounced symptom in animals with classical EAE. It manifests several days post-immunization, and is associated with disease progression (93). In chronic-progressive EAE, flaccid paralysis starts at the tail with loss of tone, proceeds to the hind and fore limbs, and could lead to death. Aside locomotor impairment, sensory and cognitive dysfunctions are also observed even before the manifestation of motor deficits (94, 95). This is in concordance with the impairment in cognitive function in MS, which is observed even at early phases of the disease, and is paralleled by demyelination and neuronal damage that affects the gray matter (96, 97).
As in the case of MS, rodents with EAE manifest pain (98–100). Pain behavior has been assessed in both chronic progressive and R-R EAE (98–100). Mechanical and thermal allodynia have been documented at onset of EAE symptoms, and concomitant with deficits in cognitive behavior (101), which has also been reported in MS patients (102). Similar to MS, pain behavior and hypersensitivity are variable among different EAE models, revealing the heterogeneity of the disease and mimicking the diversity among individuals with MS (103). Neuroinflammation and disease severity modulate mechanical hypersensitivity, in a MOG35−55-induced EAE model (104). Similar to the caudal to rostral progression of the neuroinflammatory reaction observed in the SC, ascending sensitization might manifest during EAE. Both morphological and functional changes influence the cortical synapses participating in central sensitization and pain hypersensitivity during EAE (105). An increase in immune cell infiltration, glial activation and release of soluble inflammatory mediators (106) together with alterations in glutamate and GABA levels as well as changes in neurotransmission lead to an overall increase in neuronal excitability in EAE (107, 108).
Because anxiety and depression are strongly associated with pain in individuals with MS, EAE has been widely used for the study of these comorbidities (109–111). Mice, affected by a mild form of EAE that does not substantially affect motor function, were evaluated at a later disease stage. Increased anxiety- and depression-like behavior correlated with elevated TNFα, neuronal dysfunction, synapse loss, and neuroinflammation-induced hippocampal damage (112).
EAE has been useful to unravel several cellular and molecular mechanism implicated in MS pathogenesis including the involvement of T cells and other infiltrating immune cells, and the role played by humoral components of the immune system in neurodegeneration. In fact, during the initial phase of EAE, T cells play the major role. Upon their activation in the periphery, T cells produce and secrete pro-inflammatory cytokines and cross the blood-brain barrier. The entry of T cells into the CNS is mediated by integrins and cell adhesion molecules which are upregulated. Once in the CNS, these T cells are re-activated by antigen presenting cells (113). The presentation of myelin-derived antigens by macrophages to T cells, the infiltration of additional immune cells including macrophages and B cells, and glial activation lead to a complex and extensive neuroinflammatory reaction, ultimately resulting in demyelination, loss of oligodendrocytes and neurons, and axonal degeneration (72). The cytotoxic effects of cytokines, the activation of complement proteins and the production and secretion of reactive oxygen and nitrogen species (ROS and RNS, respectively) are among molecular mechanisms that cause CNS damage (114). The free radicals have a severe and deleterious impact on neuronal metabolism, mitochondrial integrity, and energy balance leading to increased intracellular calcium (Ca2+), and eventually neuronal death (115). Because of the important role played by oxidative stress in MS, some of the latest therapies focus on reducing the deleterious effects that are the result of ROS and RNS accumulation (116). Oxidative stress does not only damage neurons, but it affects glial cells as well, with oligodendrocytes being the most sensitive cell type (117). In active lesions during the early stages of R-R MS, oxidative damage affects primarily oligodendrocytes and myelin (117, 118). The accumulation of ROS or RNS in mitochondria compromises oligodendrocyte function and impairs the differentiation of oligodendrocyte progenitor cells. This could be a mechanism underlying the impairment of remyelination (119) since mitochondria are essential for the biosynthesis of lipids to produce myelin (120, 121). Oxidative stress affects oligodendrocytes and their precursors in more than one way and therefore, could contribute directly and indirectly to MS pathophysiology (122, 123).
Following injury or disease, glia undergo morphological and functional changes switching into an active state. As indicated above, glial activation also occurs in MS and EAE, and is considered an essential determinant of disease pathology (124). In addition to myelin phagocytosis and antigen presentation, glia release pro-inflammatory and pro-nociceptive cytokines, chemokines, brain derived neurotrophic factor (BDNF), ROS, and ATP, leading to both neurotoxicity (125) and the development and maintenance of neuropathic pain (126–131). Attenuation of astrocyte and microglia activation using fingolimod has been associated with amelioration of pain hypersensitivity in mice with EAE (31).
Activated glia are also a source of glutamate. Glutamate homeostasis is impaired in MS and EAE. Glutamate levels in the cerebrospinal fluid (CSF) of individuals with MS correlate with disease severity (132). Increased serum glutamate levels and elevated glutamate in active white matter lesions have been observed in MS (133, 134). Since glutamate and glutamate receptors play essential roles in the hyperexcitability of sensory neurons and sensitization to pain, increased glutamate release by glia potentially contributes to neuropathic pain in MS/EAE. Glutamate can also induce excitotoxicity through the activation of ionotropic receptors which participate in the neurodegeneration observed in MS and EAE and other CNS disorders (135, 136). In addition to neurons, glia express ionotropic and metabotropic glutamate receptors, and therefore, their function could be modulated via direct stimulation by glutamate (137, 138).
Astrocytes are essential for the clearance of synaptic glutamate through uptake by the glutamate transporters, glutamate aspartate transporter (GLAST/EAAT1) and glutamate transporter 1 (GLT-1/EAAT2). A decrease in EAAT1 and EAAT2 levels was observed in the SC at the peak of EAE which persisted even after remission (139). A reduction of astrocyte-mediated glutamate transport could elevate glutamate levels causing neuronal hyperexcitability as well as excitotoxicity. Oligodendrocytes also express EAAT1 and EAAT2 and aberrance in glutamate uptake by oligodendrocytes also contributes to pathology in EAE/MS (140). Paradoxically, an increase in glial glutamate transporter levels in the MS optic nerve has been implicated in neuroprotection (141). A loss of EAAT1 and EAAT2 in MS lesions and an upregulation of EAAT2 in the adjacent cortex with intact myelin has been documented (142). Since both astrocytes and the white matter oligodendrocytes express glutamate transporters (143), these contradictory findings could be the result of many factors including cell type, CNS region, disease stage and whether EAE as opposed to post-mortem MS tissue were analyzed. Collectively, the studies support the notion that glial EAAT1 and EAAT2 could play a role in EAE/MS pathology. However, their precise contribution to neuropathic pain in these diseases requires further investigations.
Glutamate receptor antagonists have been assessed in EAE, primarily in the context of neuroprotection, prevention of demyelination, and improvement of motor deficits (144–148). Although glutamate receptor antagonists, including memantine (149–151) or modulators of the glutamatergic system such as ketamine (152, 153) have been used for the relief of chronic pain in various diseases or injuries, their effectiveness in the alleviation of neuropathic pain during EAE/MS has not been adequately investigated.
Ca2+, Na+, and K+ channels are essential components of pathways underlying pain mechanisms (154, 155). They have also been implicated in several aspects of MS pathophysiology (156). Voltage gated Na+ and Ca2+ channels (VGSCs and VGCCs, respectively) are expressed in sensory neurons of the DRG and in DH neurons (157, 158). Under physiological conditions, these channels participate in neuronal excitability and neurotransmitter release (159, 160). Following CNS and peripheral nervous system (PNS) injury and disease, changes that affect their expression and distribution lead to neuronal hyperexcitability, resulting in an exaggerated and repetitive response to subthreshold sensory stimuli, and increasing synaptic strength (161, 162). The increased activity of Na+ and Ca2+ channels in first order sensory neurons of the DRG promotes neurotransmitter release, which, in turn, enhances the excitatory inputs to the SC (34).
The N-type VGCC, CaV2.2, is one of the major ion channels regulating neurotransmitter release in the PNS and CNS (163). Compelling evidence indicates that CaV2.2 is involved in pain mechanisms. In fact, CaV2.2 has been extensively investigated, especially with the goal of finding pharmaceutical approaches for the treatment of chronic pain (164–166).
Several lines of evidence support the involvement of CaV2.2 in neuropathic pain.
Following CFA-induced inflammatory pain in mice, CaV2.2 mRNA and protein levels are upregulated in the lumbar DRG and correlate with the increase in thermal hyperalgesia (167). Electrophysiological recordings from the DRG showed a voltage increase in CaV2.2 currents and a rise in the frequency of spontaneous action potentials that were directly related to CaV2.2. These factors, taken together, suggested an overall enhancement of the excitability of DRG neurons (167).
Furthermore, CaV2.2 knockout mice manifest decreased pain responses in models of neuropathic and inflammatory pain (168). Blockade of CaV2.2 in nociceptors reduces chronic inflammatory pain in a mouse model of rheumatoid arthritis (169). Additionally, interference with CaV2.2 trafficking to the membrane of DRG neurons abrogates Ca2+ currents, decreases stimulus-induced pro-nociceptive neuropeptide release, and reduces the excitatory synaptic transmission in lamina II DH neurons which receive DRG afferents that express CaV2.2. These changes result in the attenuation of pain responses in several rodent models of evoked inflammatory and neuropathic pain (170). Following chronic constriction injury (CCI) of the sciatic nerve, CaV2.2 is upregulated in lamina II of the lumbar DH (171). In the lumbar L5-spinal nerve ligation (L5-SNL) model, IL-1β and IL-10, pro- and anti-nociceptive cytokines, respectively, modulate CaV2.2 expression in the DRG in opposite manner. Injury at the L5 level increases IL-1β release in the adjacent, uninjured L4 DRG which upregulates CaV2.2, resulting in neuronal hyperexcitability (172) and mechanical allodynia (173). In contrast, L5-SNL upregulates both IL-1β and IL-10 in the corresponding, injured L5 DRG and lumbar SC. However, the effects of IL-10 predominate and a significant reduction in CaV2.2 is observed (172). Collectively, the aforementioned studies illustrate the role played by CaV2.2 in pain.
Various mechanisms have been implicated in CaV2.2-mediated pain responses.
In peripheral somatosensory neurons, CaV2.2 is located close to synaptic vesicles containing pro-nociceptive neurotransmitters such as glutamate, substance P and CGRP (174). Inhibition of N-type VGCC in rat DRG neuronal cultures decreases the entry of Ca2+ and reduces neurotransmitter release (175). As indicated above (170), by regulating the release of pro-nociceptive neurotransmitters, CaV2.2 can enhance pain responses.
CaV2.2 forms a complex with NMDA receptors (NMDA-R) in the mouse and human SC (176). Following nerve injury, CaV2.2 promotes the trafficking and synaptic targeting of NMDA-R, suggesting an additional mechanism by which it could contribute to pain signaling (176).
Ectopic fiber sprouting in targets of DRG neurons, which occurs following nerve injury, has been associated with neuropathic pain (177). Inhibition of CaV2.2 activity in neurons derived from the DRG of mice manifesting CFA-induced inflammatory pain, prevented neurite outgrowth. It has been proposed that excess neurite outgrowth causes an increase in the nerve terminal density, which leads to increased nociceptive inputs and pain (167, 178).
During MS and EAE, a CaV2.2 subunit is overexpressed in active MS lesions, and MS/EAE plaques (179). It has been suggested that the overexpression of CaV2.2 impacts neuronal function and demyelination in the lesions (180, 181). Although a direct study has not yet confirmed the link between CaV2.2 and neuropathic pain in MS/EAE, the involvement of this channel in EAE pathophysiology, its role in neuropathic pain, together with the use of N-type Ca2+ channel blockers in the treatment of chronic pain in MS patients (182–184), point toward the need for further investigations to determine how CaV2.2 plays a role in EAE-associated pain.
Nav1.6 and Nav1.8 are among the VGSCs participating in inflammation and neuronal pathology in retinal ganglion cells and lumbar DRG during EAE (185–188). Nav1.6 is associated with Na+/Ca2+ exchangers (NCX). It has been proposed that in EAE, increased Nav1.6 expression prevents activity reverses the Na+/Ca2+ exchanger activity, and by doing so, promotes Ca2+ influx into neurons and causes neurodegeneration. It is possible to speculate that such interactions between Nav1.6 and the NCX also occur in sensory neurons and leads to elevated intracellular Ca2+ which, in turn, modulates pain mechanisms. Despite studies showing the role of Nav1.6 in various pain models, illustrated by examples described below, its contribution to MS- or EAE-associated pain has been underappreciated.
