 Angeliki Spathopoulou
Angeliki Spathopoulou Frank Edenhofer
Frank Edenhofer Lisa Fellner
Lisa Fellner- Department of Genomics, Stem Cell Biology and Regenerative Medicine, Institute of Molecular Biology and Center for Molecular Biosciences Innsbruck, Leopold-Franzens-University Innsbruck, Innsbruck, Austria
Parkinson's disease (PD) is a progressive, neurodegenerative disorder characterized by motor and non-motor symptoms. To date, no specific treatment to halt disease progression is available, only medication to alleviate symptoms can be prescribed. The main pathological hallmark of PD is the development of neuronal inclusions, positive for α-synuclein (α-syn), which are termed Lewy bodies (LBs) or Lewy neurites. However, the cause of the inclusion formation and the loss of neurons remain largely elusive. Various genetic determinants were reported to be involved in PD etiology, including SNCA, DJ-1, PRKN, PINK1, LRRK2, and GBA. Comprehensive insights into pathophysiology of PD critically depend on appropriate models. However, conventional model organisms fall short to faithfully recapitulate some features of this complex disease and as a matter-of-fact access to physiological tissue is limiting. The development of disease models replicating PD that are close to human physiology and dynamic enough to analyze the underlying molecular mechanisms of disease initiation and progression, as well as the generation of new treatment options, is an important and overdue step. Recently, the establishment of induced pluripotent stem cell (iPSC)-derived neural models, particularly from genetic PD-variants, developed into a promising strategy to investigate the molecular mechanisms regarding formation of inclusions and neurodegeneration. As these iPSC-derived neurons can be generated from accessible biopsied samples of PD patients, they carry pathological alterations and enable the possibility to analyze the differences compared to healthy neurons. This review focuses on iPSC models carrying genetic PD-variants of α-syn that will be especially helpful in elucidating the pathophysiological mechanisms of PD. Furthermore, we discuss how iPSC models can be instrumental in identifying cellular targets, potentially leading to the development of new therapeutic treatments. We will outline the enormous potential, but also discuss the limitations of iPSC-based α-syn models.
Introduction
Parkinson's disease (PD) is the second most common progressive neurodegenerative disorder worldwide, affecting 0.3% of the global population (1), with the majority of the cases manifesting among patients over 70 years of age (2). The main neuropathological hallmarks of PD are the loss of dopaminergic (DA) neurons in the pars compacta of the substantia nigra (SNpc) and DA terminals in the striatum (3), along with the occurrence of neuronal (4, 5) and glial (6) cytoplasmic aggregations of the misfolded protein α-synuclein (α-syn). The neuronal α-syn inclusions prevail compared to glial inclusions in PD and they are termed as Lewy bodies (LBs) and Lewy neurites (LNs) (7). PD belongs to the so-called α-synucleinopathies (ASP), also including dementia with Lewy bodies (DLB) and multiple system atrophy (MSA) [reviewed in Arnaoutoglou et al. (8) and Krismer and Wenning (9)]. In the current review, we will discuss studies employing iPSC-derived PD models, focusing especially on SNCA mutations. We will explain the generation of human neuronal cell and organoid models and furthermore discuss the advantages as well as the limitations of iPSC-derived human models.
Features and Pathology of Parkinson's Disease
The first clinical observations of the disorder were reported by James Parkinson in 1817, describing the disease as “shaking palsy,” a combination of the loss of muscular power of the limbs and their involuntarily shaking (10). Nowadays, it is well-established that the disease is characterized by a combination of both motor and non-motor symptoms. The cardinal motor symptoms are tremor, rigidity, postural instability and bradykinesia (11–16) while the non-motor features include sleep disturbances (10, 17), cognitive and neuropsychiatric abnormalities (18, 19) and dysfunctions of the autonomic nervous system (20, 21). Diagnosis of PD is challenging, especially in its early stage, as it is based on clinical symptoms only.
The degeneration of the SNpc DA neurons together with the loss of DA neuronal terminals in the striatum can lead to a decrease of dopamine of around 80%, resulting in the prominent motor symptoms typical for PD (22, 23) [for a more thorough characterization on midbrain DA neurons consult (24, 25)]. Though the exact mechanisms leading to PD still remain elusive, recent studies indicate the involvement of processes such as oxidative stress, mitochondrial dysfunction, dysregulated autophagic and proteasomal degradation of α-syn, as well as neuroinflammation (26–30). Especially, neuroinflammation driven by over-activated glial cells is considered to play a crucial role in the loss of DA cells (31). Studies using PD animal models and human post-mortem brain tissue suggest the involvement of microglial and also astroglial neuroinflammatory responses that contribute to the disease initiation and progression (32–34). Other studies proposed neuronal loss in PD to be caused by the development of α-syn aggregates following a prion-like mechanism (35–40). Kordower et al. and Li et al. report LB propagation from the PD host to grafted neurons in patients after transplantation (41, 42), indicating a prion-like, “infectious” propagation of α-syn. However, Hallet et al. report healthy and unaffected morphology of DA neurons after transplantation (43). Moreover, the prion hypothesis fails to explain the pathological findings of many post-mortem PD brain specimens, which show distinct areas where different cell types are not equally affected, indicating a selective mechanism behind the pathology (44–46). Recently, a cortical pathogenic theory was proposed by Foffani and Obeso (47). According to this hypothesis the corticostriatal activity could act as a stressor on the nigrostriatal terminals, leading to neuronal degeneration and eventually to PD onset (47). Another hypothesis suggests the involvement of the gut-brain axis in PD onset and propagation, revealing consistent alterations between the microbiome of healthy and PD individuals (48–52). Despite studies supporting all the different hypotheses described above, none of the aforementioned theories is able to explain all pathophysiological PD hallmarks.
PD is classified into idiopathic and familial forms of the disease. The cause of idiopathic PD remains unknown, even though it is assumed that it is caused by a combination of genetic and environmental factors. Several studies link exposure to different pesticides, like rotenone and paraquat (53) or the chemical compound 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (54, 55) to the onset of PD. Moreover, other parameters like the patients' diet (56), metabolism (57), inflammation (58) and exercise (59) has been suggested to be involved in the manifestation of PD.
The Pathophysiological Role of α-Synuclein
Familial PD is linked to genetic variants of PD-related risk genes, following an autosomal dominant or recessive inheritance pattern. One of the first genes identified to be connected with PD pathology was the α-syn gene (SNCA) (4). Other PD-relevant genetic mutations include variants of DJ-1, LRRK2 and PINK1 amongst others [for details see (60)]. The present review will focus on the SNCA genetic variants. The genetic PD-related risk variants of α-syn are characterized at protein level by point mutations in the N-terminal α-helices, like A30P (61, 62), A53T (63), H50Q (64), and E46K (65). In these cases, it has been reported that the mutant α-syn favors a faster formation of fibrils, which are thought to be more toxic, compared to the wild-type protein (66–69). Moreover, duplications (70) and triplications, respectively, of the SNCA gene (71) have been described in PD patients. These multiplications lead to increased levels of α-syn that subsequently result in the emergence of the disease pathology (71–73).
Mutations of the α-syn gene as well as different forms of α-syn, like protofibrils and oligomers, are considered as key players in PD pathogenesis (74, 75). α-syn, also known as the precursor protein of non-amyloid beta/A4 protein (NACP), is encoded by the SNCA gene and is a member of the synuclein protein family, together with β-synuclein (β-syn), γ-synuclein (γ-syn), also known as synoretin (76–78). At the protein level, human α-syn is 140 amino acids in length and consists of 3 structural elements, a highly conserved amphiphilic N-terminus characterized by KTKEGV motif repetitions (76, 79), the non-amyloid beta component (NAC) region, which is a hydrophobic internal region and an acidic C-terminus (80) (Figure 1). In vitro the wild-type protein does not show a fixed secondary conformation (81–84). However, the protein has the capacity to form fibrillar aggregates both in vitro and in vivo. The N-terminal domain favors an α-helical conformation, which is expected to hinder the appearance of the toxic, fibril-related β-sheet formations of the NAC region (85–87). Truncated C-terminal α-syn variants are found to be prone to fibrilization both in vitro (88–90) and in vivo (91–94) indicating a potential fibril inhibitory role of the C-terminal domain. α-syn aggregations can also be enhanced by a variety of other parameters, like phosphorylation (95), nitration and oxidation (96, 97), ionic strength (98) or acidic pH (99).
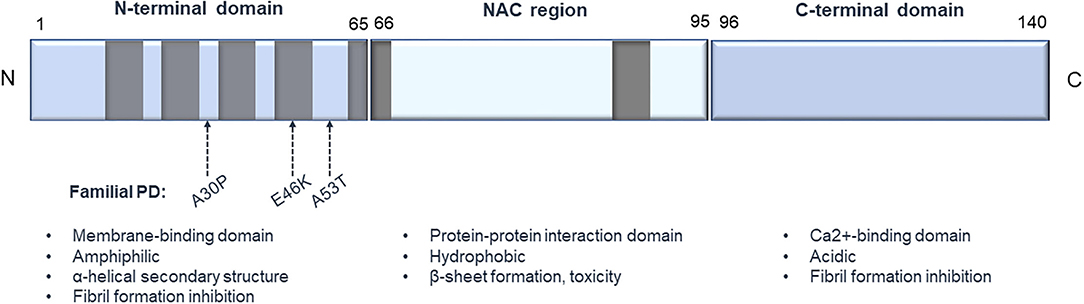
Figure 1. Structure of human α-synuclein. The protein consists of 3 main structural elements: (1) an amphiphilic N-terminus characterized by KTKEGV conserved motifs, important for binding to membranes; (2) an internal hydrophobic NAC domain that is involved in α-syn aggregation; and (3) an acidic C-terminus, which is important for Ca2+ binding. Gray boxes represent the KTKEGV motifs, and the arrows designate some of the common point mutations that are connected to PD pathology.
The physiological role of the α-syn protein is still unclear. It has been reported that α-syn is involved in the synaptic regulation and maturation (74, 100, 101). Moreover, α-syn knockout mice exhibit a DA reduction in the striatum, indicating a potential role of α-syn in DA neurotransmission (101). Decreased synaptic levels of α-syn in primary neurons led to a decrease in the levels of presynaptic proteins, like synaptophysin and synapsin I, that are important for vesicle formation (102). The protein appears to also be implicated in vesicle endocytosis and degradation (103–105). It has been shown that α-syn interacts with mitochondrial and lysosomal membranes (106, 107). Elevated levels of α-syn has been reported to lead to increased mitochondrial fragmentation in ESC-derived neurons (108). Several studies also reported the influence of α-syn in mitochondrial fusion/ fission (109, 110). The protein has also been detected in the nucleus, with recent studies reporting the effect of nuclear α-syn on transcriptional regulation (111, 112) and epigenetic modifications (113). It is apparent that despite the numerous studies in the field, the actual physiological role of α-syn remains elusive.
Generation of Midbrain DA Cells From iPSCs
Recently, induced pluripotent stem cells (iPSCs) emerged as novel tools to study human pathology in general and PD in particular. Human iPSCs were first reported by in vitro reprogramming of adult human fibroblasts, as published by Takahashi et al. (114). Like embryonic stem cells (ESCs), iPSCs are pluripotent stem cells that are able to self-renew indefinitely and differentiate into the three germ layers, endoderm, mesoderm and ectoderm (114, 115). However, in contrast to ESCs the origin of iPSCs is not the embryonic blastocyst, hence their use bypasses a major bioethical concern associated with the use of human ESCs (116). The major advantage of iPSC-derived models is that they allow the recapitulation of the patient-specific genetic background, offering a model of human origin. iPSCs are also eligible for the generation of isogenic control lines with the use of genome editing tools, like CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (117, 118). Since iPSCs can be differentiated into any cell type of interest, they allow the study of complex human disorders where patients' material is difficult to access, particularly for pathologies affecting the human brain (119–121).
