 Chen Zhang1,2
Chen Zhang1,2 Fafa Tian
Fafa Tian Xiaoyan Long
Xiaoyan Long- 1Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
Introduction: Mossy fiber sprouting (MFS) is a frequent histopathological finding in temporal lobe epilepsy (TLE) and is involved in the pathology of TLE. However, molecular signals underlying MFS remain unclear. Partitioning defective 3(Par3), atypical protein kinase C-λ(aPKC-λ), and lethal giant larvae 1(Lgl1) were involved in the neuronal polarity and axon growth. The potential roles of those proteins in MFS and epileptogenesis of TLE were investigated.
Material and Methods: The epileptic rat models were established by intracerebroventricular injection of kainic acid (KA). The degree of MFS was measured by using Timm staining, Neuronal loss and the expression aPKC-λ, Par3, and Lgl1 in hippocampus were measured by using immunohistochemistry and western blot analysis.
Results: The neuronal loss in CA3 region was observed from 3 days to 8 weeks, while the neuronal loss in the hilar region was observed from 1 to 8 weeks in experimental group. The Timm score in the CA3 region in experimental group was significantly higher than that in the control group from 2 to 8 weeks. Compared with control group, the expressions of Par3 and Lgl1 were upregulated and the expression of aPKC-λ was downregulated in the experimental groups. Positive correlation between the Par3 expression and Timm scores, and the negative correlation between the aPKC-λ expression and Timm scores in CA3 region were discovered in experimental group.
Conclusion: The findings of the present study indicated that aPKC-λ, Par3, and Lgl1 may be involved in MFS and in the epileptogenesis of temporal lobe epilepsy.
Introduction
Epilepsy is a devastating neurological disorder. There are estimated 30–40% patients with epilepsy fail to get satisfactory response to appropriate administrations of antiepileptic drugs or other treatments (1). One of the most common types of refractory epilepsy that can be found in adults is the temporal lobe epilepsy (TLE) (2). Therefore, a better understanding for the pathogenesis of TLE is urgently needed. Although exact pathogenic mechanisms of TLE are still unknown, it is generally accepted that the aberrant axonal outgrowth and the synaptic reorganization caused by Mossy Fiber Sprouting (MFS) are the causes of the imbalance between excitatory and inhibitory inputs in hippocampus, which may contribute to the epileptogenesis of TLE (3, 4). Chen et al. discovered that the Repulsive guidance molecule a (RGMa), a protein regulating axonal growth, was significantly decreased in TLE patients, and an overexpression of RGMa in the hippocampus suppresses seizures, MFS and hyperexcitability of hippocampal neurons (5). It was confirmed that a mammalian target of rapamycin (mTOR) pathway was involved in regulating axonal outgrowth (6, 7). Pun et al. selectively removed the mTOR pathway inhibitor phosphatase and tensin homolog (PTEN) gene from an adult-born granule cells, and then a mice without PTEN gene showed spontaneous seizures (8). Based on this finding, LaSarge et al. discovered that PTEN deletion in dentate granule cells resulted in aberrant axonal growth of mossy fiber and enhanced communication between CA3 area and granule cells, thus, resulting to possible promotion of the epileptogenesis of TLE (9). Those findings implicated that axonal growth may be a potential target to attenuate epileptogenesis of TLE.
Numerous studies confirmed that the Partitioning defective 3(Par3) /Partitioning defective 6(Par6)/atypical protein kinase C (aPKC) complex played a determinant role in cell polarity (10–12) and is required for axon–dendrite specification of hippocampal neurons (13–15). The aPKC family has three isoforms: aPKC-ζ, PKM-ζ and aPKC-λ in vertebrate, and Par3 proteins that interacted with aPKC-λ to achieve the function of regulating cell polarity in neuron and in neural progenitors (16–18). In embryonic hippocampal neurons, the aPKC-λ and Par3 complex has localized the presumptive axon (19), which implicated the involvement of aPKC-λ and Par3 in axon specification during the neuronal differentiation. Previous studies had discovered the long-lasting increased of mRNA expression of PKC epsilon in mossy fiber of adult rats, which followed the seizures induced by kainic acid injection (20). Moreover, Gao et al. also discovered that the PAR3 regulates CNTNAP2 spatial localization (21). CNTNAP2 has been considered a prominent disease susceptibility gene associated with epilepsy (22), and seizure was observed in Cntnap2 knockout rat (23). Those findings implied that Par3 and aPKC-λ have potentiality to be involved in epileptogenesis. However, the functions of aPKC-λ and Par3 in epileptogenesis have not yet been explored.