In neuropathic pain induced by SNL, siRNA-mediated Nav1.6 knockdown decreases the hyperexcitability of DRG neurons by reducing the frequency of the action potentials and restoring their resting potential. In addition, a decrease in sympathetic sprouting and the ectopic firing of Aβ fibers in the lumbar DRG have been documented (189). Moreover, in spared nerve injury, a model of neuropathic pain, conditional adenoviral vector (AVV)-mediated Nav1.6 knockdown in DRG neurons reduces excitability and ameliorates pain in adult mice. Furthermore, the accumulation of Nav1.6 in newly formed nodes of Ranvier, which has been implicated in pain, is mitigated by Nav1.6 knockdown (190). Accordingly, an amelioration of pain sensitivity is observed. Nav1.6 is also upregulated in a mouse model of diabetes and diabetic neuropathy (191). A gain of function mutation in Nav1.6 was found in individuals with trigeminal neuralgia. The mutated form shows decreased current threshold, increased frequency of evoked action potentials and overall potentiation of the Na+ currents with higher excitability of the trigeminal ganglion (192). Taken together, the investigations mentioned above stress the pivotal role played by Nav1.6 in pain states. The role of Nav1.6 could be a promising research direction to further explore in MS- and EAE-associated pain.
In the context of MS and EAE, NaV1.8 was first associated with alterations in the firing pattern of cerebellar Purkinje cells, causing dysfunction and disruption of motor coordination, one of the symptoms observed in EAE and MS (188). Few studies have shed light into its involvement in DRG sensitization. There is a significant increase in Nav1.8 levels in DRG neurons in CFA-induced inflammatory pain. TNFα and IL-1β directly modulate Nav1.8-like currents by activating p38/MAPK signaling pathway. This is paralleled by increased excitability of DRG neurons (160). Nav1.8 channels are expressed in more than 90% of the nociceptive neurons in the DRG (193). Since medium to large sensory neurons of the DRG are hyperexcitable at onset of EAE, and IL-1β is significantly elevated in the DRG at the same stage of the disease (194), it is possible that similar mechanisms contribute to peripheral sensitization and neuropathic pain associated with EAE. This possibility requires further investigations.
The calcium-sensitive large conductance K+ channels (BK) have recently been implicated in pain mechanisms during EAE (195). These channels are found in DRG neurons of mice with EAE and play a role in the endoplasmic reticulum (ER) stress-mediated pain response. It has been hypothesized that the prolonged neuroinflammation that occurs in the CNS during MS/EAE affects DRG neurons by conveying retrograde stress signals, which, in turn, induce ER stress (195). In fact, ER stress markers are elevated in post-mortem DRG of individuals with MS. Furthermore, 4-phenyl butyric acid (4-BPA), an inhibitor of ER stress, suppresses mechanical hypersensitivity in mice with EAE (195). The potential mechanisms by which ER stress leads to pain states in EAE have been investigated. ER stress causes variations in intracellular Ca2+ transients. Under physiological conditions, because of their Ca2+ sensitivity, BK channels respond to transient changes in Ca2+ concentrations by mediating the efflux of K+ in order to maintain the membrane potential of excitable cells. In mice affected by EAE and individuals with MS, there is a reduction in the b4 auxiliary subunit of the BK channels in DRG neurons (195). This subunit modulates the Ca2+ sensitivity of BK channels, and therefore, its electrical properties (196). The decrease in b4 subunit causes a change in BK channel physiology resulting in a higher depolarizing resting potential due to decreased BK activity and the release of K+ from neurons. An elevated depolarizing resting potential results in frequent and easier firing, ectopic discharges, and increased neurotransmitter release leading to hyperexcitability of sensory neurons. In mice with EAE, administration of 4-BPA restores the membrane potential of DRG neurons to physiological values by increasing the expression of b4 and by restoring BK channel properties.
A link between ER stress and pain behavior has been observed in other models of neuropathic pain. An increase in ER stress is recorded in the DH of rats following SNL and it corresponds with pain hypersensitivity. Inhibition of ER stress pathways in the SC alleviates pain (197, 198). ER stress in DH neurons can influence the environment and promote spinal sensitization and increased pain sensitivity (199). ROS production is an outcome of ER stress. GABAergic interneurons are sensitive to ROS. The increased production of ROS following ER stress causes long-term depression of GABAergic interneurons in the DH and impairs GABA release following SNL (197, 200). It is possible that similar mechanisms occur in EAE. In addition, astrocytes and macrophages release pro-nociceptive cytokines including IL-1β and IL-6 in response to ER stress (201, 202). These cytokines are upregulated in MS/EAE and they have been implicated in pain (130, 203). X-Box Binding Protein 1 (XBP1), a transcription factor associated with ER stress and a regulator of unfolded protein response, has been implicated in neuropathic pain (199, 204). XBP1 increases in the brain white matter in MS and in SC white matter in EAE. However, the potential contribution of XBP1 to neuropathic pain in MS/EAE has not received attention. In sum, the involvement of ER stress in pain responses in EAE or MS and its relation to BK is an intriguing research direction that warrants further studies.
Plasma membrane calcium ATPases (PMCAs) and NCX are among ion pumps and exchangers, respectively, that are modulated during MS/EAE, and implicated in pain mechanisms. Their main physiological function is to regulate intracellular Ca2+ concentrations. Both PMCA and NCX expel intracellular Ca2+. PMCAs are essential in maintaining the cytosolic Ca2+ concentrations due to their high Ca2+ affinity and low Ca2+ capacity, whereas NCX, which has low Ca2+ affinity, participates more dynamically in re-establishing Ca2+ homeostasis during large cytosolic Ca2+increases (205). Aberrant Ca2+ clearance in neurons can cause hyperactivity (206).
Studies on MOG35−55-induced chronic EAE have shown that the levels of one of the PMCA isoforms, PMCA2, which is exclusively expressed in neurons, is downregulated in the DH of mice at onset of the disease. This is paralleled by a concomitant increase in mechanical and thermal pain sensitivity (207). PMCA2 is also downregulated in motor neurons (208, 209) and photoreceptors at early stages of EAE, causing aberrance in Ca2+ signaling and contributing to synaptic dysfunction (210). Since PMCA2 is not expressed in the DRG (211), it is likely that the modulation of pain responses by PMCA2 is primarily mediated through changes in DH neurons. It has been postulated that a delay in Ca2+ clearance, as a result of decreased PMCA2 in DH neurons of EAE mice, could lead to increased intracellular Ca2+ resulting in the hyperactivation of DH neurons. Furthermore, increased intracellular Ca2+ could activate Ca2+-dependent transcription factors involved in the transcription of pro-nociceptive genes (207). It is worth noting that glutamate, which is one of the major players in the initiation and maintenance of chronic pain (138, 212, 213) and central sensitization (33), modulates PMCA2 levels in SC neurons. Kainic acid, an analog of glutamate, reduces PMCA2 levels in cultured SC neurons and administration of 2,3-dioxo-6-nitro-7-sulfamoyl-benzo quinoxaline (NBQX), an AMPA/Kainate receptor antagonist, restores PMCA2 levels in EAE mice (148). IL-1β is also a trigger that reduces PMCA2 expression by mechanisms that remain elusive (207). Further studies are needed to establish the direct link between PMCA2 and pain mechanisms in DH neurons.
NCX has been studied in animal models of neuropathic pain (214). Because of its intrinsic properties as an exchanger, this transporter allows the passage of Ca2+ and Na+ across the membrane in either direction, depending on ion gradients. In the forward mode, NCX mediates intracellular Ca2+ extrusion whereas in the reverse mode it facilitates Ca2+ influx. Reverse activity of NCX has been reported in animal models of neuropathic pain and EAE (215, 216). When the reversal of NCX activity is inhibited by pharmacological agents in rodents with peripheral nerve injury, the Ca2+ overload in the lumbar DRG is reduced and an overall decrease in pain sensitivity is observed (214).
During EAE, the number of NCX expressing neurons in the lumbar SC increases. NCX co-localizes with Nav1.6 channels in the dorsal column white matter (216). An increase in the number of Na+ channels and the consequent elevation in Na+ currents, reverses NCX activity leading to Ca2+ overload and axonal injury in the CNS during EAE (216, 217). The co-localization of Nav1.6 and NCX is also observed in cervical SC tissue of MS patients (218). NCX is expressed in cortical astrocytes where it facilitates glutamate release (219). Reactive astrocytes participate in the modulation of neuropathic pain by releasing glutamate, among other modulators of pain (220). Taken together, these investigations raise the possibility that NCX contributes, not only to axonal damage, but also to mechanisms underlying pain hypersensitivity by modulating Ca2+ signaling. This mechanism has not been adequately explored in EAE-related pain, and could be an engaging research direction.
The involvement of ion channel and exchangers in pain mechanisms has been summarized in Table 1.

Table 1. Summary of studies implicating ion channels in pain and their principal findings.
Several molecular mechanisms have been implicated in the pathogenesis and maintenance of neuropathic pain in MS/EAE. Most of these studies were undertaken to investigate molecules that been implicated at different stages of disease pathophysiology.
The Wnt signaling pathways are involved in the development of EAE-associated chronic pain by increasing the expression of pro-inflammatory and pro-nociceptive cytokines (221). The physiological roles of Wnt include CNS development (222), synaptic plasticity (223), regulation of oligodendrocyte maturation and differentiation (224) and cytokine production by cortical and SC neurons (225, 226). These functions are exerted through β-catenin-dependent and -independent pathways (227, 228). During EAE, both of these pathways are overactivated, and pharmacological inhibition of either of these pathways attenuates mechanical allodynia (221). Similar results were obtained by use of inflammatory and neuropathic pain models, supporting further the involvement of Wnt in pain mechanisms. Wnt signaling enhances synaptic plasticity and spine morphogenesis in DH neurons. This was associated with hypersensitization and chronic pain in adult mice (229).
Chemokines are potent mediators of inflammation in MS, with T-cells being the major source during the initial phase of the disease. They chemoattract leukocytes to affected sites (230). An increase in chemokines and their receptors is observed in the blood and CSF of individuals with MS (231) as well as animals affected by EAE (232).
The chemokine CX3CL1 and its receptor CX3CR1 have been implicated in pain mechanisms. In the CNS, CX3CL1 is primarily expressed in neurons, and CX3CR1, is predominantly found in microglia. The involvement of CX3CR1 in pain mechanisms is indicated by the fact that CX3CR1 knockout mice do not develop thermal hyperalgesia in an inflammatory pain model (233). Moreover, upon interaction with its ligand in the DH, CX3CR1, which is a G-coupled receptor (234), initiates a cascade of events in microglia, with ultimate production and release of pro-nociceptive mediators such as IL-1β, IL-6 and NO (235).
The link between pain and CX3CL1 was further demonstrated in a R-R EAE model. The authors showed that induction of pain by ligation of the middle, sensory branch of the trigeminal nerve during a remission, increases CX3CL1 expression in the lumbar SC leading to the recruitment of immune cells which eventually results in EAE relapse (236). Another study found that CX3CL1 and CX3CR1 are upregulated in the DRG and DH of EAE rats at the early stage of the disease, prior to demyelination and axonal injury (126). This corresponded with the onset of hypoalgesia (126). The authors postulated that the parallelism between hypoalgesia, a sensory aberrance manifesting prior to the development of hyperalgesia in MS (237), and the increase in CX3CL1 and CX3CR1 in the DRG and DH support the idea that they play a role in nociception and pain.
BDNF has been studied in the context of MS/EAE because of its involvement in MS-associated neuroinflammation (238), and the possibility of using it as a marker for disease progression (239), but its role in chronic pain has been controversial. A variety of animal models have shown both anti-nociceptive and pro-nociceptive effects of BDNF. Interestingly, ERK and c-fos, which are among the downstream signaling molecules activated by BDNF and its receptor TrkB, are considered markers of central sensitization (240). Phospho ERK (pERK) directly increases the activity of AMPA and NMDA receptors while decreasing the activity of K+ channels in the lumbar DH. The overall result is an increase in the excitability of superficial laminae neurons (241). These rapid events associated with pERK activation are followed by the slower but long-lasting effects such as the transcription of genes implicated in pain including c-fos, Cox-2 and TrkB. Thus, pERK plays a critical role in central sensitization of DH neurons (241).