The generation of iPSC-derived midbrain DA neurons is highly desired as a meaningful in vitro PD model. A human iPSC-DA model could help to study α-syn-driven neurodegeneration which is predominantly occurring in DA neurons in the SNpc in PD. Prior to the establishment of the iPSC technology, ESCs were the main source of DA neurons of human origin to study DA function and get transplantable material. The evolvement and refinement of DA differentiation methods resulted in protocols that can be classified into three main techniques, feeder-dependent methods (122), two-dimensional (2D) monolayer cultures with dual SMAD inhibition [with the use of the small molecules Noggin and SB431542 (123)], and methods that generate embryoid bodies (EBs) (124–126). First differentiation attempts involved the co-culture of ESCs with stromal feeder cells that induce ESC neuronal DA differentiation (122). However, co-culture of ESCs with feeder cells is a rather undefined process as it is difficult to recapitulate how much, and which factors the feeder cells release into the medium. Lee et al. first described a feeder-free, EB-based differentiation protocol. EBs are aggregates developing from self-assembly of iPSC that undergo gastrulation-like processes. Lee and coworkers initially obtained ~7% tyrosine hydroxylase positive (TH+) neurons, a marker indicating DA generation. After including sonic hedgehog (SHH), ascorbic acid (AA) and fibroblast growth factor 8 (FGF8) into the media, they were able to increase the yield of TH+ neurons to ~34% (124). Kawasaki et al. described a feeder-dependent DA differentiation protocol, that leads to a similar TH+ yield but is significantly simpler compared to the protocol from Lee et al. Using PA6 stromal cells they report emergence of ~30% TH+ DA neurons in a neuron-specific class III beta-tubulin (TuJ)-positive neuronal population, which comprises around 52% of total cells (122). Moreover, the differentiated neurons have been shown to integrate into the striatum of mice after implantation. Still, these co-culture systems resulted in highly heterogeneous neuronal populations, including GABAergic, cholinergic, and serotonergic neurons in TuJ-positive neuronal population, resulting in low percentages of DA neurons (122).
The increasing need for obtaining more homogeneous DA neuronal cultures was met by exposing the iPSCs in chemically defined media compositions that mimic the physiological developmental cues. One of the first approaches, developed in 2009 by Chambers et al., comprised the differentiation of iPSCs by blocking SMAD signaling with the use of two small molecule inhibitors, Noggin and SB31542 (123). A year later Fasano et al. described the derivation of floor plate (FP) origin DA neurons and proposed an early high dose SHH exposure resulting in the generation of forkhead box A2 positive (FOXA2+) cells (127). The combination of early SHH activation along with the activation of the canonical Wnt pathway, through the use of CHIR99021, a glycogen synthase kinase 3 (GSK3B) inhibitor, led to the generation of neuronal cells that were expressing various FP markers, like TH, FOXA2, LIM homeobox transcription factor 1 alpha (LMX1A), nuclear receptor NURR1 (also known as NR4A2), and paired like homeodomain 3 (PITX3) (128). Further improvements were reported by the additional supplementation of BMP5/7 that robustly increases the in vitro differentiation of human iPSCs to midbrain DA neurons up to 3-fold (129). By using further optimized DA differentiation protocols the groups of Parmar (126), Takahashi (130, 131), Barker (132), and Studer (123) were able to provide preclinical evidence that hESC-derived DA neurons are functionally equivalent to those derived from fetal tissue, supporting continued development of hESC-derived cells as a clinical approach for cell replacement treatment of PD (133). The main strategy for the generation of iPSC-derived midbrain DA neurons is illustrated in Figure 2.
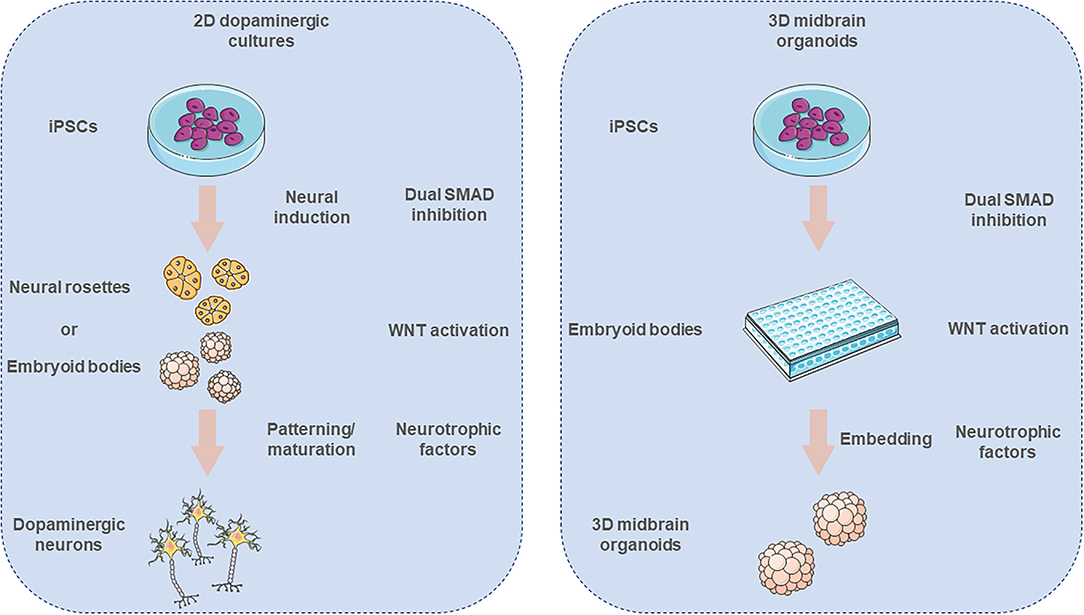
Figure 2. Derivation of in vitro iPSC-derived 2D dopaminergic neuronal cell cultures and 3D midbrain organoids. Neural induction of iPSCs is promoted via dual SMAD inhibition. Pluripotent cells form neural rosettes or embryoid bodies (EBs), that subsequently are patterned toward ventral midbrain identity. After the initial patterning the neurons are further cultured with chemically defined media in order to give rise to mature midbrain neuronal cultures. In the case of the midbrain organoid cultures, first the iPSCs are forming EBs and then get embedded into an extracellular matrix that supports the 3D growth of these organoids. The illustration was created using images from https://smart.servier.com/.
Generation of Midbrain Organoids From iPSCs
Recently, a new era of iPSC-modeling was initiated by the possibility of generating three-dimensional (3D), self-organizing cerebral organoids (134). These in vitro “mini-brains” are originally generated from iPSCs that first organize into EBs and later get embedded into an extracellular matrix hydrogel (e.g., matrigel) and differentiate toward the neuroectodermal lineage. The organoids recapitulate many key structures of the human brain, like the cortical “inside-out” architecture, including a lumen, a subventricular zone (SVZ), a cortical plate (CP) and choroid plexus-like structures. These 3D in vitro brain-like structures consist of a variety of cell types, showing also the ones found in the developing human brain. The organoid cellular composition contains neural progenitors, glial cells, intermediate progenitors as well as deep- and upper-layer neurons (135). Further developments on the organoid technology have resulted in region-specific organoids, recapitulating neuroanatomical structural features of various human brain regions including forebrain (136, 137), pallium and subpallium (138), choroid plexus (139), cerebellum (140), retina (141), hypothalamus (142), and finally midbrain (143–145). Especially, the later ones are of high relevance in the PD research field and can constitute a promising novel in vitro tool providing new insights into the early neurodevelopmental stages of the disorder (Figure 3).
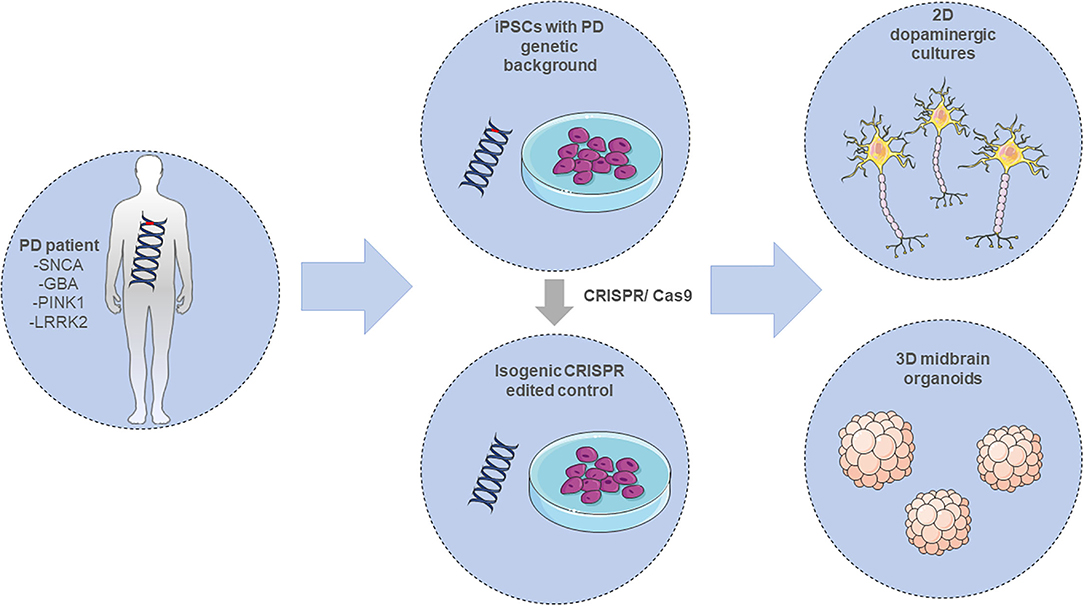
Figure 3. Modeling PD employing iPSC-derived in vitro systems. Adult differentiated cells, either from PD patients carrying a mutation in the SNCA, GBA, PINK1, or LRRK2 gene amongst others, or CRISPR-edited isogenic controls, are reprogrammed into induced pluripotent stem cells (iPSCs). These cells can be further differentiated into 2D or 3D neuronal in vitro cultures for investigating PD pathology. The illustration was created using images from https://smart.servier.com/.
Different protocols have been developed for the creation of homogeneous midbrain-like organoids (MOs). Tieng et al. described a “neurosphere” mediated method, where first the iPSCs form EBs that then get exposed to a FP-inducing medium, containing LDN193189, SB431542, SHH, FGF8, purmorphamine, and CHIR99021 and finally to a neural maturation medium. This protocol yields organoids that contain around 60% of DA neurons, in around 3 weeks (146). Alternative protocols similarly follow the EB formation method and e.g., employ the exposure of the cells to several morphogens, like Wnt activators, dual SMAD inhibitors and SHH activators (143, 147, 148). Similarly, to Tieng et al. these protocols also report high proportion of DA neurons, >60%, together with functional glial cells, astrocytes and oligodendrocytes. All protocols also result in electrophysiologically active neurons, however the protocol developed by Jo et al. was the only one that reported the identification of SNpc DA neuron-like electrophysiological patterns (149). Interestingly, Kwak et al. observed DA specific cell death after the treatment with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, indicating this 3D system as a relevant PD model (148). Even though there is a variety of protocols for deriving midbrain organoids, the usage for PD modeling is still in its infancy.
iPSC-Derived Neuronal Models to Mimic PD
Copy Number Variations of SNCA
With the establishment of DA neuronal differentiation protocols, researchers started to employ them in order to generate DA neurons with PD-related genetic backgrounds. Some of the first studies focused on the generation of DA neurons from iPSCs carrying a SNCA triplication (SNCA-tri) genetic variant. These studies revealed further insight into the occurrence of PD pathological hallmark features, like α-syn aggregation (150–157), mitochondrial dysfunction and elevated oxidative stress (150, 156, 158), as well as ER stress (154, 159).