Lethal giant larvae 1 (Lgl1) was initially known as a tumor suppressor (24) and regulator of polarization processes in variety of cells (25, 26). It has been confirmed that Lgl1 is enriched in developing axons, and upregulation of Lgl1, which has promoted the axonal growth during neuronal morphogenesis (27). Aside from this, Scott et al. found that Lgl1 protein activity is restricted by its phosphorylation by aPKC in the apical neuroblast cortex of flies (28). Nevertheless, this association between Lgl1 and epilepsy has not yet been investigated.
The effects of proteins that were involved in the neuronal polarity are not fully described in epileptogenesis of TLE. By considering the functions of aPKC-λ, Par3, and Lgl1 in neuronal polarity in hippocampal neuron, we also investigated the potential role of those proteins in development of TLE.
Materials and Methods
Ethic Statement
All animals were treated humanely. Study design and all animal experimental protocols were done in accordance with the guidelines of the National Institutes of Health, and the study were approved by the Animal Ethics Committee of Central South University (Changsha, China).
Animals and Model Establishing
Male Sprague–Dawley rats, which are between post-natal days 42 and 56, that weighed between 180 and 220 g, were purchased from the Center for Experimental Animals of Central South University (Changsha, China). The animals were housed under controlled conditions (18-25°C; 50-60% humidity; 12 h light/dark cycle) with food pellets and water available ad libitum.
A total of 120 rats were randomly divided into model group (n = 65) and control group (n = 55) and underwent a surgery for intracerebroventricular injection. Rats were anesthetized with ketamine/xylazine (100/10 mg/kg, I.P.). Rats were placed in a stereotaxic apparatus, and a guide cannula was placed into the lateral ventricle (AP = −0.8, ML = 1.4, DV = 3.3). Kainic acid dissolved in normal saline (0.5 μg/μl) was injected intracerebroventricularly (5 μl/kg, i.c.v.) through a guide cannula by a Hamilton microsyringe at an infusion rate of 0.2 μl/min. The injection usually started 1 min after microsyringe insertion, then cannula will be slowly pulled out of the brain 5 min after the drug injection. Rats in the control group were injected intracerebroventricularly with an equal dose of saline.
The KA-treated rats were observed for the occurrence of seizures immediately following the surgery, and the seizure severity was measured using Racine's scale (29). Five rats died in KA-induced SE. KA-treated rats that demonstrated seizure which reached IV levels and evolved into SE were included in the experimental group (n = 60). All saline-treated rats survived after surgery. All rats from experimental and control groups (n = 60, and 55, respectively) were sacrificed at five-time points after surgery (3 days and 1, 2, 4, 8 weeks post-surgery) for Timm staining, immunohistochemistry, and western blot analysis.
Tissue Processing
At different time points, rats were deeply anesthetized by intraperitoneal injection of ketamine/xylazine (100/10 mg/kg). Rats were perfused intracardially with 300 ml saline for immunohistochemistry or with 200 ml 0.4% sodium sulfide in 0.1 M phosphate buffer (pH 7.3) for Timm staining, then followed by application of 4% paraformaldehyde for perfusion fixation. The brains were removed, were fixed in 4% paraformaldehyde for 24 h, then underwent dehydration using 30% sucrose, then finally were cut into 20 μm coronal sections (frozen cryosections). For western blotting, Hippocampus was separated and was stored at −80°C.
Timm's Staining
In a darkroom, corresponding sections were stained for 90 min in a specific solution which was composed of 100 ml 50% gum arabic, 5 ml citrate buffer (27.2% citric acid and 31% sodium citrate), 15 ml 5.6% hydroquinone, and 0.5 ml 17% silver nitrate. After washing in water, sections were routinely dehydrated, cleaned and mounted with gum. Finally, the corresponding Timm's score for the CA3 region in the hippocampus was analyzed based on the published criteria (30).