In the DH of EAE mice, cellular markers of central sensitization, including pERK, are increased in neurons, and this is paralleled by mechanical and cold hyperalgesia (31). In R-R EAE, attenuation of BDNF-TrkB-ERK signaling in the lumbar DH, is associated with the alleviation of mechanical allodynia in the diseased mice (242). Since white blood cells of individuals with R-R MS express elevated BDNF levels (243), it is likely that immune cells infiltrating the CNS are a source of BDNF in addition to glia and neurons. In rats with EAE, BDNF in the DRG and SC was upregulated at the peak of the disease (244). In agreement, TrkB levels were elevated in the lumbar DH and associated with increased mechanical pain sensitivity in R-R EAE (242).
In the context of neuropathic pain, IL-1β, IL-6 and TNFα have been implicated in the maintenance of pain states following injury and disease (131, 245, 246). These mediators exert their actions in a synergistic manner. Upon binding to their respective receptors, they can lead to the production, secretion and modulation of additional cytokines (131). Both activated glia and infiltrating immune cells release IL-1β, IL-6 and TNFα (247, 248).
IL-1β is produced in an inactive precursor form. Cleavage of the precursor by caspase-1 produces mature IL-1β which is the active form. The binding of IL-1β to its receptor induces the activation of transcription factors such as NF-kB and this results in the further production and release of pro-inflammatory molecules including other cytokines, ROS, NO, NOS and cyclooxygenases (COXs) (249, 250). It has been proposed that in rodents experiencing inflammation-evoked hyperalgesia, IL-1β activates NF-kB in DRG and SC neurons inducing COX-2 (251). COX-2 is a potent stimulator of prostaglandins, which are lipids that exert several physiological functions including maintenance of inflammation and pain (252, 253). IL-1β also enhances mechanical and thermal pain sensitivity by perturbating the activity of glutamate receptors (254–256). IL-1 receptor I, which is expressed in nociceptors (257), colocalizes with the NR1 subunit, one of the NMDA-R subunits (246). IL-1β selectively induces the phosphorylation of NR1 subunit facilitating pain transmission (248). Furthermore, administration of IL-1 receptor antagonist (IL-1ra) significantly reduces inflammatory hyperalgesia in rats, by inhibiting NR1 phosphorylation in the SC (255).
IL-1β, IL-6 and TNFα have been investigated in MS pathology and related pain (258, 259). As stated before, the majority of the intracellular signaling pathways that lead to neuropathic pain during MS/EAE include the transcription of pro-inflammatory and pro-nociceptive cytokines such as TNFα, IL-6 and IL-1β. IL-1β is necessary for the neuroinflammatory reaction that develops during EAE as indicated by the resistance of IL-1β knockout mice to EAE (260). IL-1β has been associated with pain hypersensitivity in mice with EAE, as systemic administration of an IL-1 receptor antagonist alleviates pain (203).
The molecular mechanisms underlying pain responses in pathological conditions including EAE have been summarized in Figure 1.
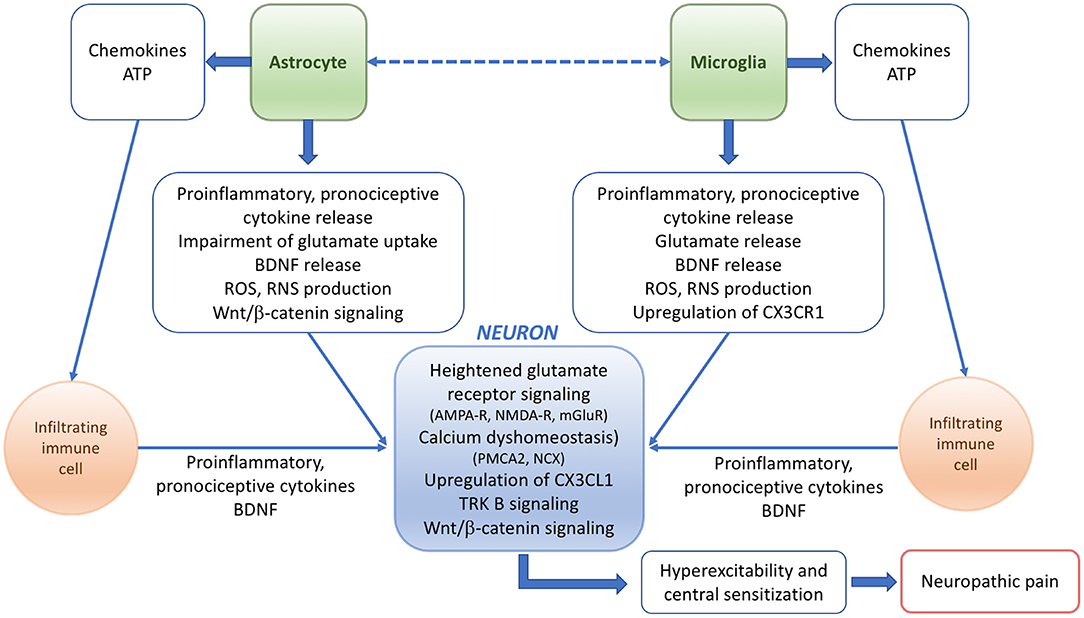
Figure 1. Scheme summarizing the potential mechanisms and players underlying the pathophysiology of neuropathic pain in EAE or MS.
In spite of a wealth of knowledge about the immune system reaction and the crosstalk between the immune and nervous systems in MS and EAE, information regarding the mechanisms underlying neuropathic pain has been limited. Given the high frequency of pain states in individuals affected by MS, it is imperative to advance the understanding of events leading to the onset and maintenance of chronic pain. Revealing the specific aberrant mechanisms and major players involved in the development and/or maintenance of pain in MS/EAE would greatly alter the pharmacological approach to its treatment, thus improving the quality of everyday life in affected individuals. Considering the type of pain experienced in MS, it is highly likely that diverse and complex mechanisms regulate pain processing. In particular, neuropathic pain remains a challenge, not only in MS but in other pathological conditions of the CNS. Whereas such mechanisms could be somehow distinct in each pathological condition, commonalties also exist. Because neuropathic pain has been extensively studied in animal models of nerve injury, findings obtained in such studies could facilitate future investigations on MS/EAE-associated pain. For example, glutamate excitotoxicity-induced death of inhibitory GABAergic neurons in the superficial DH has been implicated in the transition of acute neuropathic pain into chronic neuropathic pain following nerve injury (261). As mentioned above, a role for glutamate excitotoxicity has been shown in EAE and high glutamate levels have been reported in the CSF of individuals with MS. Therefore, mechanisms similar to those reported in nerve injury (261) could also underlie chronic neuropathic pain in EAE/MS.
Studies on EAE have also shown that demyelination and inflammation occur, not only in the CNS, but also in the PNS (262). Peripheral nerve demyelination has been reported in a subset of individuals with MS (263). This can affect the function of sensory neurons in the DRG. In fact, electrophysiological studies indicated membrane hyperexcitability of DRG sensory neurons and therefore, peripheral sensitization in EAE (195, 264).
Regarding the use of EAE for the study of pain states in MS, the multitude of EAE models that have been used in investigations, could challenge the acquisition of cohesive information (265). On the other hand, corroborating findings in more than one model is needed to determine the wider applicability of the findings to different species and experimental paradigms.
With respect to the management of chronic pain, the challenge experienced in other pathological conditions associated with neuropathic pain, are also relevant to MS. Both pharmacological and non-pharmacological approaches are used, although further studies are needed to establish the effectiveness of these therapies (69, 266, 267). Importantly, the evaluation of pharmacological therapies in EAE or other models of MS, need to be performed in a manner that can be applicable in the clinic.
EM and SE conceived and wrote this article and approved the submitted version.
The research in the authors' laboratory has been supported by the Reynolds Family Spine Laboratory Funds and the New Jersey Commission on Spinal Cord Research (Grant CSCR17IRG007 to SE).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Correale J, Gaitán MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain. (2017) 140:527–46. doi: 10.1093/brain/aww258
2. Simkins TJ, Duncan GJ, Bourdette D. Chronic demyelination and axonal degeneration in multiple sclerosis: pathogenesis and therapeutic implications. Curr Neurol Neurosci Rep. (2021) 21:26. doi: 10.1007/s11910-021-01110-5
3. Wallin MT, Culpepper WJ, Campbell JD, Nelson LM, Langer-Gould A, Marrie RA, et al. The prevalence of MS in the United States: a population-based estimate using health claims data. Neurology. (2019) 92:e1029–40. doi: 10.1212/WNL.0000000000007035
4. Harbo HF, Gold R, Tintoré M. Sex and gender issues in multiple sclerosis. Ther Adv Neurol Disord. (2013) 6:237–48. doi: 10.1177/1756285613488434
5. Kingwell E, Marriott JJ, Jetté N, Pringsheim T, Makhani N, Morrow SA, et al. Incidence and prevalence of multiple sclerosis in Europe: a systematic review. BMC Neurol. (2013) 13:128. doi: 10.1186/1471-2377-13-128
6. Cree BAC, Arnold DL, Chataway J, Chitnis T, Fox RJ, Pozo Ramajo A, et al. Secondary progressive multiple sclerosis: new insights. Neurology. (2021) 97:378–88. doi: 10.1212/WNL.0000000000012323
7. Kalia LV, O'Connor PW. Severity of chronic pain and its relationship to quality of life in multiple sclerosis. Mult Scler. (2005) 11:322–7. doi: 10.1191/1352458505ms1168oa
8. Ehde DM, Jensen MP, Engel JM, Turner JA, Hoffman AJ, Cardenas DD. Chronic pain secondary to disability: a review. Clin J Pain. (2003) 19:3–17. doi: 10.1097/00002508-200301000-00002
9. Hadjimichael O, Kerns RD, Rizzo MA, Cutter G, Vollmer T. Persistent pain and uncomfortable sensations in persons with multiple sclerosis. Pain. (2007) 127:35–41. doi: 10.1016/j.pain.2006.07.015
10. O'Connor AB, Schwid SR, Herrmann DN, Markman JD, Dworkin RH. Pain associated with multiple sclerosis: systematic review and proposed classification. Pain. (2008) 137:96–111. doi: 10.1016/j.pain.2007.08.024
11. Alschuler KN, Ehde DM, Jensen MP. The co-occurrence of pain and depression in adults with multiple sclerosis. Rehabil Psychol. (2013) 58:217–21. doi: 10.1037/a0032008
12. Marrie RA, Reingold S, Cohen J, Stuve O, Trojano M, Sorensen PS, et al. The incidence and prevalence of psychiatric disorders in multiple sclerosis: a systematic review. Mult Scler. (2015) 21:305–17. doi: 10.1177/1352458514564487
13. Solaro PC, Brichetto LG, Amato LM, Cocco LE, Colombo LB, D'aleo LG, et al. The prevalence of pain in multiple sclerosis: a multicenter cross-sectional study. Neurology. (2004) 63:919–21. doi: 10.1212/01.WNL.0000137047.85868.D6
14. Solaro C, Cella M, Signori A, Martinelli V, Radaelli M, Centonze D, et al. Identifying neuropathic pain in patients with multiple sclerosis: a cross-sectional multicenter study using highly specific criteria. J Neurol. (2018) 265:828–35. doi: 10.1007/s00415-018-8758-2
15. Truini A, Barbanti P, Pozzilli C, Cruccu G. A mechanism-based classification of pain in multiple sclerosis. J Neurol. (2013) 260:351–67. doi: 10.1007/s00415-012-6579-2
16. Kister I, Caminero AB, Herbert J, Lipton RB. Tension-type headache and migraine in multiple sclerosis. Curr Pain Headache Rep. (2010) 14:441–8. doi: 10.1007/s11916-010-0143-5
17. Nicoletti A, Patti F, Lo Fermo S, Liberto A, Castiglione A, Laisa P, et al. Headache and multiple sclerosis: a population-based case-control study in Catania, Sicily. Cephalalgia. (2008) 28:1163–9. doi: 10.1111/j.1468-2982.2008.01662.x
18. Massot C, Donze C, Guyot MA, Leteneur S. Low back pain in patients with multiple sclerosis: a systematic review and the prevalence in a French multiple sclerosis population. Rev Neurol. (2021) 177:349–58. doi: 10.1016/j.neurol.2020.07.018
19. Kassirer M. Multiple sclerosis and pain: a medical focus. Int J MS Care. (2000) 2:40–7. doi: 10.7224/1537-2073-2.3.40
20. Patti F, Nicoletti A, Pappalardo A, Castiglione A, Lo Fermo S, Messina S, et al. Frequency and severity of headache is worsened by Interferon-β therapy in patients with multiple sclerosis. Acta Neurol Scand. (2012) 125:91–5. doi: 10.1111/j.1600-0404.2011.01532.x
21. Browne TJ, Hughes DI, Dayas CV, Callister RJ, Graham BA. Projection neuron axon collaterals in the dorsal horn: placing a new player in spinal cord pain processing. Front Physiol. (2020) 11:560802. doi: 10.3389/fphys.2020.560802
22. Dworsky-Fried Z, Faig CA, Vogel HA, Kerr BJ, Taylor AMW. Central amygdala inflammation drives pain hypersensitivity and attenuates morphine analgesia in experimental autoimmune encephalomyelitis. Pain. (2021). doi: 10.1097/j.pain.0000000000002307. [Epub ahead of print].