Byers et al. report that PD midbrain DA neurons overexpressed oxidative stress related markers, like heme oxygenase 2 (HMOX2), when compared to healthy controls. The disease related neurons appeared to be more vulnerable to oxidative stress and they showed increased cell death (150). Mitochondrial dysfunction accompanied by increased oxidative stress was also observed by Zambon et al. The group reported decreased cellular respiration accompanied by abnormalities in the mitochondrial membrane potential that could be induced by the reduced phosphorylation of Dynamin-related protein 1 (DRP1Ser616), a GTPase important for the mitochondrial morphology (156). Along the same lines, Ludtmann et al. observed mitochondrial accumulation of α-syn oligomers. The oligomers selectively oxidized mitochondrial proteins, e.g., the ATP synthase among others, an event that induced the early opening of the mitochondrial permeability transition pore (MPTP) that eventually led to mitochondrial swelling and increased cell death. These pathological effects were reversed after the inhibition of MPTP with the use of cyclosporin A (158).
It is considered that lysosomal function plays a crucial role in PD pathology. There is substantial evidence of an association between synucleinopathies and Gaucher disease, a lysosomal storage disorder (LSD) that is characterized by mutations in the glucocerebrosidase (GBA) gene (160, 161). In fact, mutations in the GBA gene in mice have been reported to result in accumulation of α-syn and finally lead to neurotoxicity. Moreover, the aggregations result in further inhibition of the normal GBA lysosomal function (161). This connection indicates that the impairment in the physiological lysosomal function reinforces the accumulation of α-syn, and thus contributes to the PD pathology (162). Mazzulli et al. confirmed in an in vitro cell culture model that iPSC-derived midbrain DA neurons, carrying a SNCA triplication or an A53T point mutation, exhibited reduced lysosomal function leading to elevated α-syn accumulation. Furthermore, a disruption in the subcellular protein sorting of the Rab1a protein, a GTPase with important ER-Golgi vesicular transport function, was reported, resulting in reduced hydrolase trafficking and eventually fragmentation of the Golgi apparatus. These pathological findings could be reversed with the overexpression of Rab1a in DA neurons (154). In a later study the same group showed the restoration of the lysosomal activity and the subsequent reduction of α-syn accumulation using a small molecule modulating the function of the lysosomal β-glucocerebrosidase enzyme (GCase) (163).
In the nuclei of SNCA-tri neurons perturbations of the normal cellular morphology were reported. Impairments of the nuclear envelope in PD neurons were shown along with abnormalities in the nuclear protein transport after the interaction of α-syn with Ras-related nuclear protein (RAN) (164). Similar findings were also described by Vasquez et al. in a study that employed SNCA-tri neural progenitor cells (NPCs). The group confirmed the nuclear localization of α-syn and its association with chromatin. The aggregates induced DNA damage and ultimately, increased cell death was reported (165).
Following the general developmental impairment that characterize pathological features of PD, studies reported a maturation delay of diseased neurons after comparing their transcriptomic profile with either healthy controls or primary midbrain DA neurons. Oliveira et al. reported the decreased expression of many genes that are implicated in neuronal differentiation processes (152). A year later, Xia et al. confirmed this finding. Among others, they described the downregulation of 37 genes in the PD midbrain DA neuronal cohort. The list included genes that are important for the DA neuronal identity, like TH and LMX1B, genes that are involved in neuronal maturation, as well as genes that are considered as risk genes for neurological pathologies (166). Further studies described developmental and morphological neuronal abnormalities, such as aberrant neurite morphologies showing reduced lengths and irregularities of the membrane integrity. Furthermore, Lin et al. reported that the SNCA-tri neurons exhibited decreased firing rates and complete absence of synchronized neuronal firing, indicating a decline in normal neuronal activity and connectivity (153). SNCA-tri neurons with impaired neurites were also reported by a study of Siddu et al. The number of healthy neurites was ameliorated after treatment with cysteamine that additionally reduced the levels of the α-syn aggregates (157). Cysteamine is an aminothiol derivative that is physiologically produced from the degradation of coenzyme A (167, 168). Notably, cysteamine exhibits a protective role against DNA damage when induced by irradiation since it acts as a reactive oxygen species (ROS) scavenger (169). Moreover, the compound has been used in multiple Huntington's disease (HD) preclinical studies with promising results, including prolonged mice survival (170–172). Siddu et al., after having additional encouraging results on mouse studies, proposed that cysteamine could be considered as a potential future drug candidate for PD (157).
Since PD is an age-related neurodegenerative disorder, mimicking the aging aspect plays a crucial role in the disease modeling. One of the limitations of the iPSC models is the rejuvenation of the cells during their reprogramming. In an auspicious study led by Chiba-Falek, the scientists bypassed this drawback with the generation of aged NPCs without the ectopic expression of genes or the use of toxins. In detail, the aged NPCs were derived after extensive passaging, 14–16 times. The aging phenotype was confirmed by assessing the expression of heterochromatin markers (e.g., histone 3 lysine 9 trimethylation, H3K9me3), by evaluating the nuclear membrane structure (e.g., stainings with lamin A/C), DNA damage (with cellular stainings of heterochromatin protein 1 γ, HP1γ) and the evaluation of the global methylation. The aged NPCs expressed age-related markers and gave rise to midbrain DA neurons that attained the aged phenotype. Notably, even the young SNCA-tri neurons, derived from young NPCs, showed increased aging-related markers when compared to control neurons (173).
The transfer of α-syn aggregates from SNCA-tri neurons to neighboring wild type neurons has been reported by Reyes et al., indicating a potential proof of the prion hypothesis for PD pathogenesis (174). Prions are defined by their “protein-only” composition that is—though devoid of nucleic acids—able to carry infectivity by direct protein-protein contact (175). This co-culture system can also be used as a small molecule screening system (174).
Besides iPSC models that are based on the triplication of the α-syn gene, some others carry SNCA duplications. This genetic background also led to the generation of neurons that exhibited hallmark pathological PD features, in a lesser extent compared to the triplications, like α-syn aggregations that specifically accumulated in midbrain DA neurons, mitochondrial dysfunction and energy deficits, synaptic loss, ROS accumulation, protein nitration and ultimately increased cell death (155, 176).
Taken together, multiplication of the SNCA locus, either triplication or duplication, appears adequate to initiate PD pathology (177, 178). The SNCA copy number variation (CNV) leads to PD symptom manifestation, like α-syn aggregation, lysosomal and mitochondrial impairments and accelerated neuronal cell death. Therefore, the SNCA multiplication iPSC-derived neuronal models appear to be useful in mimicking PD pathology. They offer valuable, new insights into the pathological pathways of PD initiation and progression and can also be used as drug screening platforms. The relevant studies and their main findings are summarized in Table 1.
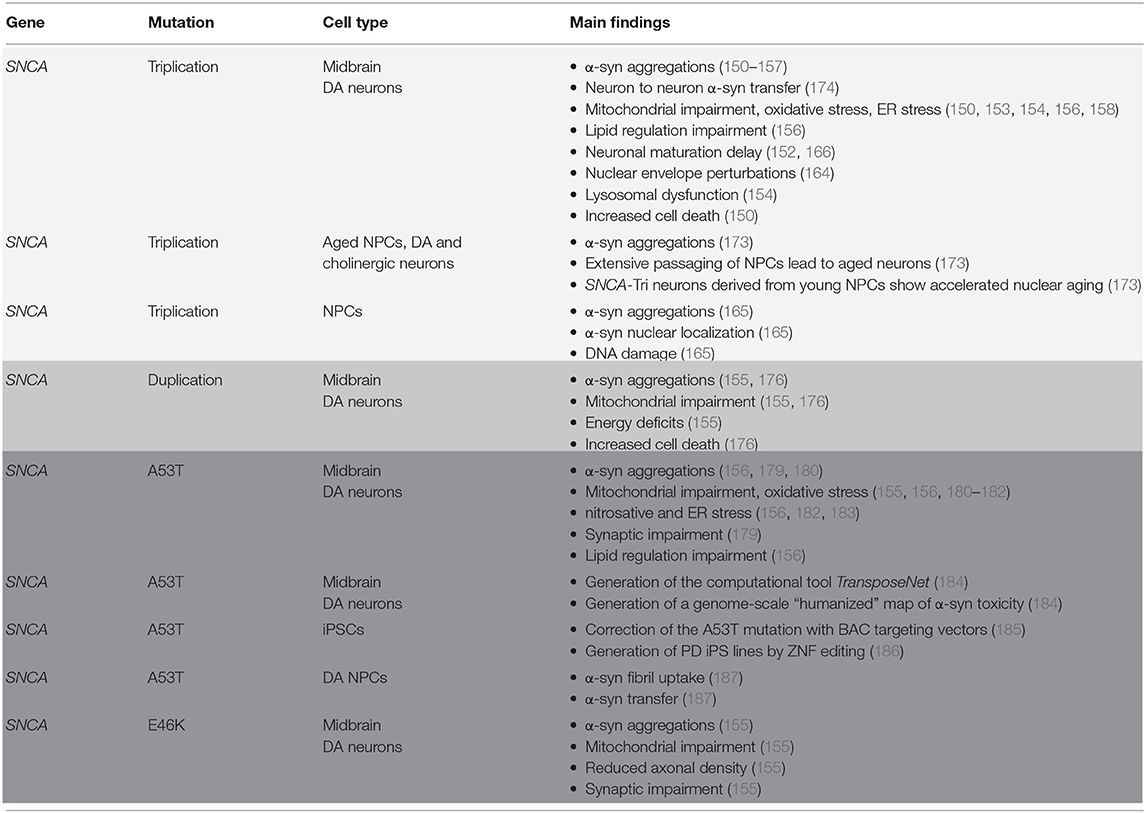
Table 1. Summary on 2D models using iPSC and iPSC-generated neuronal cells having a PD-SNCA-related genetic background.
SNCA Point Mutations
PD is also associated with various point mutations in the SNCA gene, with A53T and E46K being the most extensively studied ones. Like for CNVs iPSC-derived neuronal models based on SNCA point mutations do also result in α-syn aggregation (155, 156, 179, 180). Fernandes et al. showed in A53T-mutant midbrain DA neurons dysregulated expression of genes, related to chromatin organization and histone modifications. They also demonstrated a repression of oxidative phosphorylation and a reduction in the cholesterol biosynthesis (182). Studies of multiple research groups also report ER and mitochondrial stress linked to these point mutations. Interestingly, Ryan et al. described impaired and fragmented mitochondria that were also observed in control neurons after co-culture with SNCA-A53T neurons, indicating the transfer of α-syn fibrils of the pathogenic neurons to the isogenic control. This cell-to-cell transfer was inhibited by a monoclonal antibody directed against α-syn. Moreover, the same study identified that treatment with cardiolipin, a group of phospholipids that account for around 20% of the inner mitochondria membrane phospholipid mass, reduces α-syn accumulation at the mitochondrial outer membrane (180). A study on A53T-NPCs revealed lysosomal storage of α-syn aggregations and cell-to-cell transfer. The aggregates were found to be accumulated in tunneling nanotubule (TNT)-like structures (187). Kouroupi et al. described synaptic impairments that were reversed after treatment with a de novo designed small molecule, inhibiting the accumulation of α-syn (179). Khurana et al. developed an in silico tool, the so-called TransposeNet, a computational method that can interchange molecular interactions across species. It allows the generation of genome-scale maps of α-syn toxicity in yeast that then could reveal links to PD-relevant genes and to candidates targeting α-syn toxicity in humans. These then can be tested in vitro e.g., in patient-derived neurons (184).