Immunohistochemistry
The tissue sections were incubated at room temperature with 3% H2O2 solution for 20 min to block the endogenous peroxidase activity. After being washed three times with PBS, the sections were pre-incubated in PBS containing 0.1%Triton X-100 and 5% normal goat-serum for 30 min, then incubated again overnight with the following primary antibodies at 4°C: rabbit anti-aPKC-λ (1:50, Santa cruz), rabbit anti-PAR3(1:300, Abcam), rabbit anti-LGL1(1:40, Santa Cruz), mouse anti- NeuN(1:1,500, Chemicon). After being washed by PBS, the sections were re-incubated with biotin-labeled anti-rabbit or anti-mouse immunoglobulin (Ig) G secondary antibodies which were raised in a goat (1:200, Cwbiotech) at room temperature for 2 h. Afterwards, these were washed in PBS and incubated in avidin-biotin-horseradish peroxidase complex (1:200, Vector laboratories) at room temperature for another 2 h. Finally, the sections were processed with 3,3′-diaminobenzidine tetrahydrochloride solution (Zsbio) for visualization. Following this, the slides were routinely washed, dehydrated, and mounted. Mean optical density (OD) was calculated by Image pro plus 6. The total number of NeuN(+) cells in CA3 and hilar region of five sections, which were randomly selected from every rat, were calculated at 40× magnification of the microscope.
Western Blotting
Proteins were extracted from hippocampal tissues using RIPA lysate buffer (Beyotime), and then concentrations of proteins were measured using bicinchoninic acid (BCA) Protein Assay Kit (Beyotime). Proteins were separated by using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose membranes. Membranes. which had been washed by using Tris Buffered Saline Tween (TBST) and had been blocked with 5% skimmed milk in TBST (room temperature, 2 h), were incubated at 4°C overnight with primary antibodies. The primary antibodies included rabbit anti-aPKC-λ (1:1,000, Santa Cruz), rabbit anti-PAR3 (1:1,000, Abcam), rabbit anti-LGL1 (1:100, Santa Cruz), and rabbit anti-GAPDH (1:10,000, Proteintech). Unbound antibodies were washed by TBST, then the membranes were subsequently incubated with HRP-labeled goat antirabbit IgG secondary antibodies (1:200, Beyotime) for 1 h. The immunoreactive bands were visualized by using chemiluminescence (ECL) kit (Beyotime), and the optical density (OD) of these bands were quantified by using the Image J software.
Statistical Analysis
All values were expressed as means ± standard deviations. All statistical analyses were performed using SPSS 21.0 for Windows (SPSS Inc., Chicago, IL, USA). Comparisons between two groups were performed by using two-sample t test (parametric) and Mann Whitney U test (non-parametric). Multiple comparisons were performed by using one-way analysis of variance and had been followed, respectively, by three more tests; the Least Significant Difference post-hoc test (parametric), the Kruskal-Wallis test, and the Bonferroni Procedure (non-parametric). The correlation of MFS severity with expression levels of aPKC-λ, Par3, and Lgl1 were investigated by using Spearman rank correlation coefficient. All tests were two-sided and statistical significance was determined at p < 0.05.
Results
KA-Induced SE Lead to Neuronal Loss and MFS in Hippocampus
The number of NeuN-positive cells, which was measured by using NeuN immunohistochemistry in CA3 and hilar regions, was counted to investigate the neuronal death in hippocampus (Figure 1). Compared with control group, the number of NeuN-positive cells in CA3 region decreased significantly from 3 days to 8 weeks (p < 0.001 for all, Figure 1G), and the number of NeuN-positive cells in hilar region decreased significantly from 1 to 8 weeks in experimental group (p < 0.01 for all, Figure 1H).