23. Zhuo M. Descending facilitation. Mol Pain. (2017) 13:1744806917699212. doi: 10.1177/1744806917699212
24. Jiang YY, Shao S, Zhang Y, Zheng J, Chen X, Cui S, et al. Neural pathways in medial septal cholinergic modulation of chronic pain: distinct contribution of the anterior cingulate cortex and ventral hippocampus. Pain. (2018) 159:1550–61. doi: 10.1097/j.pain.0000000000001240
25. Mills EP, Di Pietro F, Alshelh Z, Peck CC, Murray GM, Vickers ER, et al. Brainstem pain-control circuitry connectivity in chronic neuropathic pain. J Neurosci. (2018) 38:465–73. doi: 10.1523/JNEUROSCI.1647-17.2017
26. St John Smith E. Advances in understanding nociception and neuropathic pain. J Neurol. (2018) 265:231–8. doi: 10.1007/s00415-017-8641-6
27. Boadas-Vaello P, Castany S, Homs J, Álvarez-Pérez B, Deulofeu M, Verdú E. Neuroplasticity of ascending and descending pathways after somatosensory system injury: reviewing knowledge to identify neuropathic pain therapeutic targets. Spinal Cord. (2016) 54:330–40. doi: 10.1038/sc.2015.225
28. Sanzarello I, Merlini L, Rosa MA, Perrone M, Frugiuele J, Borghi R, et al. Central sensitization in chronic low back pain: a narrative review. J Back Musculoskelet Rehabil. (2016) 29:625–33. doi: 10.3233/BMR-160685
29. Arendt-Nielsen L, Morlion B, Perrot S, Dahan A, Dickenson A, Kress HG, et al. Assessment and manifestation of central sensitisation across different chronic pain conditions. Eur J Pain. (2018) 22:216–41. doi: 10.1002/ejp.1140
30. Zhang A, Lee YC. Mechanisms for joint pain in Rheumatoid Arthritis (RA): from cytokines to central Sensitization. Curr Osteoporos Rep. (2018) 16:603–10. doi: 10.1007/s11914-018-0473-5
31. Doolen S, Iannitti T, Donahue RR, Shaw BC, Grachen CM, Taylor BK. Fingolimod reduces neuropathic pain behaviors in a mouse model of multiple sclerosis by a sphingosine-1 phosphate receptor 1-dependent inhibition of central sensitization in the dorsal horn. Pain. (2018) 159:224–38. doi: 10.1097/j.pain.0000000000001106
32. van den Broeke EN. Central sensitization and pain hypersensitivity: some critical considerations. F1000Res. (2018) 7:1325. doi: 10.12688/f1000research.15956.2
33. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. (2009) 10:895–926. doi: 10.1016/j.jpain.2009.06.012
34. Baron R, Hans G, Dickenson AH. Peripheral input and its importance for central sensitization. Ann Neurol. (2013) 74:630–6. doi: 10.1002/ana.24017
35. Tao YX. AMPA receptor trafficking in inflammation-induced dorsal horn central sensitization. Neurosci Bull. (2012) 28:111–20. doi: 10.1007/s12264-012-1204-z
36. Qu XX, Cai J, Li MJ, Chi YN, Liao FF, Liu FY, et al. Role of the spinal cord NR2B-containing NMDA receptors in the development of neuropathic pain. Exp Neurol. (2009) 215:298–307. doi: 10.1016/j.expneurol.2008.10.018
37. Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. (2008) 28:5189–94. doi: 10.1523/JNEUROSCI.3338-07.2008
38. Tang J, Bair M, Descalzi G. Reactive astrocytes: critical players in the development of chronic pain. Front Psychiatry. (2021) 12:682056. doi: 10.3389/fpsyt.2021.682056
39. Ossipov MH, Morimura K, Porreca F. Descending pain modulation and chronification of pain. Curr Opin Support Palliat Care. (2014) 8:143–51. doi: 10.1097/SPC.0000000000000055
40. Papadopoulou A, Naegelin Y, Weier K, Amann M, Hirsch J, von Felten S, et al. MRI characteristics of periaqueductal lesions in multiple sclerosis. Mult Scler Relat Disord. (2014) 3:542–51. doi: 10.1016/j.msard.2014.01.001
41. François A, Low SA, Sypek EI, Christensen AJ, Sotoudeh C, Beier KT, et al. A brainstem-spinal cord inhibitory circuit for mechanical pain modulation by GABA and enkephalins. Neuron. (2017) 93:822–39. doi: 10.1016/j.neuron.2017.01.008
42. Wei F, Guo W, Zou S, Ren K, Dubner R. Supraspinal glial–neuronal interactions contribute to descending pain facilitation. J Neurosci. (2008) 28:10482–95. doi: 10.1523/JNEUROSCI.3593-08.2008
43. Iyengar S, Ossipov MH, Johnson KW. The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain. (2017) 158:543–59. doi: 10.1097/j.pain.0000000000000831
45. Gautam M, Prasoon P, Kumar R, Reeta KH, Kaler S, Ray SB. Role of neurokinin type 1 receptor in nociception at the periphery and the spinal level in the rat. Spinal Cord. (2016) 54:172–82. doi: 10.1038/sc.2015.206
46. Barker PA, Mantyh P, Arendt-Nielsen L, Viktrup L, Tive L. Nerve growth factor signaling and its contribution to pain. J Pain Res. (2020) 13:1223–41. doi: 10.2147/JPR.S247472
47. Iyengar S, Johnson KW, Ossipov MH, Aurora SK. CGRP and the trigeminal system in migraine. Headache. (2019) 59:659–81. doi: 10.1111/head.13529
48. Cao DL, Qian B, Zhang ZJ, Gao YJ, Wu XB. Chemokine receptor CXCR2 in dorsal root ganglion contributes to the maintenance of inflammatory pain. Brain Res Bull. (2016) 127:219–25. doi: 10.1016/j.brainresbull.2016.09.016
49. Salzer I, Ray S, Schicker K, Boehm S. Nociceptor signalling through ion channel regulation via GPCRs. Int J Mol Sci. (2019) 20:2488. doi: 10.3390/ijms20102488
50. Bhave G, Gereau R. Posttranslational mechanisms of peripheral sensitization. J Neurobiol. (2004) 61:88–106. doi: 10.1002/neu.20083
51. Schaible HG, Ebersberger A, Natura G. Update on peripheral mechanisms of pain: beyond prostaglandins and cytokines. Arthritis Res Ther. (2011) 13:210. doi: 10.1186/ar3305
52. Khan N, Smith MT. Multiple sclerosis-induced neuropathic pain: pharmacological management and pathophysiological insights from rodent EAE models. Inflammopharmacology. (2014) 22:1–22. doi: 10.1007/s10787-013-0195-3
53. Morin C, Bushnell MC, Luskin MB, Craig AD. Disruption of thermal perception in a multiple sclerosis patient with central pain. Clin J Pain. (2002) 18:191–5. doi: 10.1097/00002508-200205000-00008
54. Samkoff LM, Daras M, Tuchman AJ, Koppel BS. Amelioration of refractory dysesthetic limb pain in multiple sclerosis by gabapentin. Neurology. (1997) 49:304–5. doi: 10.1212/WNL.49.1.304
55. Di Stefano G, Maarbjerg S, Truini A. Trigeminal neuralgia secondary to multiple sclerosis: from the clinical picture to the treatment options. J Headache Pain. (2019) 20:20. doi: 10.1186/s10194-019-0969-0
56. Khare S, Seth D. Lhermitte's sign: the current status. Ann Indian Acad Neurol. (2015) 18:154–6. doi: 10.4103/0972-2327.150622
57. Teoli D, Rocha Cabrero F, Ghassemzadeh S. Lhermitte Sign. StatPearls Treasure Island (FL): StatPearls Publishing (2021).
58. Al-Araji AH, Oger J. Reappraisal of lhermitte's sign in multiple sclerosis. Mult Scler. (2005) 11:398–402. doi: 10.1191/1352458505ms1177oa
59. Solaro C, Messmer Uccelli M. Pharmacological management of pain in patients with multiple sclerosis. Drugs. (2010) 70:1245–54. doi: 10.2165/11537930-000000000-00000
60. Solaro C, Brichetto G, Battaglia MA, Messmer Uccelli M, Mancardi GL. Antiepileptic medications in multiple sclerosis: adverse effects in a three-year follow-up study. Neurol Sci. (2005) 25:307–10. doi: 10.1007/s10072-004-0362-9
61. Sadiq SA, Poopatana CA. Intrathecal baclofen and morphine in multiple sclerosis patients with severe pain and spasticity. J Neurol. (2007) 254:1464–5. doi: 10.1007/s00415-007-0566-z
62. Mathieson S, Lin CWC, Underwood M, Eldabe S. Pregabalin and gabapentin for pain. BMJ. (2020) 369:m1315. doi: 10.1136/bmj.m1315
63. Murphy KL, Bethea JR, Fischer R. Neuropathic pain in multiple sclerosis—current therapeutic intervention and future treatment perspectives. In: Zagon IS, McLaughlin PJ, editors. Multiple Sclerosis: Perspectives in Treatment and Pathogenesis. Brisbane, QLD: Codon Publications Copyright: The Authors (2017). p. 141.
64. Cristino L, Bisogno T, Di Marzo V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat Rev Neurol. (2020) 16:9–29. doi: 10.1038/s41582-019-0284-z
65. Abboud H, Hill E, Siddiqui J, Serra A, Walter B. Neuromodulation in multiple sclerosis. Mult Scler. (2017) 23:1663–76. doi: 10.1177/1352458517736150
66. Knotkova H, Hamani C, Sivanesan E, Le Beuffe MFE, Moon JY, Cohen SP, et al. Neuromodulation for chronic pain. Lancet. (2021) 397:2111–24. doi: 10.1016/S0140-6736(21)00794-7
67. Young J, Zoghi M, Khan F, Galea MP. The effect of transcranial direct current stimulation on chronic neuropathic pain in patients with multiple sclerosis: randomized controlled trial. Pain Medicine. (2020) 21:3451–7. doi: 10.1093/pm/pnaa128
68. Klein J, Siepmann T, Schackert G, Ziemssen T, Juratli TA. Peripheral nerve field stimulation in medically refractory trigeminal neuralgia attributed to multiple sclerosis. J Neurosurg. (2020) 134:1244–50. doi: 10.3171/2019.12.JNS192261
69. Berra E, Bergamaschi R, De Icco R, Dagna C, Perrotta A, Rovaris M, et al. The effects of transcutaneous spinal direct current stimulation on neuropathic pain in multiple sclerosis: clinical and neurophysiological assessment. Front Hum Neurosci. (2019) 13:31. doi: 10.3389/fnhum.2019.00031
70. Aboud T, Schuster NM. Pain management in multiple sclerosis: a review of available treatment options. Curr Treat Options Neurol. (2019) 21:62. doi: 10.1007/s11940-019-0601-2
71. Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. (2006) 60:12–21. doi: 10.1002/ana.20913
72. Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. (2006) 129(Pt 8):1953–71. doi: 10.1093/brain/awl075
73. Hart BAT, Laman JD, Bauer J, Blezer E, van Kooyk Y, Hintzen RQ. Modelling of multiple sclerosis: lessons learned in a non-human primate. Lancet Neurol. (2004) 3:588–97. doi: 10.1016/S1474-4422(04)00879-8
74. Steinman L, Zamvil SS. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. (2005) 26:565–71. doi: 10.1016/j.it.2005.08.014
75. Voorthuis JAC, Uitdehaag BMJ, Groot CJA, Goede PH, Meide PHVD, Dijkstra CD. Suppression of experimental allergic encephalomyelitis by intraventricular administration of interferon-gamma in Lewis rats. Clin Exp Immunol. (1990) 81:183–8. doi: 10.1111/j.1365-2249.1990.tb03315.x
76. Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Extended use of glatiramer acetate (Copaxone) is well tolerated and maintains its clinical effect on multiple sclerosis relapse rate and degree of disability. Copolymer 1 multiple sclerosis study group. Neurology. (1998) 50:701–8. doi: 10.1212/WNL.50.3.701
77. Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. (2003) 197:1073–81. doi: 10.1084/jem.20021603
78. Friese MA, Montalban X, Willcox N, Bell JI, Martin R, Fugger L. The value of animal models for drug development in multiple sclerosis. Brain. (2006) 129(Pt 8):1940–52. doi: 10.1093/brain/awl083
79. Rangachari M, Kuchroo VK. Using EAE to better understand principles of immune function and autoimmune pathology. J Autoimmun. (2013) 45:31–9. doi: 10.1016/j.jaut.2013.06.008
80. Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. (2011) 164:1079–106. doi: 10.1111/j.1476-5381.2011.01302.x
81. Burrows DJ, McGown A, Jain SA, De Felice M, Ramesh TM, Sharrack B, et al. Animal models of multiple sclerosis: from rodents to zebrafish. Mult Scler. (2019) 25:306–24. doi: 10.1177/1352458518805246
82. Procaccini C, De Rosa V, Pucino V, Formisano L, Matarese G. Animal models of Multiple Sclerosis. Eur J Pharmacol. (2015) 759:182–91. doi: 10.1016/j.ejphar.2015.03.042
83. Lebar R, Lubetzki C, Vincent C, Lombrail P, Boutry JM. The M2 autoantigen of central nervous system myelin, a glycoprotein present in oligodendrocyte membrane. Clin Exp Immunol. (1986) 66:423–34.