PD is considered a complex disorder, influenced by a combination of both genetic and environmental factors. Recently, an intriguing study performed by Stykel et al. proved a genetic-environmental interaction by inhibiting the anterograde mitochondrial transport in SNCA-A53T harboring DA neurons. When exposed to the agrochemicals rotenone, or paraquat and manet, the mutated DA neurons showed a change in the nitration of α-tubulin (α-Tub), which led to altered microtubule architecture and eventually to the arrest of the anterograde mitochondrial transport toward the axon terminal (181). In contrast, the isogenic control neurons showed only minor effects to the exposure. As shown before, defects in mitochondrial transport are critical for the neuronal physiology (188). Therefore, Stykel et al. showed an additive effect of the environmental toxins, paraquat, manet and rotenone with the PD susceptible genetic background, that could lead to an earlier PD onset. The use of the nitric oxide biosynthesis inhibitor, Nω-nitro-L-arginine methyl ester, reversed these effects (181).
In conclusion, like the SNCA CNV models, point mutations also lead to the manifestation of PD hallmarks, ranging from α-syn aggregation to ER and mitochondrial stress. Though, a particular characteristic of the point mutation models is the wide variety of pathological observations. The relevant studies and their main findings are summarized in Table 1.
Notably, all 2D DA midbrain models involving SNCA triplication, duplication, or point mutations (A53T and E46K) reproduced the ability to aggregate α-syn (150– 153, 155–157, 163, 176, 179, 180), which further led to mitochondrial impairment (150, 155, 156, 158, 176). Furthermore, DA midbrain neuronal cells showing the SNCA triplication and the point mutation A53T genetic background revealed oxidative stress, ER stress and lipid dysregulation (150, 156, 158, 182), whereas increased cell death was found in the SNCA triplication and duplication DA midbrain neuronal cell lines (150, 176). Neuronal maturation delay in iPSC-derived DA midbrain neurons was shown in two separate studies using the SNCA triplication background (152, 166). Additional features or impairments were found in the different models, yet most of them need further verification.
The generation of 3D iPSC derived in vitro models allowed to bypass many of the limitations of the 2D counterpart. More specifically, the organoid models recapitulate more precisely the in vivo physiology and development, features that are not possible to be modeled in a simpler 2D cell culture model. Furthermore, organoids offer a higher cellular complexity, they allow complex cell-to-cell interactions and recapitulate the spatial architectural features of human development (189, 190). Nevertheless, 2D models are still useful and well-established providing easier and less complicated data interpretation.
Given the recent development of the 3D neuronal organoid technology, not many studies employed organoids derived from iPSC lines expressing SNCA mutations, so far. Based on previous studies Jo et al. generated SNCA-triplication and GBA1 knock-out midbrain organoids that successfully exhibited α-syn aggregation, LB generation and DA neuronal loss (149). A very recent study focuses on the establishment of SNCA-triplication midbrain organoids employing CRISPR/Cas9 edited isogenic controls. This study also stated the neuronal and glial accumulation of α-syn aggregates that interestingly increased in organoids of higher age, recapitulating the physiological age-dependency of PD pathology (191). Smits et al. focused on the establishment of LRRK2-G2019S midbrain organoids. These PD midbrain organoids exhibit disease-related phenotypes, like impaired neuronal complexity and interestingly, they showed a decreased number of DA neurons, even though there was an excess of FOXA2+ progenitors, indicating a developmental impairment in the DA neuronal differentiation (147).
Aside from the SNCA gene variants, PD GWAS studies identified a plethora of disease-associated risk variants that still need further functional studies, focusing on the underlying pathological mechanisms (192). Therefore, the generation of different disease-relevant 3D models, but also 2D models, recapitulating the genetic background of PD, will be of great significance in order to get more insights into PD initiation and progression.
Discussion and Conclusion
PD is a neurodegenerative disorder affecting a substantial fraction of the global population. The disease is both, devastating for the patients' life quality, and also a big burden for the global healthcare system. Due to the restricted access to PD-affected human brain tissue there is a fundamental need for reliable in vivo and in vitro models. The current in vivo models employ a wide range of animals including rodents, non-mammalian species and non-human primates (NHP). Transgenic animal models, especially mice, have been extensively used for studying PD, since they do not only enable insights into the pathogenesis of PD, but also can be employed for the identification and validation of new potential therapeutics (193, 194). Reported mouse models include transgenic α-syn models and transgenic animals with mutations in the LRRK, PINK1 and DJ-1 gene (194). In vitro PD cellular models include the use of various cells lines, both primary and immortalized. Among the most widely used lines for PD modeling are the neuroblastoma immortalized cell line SH-SY5Y (195), the Lund human mesencephalic immortalized cell line (LUHMES) (196), the rat N27 immortalized mesencephalic cells and primary DA cells (197). The dopamine synthesis and metabolism machinery of these lines turns them into suitable PD models.
Here, we highlighted the importance of developing complementary in vitro iPSC-derived neuronal models for studying particularly the involvement of α-syn in PD pathology. These models can offer valuable insights into the pathology mechanisms, but they could also serve as platforms for drug screening. The iPSC technology allows the generation of models of human origin, carrying the patient's genetic background, which makes them a useful tool for getting insights into the pathological disease mechanisms. Furthermore, these systems are readily available, for instance through various biobanks [among others the Corriel (https://www.coriell.org/1/Browse/Biobanks) and the European Bank for induced pluripotent Stem Cells (EBiSC)], as well as through various commercial sources, and well-established for disease modeling, as it is apparent from the numerous studies employing iPSCs for this purpose (120).
PD is a complex multifactorial disorder. iPSCs can be employed for addressing several pathophysiological mechanisms and life-style parameters that are implicated in the disorder. For example, there are studies deciphering the implication of inflammation in PD, employing iPSC-derived macrophages (198) and microglial cells (199). The influence of metabolism and diet were addressed in metabolomic studies (200–202) and studies that control the nutrient composition of the culture medium (203). However, other aspects, like the effect of exercise in disease progression are hard to be modeled in an in vitro system and therefore an in vivo mouse model would be the appropriate system for this scientific question.
However, though iPSC are powerful tools in generating patient-specific diseased cells, they exhibit several limitations. Genomic instability and epigenetic aberrations potentially induced by reprogramming need to be carefully monitored (204, 205). Moreover, iPSC reprogramming involves a global epigenetic reset representing an undesired cell rejuvenation process if it comes to study age-related disorders (206). Furthermore, for a clinical application of iPSC-derived cells the potential tumorigenicity and immunogenicity need to be addressed (207–209). Finally, since iPSCs carry the patient's genetic background, special attention needs to be dedicated into the proper handling of the personal, genomic data (210, 211).
To sum up, iPSC-derived SNCA neuronal models are either focusing on SNCA gene multiplications or mimic the SNCA disease-related point mutations. Even though these models partly enable the recapitulation of PD hallmark pathological features, like α-syn aggregation, mitochondrial stress, lysosomal dysfunction, ER stress, DNA damage and accelerated cell death, none of them managed to demonstrate the combination of all these pathological characteristics. Additionally, some of the studies confirmed the transfer of α-syn fibrils from SNCA-tri neurons to healthy controls. Despite the lack of an ultimate iPSC-derived PD model that demonstrates holistically the pathological PD hallmarks, these studies using iPSC-derived PD models identified significant findings regarding the pathological pathway involved in PD initiation and progression.
However, iPSC-derived PD models are poor in modeling aging, due to the rejuvenation of the cells during the reprogramming (206, 212, 213). In fact, PD is an age-related pathology that manifests mostly in patients with higher age. Some studies addressed this issue by artificial aging, e.g., by extensive NPC passaging initiating the expression of age-related markers and later differentiating them into neurons (173). The iPSC rejuvenation could also be bypassed by transdifferentiation, also known as direct conversion, a technique that differentiates the adult cells into the cell type of interest, without passing through the reprogrammed state. This way, the cells retain their age and could provide a better suited model for studying late onset disorders (214). In the context of PD, adult dermal fibroblasts have been directly converted into DA neurons through the overexpression of a cocktail of transcriptional factors (215, 216). Another study, has also generated DA neurons by directly converting astrocytes (217).
Even though, direct conversion into DA neurons is a great tool, the generated neuronal population is post-mitotic. Therefore, there are several concerns about the efficacy and the survival of the neuronal populations in cases of transplantation for therapeutic purposes. Another promising alternative is the use of NPCs. NPCs are multipotent progenitor cells that can give rise to neurons, oligodendrocytes and astrocytes (218–221) and novel reprogramming paradigms allow direct conversion of patient-specific cells into NPCs (221–223). NPCs combine a series of interesting features, like the partial maintenance of their aging signatures, such as induced neurons. They are also safer for cell therapy since they are not tumorigenic, in contrast to iPSCs. More importantly they are expandable, making them ultimately a promising therapeutical approach.
The exogenous administration of α-syn pre-formed fibrils is another alternative approach in PD modeling. There is extensive use of this approach in animal models, including mice and non-human primates (224). Although, many insights were gained from these in vivo studies, the animal models do not recapitulate the full spectrum of the human PD pathology. Therefore, translating the results of the neuronal reactions to extracellular human α-syn from animals to humans is a difficult task. The same approach has recently been used in an iPSC-derived neuronal system. The study established 2D cortical neuronal networks using microfluidic devices. The healthy neurons of the one side of the microfluidic device were incubated with α-syn fibrils and the researchers assessed the uptake, the intracellular fibril transfer, and the cell-to-cell fibril transfer. They reported a prion-like α-syn fibril transfer between the two neuronal populations. Moreover, the α-syn aggregates led progressively to the appearance of PD pathological features (225).
Conclusively, it is apparent that there are exciting new developments regarding the PD research field, both in basic research and in the applied clinical field. Many emerging new technologies, e.g., midbrain organoids and multi-omics data, promise to give invaluable new insights to the research community. Based on the literature though, it is evident that there is a need for additional studies employing these new models. The upcoming years will bring a great influx of exciting new data that hopefully will lead to better understanding of the PD pathology helping to generate new treatment options that will ameliorate the patients' lives or even stop the progression of the disease.
Author Contributions
AS wrote the initial draft of this review. LF and FE conceptualized the review and reviewed the final manuscript. All authors were involved in revising and editing the manuscript, and they all read and approved the final version of the manuscript.