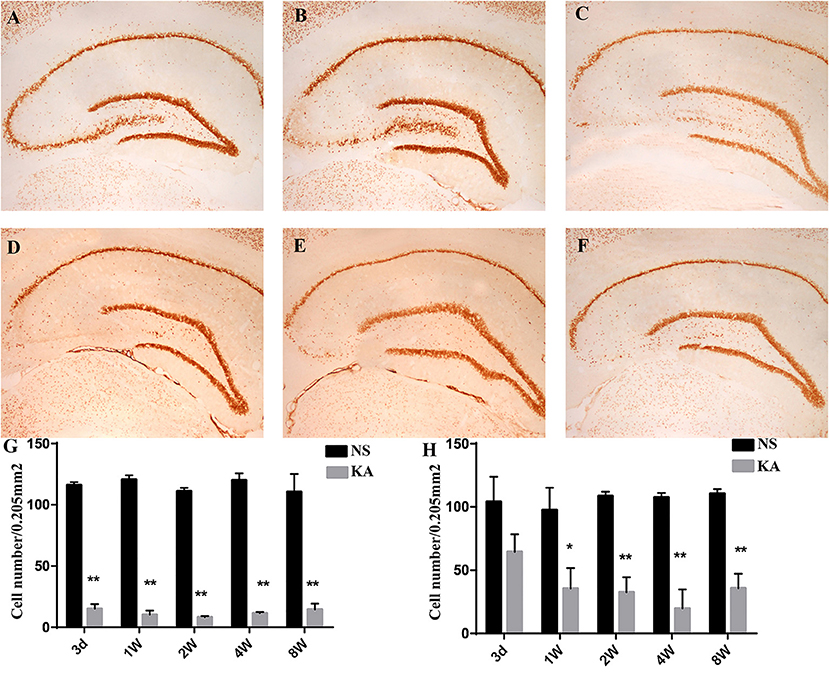
Figure 1. NeuN staining in hippocampus in the control and experimental groups. There is no obvious neuronal loss in control group (A); NeuN staining at five different time points after injection of kainic acid in experimental group: 3 d (B), 1 w (C), 2 w (D), 4 w (E), 8 w (F). The number of NeuN-positive cells measured using NeuN immunohistochemistry in CA3 (G) and hilar regions (H). The data are expressed as the mean ± SD. *P < 0.05, compared with the control; and **P < 0.01, compared with the control; KA, kainic acid; NS, normal saline.
Timm staining was used to evaluate the aberrant mossy fiber reorganization in CA3 region (Figure 2), which was graded by Timm scores (a scale of 0–5). In control group, extremely slight MFS appeared in CA3 region from 1 to 8 weeks, and there was no significant difference in Timm scores between all time points (P > 0.05). Compared with control group, Timm scores increased significantly from 2 to 8 weeks in experimental group (P < 0.05 for all, Figure 2G).
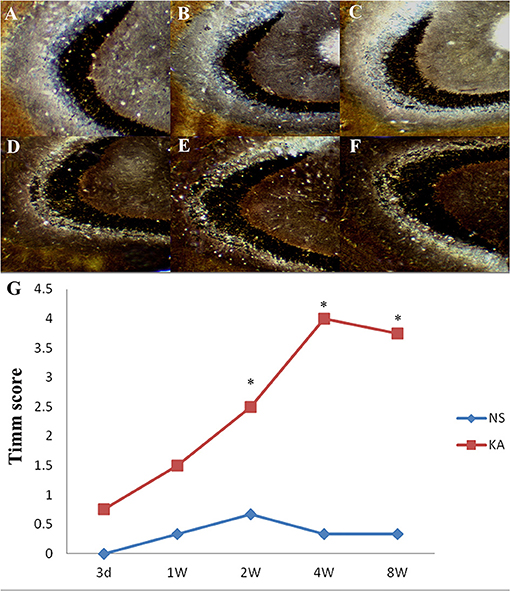
Figure 2. Timm staining in the CA3 region in the control and experimental groups. There is no obvious MFS in control group (A); Timm staining at 5 different time points after injection of kainic acid in experimental group: 3 d (B), 1 w (C), 2 w (D), 4 w (E), 8 w (F). (G) Time-dependent changes of Timm scores in the CA3 region in the experimental (n = 4 at each time point) and control group (n = 3 at each time point). *P < 0.05, compared with the control; KA, kainic acid; NS, normal saline.
Expression of aPKC-λ, Par3, and Lgl1 in Hippocampus
There is no significant difference among the all time points with respect to the expression of aPKC-λ, Par3 and Lgl1 in CA3, and hilar region in control group.
Compared with control group, the expression of Par3 in CA3 region decreased significantly in 3 days and increased from 2 weeks to 8 weeks (p < 0.05 for all, Figures 3, 5A), while the expression of Par3 in hilar region decreased significantly in 1 week and increased from 2 weeks to 8 weeks in experimental group (p < 0.01 for all, Figures 3, 5B).