84. Zamvil SS, Nelson PA, Mitchell DJ, Knobler RL, Fritz RB, Steinman L. Encephalitogenic T cell clones specific for myelin basic protein. An unusual bias in antigen recognition. J Exp Med. (1985) 162:2107–24. doi: 10.1084/jem.162.6.2107
85. Yokote H, Miyake S, Croxford JL, Oki S, Mizusawa H, Yamamura T, et al. Cell-dependent amelioration of a mouse model of multiple sclerosis by altering gut flora. Am J Pathol. (2008) 173:1714–23. doi: 10.2353/ajpath.2008.080622
86. Ben-Nun A, Kaushansky N, Kawakami N, Krishnamoorthy G, Berer K, Liblau R, et al. From classic to spontaneous and humanized models of multiple sclerosis: impact on understanding pathogenesis and drug development. J Autoimmun. (2014) 54:33–50. doi: 10.1016/j.jaut.2014.06.004
87. Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod'homme T, Varrin-Doyer M, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. (2013) 210:2921–37. doi: 10.1084/jem.20130699
88. Krishnamoorthy G, Lassmann H, Wekerle H, Holz A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest. (2006) 116:2385–92. doi: 10.1172/JCI28330
89. Faber H, Kurtoic D, Krishnamoorthy G, Weber P, Pütz B, Müller-Myhsok B, et al. Gene expression in spontaneous experimental autoimmune encephalomyelitis is linked to human multiple sclerosis risk genes. Front Immunol. (2020) 11:2165. doi: 10.3389/fimmu.2020.02165
90. Pöllinger B, Krishnamoorthy G, Berer K, Lassmann H, Bösl MR, Dunn R, et al. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med. (2009) 206:1303–16. doi: 10.1084/jem.20090299
91. Bjartmar C, Kidd G, Mörk S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. (2000) 48:893–901.
92. Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev. (2012) 248:205–15. doi: 10.1111/j.1600-065X.2012.01126.x
93. Takeuchi C, Yamagata K, Takemiya T. Variation in experimental autoimmune encephalomyelitis scores in a mouse model of multiple sclerosis. World J Neurol. (2013) 3:56–61. doi: 10.5316/wjn.v3.i3.56
94. Acharjee S, Nayani N, Tsutsui M, Hill MN, Ousman SS, Pittman QJ. Altered cognitive-emotional behavior in early experimental autoimmune encephalitis–cytokine and hormonal correlates. Brain Behav Immun. (2013) 33:164–72. doi: 10.1016/j.bbi.2013.07.003
95. Dutra RC, Moreira ELG, Alberti TB, Marcon R, Prediger RD, Calixto JB. Spatial reference memory deficits precede motor dysfunction in an experimental autoimmune encephalomyelitis model: The role of kallikrein–kinin system. Brain Behav Immun. (2013) 33:90–101. doi: 10.1016/j.bbi.2013.06.002
96. Riccitelli G, Rocca MA, Pagani E, Rodegher ME, Rossi P, Falini A, et al. Cognitive impairment in multiple sclerosis is associated to different patterns of gray matter atrophy according to clinical phenotype. Hum Brain Mapp. (2011) 32:1535–43. doi: 10.1002/hbm.21125
97. Migliore S, Ghazaryan A, Simonelli I, Pasqualetti P, Squitieri F, Curcio G, et al. Cognitive impairment in relapsing-remitting multiple sclerosis patients with very mild clinical disability. Behav Neurol. (2017) 2017:7404289. doi: 10.1155/2017/7404289
98. Olechowski JC, Truong JJ, Kerr JB. Neuropathic pain behaviours in a chronic-relapsing model of experimental autoimmune encephalomyelitis (EAE). Pain. (2009) 141:156–64. doi: 10.1016/j.pain.2008.11.002
99. Olechowski JC, Parmar JA, Miller JB, Stephan JJ, Tenorio JG, Tran JK, et al. A diminished response to formalin stimulation reveals a role for the glutamate transporters in the altered pain sensitivity of mice with experimental autoimmune encephalomyelitis (EAE). Pain. (2010) 149:565–72. doi: 10.1016/j.pain.2010.03.037
100. Lisi L, Navarra P, Cirocchi R, Sharp A, Stigliano E, Feinstein DL, et al. Rapamycin reduces clinical signs and neuropathic pain in a chronic model of experimental autoimmune encephalomyelitis. J Neuroimmunol. (2012) 243:43–51. doi: 10.1016/j.jneuroim.2011.12.018
101. Olechowski CJ, Tenorio G, Sauve Y, Kerr BJ. Changes in nociceptive sensitivity and object recognition in experimental autoimmune encephalomyelitis (EAE). Exp Neurol. (2013) 241:113–21. doi: 10.1016/j.expneurol.2012.12.012
102. Damasceno A, Damasceno B, Cendes F. Brain cortical lesion load is related to cognitive dysfunction in multiple sclerosis patients. Neurology. (2012) 78. doi: 10.1212/WNL.78.1_MeetingAbstracts.P03.079
103. Lu J, Kurejova M, Wirotanseng LN, Linker RA, Kuner R, Tappe-Theodor A. Pain in experimental autoimmune encephalitis: a comparative study between different mouse models. J Neuroinflammation. (2012) 9:233. doi: 10.1186/1742-2094-9-233
104. Segal JP, Bannerman CA, Silva JR, Haird CM, Baharnoori M, Gilron I, et al. Chronic mechanical hypersensitivity in experimental autoimmune encephalomyelitis is regulated by disease severity and neuroinflammation. Brain Behav Immun. (2020) 89:314–25. doi: 10.1016/j.bbi.2020.07.010
105. Potter LE, Paylor JW, Suh JS, Tenorio G, Caliaperumal J, Colbourne F, et al. Altered excitatory-inhibitory balance within somatosensory cortex is associated with enhanced plasticity and pain sensitivity in a mouse model of multiple sclerosis. J Neuroinflammation. (2016) 13:142. doi: 10.1186/s12974-016-0609-4
106. Duffy SS, Perera CJ, Makker PGS, Lees JG, Carrive P, Moalem-Taylor G. Peripheral and central neuroinflammatory changes and pain behaviors in an animal model of multiple sclerosis. Front Immunol. (2016) 7:369. doi: 10.3389/fimmu.2016.00369
107. Gao F, Yin X, Edden RAE, Evans AC, Xu J, Cao G, et al. Altered hippocampal GABA and glutamate levels and uncoupling from functional connectivity in multiple sclerosis. Hippocampus. (2018) 28:813–23. doi: 10.1002/hipo.23001
108. Falco A, Pennucci R, Brambilla E, de Curtis I. Reduction in parvalbumin-positive interneurons and inhibitory input in the cortex of mice with experimental autoimmune encephalomyelitis. Exp Brain Res. (2014) 232:2439–49. doi: 10.1007/s00221-014-3944-7
109. Duarte-Silva E, Macedo D, Maes M, Peixoto CA. Novel insights into the mechanisms underlying depression-associated experimental autoimmune encephalomyelitis. Prog Neuropsychopharmacol Biol Psychiatry. (2019) 93:1–10. doi: 10.1016/j.pnpbp.2019.03.001
110. Peres DS, Theisen MC, Fialho MFP, Dalenogare DP, Rodrigues P, Kudsi SQ, et al. TRPA1 involvement in depression- and anxiety-like behaviors in a progressive multiple sclerosis model in mice. Brain Res Bull. (2021) 175:1–15. doi: 10.1016/j.brainresbull.2021.07.011
111. Haji N, Mandolesi G, Gentile A, Sacchetti L, Fresegna D, Rossi S, et al. TNF-α-mediated anxiety in a mouse model of multiple sclerosis. Exp Neurol. (2012) 237:296–303. doi: 10.1016/j.expneurol.2012.07.010
112. Peruga I, Hartwig S, Thöne J, Hovemann B, Gold R, Juckel G, et al. Inflammation modulates anxiety in an animal model of multiple sclerosis. Behav Brain Res. (2011) 220:20–9. doi: 10.1016/j.bbr.2011.01.018
113. Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KHG. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. (2010) 162:1–11. doi: 10.1111/j.1365-2249.2010.04143.x
114. Ortiz GG, Pacheco-Moisés FP, Bitzer-Quintero OK, Ramírez-Anguiano AC, Flores-Alvarado LJ, Ramírez-Ramírez V, et al. Immunology and oxidative stress in multiple sclerosis: clinical and basic approach. Clin Dev Immunol. (2013) 2013:708659. doi: 10.1155/2013/708659
115. Chen X, Guo C, Kong J. Oxidative stress in neurodegenerative diseases. Neural Regen Res. (2012) 7:376–85. doi: 10.3969/j.issn.1673-5374.2012.05.009
116. Witherick J, Wilkins A, Scolding N, Kemp K. Mechanisms of oxidative damage in multiple sclerosis and a cell therapy approach to treatment. Autoimmune Dis. (2010) 2011:164608. doi: 10.4061/2011/164608
117. Lassmann H, van Horssen J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim Biophys Acta. (2016) 1862:506–10. doi: 10.1016/j.bbadis.2015.09.018
118. Haider L, Fischer MT, Frischer JM, Bauer J, Höftberger R, Botond G, et al. Oxidative damage in multiple sclerosis lesions. Brain. (2011) 134(Pt 7):1914–24. doi: 10.1093/brain/awr128
119. Spaas J, van Veggel L, Schepers M, Tiane A, van Horssen J, Wilson DM, et al. Oxidative stress and impaired oligodendrocyte precursor cell differentiation in neurological disorders. Cell Mol Life Sci. (2021) 78:4615–37. doi: 10.1007/s00018-021-03802-0
120. Meyer N, Rinholm JE. Mitochondria in myelinating oligodendrocytes: slow and out of breath? Metabolites. (2021) 11:359. doi: 10.3390/metabo11060359
121. Rinholm JE, Vervaeke K, Tadross MR, Tkachuk AN, Kopek BG, Brown TA, et al. Movement and structure of mitochondria in oligodendrocytes and their myelin sheaths. Glia. (2016) 64:810–25. doi: 10.1002/glia.22965
122. Adamczyk B, Adamczyk-Sowa M. New insights into the role of oxidative stress mechanisms in the pathophysiology and treatment of multiple sclerosis. Oxid Med Cell Longev. (2016) 2016:1973834. doi: 10.1155/2016/1973834
123. Ohl K, Tenbrock K, Kipp M. Oxidative stress in multiple sclerosis: central and peripheral mode of action. Exp Neurol. (2016) 277:58–67. doi: 10.1016/j.expneurol.2015.11.010
124. Zia S, Rawji KS, Michaels NJ, Burr M, Kerr BJ, Healy LM, et al. Microglia diversity in health and multiple sclerosis. Front Immunol. (2020) 11:588021. doi: 10.3389/fimmu.2020.588021
125. Robinson RR, Dietz AK, Maroof AM, Asmis R, Forsthuber TG. The role of glial–neuronal metabolic cooperation in modulating progression of multiple sclerosis and neuropathic pain. Immunotherapy. (2019) 11:129–47. doi: 10.2217/imt-2018-0153
126. Zhu W, Acosta C, MacNeil B, Cortes C, Intrater H, Gong Y, et al. Elevated expression of fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) in the dorsal root ganglia and spinal cord in experimental autoimmune encephalomyelitis: implications in multiple sclerosis-induced neuropathic pain. Biomed Res Int. (2013) 2013:480702. doi: 10.1155/2013/480702
127. Tsuda M, Inoue K. Neuron–microglia interaction by purinergic signaling in neuropathic pain following neurodegeneration. Neuropharmacology. (2016) 104:76–81. doi: 10.1016/j.neuropharm.2015.08.042
128. Di Virgilio F, Sarti AC. Microglia P2X4 receptors as pharmacological targets for demyelinating diseases. EMBO Mol Med. (2018) 10:e9369. doi: 10.15252/emmm.201809369