Funding
This work was supported by the Austrian Science Fund (FWF, I 4791-B and SFB F7810), the German Research Foundation (DFG; ED 79/4-1) to FE, and by the European Union's Horizon 2020 Marie Sklodowska-Curie research grant No. 847681 (ARDRE).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lee A, Gilbert RM. Epidemiology of Parkinson disease. Neurol Clin. (2016) 34:955–65. doi: 10.1016/j.ncl.2016.06.012
2. Twelves D, Perkins KSM, Counsell C. Systematic review of incidence studies of Parkinson's disease. Mov Disord. (2003) 18:19–31. doi: 10.1002/mds.10305
3. Fearnley JM, Lees AJ. Aging and parkinson's disease: Substantia nigra regional selectivity. Brain. (1991) 114:2283–301. doi: 10.1093/brain/114.5.2283
4. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. (1997) 276:2045–7. doi: 10.1126/science.276.5321.2045
5. Spillantini MG, Anthony Crowther R, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neurosci Lett. (1998) 251:205–8. doi: 10.1016/S0304-3940(98)00504-7
6. Arai T, Uéda K, Ikeda K, Akiyama H, Haga C, Kondo H, et al. Argyrophilic glial inclusions in the midbrain of patients with Parkinson's disease and diffuse Lewy body disease are immunopositive for NACP/α-synuclein. Neurosci Lett. (1999) 259:83–6. doi: 10.1016/S0304-3940(98)00890-8
7. Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies [8]. Nature. (1997) 388:839–40. doi: 10.1038/42166
8. Arnaoutoglou NA, O'Brien JT, Underwood BR. Dementia with Lewy bodies - from scientific knowledge to clinical insights. Nat Rev Neurol. (2019) 15:103–12. doi: 10.1038/s41582-018-0107-7
9. Krismer F, Wenning GK. Multiple system atrophy: Insights into a rare and debilitating movement disorder. Nat Rev Neurol. (2017) 13:232–43. doi: 10.1038/nrneurol.2017.26
10. Parkinson J. An Essay on Shaking Palsy in Sherwood, Neely and Jones. London: Sherwood, Neely and Jones (1817).
11. Berardelli A, Rothwell JC, Thompson PD, Hallett M. Pathophysiology of bradykinesia in parkinson's disease. Brain. (2001) 124:2131–46. doi: 10.1093/brain/124.11.2131
12. Winkler AS, Reuter I, Harwood G, Chaudhuri KR. The frequency and significance of “striatal toe” in parkinsonism. Park Relat Disord. (2002) 9:97–101. doi: 10.1016/S1353-8020(02)00010-X
13. Ashour R, Jankovic J. Joint and skeletal deformities in Parkinson's disease, multiple system atrophy, and progressive supranuclear palsy. Mov Disord. (2006) 21:1856–63. doi: 10.1002/mds.21058
14. Broussolle E, Krack P, Thobois S, Xie-Brustolin J, Pollak P, Goetz CG. Contribution of Jules Froment to the study of parkinsonian rigidity. Mov Disord. (2007) 22:909–14. doi: 10.1002/mds.21484
15. Shahed J, Jankovic J. Exploring the relationship between essential tremor and Parkinson's disease. Park Relat Disord. (2007) 13:67–76. doi: 10.1016/j.parkreldis.2006.05.033
16. Jankovic J. Parkinson's disease: Clinical features and diagnosis. J Neurol Neurosurg Psychiatry. (2008) 79:368–76. doi: 10.1136/jnnp.2007.131045
17. Xu Z, Anderson KN, Saffari SE, Lawson RA, Chaudhuri KR, Brooks D, et al. Progression of sleep disturbances in Parkinson's disease: a 5-year longitudinal study. J Neurol. (2021) 268:312–20. doi: 10.1007/s00415-020-10140-x
18. Aarsland D, Brønnick K, Ehrt U, De Deyn PP, Tekin S, Emre M, et al. Neuropsychiatric symptoms in patients with Parkinson's disease and dementia: frequency, profile and associated care giver stress. J Neurol Neurosurg Psychiatry. (2007) 78:36–42. doi: 10.1136/jnnp.2005.083113
19. Ravina B, Camicioli R, Como PG, Marsh L, Jankovic J, Weintraub D, et al. The impact of depressive symptoms in early Parkinson disease. Neurology. (2007) 69:342–7. doi: 10.1212/01.wnl.0000268695.63392.10
20. Swinn L, Schrag A, Viswanathan R, Bloem BR, Lees A, Quinn N. Sweating dysfunction in Parkinson's disease. Mov Disord. (2003) 18:1459–63. doi: 10.1002/mds.10586
21. Allcock LM, Ullyart K, Kenny RA, Burn DJ. Frequency of orthostatic hypotension in a community based cohort of patients with Parkinson's disease. J Neurol Neurosurg Psychiatry. (2004) 75:1470–1. doi: 10.1136/jnnp.2003.029413
22. Uhl GR. Dopamine compartmentalization, selective dopaminergic vulnerabilities in Parkinson's disease and therapeutic opportunities. Ann Clin Transl Neurol. (2019) 6:406–15. doi: 10.1002/acn3.707
23. Sackner-Bernstein J. Estimates of intracellular dopamine in Parkinson's disease: A systematic review and meta-analysis. J Parkinsons Dis. (2021) 11:1011–18. doi: 10.3233/JPD-212715
24. Hegarty SV, Sullivan AM, O'Keeffe GW. Midbrain dopaminergic neurons: A review of the molecular circuitry that regulates their development. Dev Biol. (2013) 379:123–38. doi: 10.1016/j.ydbio.2013.04.014
25. Luo SX, Huang EJ. Dopaminergic neurons and brain reward pathways: from neurogenesis to circuit assembly. Am J Pathol. (2016) 186:478. doi: 10.1016/j.ajpath.2015.09.023
26. Bentea E, Verbruggen L, Massie A. The proteasome inhibition model of Parkinson's disease. J Parkinsons Dis. (2017) 7:31–63. doi: 10.3233/JPD-160921
27. Lee Y, Lee S, Chang SC, Lee J. Significant roles of neuroinflammation in Parkinson's disease: therapeutic targets for PD prevention. Arch Pharmacal Res. (2019) 42:416–25. doi: 10.1007/s12272-019-01133-0
28. Chang KH, Chen CM. The role of oxidative stress in Parkinson's disease. Antioxidants. (2020) 9:597. doi: 10.3390/antiox9070597
29. Hou X, Watzlawik JO, Fiesel FC, Springer W. Autophagy in Parkinson's disease. J Mol Biol. (2020) 432:2651–72. doi: 10.1016/j.jmb.2020.01.037
30. Prasuhn J, Davis RL, Kumar KR. Targeting mitochondrial impairment in Parkinson's disease: challenges and opportunities. Front Cell Dev Biol. (2021) 8:1704. doi: 10.3389/fcell.2020.615461
31. Brück D, Wenning GK, Stefanova N, Fellner L. Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol Dis. (2016) 85:262–74. doi: 10.1016/j.nbd.2015.03.003
32. Bartels T, De Schepper S, Hong S. Microglia modulate neurodegeneration in Alzheimer's and Parkinson's diseases. Science. (2020) 370:66–9. doi: 10.1126/science.abb8587
33. Kam TI, Hinkle JT, Dawson TM, Dawson VL. Microglia and astrocyte dysfunction in parkinson's disease. Neurobiol Dis. (2020) 144:105028. doi: 10.1016/j.nbd.2020.105028
34. Lazdon E, Stolero N, Frenkel D. Microglia and Parkinson's disease: footprints to pathology. J. Neural Transm. (2020) 127:149–158. doi: 10.1007/s00702-020-02154-6
35. Braak H, Del Tredici K, Rüb U, De Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003) 24:197–211. doi: 10.1016/S0197-4580(02)00065-9
36. Brundin P, Li JY, Holton JL, Lindvall O, Revesz T. Research in motion: The enigma of Parkinson's disease pathology spread. Nat Rev Neurosci. (2008) 9:741–5. doi: 10.1038/nrn2477
37. Steiner JA, Quansah E, Brundin P. The concept of alpha-synuclein as a prion-like protein: ten years after. Cell Tissue Res. (2018) 373:161–73. doi: 10.1007/s00441-018-2814-1
38. Kim S, Kwon SH, Kam TI, Panicker N, Karuppagounder SS, Lee S, et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson's disease. Neuron. (2019) 103:627–41. doi: 10.1016/j.neuron.2019.05.035
39. Ma J, Gao J, Wang J, Xie A. Prion-like mechanisms in Parkinson's disease. Front Neurosci. (2019) 13:552. doi: 10.3389/fnins.2019.00552
40. Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, et al. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci. (2020) 23:327–36. doi: 10.1038/s41593-020-0589-7
41. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. (2008) 14:504–6. doi: 10.1038/nm1747
42. Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. (2008) 14:501–3. doi: 10.1038/nm1746
43. Hallett PJ, Cooper O, Sadi D, Robertson H, Mendez I, Isacson O. Long-term health of dopaminergic neuron transplants in Parkinson's disease patients. Cell Rep. (2014) 7:1755–61. doi: 10.1016/j.celrep.2014.05.027
44. Halliday G, McCann H, Shepherd C. Evaluation of the Braak hypothesis: How far can it explain the pathogenesis of Parkinson's disease? Expert Rev Neurother. (2012) 12:673–86. doi: 10.1586/ern.12.47
45. James Surmeier D, Obeso JA, Halliday GM. Parkinson's disease is not simply a prion disorder. J Neurosci. (2017) 37:9799–807. doi: 10.1523/JNEUROSCI.1787-16.2017
46. Heras-Garvin A, Stefanova N. From synaptic protein to prion: the long and controversial journey of α-synuclein. Front Synaptic Neurosci. (2020) 12:42. doi: 10.3389/fnsyn.2020.584536
47. Foffani G, Obeso JA. A cortical pathogenic theory of Parkinson's disease. Neuron. (2018) 99:1116–28. doi: 10.1016/j.neuron.2018.07.028
48. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell. (2016) 167:1469–80.e12. doi: 10.1016/j.cell.2016.11.018
49. Ulusoy A, Phillips RJ, Helwig M, Klinkenberg M, Powley TL, Di Monte DA. Brain-to-stomach transfer of α-synuclein via vagal preganglionic projections. Acta Neuropathol. (2017) 133:381–93. doi: 10.1007/s00401-016-1661-y
50. Chao YX, Gulam MY, Chia NSJ, Feng L, Rotzschke O, Tan EK. Gut-brain axis: potential factors involved in the pathogenesis of Parkinson's disease. Front Neurol. (2020) 11:849. doi: 10.3389/fneur.2020.00849
51. Maiuolo J, Gliozzi M, Musolino V, Carresi C, Scarano F, Nucera S, et al. The contribution of gut microbiota-brain axis in the development of brain disorders. Front Neurosci. (2021) 15:170. doi: 10.3389/fnins.2021.616883
52. Wallen ZD, Stone WJ, Factor SA, Molho E, Zabetian CP, Standaert DG, et al. Exploring human-genome gut-microbiome interaction in Parkinson's disease. NPJ Park Dis. (2021) 7:1–11. doi: 10.1038/s41531-021-00218-2
53. Tanner CM, Kame F, Ross GW, Hoppin JA, Goldman SM, Korell M, et al. Rotenone, paraquat, Parkinson's disease. Environ Health Perspect. (2011) 119:866–72. doi: 10.1289/ehp.1002839
54. William Langston J, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. (1983) 219:979–80. doi: 10.1126/science.6823561
55. Gasser T. Molecular pathogenesis of Parkinson disease: Insights from genetic studies. Expert Rev Mol Med. (2009) 11:22. doi: 10.1017/S1462399409001148
56. Jackson A, Forsyth CB, Shaikh M, Voigt RM, Engen PA, Ramirez V, et al. Diet in Parkinson's disease: critical role for the microbiome. Front Neurol. (2019) 10:1245. doi: 10.3389/fneur.2019.01245
57. Bedarf JR, Hildebrand F, Coelho LP, Sunagawa S, Bahram M, Goeser F, et al. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson's disease patients. Genome Med. (2017) 9:1–13. doi: 10.1186/s13073-017-0428-y
58. Pajares MI, Rojo A, Manda G, Boscá L, Cuadrado A. Inflammation in Parkinson's disease: mechanisms and therapeutic implications. Cells. (2020) 9:1687. doi: 10.3390/cells9071687
59. Carvalho AO, de, Filho ASS, Murillo-Rodriguez E, Rocha NB, Carta MG, et al. Physical exercise For Parkinson's disease: clinical and experimental evidence. Clin Pract Epidemiol Ment Health. (2018) 14:89–98. doi: 10.2174/1745017901814010089