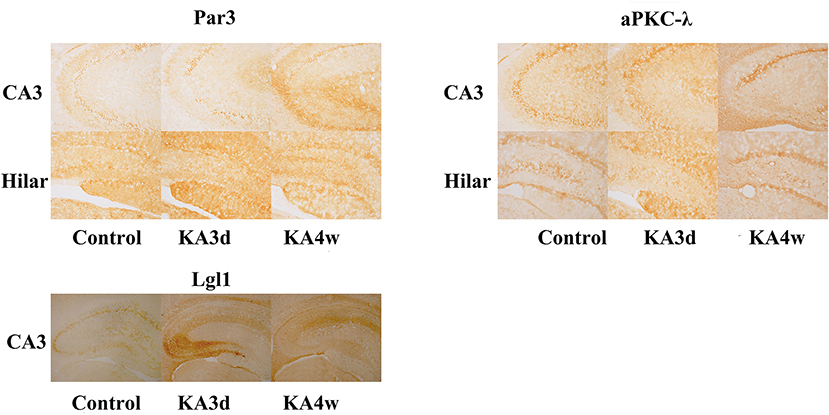
Figure 3. Expression of Par3, aPKC-λ and Lgl1 in the CA3 and hilar region by immunohistochemistry.
Compared with control group, the expression of aPKC-λ in CA3 region decreased significantly at all time points (p < 0.01 for all, Figures 3, 5C), and the expression of aPKC-λ in hilar region decreased significantly from 2 weeks to 8 weeks in experimental group (p < 0.01 for all, Figures 3, 5D).
The expression of Lgl1 was mainly observed in the cytoplasm in the CA3 region in control group, but the expression of Lgl1 mainly appeared in the axon terminal at CA3 region from experimental group. Compared with control group, the expression of LGL1 in CA3 region increased significantly from 3 days to 4 weeks in experimental group (p < 0.05 for all, Figures 3, 5E).
The expression of Par3, aPKC-λ, and Lgl1 in hippocampus have been measured by western blot analysis, and multiple comparisons of western blot results were performed (Figures 4, 5). In experimental group, the expression of Par3 in hippocampus increased at 4 weeks (p < 0.05, Figure 5F); while the expression of Lgl1 in hippocampus increased significantly from 3 days to 2 weeks (p < 0.05 for all, Figure 5G), lastly, the expression of aPKC-λ in hippocampus decreased at 4 weeks (p < 0.05, Figure 5H).
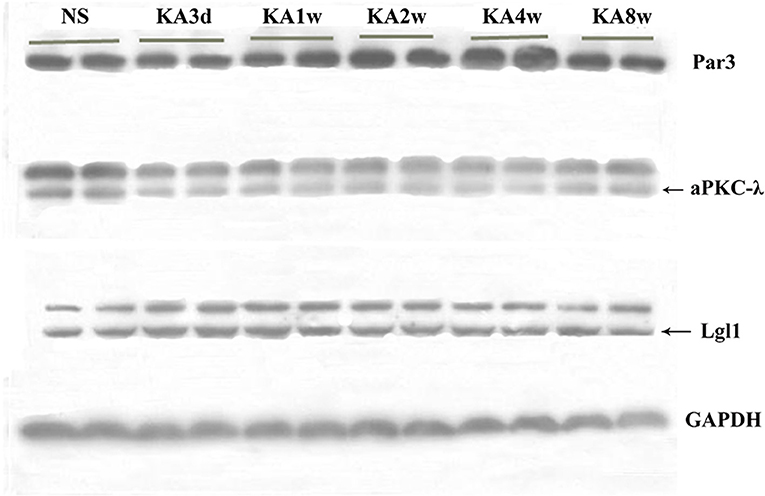
Figure 4. Expression of Par3, aPKC-λ and Lgl1 in hippocampus between experimental and control group by western blot.