129. Pegoretti V, Swanson KA, Bethea JR, Probert L, Eisel ULM, Fischer R. Inflammation and oxidative stress in multiple sclerosis: consequences for therapy development. Oxid Med Cell Longev. (2020) 2020:7191080. doi: 10.1155/2020/7191080
130. Serizawa K, Tomizawa-Shinohara H, Magi M, Yogo K, Matsumoto Y. Anti-IL-6 receptor antibody improves pain symptoms in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. (2018) 319:71–9. doi: 10.1016/j.jneuroim.2018.03.017
131. Zhang JM, An J. Cytokines, inflammation, and pain. Int Anesthesiol Clin. (2007) 45:27–37. doi: 10.1097/AIA.0b013e318034194e
132. Sarchielli P, Greco L, Floridi A, Floridi A, Gallai V. Excitatory amino acids and multiple sclerosis: evidence from cerebrospinal fluid. Arch Neurol. (2003) 60:1082–8. doi: 10.1001/archneur.60.8.1082
133. Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain. (2005) 128(Pt 5):1016–25. doi: 10.1093/brain/awh467
134. Al Gawwam G, Sharquie IK. Serum glutamate is a predictor for the diagnosis of multiple sclerosis. Sci World J. (2017) 2017:9320802. doi: 10.1155/2017/9320802
135. Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. (2000) 6:67–70. doi: 10.1038/71555
136. Rodriguez-Chavez V, Moran J, Molina-Salinas G, Zepeda Ruiz WA, Rodriguez MC, Picazo O, et al. Participation of glutamatergic ionotropic receptors in excitotoxicity: the neuroprotective role of prolactin. Neuroscience. (2021) 461:180–93. doi: 10.1016/j.neuroscience.2021.02.027
137. Spampinato SF, Copani A, Nicoletti F, Sortino MA, Caraci F. Metabotropic glutamate receptors in glial cells: a new potential target for neuroprotection? Front Mol Neurosci. (2018) 11:414. doi: 10.3389/fnmol.2018.00414
138. Pereira V, Goudet C. Emerging trends in pain modulation by metabotropic glutamate receptors. Front Mol Neurosci. (2019) 11:464. doi: 10.3389/fnmol.2018.00464
139. Ohgoh M, Hanada T, Smith T, Hashimoto T, Ueno M, Yamanishi Y, et al. Altered expression of glutamate transporters in experimental autoimmune encephalomyelitis. J Neuroimmunol. (2002) 125:170–8. doi: 10.1016/S0165-5728(02)00029-2
140. Pitt D, Nagelmeier IE, Wilson HC, Raine CS. Glutamate uptake by oligodendrocytes: implications for excitotoxicity in multiple sclerosis. Neurology. (2003) 61:1113–20. doi: 10.1212/01.WNL.0000090564.88719.37
141. Vallejo-Illarramendi A, Domercq M, Pérez-Cerdá F, Ravid R, Matute C. Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol Dis. (2006) 21:154–64. doi: 10.1016/j.nbd.2005.06.017
142. Vercellino M, Merola A, Piacentino C, Votta B, Capello E, Mancardi GL, et al. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol. (2007) 66:732–9. doi: 10.1097/nen.0b013e31812571b0
143. Werner P, Pitt D, Raine CS. Multiple sclerosis: Altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. (2001) 50:169–80. doi: 10.1002/ana.1077
144. Grasselli G, Rossi S, Musella A, Gentile A, Loizzo S, Muzio L, et al. Abnormal NMDA receptor function exacerbates experimental autoimmune encephalomyelitis. Br J Pharmacol. (2013) 168:502–17. doi: 10.1111/j.1476-5381.2012.02178.x
145. Macrez R, Ortega MC, Bardou I, Mehra A, Fournier A, Van der Pol SMA, et al. Neuroendothelial NMDA receptors as therapeutic targets in experimental autoimmune encephalomyelitis. Brain. (2016) 139:2406–19. doi: 10.1093/brain/aww172
146. Evonuk KS, Moseley CE, Doyle RE, Weaver CT, Desilva TM. Determining immune system suppression versus CNS protection for pharmacological interventions in autoimmune demyelination. J Vis Exp. (2016) 54348. doi: 10.3791/54348
147. Sühs KW, Fairless R, Williams SK, Heine K, Cavalié A, Diem R. N-Methyl-d-aspartate receptor blockade is neuroprotective in experimental autoimmune optic neuritis. J Neuropathol Exp Neurol. (2014) 73:507–18. doi: 10.1097/NEN.0000000000000073
148. Kurnellas M, Li H, Jain M, Giraud S, Nicot A, Ratnayake A, et al. Reduced expression of plasma membrane calcium ATPase 2 and collapsin response mediator protein 1 promotes death of spinal cord neurons. Cell Death Differ. (2010) 17:1501–10. doi: 10.1038/cdd.2010.54
149. Kurian R, Raza K, Shanthanna H. A systematic review and meta-analysis of memantine for the prevention or treatment of chronic pain. Eur J Pain. (2019) 23:1234–50. doi: 10.1002/ejp.1393
150. Alomar SY, Gheit REAE, Enan ET, El-Bayoumi KS, Shoaeir MZ, Elkazaz AY, et al. Novel mechanism for memantine in attenuating diabetic neuropathic pain in mice via downregulating the spinal HMGB1/TRL4/NF-kB inflammatory axis. Pharmaceuticals. (2021) 14:307. doi: 10.3390/ph14040307
151. Aiyer R, Mehta N, Gungor S, Gulati A. A systematic review of NMDA receptor antagonists for treatment of neuropathic pain in clinical practice. Clin J Pain. (2018) 34:450–67. doi: 10.1097/AJP.0000000000000547
152. Tai WL, Sun L, Li H, Gu P, Joosten EA, Cheung CW. Additive effects of environmental enrichment and ketamine on neuropathic pain relief by reducing glutamatergic activation in spinal cord injury in rats. Front Neurosci. (2021) 15: 635187. doi: 10.3389/fnins.2021.635187
153. Velzen Mv, Dahan JDC, van Dorp ELA, Mogil JS, Hooijmans CR, Dahan A. Efficacy of ketamine in relieving neuropathic pain: a systematic review and meta-analysis of animal studies. Pain. (2021) 162:2320–30. doi: 10.1097/j.pain.0000000000002231
154. Tsantoulas C, McMahon SB. Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci. (2014) 37:146–58. doi: 10.1016/j.tins.2013.12.002
155. Devor M. Sodium channels and mechanisms of neuropathic pain. J Pain. (2006) 7(1 Suppl. 1):S3–12. doi: 10.1016/j.jpain.2005.09.006
156. Bittner S, Meuth SG. Targeting ion channels for the treatment of autoimmune neuroinflammation. Ther Adv Neurol Disord. (2013) 6:322–36. doi: 10.1177/1756285613487782
157. Ho C, O'Leary ME. Single-cell analysis of sodium channel expression in dorsal root ganglion neurons. Mol Cell Neurosci. (2011) 46:159–66. doi: 10.1016/j.mcn.2010.08.017
158. Park J, Luo ZD. Calcium channel functions in pain processing. Channels. (2010) 4:510–7. doi: 10.4161/chan.4.6.12869
159. Dolphin AC. Functions of presynaptic voltage-gated calcium channels. Function. (2021) 2:zqaa027. doi: 10.1093/function/zqaa027
160. Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The role of voltage-gated sodium channels in pain signaling. Physiol Rev. (2019) 99:1079–151. doi: 10.1152/physrev.00052.2017
161. Ma RSY, Kayani K, Whyte-Oshodi D, Whyte-Oshodi A, Nachiappan N, Gnanarajah S, et al. Voltage gated sodium channels as therapeutic targets for chronic pain. J Pain Res. (2019) 12:2709–22. doi: 10.2147/JPR.S207610
162. Youn DH, Gerber G, Sather WA. Ionotropic glutamate receptors and voltage-gated Ca2? channels in long-term potentiation of spinal dorsal horn synapses and pain hypersensitivity. Neural Plast. (2013) 2013:654257. doi: 10.1155/2013/654257
163. Dolphin AC, Lee A. Presynaptic calcium channels: specialized control of synaptic neurotransmitter release. Nat Rev Neurosci. (2020) 21:213–29. doi: 10.1038/s41583-020-0278-2
164. Winquist RJ, Pan JQ, Gribkoff VK. Use-dependent blockade of Cav2. 2 voltage-gated calcium channels for neuropathic pain. Biochem Pharmacol. (2005) 70:489–99. doi: 10.1016/j.bcp.2005.04.035
165. Patel R, Montagut-Bordas C, Dickenson AH. Calcium channel modulation as a target in chronic pain control. Br J Pharmacol. (2018) 175:2173–84. doi: 10.1111/bph.13789
166. Adams DJ, Berecki G. Mechanisms of conotoxin inhibition of N-type (Ca(v)2. 2) calcium channels. Biochim Biophys Acta. (2013) 1828:1619–28. doi: 10.1016/j.bbamem.2013.01.019
167. Pitake S, Middleton LJ, Abdus-Saboor I, Mishra SK. Inflammation induced sensory nerve growth and pain hypersensitivity requires the N-type calcium channel Cav2.2. Front Neurosci. (2019) 13:1009. doi: 10.3389/fnins.2019.01009
168. Saegusa H, Kurihara T, Zong S, Kazuno A, Matsuda Y, Nonaka T, et al. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. EMBO J. (2001) 20:2349–56. doi: 10.1093/emboj/20.10.2349
169. Baddack U, Frahm S, Antolin-Fontes B, Grobe J, Lipp M, Müller G, et al. Suppression of peripheral pain by blockade of voltage-gated calcium 2. 2 Channels in nociceptors induces RANKL and impairs recovery from inflammatory arthritis in a mouse model. Arthritis Rheumatol. (2015) 67:1657–67. doi: 10.1002/art.39094
170. Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat Med. (2011) 17:822–9. doi: 10.1038/nm.2345
171. Cizkova D, Marsala J, Lukacova N, Marsala M, Jergova S, Orendacova J, et al. Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Exp Brain Res. (2002) 147:456–63. doi: 10.1007/s00221-002-1217-3
172. Yang J, Xie MX, Hu L, Wang XF, Mai JZ, Li YY, et al. Upregulation of N-type calcium channels in the soma of uninjured dorsal root ganglion neurons contributes to neuropathic pain by increasing neuronal excitability following peripheral nerve injury. Brain Behav Immun. (2018) 71:52–65. doi: 10.1016/j.bbi.2018.04.016
173. Li Y, Dorsi MJ, Meyer RA, Belzberg AJ. Mechanical hyperalgesia after an L5 spinal nerve lesion in the rat is not dependent on input from injured nerve fibers. Pain. (2000) 85:493–502. doi: 10.1016/S0304-3959(00)00250-5
174. Stevens EB, Stephens GJ. Recent advances in targeting ion channels to treat chronic pain. Br J Pharmacol. (2018) 175:2133–7. doi: 10.1111/bph.14215
175. Gerevich Z, Borvendeg SJ, Schröder W, Franke H, Wirkner K, Nörenberg W, et al. Inhibition of N-type voltage-activated calcium channels in rat dorsal root ganglion neurons by P2Y receptors is a possible mechanism of ADP-induced analgesia. J Neurosci. (2004) 24:797–807. doi: 10.1523/JNEUROSCI.4019-03.2004
176. Chen J, Li L, Chen SR, Chen H, Xie JD, Sirrieh RE, et al. The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep. (2018) 22:2307–21. doi: 10.1016/j.celrep.2018.02.021
177. Yen L, Bennett G. Ribeiro-da-Silva A. Sympathetic sprouting and changes in nociceptive sensory innervation in the glabrous skin of the rat hind paw following partial peripheral nerve injury. J Comp Neurol. (2006) 495:679–90. doi: 10.1002/cne.20899