60. Poewe W, Seppi K, Tanner C, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Prim. (2017) 3:1–21. doi: 10.1038/nrdp.2017.13
61. El-Agnaf OM, Jakes R, Curran MD, Wallace A. Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of alpha-synuclein protein implicated in Parkinson's disease. FEBS Lett. (1998) 440:67–70. doi: 10.1016/S0014-5793(98)01419-7
62. Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. (1998) 18:106–8. doi: 10.1038/ng0298-106
63. Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. (1998) 4:1318–20. doi: 10.1038/3311
64. Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord. (2013) 28:811–3. doi: 10.1002/mds.25421
65. Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of α-synuclein causes Parkinson and lewy body dementia. Ann Neurol. (2004) 55:164–73. doi: 10.1002/ana.10795
66. Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B. The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci. (2000) 20:6048–54. doi: 10.1523/JNEUROSCI.20-16-06048.2000
67. Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, et al. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett. (2004) 576:363–8. doi: 10.1016/j.febslet.2004.09.038
68. Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, et al. The E46K mutation in alpha-synuclein increases amyloid fibril formation. J Biol Chem. (2005) 280:7800–7. doi: 10.1074/jbc.M411638200
69. Khalaf O, Fauvet B, Oueslati A, Dikiy I, Mahul-Mellier AL, Ruggeri FS, et al. The H50Q mutation enhances αα-synuclein aggregation, secretion, and toxicity. J Biol Chem. (2014) 289:21856–76. doi: 10.1074/jbc.M114.553297
70. Nishioka K, Hayashi S, Farrer MJ, Singleton AB, Yoshino H, Imai H, et al. Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson's disease. Ann Neurol. (2006) 59:298–309. doi: 10.1002/ana.20753
71. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. (2003) 302:841. doi: 10.1126/science.1090278
72. Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. (2004) 364:1167–9. doi: 10.1016/S0140-6736(04)17103-1
73. Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol. (2004) 55:174–9. doi: 10.1002/ana.10846
74. Bridi JC, Hirth F. Mechanisms of α-Synuclein induced synaptopathy in parkinson's disease. Front Neurosci. (2018) 12:80. doi: 10.3389/fnins.2018.00080
75. Chakraborty R, Chattopadhyay K. Cryo-electron microscopy uncovers key residues within the core of alpha-synuclein fibrils. ACS Chem Neurosci. (2019) 10:1135–6. doi: 10.1021/acschemneuro.9b00090
76. Maroteaux L, Campanelli JT, Scheller RH. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. (1988) 8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988
77. Clayton DF, George JM. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. (1998) 21:249–54. doi: 10.1016/S0166-2236(97)01213-7
79. Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA. (1993) 90:11282–6. doi: 10.1073/pnas.90.23.11282
81. Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT. NACP. a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. (1996) 35:13709–15. doi: 10.1021/bi961799n
82. van Rooijen BD, van Leijenhorst-Groener KA, Claessens MMAE, Subramaniam V. Tryptophan fluorescence reveals structural features of alpha-synuclein oligomers. J Mol Biol. (2009) 394:826–33. doi: 10.1016/j.jmb.2009.10.021
83. Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. (2011) 477:107–10. doi: 10.1038/nature10324
84. Burré J, Vivona S, Diao J, Sharma M, Brunger AT, Südhof TC. Properties of native brain α-synuclein. Nature. (2013) 498:4–6. doi: 10.1038/nature12125
85. Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, et al. Structure of the toxic core of α-synuclein from invisible crystals. Nature. (2015) 525:486. doi: 10.1038/nature15368
86. Ma L, Yang C, Zhang X, Li Y, Wang S, Zheng L, et al. C-terminal truncation exacerbates the aggregation and cytotoxicity of α-Synuclein: A vicious cycle in Parkinson's disease. Biochim Biophys Acta – Mol Basis Dis. (2018) 1864:3714–25. doi: 10.1016/j.bbadis.2018.10.003
87. Stephens AD, Zacharopoulou M, Moons R, Fusco G, Seetaloo N, Chiki A, et al. Extent of N-terminus exposure of monomeric alpha-synuclein determines its aggregation propensity. Nat Commun. (2020) 11:1–15. doi: 10.1038/s41467-020-16564-3
88. Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease-linked mutations. Proc Natl Acad Sci USA. (2005) 102:2162–7. doi: 10.1073/pnas.0406976102
89. Iyer A, Roeters SJ, Kogan V, Woutersen S, Claessens MMAE, et al. C-terminal truncated α-synuclein fibrils contain strongly twisted β-sheets. J Am Chem Soc. (2017) 139:15392–400. doi: 10.1021/jacs.7b07403
90. Sorrentino ZA, Giasson BI. The emerging role of α-synuclein truncation in aggregation and disease. J Biol Chem. (2020) 295:10224–44. doi: 10.1074/jbc.REV120.011743
91. Gai WP, Power JH, Blumbergs PC, Culvenor JG, Jensen PH. Alpha-synuclein immunoisolation of glial inclusions from multiple system atrophy brain tissue reveals multiprotein components. J Neurochem. (1999) 73:2093–100. doi: 10.1046/j.1471-4159.1999.02093.x
92. Prasad K, Beach TG, Hedreen J, Richfield EK. Critical role of truncated α-synuclein and aggregates in Parkinson's disease and incidental Lewy body disease. Brain Pathol. (2012) 22:811–25. doi: 10.1111/j.1750-3639.2012.00597.x
93. Sorrentino ZA, Vijayaraghavan N, Gorion KM, Riffe CJ, Strang KH, Caldwell J, et al. Physiological C-terminal truncation of α-synuclein potentiates the prion-like formation of pathological inclusions. J Biol Chem. (2018) 293:18914. doi: 10.1074/jbc.RA118.005603
94. McGlinchey RP, Lacy SM, Huffer KE, Tayebi N, Sidransky E, Lee JC. C-terminal α-synuclein truncations are linked to cysteine cathepsin activity in Parkinson's disease. J Biol Chem. (2019) 294:9973–84. doi: 10.1074/jbc.RA119.008930
95. Zhang J, Li X, Li J. The roles of post-translational modifications on α-synuclein in the pathogenesis of Parkinson's diseases. Front Neurosci. (2019) 13:381. doi: 10.3389/fnins.2019.00381
96. He Y, Yu Z, Chen S. Alpha-synuclein nitration and its implications in Parkinson's disease. ACS Chem Neurosci. (2019) 10:777–82. doi: 10.1021/acschemneuro.8b00288
97. Ponzini E, De Palma A, Cerboni L, Natalello A, Rossi R, Moons R, et al. Methionine oxidation in -synuclein inhibits its propensity for ordered secondary structure. J Biol Chem. (2019) 294:5657–65. doi: 10.1074/jbc.RA118.001907
98. Roeters SJ, Iyer A, Pletikapiä G, Kogan V, Subramaniam V, Woutersen S. Evidence for intramolecular antiparallel beta-sheet structure in alpha-synuclein fibrils from a combination of two-dimensional infrared spectroscopy and atomic force microscopy. Sci Rep. (2017) 7:1–11. doi: 10.1038/srep41051
99. Lv Z, Krasnoslobodtsev AV, Zhang Y, Ysselstein D, Rochet JC, Blanchard SC, et al. Effect of acidic pH on the stability of α-synuclein dimers. Biopolymers. (2016) 105:715–24. doi: 10.1002/bip.22874
100. Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. (2000) 25:239–52. doi: 10.1016/S0896-6273(00)80886-7
101. Burré J. The synaptic function of α-synuclein. J Parkinsons Dis. (2015) 5:699–713. doi: 10.3233/JPD-150642
102. Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. (2000) 20:3214–20. doi: 10.1523/JNEUROSCI.20-09-03214.2000
103. Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, et al. α-Synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. (2006) 26:11915–22. doi: 10.1523/JNEUROSCI.3821-06.2006
104. Froula JM, Henderson BW, Gonzalez JC, Vaden JH, Mclean JW, Wu Y, et al. α-Synuclein fibril-induced paradoxical structural and functional defects in hippocampal neurons. Acta Neuropathol Commun. (2018) 6:35. doi: 10.1186/s40478-018-0537-x
105. Sun J, Wang L, Bao H, Premi S, Das U, Chapman ER, et al. Functional cooperation of α-synuclein and VAMP2 in synaptic vesicle recycling. Proc Natl Acad Sci USA. (2019) 116:11113–5. doi: 10.1073/pnas.1903049116
106. Vicario M, Cieri D, Brini M, Cal,ì T. The close encounter between alpha-synuclein and mitochondria. Front Neurosci. (2018) 12:388. doi: 10.3389/fnins.2018.00388
107. Wildburger NC, Hartke AS, Schidlitzki A, Richter F. Current evidence for a bidirectional loop between the lysosome and alpha-synuclein proteoforms. Front Cell Dev Biol. (2020) 8:1372. doi: 10.3389/fcell.2020.598446
108. Devoto VMP, Dimopoulos N, Alloatti M, Pardi MB, Saez TM, Otero MG, et al. αSynuclein control of mitochondrial homeostasis in human-derived neurons is disrupted by mutations associated with Parkinson's disease. Sci Rep. (2017) 7:1–13. doi: 10.1038/s41598-017-05334-9
109. Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. (2010) 29:3571–89. doi: 10.1038/emboj.2010.223
110. Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J Biol Chem. (2011) 286:20710–26. doi: 10.1074/jbc.M110.213538
111. Pinho R, Paiva I, Jerčić KG, Fonseca-Ornelas L, Gerhardt E, Fahlbusch C, et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum Mol Genet. (2019) 28:31–50. doi: 10.1093/hmg/ddy326
112. Davidi D, Schechter M, Elhadi SA, Matatov A, Nathanson L, Sharon R. α-Synuclein translocates to the nucleus to activate retinoic-acid-dependent gene transcription. iScience. (2020) 23:100910. doi: 10.1016/j.isci.2020.100910
113. Surguchev AA, Surguchov A. Synucleins and gene expression: Ramblers in a crowd or cops regulating traffic? Front Mol Neurosci. (2017) 10:224. doi: 10.3389/fnmol.2017.00224
114. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. (2007) 131:861–72. doi: 10.1016/j.cell.2007.11.019
115. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
116. Pappas JJ, Yang PC. Human ESC vs. iPSC-pros and cons. J Cardiovasc Transl Res. (2008) 1:96–9. doi: 10.1007/s12265-008-9032-2
117. Bassett AR. Editing the genome of hiPSC with CRISPR/Cas9: disease models. Mamm Genome. (2017) 28:348–64. doi: 10.1007/s00335-017-9684-9
118. Ben Jehuda R, Shemer Y, Binah O. Genome editing in induced pluripotent stem cells using CRISPR/Cas9. Stem Cell Rev. (2018) 14:323–36. doi: 10.1007/s12015-018-9811-3
119. Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: A decade of progress. Nat Rev Drug Discov. (2017) 16:115–30. doi: 10.1038/nrd.2016.245
120. Rowe RG, Daley GQ. Induced pluripotent stem cells in disease modelling and drug discovery. Nat Rev Genet. (2019) 20:377–88. doi: 10.1038/s41576-019-0100-z
121. Chang CY, Ting HC, Liu CA, Su HL, Chiou TW, Lin SZ, et al. Induced pluripotent stem cell (iPSC)-based neurodegenerative disease models for phenotype recapitulation and drug screening. Molecules. (2020) 25:2000. doi: 10.3390/molecules25082000
122. Kawasaki H, Mizuseki K, Nishikawa S, Kaneko S, Kuwana Y, Nakanishi S, et al. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron. (2000) 28:31–40. doi: 10.1016/S0896-6273(00)00083-0
123. Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. (2009) 27:275–80. doi: 10.1038/nbt.1529
124. Lee SH, Lumelsky N, Studer L, Auerbach JM, McKay RD. Efficient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat Biotechnol. (2000) 18:675–9. doi: 10.1038/76536
125. Zhang SC, Wernig M, Duncan ID, Brüstle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. (2001) 19:1129–33. doi: 10.1038/nbt1201-1129
126. Kirkeby A, Grealish S, Wolf DA, Nelander J, Wood J, Lundblad M, et al. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. (2012) 1:703–14. doi: 10.1016/j.celrep.2012.04.009
127. Fasano CA, Chambers SM, Lee G, Tomishima MJ, Studer L. Efficient derivation of functional floor plate tissue from human embryonic stem cells. Cell Stem Cell. (2010) 6:336–47. doi: 10.1016/j.stem.2010.03.001
128. Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature. (2011) 480:547–51. doi: 10.1038/nature10648
129. Jovanovic VM, Salti A, Tilleman H, Zega K, Jukic MM, Zou H, et al. BMP/SMAD pathway promotes neurogenesis of midbrain dopaminergic neurons in vivo and in human induced pluripotent and neural stem cells. J Neurosci. (2018) 38:1662–76. doi: 10.1523/JNEUROSCI.1540-17.2018
130. Morizane A, Doi D, Kikuchi T, Nishimura K, Takahashi J. Small-molecule inhibitors of bone morphogenic protein and activin/nodal signals promote highly efficient neural induction from human pluripotent stem cells. J Neurosci Res. (2011) 89:117–26. doi: 10.1002/jnr.22547
131. Doi D, Magotani H, Kikuchi T, Ikeda M, Hiramatsu S, Yoshida K, et al. Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson's disease. Nat Commun. (2020) 11:1–14. doi: 10.1038/s41467-020-17165-w
132. Barker RA, Farrell K, Guzman NV, He X, Lazic SE, Moore S, et al. Designing stem-cell-based dopamine cell replacement trials for Parkinson's disease. Nat Med. (2019) 257:1045–53. doi: 10.1038/s41591-019-0507-2
133. Barker RA, Parmar M, Studer L, Takahashi J. Human trials of stem cell-derived dopamine neurons for parkinson's disease: dawn of a new era. Cell Stem Cell. (2017) 21:569–73. doi: 10.1016/j.stem.2017.09.014
134. Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. (2014) 9:2329–40. doi: 10.1038/nprot.2014.158
135. Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Yang SM, Berger DR, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. (2017) 545:48–53. doi: 10.1038/nature22047
136. Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc Natl Acad Sci USA. (2013) 110:20284–9. doi: 10.1073/pnas.1315710110
137. Sloan SA, Andersen J, Paşca AM, Birey F, Paşca SP. Generation and assembly of human brain region-specific three-dimensional cultures. Nat Protoc. (2018) 13:2062–85. doi: 10.1038/s41596-018-0032-7
138. Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, et al. Assembly of functionally integrated human forebrain spheroids. Nature. (2017) 545:54–9. doi: 10.1038/nature22330
139. Pellegrini L, Bonfio C, Chadwick J, Begum F, Skehel M, Lancaster MA. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science. (2020) 369:eaaz5626. doi: 10.1126/science.aaz5626
140. Muguruma K, Nishiyama A, Kawakami H, Hashimoto K, Sasai Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. (2015) 10:537–50. doi: 10.1016/j.celrep.2014.12.051
141. Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell. (2012) 10:771–85. doi: 10.1016/j.stem.2012.05.009
142. Qian X, Jacob F, Song MM, Nguyen HN, Song H, Ming GL. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nat Protoc. (2018) 13:565–80. doi: 10.1038/nprot.2017.152
143. Jo J, Xiao Y, Sun AX, Cukuroglu E, Tran HD, Göke J, et al. Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell. (2016) 19:248–57. doi: 10.1016/j.stem.2016.07.005
144. Monzel AS, Smits LM, Hemmer K, Hachi S, Moreno EL, van Wuellen T, et al. Derivation of human midbrain-specific organoids from neuroepithelial stem cells. Stem Cell Rep. (2017) 8:1144–54. doi: 10.1016/j.stemcr.2017.03.010
145. Smits LM, Magni S, Kinugawa K, Grzyb K, Luginbühl J, Sabate-Soler S, et al. Single-cell transcriptomics reveals multiple neuronal cell types in human midbrain-specific organoids. Cell Tissue Res. (2020) 382:463–76. doi: 10.1007/s00441-020-03249-y
146. Tieng V, Stoppini L, Villy S, Fathi M, Dubois-Dauphin M, Krause KH. Engineering of midbrain organoids containing long-lived dopaminergic neurons. Stem Cells Dev. (2014) 23:1535–47. doi: 10.1089/scd.2013.0442
147. Smits LM, Reinhardt L, Reinhardt P, Glatza M, Monzel AS, Stanslowsky N, et al. Modeling Parkinson's disease in midbrain-like organoids. NPJ Park Dis. (2019) 5:1–8. doi: 10.1038/s41531-019-0078-4
148. Kwak TH, Kang JH, Hali S, Kim J, Kim KP, Park C, et al. Generation of homogeneous midbrain organoids with in vivo-like cellular composition facilitates neurotoxin-based Parkinson's disease modeling. Stem Cells. (2020) 38:727–40. doi: 10.1002/stem.3163
149. Jo J, Yang L, Tran HD, Yu W, Sun AX, Chang YY, et al. Lewy body-like inclusions in human midbrain organoids carrying glucocerebrosidase and α-synuclein mutations. Ann Neurol. (2021) 90:490–505. doi: 10.1002/ana.26166
150. Byers B, Cord B, Nguyen HN, Schüle B, Fenno L, Lee PC, et al. SNCA triplication parkinson's patient's iPSC-Derived DA neurons accumulate α-Synuclein and are susceptible to oxidative stress. PLoS ONE. (2011) 6:e26159. doi: 10.1371/journal.pone.0026159
151. Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, et al. Parkinson's disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat Commun. (2011) 2:1–10. doi: 10.1038/ncomms1453
152. Oliveira LMA, Falomir-Lockhart LJ, Botelho MG, Lin KH, Wales P, Koch JC, et al. Elevated α-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson's patient-derived induced pluripotent stem cells. Cell Death Dis. (2015) 6:1994. doi: 10.1038/cddis.2015.318
153. Lin L, Göke J, Cukuroglu E, Dranias MR, VanDongen AMJ, Stanton LW. Molecular features underlying neurodegeneration identified through in vitro modeling of genetically diverse parkinson's disease patients. Cell Rep. (2016) 15:2411–26. doi: 10.1016/j.celrep.2016.05.022
154. Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci USA. (2016) 113:1931–6. doi: 10.1073/pnas.1520335113
155. Prots I, Grosch J, Brazdis RM, Simmnacher K, Veber V, Havlicek S, et al. α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc Natl Acad Sci USA. (2018) 115:7813–8. doi: 10.1073/pnas.1713129115
156. Zambon F, Cherubini M, Fernandes HJR, Lang C, Ryan BJ, Volpato V, et al. Cellular α-synuclein pathology is associated with bioenergetic dysfunction in Parkinson's iPSC-derived dopamine neurons. Hum Mol Genet. (2019) 28:2001–13. doi: 10.1093/hmg/ddz038
157. Siddu A, David LS, Lauinger N, Chen X, Saint-Pierre M, Alpaugh M, et al. Beneficial effects of cysteamine in Thy1-α-Syn mice and induced pluripotent stem cells with a SNCA gene triplication. Neurobiol Dis. (2020) 145:1–17. doi: 10.1016/j.nbd.2020.105042
158. Ludtmann MHR, Angelova PR, Horrocks MH, Choi ML, Rodrigues M, Baev AY, et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson's disease. Nat Commun. (2018) 9:1–16. doi: 10.1038/s41467-018-04422-2
159. Heman-Ackah SM, Manzano R, Hoozemans JJM, Scheper W, Flynn R, Haerty W, et al. Alpha-synuclein induces the unfolded protein response in Parkinson's disease SNCA triplication iPSC-derived neurons. Hum Mol Genet. (2017) 26:4441–50. doi: 10.1093/hmg/ddx331
160. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. (2009) 361:1651–61. doi: 10.1056/NEJMoa0901281
161. Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. (2011) 146:37–52. doi: 10.1016/j.cell.2011.06.001
162. Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. (2013) 19:983–97. doi: 10.1038/nm.3232
163. Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF, et al. Activation of β-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson's patient midbrain neurons. J. Neurosci. (2016) 36:7693–706. doi: 10.1523/JNEUROSCI.0628-16.2016
164. Chen V, Moncalvo M, Tringali D, Tagliafierro L, Shriskanda A, Ilich E, et al. The mechanistic role of alpha-synuclein in the nucleus: Impaired nuclear function caused by familial Parkinson's disease SNCA mutations. Hum Mol Genet. (2020) 29:3107–21. doi: 10.1093/hmg/ddaa183
165. Vasquez V, Mitra J, Hegde PM, Pandey A, Sengupta S, Mitra S, et al. Chromatin-bound oxidized α-synuclein causes strand breaks in neuronal genomes in in vitro models of Parkinson's disease. J Alzheimer's Dis. (2017) 60:133–50. doi: 10.3233/JAD-170342
166. Xia N, Zhang P, Fang F, Wang Z, Rothstein M, Angulo B, et al. Transcriptional comparison of human induced and primary midbrain dopaminergic neurons. Sci Rep. (2016) 6:1–9. doi: 10.1038/srep20270
167. Pitari G, Maurizi G, Flati V, Ursini CL, Spera L, Duprè S, et al. Enzymatic synthesis of S-aminoethyl-l-cysteine from pantetheine. BBA – Gen Subj. (1992) 1116:27–33. doi: 10.1016/0304-4165(92)90124-D
168. Cicchetti F, David LS, Siddu A, Denis HL. Cysteamine as a novel disease-modifying compound for Parkinson's disease: Over a decade of research supporting a clinical trial. Neurobiol Dis. (2019) 130:104530. doi: 10.1016/j.nbd.2019.104530
169. Gibrat C, Cicchetti F. Potential of cystamine and cysteamine in the treatment of neurodegenerative diseases. Prog Neuro-Psychopharmacology Biol Psychiatry. (2011) 35:380–9. doi: 10.1016/j.pnpbp.2010.11.023
170. Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, et al. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med. (2002) 8:143–9. doi: 10.1038/nm0202-143
171. Apostol BL, Kazantsev A, Raffioni S, Illes K, Pallos J, Bodai L, et al. A cell-based assay for aggregation inhibitors as therapeutics of polyglutamine-repeat disease and validation in Drosophila. Proc Natl Acad Sci USA. (2003) 100:5950–5. doi: 10.1073/pnas.2628045100
172. Bailey CDC, Johnson GVW. The protective effects of cystamine in the R6/2 Huntington's disease mouse involve mechanisms other than the inhibition of tissue transglutaminase. Neurobiol Aging. (2006) 27:871–9. doi: 10.1016/j.neurobiolaging.2005.04.001
173. Tagliafierro L, Zamora ME, Chiba-Falek O. Multiplication of the SNCA locus exacerbates neuronal nuclear aging. Hum Mol Genet. (2019) 28:407–21. doi: 10.1093/hmg/ddy355
174. Reyes JF, Olsson TT, Lamberts JT, Devine MJ, Kunath T, Brundin P. A cell culture model for monitoring α-synuclein cell-to-cell transfer. Neurobiol Dis. (2015) 77:266–75. doi: 10.1016/j.nbd.2014.07.003
175. Prusiner SB, Hadlow WJ, Eklund CM, Race RE. Sedimentation properties of the scrapie agent. Proc Natl Acad Sci USA. (1977) 74:4656–60. doi: 10.1073/pnas.74.10.4656