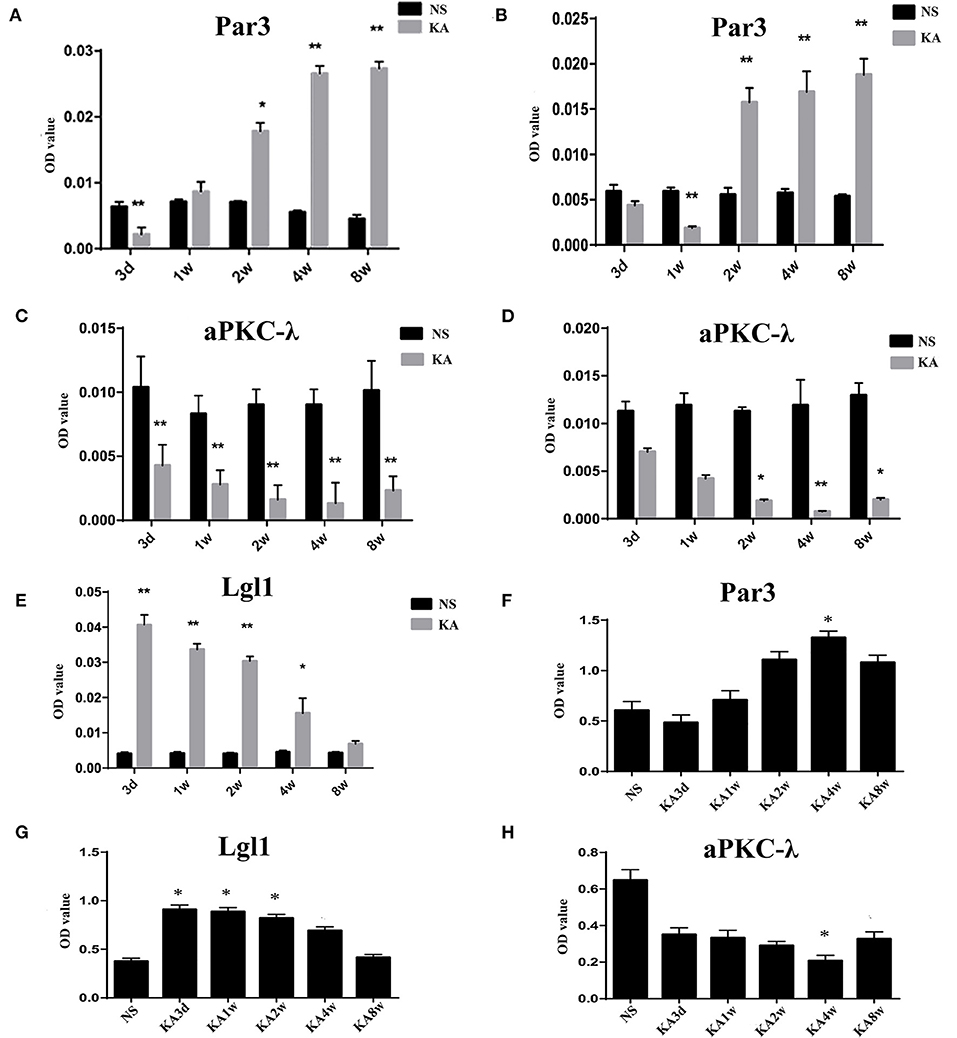
Figure 5. Comparison of Par3, aPKC-λ and Lgl1 in CA3 region (A,C,E) and hilar region (B,D) between experimental and control group. (F–H) Comparison of Par3, aPKC-λ and Lgl1 in hippocampus between experimental and control group.*P < 0.05, compared with the control; and **P < 0.01, compared with the control; KA, kainic acid; NS, normal saline.
In addition, there is a positive correlation between the expression of Par3 and Timm scores (r = 0.903, P < 0.01, Figure 6) and a negative correlation between the expression of aPKC-λ and Timm scores (r = −0.785, P < 0.01, Figure 6) in CA3 region in the experimental group. No correlation between the expression of Lgl1 and Timm scores (r = −0.405, P > 0.05) was observed in experimental group.
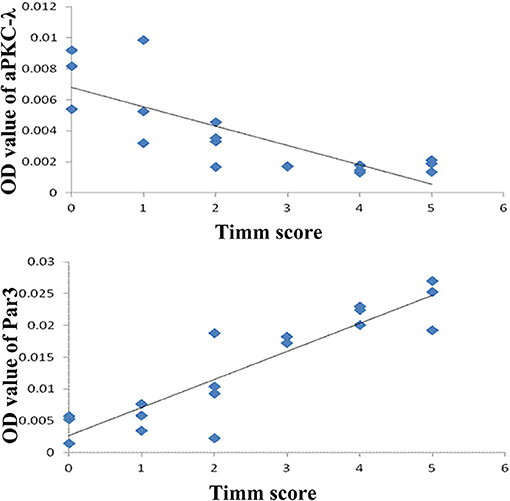
Figure 6. Correlation between Timm score and expression of Par3 and aPKC-λ.