178. Park SH, Eber MR, Fonseca MM, Patel CM, Cunnane KA, Ding H, et al. Usefulness of the measurement of neurite outgrowth of primary sensory neurons to study cancer-related painful complications. Biochem Pharmacol. (2021) 188:114520. doi: 10.1016/j.bcp.2021.114520
179. Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, et al. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol. (2000) 157:267–76. doi: 10.1016/S0002-9440(10)64537-3
180. Enders M, Heider T, Ludwig A, Kuerten S. Strategies for neuroprotection in multiple sclerosis and the role of calcium. Int J Mol Sci. (2020) 21:1663. doi: 10.3390/ijms21051663
181. Kornek B, Storch MK, Bauer J, Djamshidian A, Weissert R, Wallstroem E, et al. Distribution of a calcium channel subunit in dystrophic axons in multiple sclerosis and experimental autoimmune encephalomyelitis. Brain. (2001) 124(Pt 6):1114–24. doi: 10.1093/brain/124.6.1114
182. Silva RBM, Greggio S, Venturin GT, da Costa JC, Gomez MV, Campos MM. Beneficial effects of the calcium channel blocker CTK 01512-2 in a mouse model of multiple sclerosis. Mol Neurobiol. (2018) 55:9307–27. doi: 10.1007/s12035-018-1049-1
183. Tokuhara N, Namiki K, Uesugi M, Miyamoto C, Ohgoh M, Ido K, et al. N-type calcium channel in the pathogenesis of experimental autoimmune encephalomyelitis. J Biol Chem. (2010) 285:33294–306. doi: 10.1074/jbc.M109.089805
184. Hundehege P, Fernandez-Orth J, Römer P, Ruck T, Müntefering T, Eichler S, et al. Targeting voltage-dependent calcium channels with pregabalin exerts a direct neuroprotective effect in an animal model of multiple sclerosis. Neurosignals. (2018) 26:77–93. doi: 10.1159/000495425
185. Alrashdi B, Dawod B, Schampel A, Tacke S, Kuerten S, Marshall JS, et al. Nav1. 6 promotes inflammation and neuronal degeneration in a mouse model of multiple sclerosis. J Neuroinflammation. (2019) 16:215. doi: 10.1186/s12974-019-1622-1
186. Stevens M, Timmermans S, Bottelbergs A, Hendriks JJ, Brône B, Baes M, et al. Block of a subset of sodium channels exacerbates experimental autoimmune encephalomyelitis. J Neuroimmunol. (2013) 261:21–8. doi: 10.1016/j.jneuroim.2013.04.012
187. Giacoppo S, Iori R, Bramanti P, Mazzon E. Topical moringin-cream relieves neuropathic pain by suppression of inflammatory pathway and voltage-gated ion channels in murine model of multiple sclerosis. Mol Pain. (2017) 13:1744806917724318. doi: 10.1177/1744806917724318
188. Shields S, Cheng X, Gasser A, Saab C, Tyrrell L, Eastman E, et al. A channelopathy contributes to cerebellar dysfunction in a model of multiple sclerosis. Ann Neurol. (2012) 71:186–94. doi: 10.1002/ana.22665
189. Xie W, Strong JA, Zhang JM. Local knockdown of the NaV1.6 sodium channel reduces pain behaviors, sensory neuron excitability, and sympathetic sprouting in rat models of neuropathic pain. Neuroscience. (2015) 291:317–30. doi: 10.1016/j.neuroscience.2015.02.010
190. Chen L, Huang J, Zhao P, Persson AK, Dib-Hajj FB, Cheng X, et al. Conditional knockout of Na(V)1.6 in adult mice ameliorates neuropathic pain. Sci Rep. (2018) 8:3845. doi: 10.1038/s41598-018-22216-w
191. Ren YS, Qian NS, Tang Y, Liao YH, Yang YL, Dou KF, et al. Sodium channel Nav1.6 is up-regulated in the dorsal root ganglia in a mouse model of type 2 diabetes. Brain Res Bull. (2012) 87:244–9. doi: 10.1016/j.brainresbull.2011.10.015
192. Tanaka BS, Zhao P, Dib-Hajj FB, Morisset V, Tate S, Waxman SG, et al. A gain-of-function mutation in Nav1.6 in a case of trigeminal neuralgia. Mol Med. (2016) 22:338–48. doi: 10.2119/molmed.2016.00131
193. Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, et al. Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain. (2012) 153:2017–30. doi: 10.1016/j.pain.2012.04.022
194. Yousuf MS, Noh MC, Friedman TN, Zubkow K, Johnson JC, Tenorio G, et al. Sensory neurons of the dorsal root ganglia become hyperexcitable in a T-cell-mediated MOG-EAE model of multiple sclerosis. eNeuro. (2019) 6:ENEURO.0024-19.2019. doi: 10.1523/ENEURO.0024-19.2019
195. Yousuf MS, Samtleben S, Lamothe SM, Friedman TN, Catuneanu A, Thorburn K, et al. Endoplasmic reticulum stress in the dorsal root ganglia regulates large-conductance potassium channels and contributes to pain in a model of multiple sclerosis. FASEB J. (2020) 34:12577–98. doi: 10.1096/fj.202001163R
196. Contreras GF, Neely A, Alvarez O, Gonzalez C, Latorre R. Modulation of BK channel voltage gating by different auxiliary β subunits. Proc Natl Acad Sci U S A. (2012) 109:18991–6. doi: 10.1073/pnas.1216953109
197. Ge Y, Jiao Y, Li P, Xiang Z, Li Z, Wang L, et al. Coregulation of endoplasmic reticulum stress and oxidative stress in neuropathic pain and disinhibition of the spinal nociceptive circuitry. Pain. (2018) 159:894–906. doi: 10.1097/j.pain.0000000000001161
198. Liu Y, Wang S, Wang Z, Ding M, Li X, Guo J, et al. Dexmedetomidine alleviated endoplasmic reticulum stress via inducing ER-phagy in the spinal cord of neuropathic pain model. Front Neurosci. (2020) 14:90. doi: 10.3389/fnins.2020.00090
199. Zhang E, Yi MH, Shin N, Baek H, Kim S, Kim E, et al. Endoplasmic reticulum stress impairment in the spinal dorsal horn of a neuropathic pain model. Sci Rep. (2015) 5:11555. doi: 10.1038/srep11555
200. Bittar A, Jun J, La JH, Wang J, Leem JW, Chung JM. Reactive oxygen species affect spinal cell type-specific synaptic plasticity in a model of neuropathic pain. Pain. (2017) 158:2137–46. doi: 10.1097/j.pain.0000000000001014
201. Liu D, Zhou Y, Peng Y, Su P, Li Z, Xu Q, et al. Endoplasmic reticulum stress in spinal cord contributes to the development of morphine tolerance. Front Mol Neurosci. (2018) 11:72. doi: 10.3389/fnmol.2018.00072
202. Sanchez CL, Sims SG, Nowery JD, Meares GP. Endoplasmic reticulum stress differentially modulates the IL-6 family of cytokines in murine astrocytes and macrophages. Sci Rep. (2019) 9:14931. doi: 10.1038/s41598-019-51481-6
203. Rodrigues D, Leles B, Costa V, Miranda A, Cisalpino D, Gomes D, et al. IL-1β is involved with the generation of pain in experimental autoimmune encephalomyelitis. Mol Neurobiol. (2016) 53:6540–7. doi: 10.1007/s12035-015-9552-0
204. Chopra S, Giovanelli P, Alvarado-Vazquez PA, Alonso S, Song M, Sandoval TA, et al. IRE1α-XBP1 signaling in leukocytes controls prostaglandin biosynthesis and pain. Science. (2019) 365:eaau6499. doi: 10.1126/science.aau6499
205. Blaustein MP, Juhaszova M, Golovina VA, Church P, Stanley E. Na/Ca exchanger and PMCA localization in neurons and astrocytes. Funct Implications. (2002) 976:356–66. doi: 10.1111/j.1749-6632.2002.tb04762.x
206. Lerdkrai C, Asavapanumas N, Brawek B, Kovalchuk Y, Mojtahedi N, Olmedillas Del Moral M, et al. Intracellular Ca(2+) stores control in vivo neuronal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. (2018) 115:1279–88. doi: 10.1073/pnas.1714409115
207. Mirabelli E, Ni L, Li L, Acioglu C, Heary RF, Elkabes S. Pathological pain processing in mouse models of multiple sclerosis and spinal cord injury: contribution of plasma membrane calcium ATPase 2 (PMCA2). J Neuroinflammation. (2019) 16:207. doi: 10.1186/s12974-019-1585-2
208. Nicot A, Kurnellas M, Elkabes S. Temporal pattern of plasma membrane calcium ATPase 2 expression in the spinal cord correlates with the course of clinical symptoms in two rodent models of autoimmune encephalomyelitis. Eur J Neurosci. (2005) 21:2660–70. doi: 10.1111/j.1460-9568.2005.04086.x
209. Nicot A, Ratnakar PV, Ron Y, Chen C, Elkabes S. Regulation of gene expression in experimental autoimmune encephalomyelitis indicates early neuronal dysfunction. Brain. (2003) 126:398–412. doi: 10.1093/brain/awg041
210. Mukherjee A, Katiyar R, Dembla E, Dembla M, Kumar P, Belkacemi A, et al. Disturbed presynaptic Ca2+ signaling in photoreceptors in the EAE mouse model of multiple sclerosis. iScience. (2020) 23:101830. doi: 10.1016/j.isci.2020.101830
211. Khariv V, Ni L, Ratnayake A, Sampath S, Lutz BM, Tao XX, et al. Impaired sensitivity to pain stimuli in plasma membrane calcium ATPase 2 (PMCA2) heterozygous mice: a possible modality- and sex-specific role for PMCA2 in nociception. FASEB J. (2017) 31:224–37. doi: 10.1096/fj.201600541r
212. Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. (2003) 23:2899–910. doi: 10.1523/JNEUROSCI.23-07-02899.2003
213. Osikowicz M, Mika J, Przewlocka B. The glutamatergic system as a target for neuropathic pain relief. Exp Physiol. (2013) 98:372–84. doi: 10.1113/expphysiol.2012.069922
214. Huang Y, Wen LL, Xie JD, Ouyang HD, Chen DT, Zeng WA. Antinociceptive effectiveness of the inhibition of NCX reverse-mode action in rodent neuropathic pain model. Mol Pain. (2019) 15:1744806919864511. doi: 10.1177/1744806919864511
215. Kuroda H, Sobhan U, Sato M, Tsumura M, Ichinohe T, Tazaki M, et al. Sodium-calcium exchangers in rat trigeminal ganglion neurons. Mol Pain. (2013) 9:22. doi: 10.1186/1744-8069-9-22
216. Craner MJ, Hains BC, Lo AC, Black JA, Waxman SG. Co-localization of sodium channel Nav1.6 and the sodium–calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain. (2004) 127:294–303. doi: 10.1093/brain/awh032
217. Rush AM, Dib-Hajj SD, Waxman SG. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J Physiol. (2005) 564(Pt 3):803–15. doi: 10.1113/jphysiol.2005.083089
218. Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1. 2 and Nav16 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A. (2004) 101:8168–73. doi: 10.1073/pnas.0402765101
219. Reyes RC, Verkhratsky A, Parpura V. Plasmalemmal Na+/Ca2+ exchanger modulates Ca2+-dependent exocytotic release of glutamate from rat cortical astrocytes. ASN Neuro. (2012) 4:e00075. doi: 10.1042/AN20110059
220. Li T, Chen X, Zhang C, Zhang Y, Yao W. An update on reactive astrocytes in chronic pain. J Neuroinflammation. (2019) 16:140. doi: 10.1186/s12974-019-1524-2
221. Yuan S, Shi Y, Tang SJ. Wnt signaling in the pathogenesis of multiple sclerosis-associated chronic pain. J Neuroimmune Pharmacol. (2012) 7:904–13. doi: 10.1007/s11481-012-9370-3
222. Rosso S, Inestrosa N, Rosso S. WNT signaling in neuronal maturation and synaptogenesis. Front Cell Neurosci. (2013) 7:103. doi: 10.3389/fncel.2013.00103