176. Brazdis RM, Alecu JE, Marsch D, Dahms A, Simmnacher K, Lörentz S, et al. Demonstration of brain region-specific neuronal vulnerability in human iPSC-based model of familial Parkinson's disease. Hum Mol Genet. (2021) 29:1180–91. doi: 10.1093/hmg/ddaa039
177. Eriksen JL, Przedborski S, Petrucelli L. Gene dosage and pathogenesis of Parkinson's disease. Trends Mol Med. (2005) 11:91–6. doi: 10.1016/j.molmed.2005.01.001
178. Ross OA, Braithwaite AT, Skipper LM, Kachergus J, Hulihan MM, Middleton FA, et al. Genomic investigation of α-synuclein multiplication and parkinsonism. Ann Neurol. (2008) 63:743–50. doi: 10.1002/ana.21380
179. Kouroupi G, Taoufik E, Vlachos IS, Tsioras K, Antoniou N, Papastefanaki F, et al. Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson's disease. Proc Natl Acad Sci USA. (2017) 114:3679–88. doi: 10.1073/pnas.1617259114
180. Ryan T, Bamm VV, Stykel MG, Coackley CL, Humphries KM, Jamieson-Williams R, et al. Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat Commun. (2018) 9:1–17. doi: 10.1038/s41467-018-03241-9
181. Stykel MG, Humphries K, Kirby MP, Czaniecki C, Wang T, Ryan T, et al. Nitration of microtubules blocks axonal mitochondrial transport in a human pluripotent stem cell model of Parkinson's disease. FASEB J. (2018) 32:5350–64. doi: 10.1096/fj.201700759RR
182. Fernandes HJR, Patikas N, Foskolou S, Field SF, Park JE, Byrne ML, et al. Single-cell transcriptomics of parkinson's disease human in vitro models reveals dopamine neuron-specific stress responses. Cell Rep. (2020) 33:108263. doi: 10.1016/j.celrep.2020.108263
183. Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, et al. Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science. (2013) 342:983–7. doi: 10.1126/science.1245296
184. Khurana V, Peng J, Chung CY, Auluck PK, Fanning S, Tardiff DF, et al. Genome-scale networks link neurodegenerative disease genes to α-synuclein through specific molecular pathways. Cell Syst. (2017) 4:157–70. doi: 10.1016/j.cels.2016.12.011
185. Lee SY, Jeong SK, Kim J, Chung SK. Generation of gene-corrected iPSC line from Parkinson's disease patient iPSC line with alpha-SNCA A53T mutation. Stem Cell Res. (2018) 30:145–9. doi: 10.1016/J.SCR.2018.06.002
186. Soldner F, Laganière J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset parkinson point mutations. Cell. (2011) 146:318–31. doi: 10.1016/j.cell.2011.06.019
187. Grudina C, Kouroupi G, Nonaka T, Hasegawa M, Matsas R, Zurzolo C. Human NPCs can degrade α-syn fibrils and transfer them preferentially in a cell contact-dependent manner possibly through TNT-like structures. Neurobiol Dis. (2019) 132:104609. doi: 10.1016/j.nbd.2019.104609
188. Li Z, Okamoto KI, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. (2004) 119:873–87. doi: 10.1016/j.cell.2004.11.003
189. Jensen C, Teng Y. Is it time to start transitioning from 2D to 3D cell culture? Front Mol Biosci. (2020) 7:33. doi: 10.3389/fmolb.2020.00033
190. Kim J, Koo BK, Knoblich JA. Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol. (2020) 21:571–84. doi: 10.1038/s41580-020-0259-3
191. Mohamed N.-V., Sirois J, Ramamurthy J, Mathur M, Lépine P, et al. Midbrain organoids with an SNCA gene triplication model key features of synucleinopathy. Brain Commun. (2021) 3:223. doi: 10.1093/braincomms/fcab223
192. Gallagher MD, Chen-Plotkin AS. The post-GWAS Era: From association to function. Am J Hum Genet. (2018) 102:717–30. doi: 10.1016/j.ajhg.2018.04.002
193. Konnova EA, Swanberg M. Animal Models of Parkinson's Disease in Parkinson's Disease: Pathogenesis and Clinical Aspects (2018). doi: 10.15586/codonpublications.parkinsonsdisease.2018.ch5
194. Kin K, Yasuhara T, Kameda M, Date I. Animal models for Parkinson's disease research: trends in the 2000s. Int J Mol Sci. (2019) 20:5402. doi: 10.3390/ijms20215402
195. Xicoy H, Wieringa B, Martens GJM. The SH-SY5Y cell line in Parkinson's disease research: a systematic review. Mol Neurodegener. (2017) 12:1–11. doi: 10.1186/s13024-017-0149-0
196. Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P. Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J Biol Chem. (2002) 277:38884–94. doi: 10.1074/jbc.M205518200
197. Ferrari E, Cardinale A, Picconi B, Gardoni F. From cell lines to pluripotent stem cells for modelling Parkinson's disease. J Neurosci Methods. (2020) 340:108741. doi: 10.1016/j.jneumeth.2020.108741
198. Haenseler W, Zambon F, Lee H, Vowles J, Rinaldi F, Duggal G, et al. Excess α-synuclein compromises phagocytosis in iPSC-derived macrophages. Sci Rep. (2017) 7:1–11. doi: 10.1038/s41598-017-09362-3
199. Badanjak K, Mulica P, Smajic S, Delcambre S, Tranchevent LC, et al. iPSC-derived microglia as a model to study inflammation in idiopathic Parkinson's disease. Front Cell Dev Biol. (2021) 9:740758. doi: 10.3389/fcell.2021.740758
200. Fanning S, Haque A, Imberdis T, Baru V, Barrasa MI, Nuber S, et al. Lipidomic analysis of α-synuclein neurotoxicity identifies stearoyl CoA desaturase as a target for parkinson treatment. Mol Cell. (2019) 73:1001–14. doi: 10.1016/j.molcel.2018.11.028
201. Sonninen TM, Hämäläinen RH, Koskuvi M, Oksanen M, Shakirzyanova A, Wojciechowski S, et al. Metabolic alterations in Parkinson's disease astrocytes. Sci Rep. (2020) 10:1–14. doi: 10.1038/s41598-020-71329-8
202. Okarmus J, Havelund JF, Ryding M, Schmidt SI, Bogetofte H, Heon-Roberts R, et al. Identification of bioactive metabolites in human iPSC-derived dopaminergic neurons with PARK2 mutation: Altered mitochondrial and energy metabolism. Stem Cell Rep. (2021) 16:1510–26. doi: 10.1016/j.stemcr.2021.04.022
203. Ng KM, Lau YM, Dhandhania V, Cai ZJ, Lee YK, Lai WH, et al. Empagliflozin ammeliorates high glucose induced-cardiac dysfuntion in human iPSC-derived cardiomyocytes. Sci Rep. (2018) 8:1–13. doi: 10.1038/s41598-018-33293-2
204. Weissbein U, Benvenisty N, Ben-David U. Quality control: Genome maintenance in pluripotent stem cells. J Cell Biol. (2014) 204:153–63. doi: 10.1083/jcb.201310135
205. Bar S, Benvenisty N. Epigenetic aberrations in human pluripotent stem cells. EMBO J. (2019) 38:10103–4. doi: 10.15252/embj.2018101033
206. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. (2013) 14:1–20. doi: 10.1186/gb-2013-14-10-r115
207. Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature. (2017) 545:229–33. doi: 10.1038/nature22312
208. Yamanaka S. Pluripotent stem cell-based cell therapy-promise and challenges. Cell Stem Cell. (2020) 27:523–31. doi: 10.1016/j.stem.2020.09.014
209. Lezmi E, Benvenisty N. Identification of cancer-related mutations in human pluripotent stem cells using RNA-seq analysis. Nat. Protoc. (2021) 16:4522–37. doi: 10.1038/s41596-021-00591-5
210. Zheng YL. Some ethical concerns about human induced pluripotent stem cells. Sci Eng Ethics. (2015) 22:1277–84. doi: 10.1007/s11948-015-9693-6
211. Assen LS, Jongsma KR, Isasi R, Tryfonidou MA, Bredenoord AL. Recognizing the ethical implications of stem cell research: A call for broadening the scope. Stem Cell Rep. (2021) 16:1656–61. doi: 10.1016/j.stemcr.2021.05.021
212. Marion RM, Strati K, Li H, Tejera A, Schoeftner S, Ortega S, et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. (2009) 4:141–54. doi: 10.1016/j.stem.2008.12.010
213. Lo Sardo V, Ferguson W, Erikson GA, Topol EJ, Baldwin KK, Torkamani A. The effect of aging on human induced pluripotent stem cells. Nat Biotechnol. (2017) 35:69. doi: 10.1038/nbt.3749
214. Cieślar-Pobuda A, Knoflach V, Ringh MV, Stark J, Likus W, Siemianowicz K, et al. Transdifferentiation and reprogramming: Overview of the processes, their similarities and differences. Biochim Biophys Acta Mol Cell Res. (2017) 1864:1359–69. doi: 10.1016/j.bbamcr.2017.04.017
215. Caiazzo M, Dell'Anno MT, Dvoretskova E, Lazarevic D, Taverna S, Leo D, et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature. (2011) 476:224–7. doi: 10.1038/nature10284
216. Pfisterer U, Kirkeby A, Torper O, Wood J, Nelander J, Dufour A, et al. Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci USA. (2011) 108:10343–8. doi: 10.1073/pnas.1105135108
217. Addis RC, Hsu FC, Wright RL, Dichter MA, Coulter DA, Gearhart JD. Efficient conversion of astrocytes to functional midbrain dopaminergic neurons using a single polycistronic vector. PLoS ONE. (2011) 6:28719–22. doi: 10.1371/journal.pone.0028719
218. Han DW, Tapia N, Hermann A, Hemmer K, Höing S, Araúzo-Bravo MJ, et al. Direct reprogramming of fibroblasts into neural stem cells by defined factors. Cell Stem Cell. (2012) 10:465–72. doi: 10.1016/j.stem.2012.02.021
219. Hermann A, Storch A. Induced neural stem cells (iNSCs) in neurodegenerative diseases. J Neural Transm. (2013) 120:19–25. doi: 10.1007/s00702-013-1042-9
220. Lee M, Sim H, Ahn H, Ha J, Baek A, Jeon YJ, et al. Direct reprogramming to human induced neuronal progenitors from fibroblasts of familial and sporadic parkinson's disease patients. Int J Stem Cells. (2019) 12:474–83. doi: 10.15283/ijsc19075
221. Thier MC, Hommerding O, Panten J, Pinna R, García-González D, Berger T, et al. Identification of embryonic neural plate border stem cells and their generation by direct reprogramming from adult human blood cells. Cell Stem Cell. (2019) 24:166–82.e13. doi: 10.1016/j.stem.2018.11.015
222. Meyer S, Wörsdörfer P, Günther K, Thier M, Edenhofer F. Derivation of adult human fibroblasts and their direct conversion into expandable neural progenitor cells. J Vis Exp. (2015) 2015:e52831. doi: 10.3791/52831
223. Erharter A, Rizzi S, Mertens J, Edenhofer F. Take the shortcut - direct conversion of somatic cells into induced neural stem cells and their biomedical applications. FEBS Lett. (2019) 593:3353–69. doi: 10.1002/1873-3468.13656
224. Gómez-Benito M, Granado N, García-Sanz P, Michel A, Dumoulin M, Moratalla R. Modeling Parkinson's disease with the alpha-synuclein protein. Front Pharmacol. (2020) 11:356. doi: 10.3389/fphar.2020.00356
Keywords: induced pluripotent stem cells, α-synuclein, Parkinson's disease, neurons, SNCA triplication, SNCA duplication, A53T SNCA point mutation
Citation: Spathopoulou A, Edenhofer F and Fellner L (2022) Targeting α-Synuclein in Parkinson's Disease by Induced Pluripotent Stem Cell Models. Front. Neurol. 12:786835. doi: 10.3389/fneur.2021.786835
Received: 30 September 2021; Accepted: 24 December 2021;
Published: 25 January 2022.
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Jordi Bové, Vall d'Hebron Research Institute (VHIR), SpainNicolas Casadei, University Hospital and Faculty of Medicine, University of Tübingen, Germany
Copyright © 2022 Spathopoulou, Edenhofer and Fellner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa Fellner, bGlzYS5mZWxsbmVyQHVpYmsuYWMuYXQ=