Discussion
MFS, a frequent histopathological finding in TLE (31), has preceded the appearance of spontaneous seizures in PTZ kindling rat model of epilepsy (32). It has been found that severity of MFS is associated with susceptibility of spontaneous seizures (33, 34). Furthermore, it was confirmed that mTOR pathway was involved in regulating axonal outgrowth (7). In the study conducted by Pun et al., the mice presented MFS and a spontaneous recurrent seizure by deleting PTEN gene that regulates the mTOR pathway (8). All those studies supported the hypothesis that MFS was involved in the mechanism of epileptogenesis of TLE (35–37). Moreover, several studies had discovered that severity of MFS and spontaneous seizures could be relieved by regulating the expression of specific proteins involved in axonal growth (5, 38, 39), which means that further studies are required to explore the role of other proteins that are regulating axonal growth in the mechanism of epileptogenesis of TLE.
Numerous studies confirmed that only two kinds of protein, kinase C (aPKC) isoforms (aPKC-λ and PKM-ζ), were expressed in rat hippocampal neurons, and aPKC-λ-Par3 complex promotes the axon specification, while PKM-ζ competes with aPKC-λ for binding to Par3 and PKMζ-PAR3 complex inhibits axon formation (19, 40), and the upregulating of aPKC-λ or the silencing of PKM-ζ has caused supernumerary axon growth in hippocampal neurons (19). On the contrary, Buchser et al. discovered that the overexpression of aPKC-λ has inhibited neuronal growth and axon formation (41). However, Yamanaka et al. observed that Neuronal deletion of aPKC-λ does not affect distribution of neural structures including dendrites, axons and synapses in mouse brain cortex (42). Those findings indicated that the function of aPKC-λ in axon formation and its growth in neurons are controversial. In the present study, the decrease in aPKC-λ expression at hippocampus and negative correlation between aPKC-λ expression, and the severity of MFS were observed in experimental group. Those findings imply that the condition-dependent effect of aPKC-λ in morphology of axon and aPKC-λ may suppress axonal formation and growth in hippocampus under certain pathological condition (such as epileptogenesis). Besides, effects of aPKC-λ in MFS and epileptogenesis of TLE are still open to debate and further research.
In the present study, Par3 and Lgl 1 increased during epileptogenesis with presence of MFS. Par3 is established as an interaction partner of full-length aPKC isoforms and is involved in neuronal polarity and axon-dendrite differentiation (10–12). It has been demonstrated that the increase in axonal concentration of Par3 could be induced by nerve growth factor or netrin-1 stimulation, which is known to promote axonal growth (43), and the Par3 complex is localized at the presumptive axon in embryonic hippocampal neurons (19). Those findings indicated Par3 may display facilitative effort on MFS and may aggravate epileptogenesis. Moreover, Par3 may indirectly contribute to epileptogenesis through CNTNAP2, which has been considered a prominent disease susceptibility gene associated with epilepsy (21, 22).
Lgl1 is well-known to be a cytoskeletal protein and regulates establishment of polarity in many cell types. It has been confirmed that Lgl1 is enriched in developing axons, and its upregulation promoted the axonal growth (27), which is consistent with findings in present study.
In the present study, the expression of Par 3 in CA3 decreased at 3 days and increased from 2 weeks, then peaked at 8 weeks following KA administration. Conversely, the expression of Lgl1 in CA3 peaked at 3days and declined from 1 week. It reached the lowest level at 8 weeks following KA administration. Those opposite observations may result from antagonism between Par3 and Lgl1 in a cell polarity. Previous studies have confirmed that Par3 and Lgl1/2 all binded directly to Par6 (28, 44–47). However, the binding of Par3 and Lgl to Par6 appeared to be mutually exclusive in establishing and maintaining cell polarization (45–47). Moreover, Wang et al. found that Par3 and Lgl antagonized each other in modulating myosin II activation during cell–cell contact formation (48). Our finding indicated that increase in Par3 and Lgl1 at different phase of epileptogenesis may promote aberrant axonal growth to form excitatory neural connections and aggravate epileptogenesis.
Numerous studies confirmed Par 3 (46) and Lgl (45, 49) could be phosphorylated by aPKC in many cell types. Bailey et al. found that phosphorylation by aPKC prevented lgl from the apical cell cortex in S2 cell system (49). Yamanaka et al. found that activation of aPKC in the Par6 complex resulted in the phosphorylation of Lgl and its dissociation from the cortex in epithelial cell (50). In addition, it has been proposed that the release of Par3 from the Par6/aPKC complex was a result from aPKC-mediated phosphorylation on Par3 via conserved region 3 (51–53). Those findings indicated that the subcellular localizations of lgl and Par3 were regulated by phosphorylation by aPKC, which may have been a possible reason for the expression of aPKC to change in the opposite direction in CA3 and in hilar regions during epileptogenesis compared with lgl and Par3.