223. Tang SJ. Synaptic activity-regulated Wnt signaling in synaptic plasticity, glial function and chronic pain. CNS Neurol Disord Drug Targets. (2014) 13:737–44. doi: 10.2174/1871527312666131223114457
224. Dai ZM, Sun S, Wang C, Huang H, Hu X, Zhang Z, et al. Stage-specific regulation of oligodendrocyte development by Wnt/β-catenin signaling. J Neurosci. (2014) 34:8467–73. doi: 10.1523/JNEUROSCI.0311-14.2014
225. Li B, Zhong L, Yang X, Andersson T, Huang M, Tang SJ. WNT5A signaling contributes to Aβ-induced neuroinflammation and neurotoxicity. PLoS ONE. (2011) 6:e22920. doi: 10.1371/journal.pone.0022920
226. Zhang YK, Huang ZJ, Liu S, Liu YP, Song AA, Song XJ, et al. WNT signaling underlies the pathogenesis of neuropathic pain in rodents. J Clin Invest. (2013) 123:2268–86. doi: 10.1172/JCI65364
227. Kohn AD, Moon RT. Wnt and calcium signaling: β-Catenin-independent pathways. Cell Calcium. (2005) 38:439–46. doi: 10.1016/j.ceca.2005.06.022
228. He CW, Liao CP, Pan CL. Wnt signalling in the development of axon, dendrites and synapses. Open Biol. (2018) 8:180116. doi: 10.1098/rsob.180116
229. Simonetti M, Kuner R. Spinal Wnt5a plays a key role in spinal dendritic spine remodeling in neuropathic and inflammatory pain models and in the proalgesic effects of peripheral Wnt3a. J Neurosci. (2020) 40:6664–77. doi: 10.1523/JNEUROSCI.2942-19.2020
230. Cheng W, Chen G. Chemokines and chemokine receptors in multiple sclerosis. Mediators Inflamm. (2014) 2014:659206. doi: 10.1155/2014/659206
231. Mahad DJ, Howell SJ, Woodroofe MN. Expression of chemokines in the CSF and correlation with clinical disease activity in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry. (2002) 72:498–502. doi: 10.1136/jnnp.72.4.498
232. Borjini N, Fernández M, Giardino L, Calzà L. Cytokine and chemokine alterations in tissue, CSF, and plasma in early presymptomatic phase of experimental allergic encephalomyelitis (EAE), in a rat model of multiple sclerosis. J Neuroinflammation. (2016) 13:291. doi: 10.1186/s12974-016-0757-6
233. Staniland AA, Clark AK, Wodarski R, Sasso O, Maione F, D'Acquisto F, et al. Reduced inflammatory and neuropathic pain and decreased spinal microglial response in fractalkine receptor (CX3CR1) knockout mice. J Neurochem. (2010) 114:1143–57. doi: 10.1111/j.1471-4159.2010.06837.x
234. Cardona SM, Kim SV, Church KA, Torres VO, Cleary IA, Mendiola AS, et al. Role of the fractalkine receptor in CNS autoimmune inflammation: new approach utilizing a mouse model expressing the human CX3CR1(I249/M280) variant. Front Cell Neurosci. (2018) 12:365. doi: 10.3389/fncel.2018.00365
235. Milligan E, Zapata V, Schoeniger D, Chacur M, Green P, Poole S, et al. An initial investigation of spinal mechanisms underlying pain enhancement induced by fractalkine, a neuronally released chemokine. Eur J Neurosci. (2005) 22:2775–82. doi: 10.1111/j.1460-9568.2005.04470.x
236. Arima Y, Kamimura D, Atsumi T, Harada M, Kawamoto T, Nishikawa N, et al. A pain-mediated neural signal induces relapse in murine autoimmune encephalomyelitis, a multiple sclerosis model. Elife. (2015) 4:e08733. doi: 10.7554/eLife.08733
237. Aicher SA, Silverman MB, Winkler CW, Bebo BF. Hyperalgesia in an animal model of multiple sclerosis. Pain. (2004) 110:560–70. doi: 10.1016/j.pain.2004.03.025
238. Nociti V. What is the role of brain derived neurotrophic factor in multiple sclerosis neuroinflammation? Neuroimmunol Neuroinflammation. (2020) 7:291–9. doi: 10.20517/2347-8659.2020.25
239. Oraby MI, El Masry HA, Abd El Shafy SS, Abdul Galil EM. Serum level of brain-derived neurotrophic factor in patients with relapsing–remitting multiple sclerosis: a potential biomarker for disease activity. Egypt J Neurol, Psychiatry Neurosurg. (2021) 57:40. doi: 10.1186/s41983-021-00296-2
240. Gao YJ, Ji RR. c-Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? Open Pain J. (2009) 2:11–7. doi: 10.2174/1876386300902010011
241. Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der Meer C, et al. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. (2004) 24:8310–21. doi: 10.1523/JNEUROSCI.2396-04.2004
242. Khan N, Gordon R, Woodruff TM, Smith MT. Antiallodynic effects of alpha lipoic acid in an optimized RR-EAE mouse model of MS-neuropathic pain are accompanied by attenuation of upregulated BDNF-TrkB-ERK signaling in the dorsal horn of the spinal cord. Pharmacol Res Perspect. (2015) 3:e00137. doi: 10.1002/prp2.137
243. Gielen A, Khademi M, Muhallab S, Olsson T, Piehl F. Increased brain-derived neurotrophic factor expression in white blood cells of relapsing-remitting multiple sclerosis patients. Scand J Immunol. (2003) 57:493–7. doi: 10.1046/j.1365-3083.2003.01260.x
244. Zhu W, Frost EE, Begum F, Vora P, Au K, Gong Y, et al. The role of dorsal root ganglia activation and brain-derived neurotrophic factor in multiple sclerosis. J Cell Mol Med. (2012) 16:1856–65. doi: 10.1111/j.1582-4934.2011.01481.x
245. Zhou YQ, Liu Z, Liu ZH, Chen SP, Li M, Shahveranov A, et al. Interleukin-6: an emerging regulator of pathological pain. J Neuroinflammation. (2016) 13:141. doi: 10.1186/s12974-016-0607-6
246. Ren K, Torres R. Role of interleukin-1β during pain and inflammation. Brain Res Rev. (2009) 60:57–64. doi: 10.1016/j.brainresrev.2008.12.020
247. Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci. (2009) 29:14415–22. doi: 10.1523/JNEUROSCI.4569-09.2009
248. Guo W, Wang H, Watanabe M, Shimizu K, Zou S, Lagraize SC, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. (2007) 27:6006–18. doi: 10.1523/JNEUROSCI.0176-07.2007
249. Stuart MA, Pippa JT, Nancy JR. Interleukin-1 and neuronal injury. Nat Rev Immunol. (2005) 5:629–40. doi: 10.1038/nri1664
250. Gajtkó A, Bakk E, Hegedus K, Ducza L, Holló K. IL-1β induced cytokine expression by spinal astrocytes can play a role in the maintenance of chronic inflammatory pain. Front Physiol. (2020) 11:543331. doi: 10.3389/fphys.2020.543331
251. Lee KM, Kang BS, Lee HL, Son SJ, Hwang SH, Kim DS, et al. Spinal NF-kB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity. Eur J Neurosci. (2004) 19:3375–81. doi: 10.1111/j.0953-816X.2004.03441.x
252. Simon LS. Role and regulation of cyclooxygenase-2 during inflammation. Am J Med. (1999) 106(5b):37S−42. doi: 10.1016/S0002-9343(99)00115-1
253. Famitafreshi H, Karimian M. Prostaglandins as the agents that modulate the course of brain disorders. Degener Neurol Neuromuscul Dis. (2020) 10:1–13. doi: 10.2147/DNND.S240800
254. Liu T, Jiang CY, Fujita T, Luo SW, Kumamoto E. Enhancement by interleukin-1β of AMPA and NMDA receptor-mediated currents in adult rat spinal superficial dorsal horn neurons. Mol Pain. (2013) 9:16. doi: 10.1186/1744-8069-9-16
255. Zhang MRX, Li MA, Liu MB, Wang ML, Ren MK, Zhang MH, et al. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain. (2008) 135:232–9. doi: 10.1016/j.pain.2007.05.023
256. Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, et al. Nociceptors are interleukin-1beta sensors. J Neurosci. (2008) 28:14062–73. doi: 10.1523/JNEUROSCI.3795-08.2008
257. Obreja O, Rathee P, Lips K, Distler C, Kress M. IL-1 beta potentiates heat-activated currents in rat sensory neurons: involvement of IL-1RI, tyrosine kinase, and protein kinase C. FASEB Journal. (2002) 16:1497–503. doi: 10.1096/fj.02-0101com
258. Sharief M, Hentges R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med. (1991) 325:467–72. doi: 10.1056/NEJM199108153250704
259. Göbel K, Ruck T, Meuth SG. Cytokine Signaling in Multiple Sclerosis: Lost in Translation. London, England: SAGE Publications (2018). p. 432–9.
260. Paré A, Mailhot B, Lévesque SA, Juzwik C, Ignatius Arokia Doss PM, Lécuyer M-A, et al. IL-1β enables CNS access to CCR2hi monocytes and the generation of pathogenic cells through GM-CSF released by CNS endothelial cells. Proc Natl Acad Sci U S A. (2018) 115:E1194–203. doi: 10.1073/pnas.1714948115
261. Inquimbert P, Moll M, Latremoliere A, Tong CK, Whang J, Sheehan GF, et al. NMDA receptor activation underlies the loss of spinal dorsal horn neurons and the transition to persistent pain after peripheral nerve injury. Cell Rep. (2018) 23:2678–89. doi: 10.1016/j.celrep.2018.04.107
262. Pender MP, Tabi Z, Nguyen KB, McCombe PA. The proximal peripheral nervous system is a major site of demyelination in experimental autoimmune encephalomyelitis induced in the Lewis rat by a myelin basic protein-specific T cell clone. Acta Neuropathol. (1995) 89:527–31. doi: 10.1007/BF00571507
263. Misawa S, Kuwabara S, Mori M, Hayakawa S, Sawai S, Hattori T. Peripheral nerve demyelination in multiple sclerosis. Clin Neurophysiol. (2008) 119:1829–33. doi: 10.1016/j.clinph.2008.04.010
264. Teixeira NB, Picolo G, Giardini AC, Boumezbeur F, Pottier G, Kuhnast B, et al. Alterations of peripheral nerve excitability in an experimental autoimmune encephalomyelitis mouse model for multiple sclerosis. J Neuroinflammation. (2020) 17:266. doi: 10.1186/s12974-020-01936-9
265. Baker D, Gerritsen W, Rundle J, Amor S. Critical appraisal of animal models of multiple sclerosis. Mult Scler. (2011) 17:647–57. doi: 10.1177/1352458511398885
266. Zucchella C, Mantovani E, De Icco R, Tassorelli C, Sandrini G, Tamburin S. Non-invasive brain and spinal stimulation for pain and related symptoms in multiple sclerosis: a systematic review. Front Neurosci. (2020) 14:547069. doi: 10.3389/fnins.2020.547069
Keywords: experimental autoimmune encephalomyelitis (EAE), glia and neuroinflammation, pain, ion channel, ion pumps, ion exchangers/transporters
Citation: Mirabelli E and Elkabes S (2021) Neuropathic Pain in Multiple Sclerosis and Its Animal Models: Focus on Mechanisms, Knowledge Gaps and Future Directions. Front. Neurol. 12:793745. doi: 10.3389/fneur.2021.793745
Received: 12 October 2021; Accepted: 17 November 2021;
Published: 16 December 2021.
Edited by:
David Henrique Rodrigues, Juiz de Fora Federal University, BrazilReviewed by:
Cyril Rivat, Université de Montpellier, FranceCopyright © 2021 Mirabelli and Elkabes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stella Elkabes, c3RlbGxhLmVsa2FiZXNAcnV0Z2Vycy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.