In the present study, the neuronal loss in CA3 and hilar region demonstrated by a significant decrease in the number of NeuN-positive cells is observed in the course of epileptogenesis of TLE, which is consistent with previous studies (54, 55). With the neuronal loss in CA3 and hilar region, the loss of connecting targets resulted in the abnormal growth of mossy fiber, and the positive correlation between the degree of neuron loss and extent of MFS has been confirmed in TLE model and patients (56, 57), which indicated that neuronal loss may promote the effect of MFS in epileptogenesis of TLE.
Conclusions
The findings of this study indicated, for the first time, that aPKC-λ, Par3, and Lgl1 may be involved in MFS and epileptogenesis of TLE. Further studies are required to explore the effects of those proteins mediating cell polarity in MFS and epileptogenesis of TLE, which may lead to more interventions that can prevent or relieve the recurrent spontaneous seizures.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Research Ethics Committee of the Xiangya Hospital.
Author Contributions
CZ, ZT, and XL: conceptualization. ZT and CZ: methodology. CZ, ZT, and JD: investigation. CZ: formal analysis, resources, and writing—original draft. FT and XL: writing—review and editing and supervision. All authors have read and approved the manuscript and had full access to all the data in the study, took responsibility for the integrity of the data, and the accuracy of the data analysis.
Funding
This work was supported by Public Health Program of Hunan Provincial Department of Finance, China (Xiangcaishezhi No. 2020-46, Xiangcaiqizhi No. 2015-122, and Xiangcaijiaozhi No. 2010-216).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank all staffs of experimental animal facility of Central South University for their assistance.
References
1. Laxer KD, Trinka E, Hirsch LJ, Cendes F, Langfitt J, Delanty N, et al. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. (2014) 37:59–70. doi: 10.1016/j.yebeh.2014.05.031
2. Falconer MA, Serafetinides EA, Corsellis JA. Etiology and pathogenesis of temporal lobe epilepsy. Arch Neurol. (1964) 10:233–48. doi: 10.1001/archneur.1964.00460150003001
3. Godale CM, Danzer SC. Signaling pathways and cellular mechanisms regulating mossy fiber sprouting in the development of epilepsy. Front Neurol. (2018) 9:298. doi: 10.3389/fneur.2018.00298
4. Parent JM, Murphy GG. Mechanisms and functional significance of aberrant seizure-induced hippocampal neurogenesis. Epilepsia. (2008) 49:19–25. doi: 10.1111/j.1528-1167.2008.01634.x
5. Chen L, Gao B, Fang M, Li J, Mi X, Xu X, et al. Lentiviral vector-induced overexpression of rgma in the hippocampus suppresses seizures and mossy fiber sprouting. Mol Neurobiol. (2017) 54:1379–91. doi: 10.1007/s12035-016-9744-2
6. Huang S, Ge X, Yu J, Han Z, Yin Z, Li Y, et al. Increased miR-124-3p in microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via their transfer into neurons. FASEB J. (2018) 32:512–28. doi: 10.1096/fj.201700673r
7. Zhu H, Wang Y, Yang X, Wan G, Qiu Y, Ye X, et al. Catalpol improves axonal outgrowth and reinnervation of injured sciatic nerve by activating Akt/mTOR pathway and regulating BDNF and PTEN expression. Am J Transl Res. (2019) 11:1311–26.
8. Pun RYK, Rolle IJ, LaSarge CL, Hosford BE, Rosen JM, Uhl JD, et al. Excessive activation of mtor in postnatally generated granule cells is sufficient to cause epilepsy. Neuron. (2012) 75:1022–34. doi: 10.1016/j.neuron.2012.08.002
9. LaSarge CL, Santos VR, Danzer SC. PTEN deletion from adult-generated dentate granule cells disrupts granule cell mossy fiber axon structure. NEUROBIOL DIS. (2015) 75:142–50. doi: 10.1016/j.nbd.2014.12.029
10. Goldstein B, Macara IG. The PAR proteins: fundamental players in animal cell polarization. Dev Cell. (2007) 13:609–22. doi: 10.1016/j.devcel.2007.10.007
11. McCaffrey LM, Macara IG. Signaling pathways in cell polarity. Cold Spring Harb Perspect Biol. (2012) 4:a009654. doi: 10.1101/cshperspect.a009654
12. Thompson BJ. Cell polarity: models and mechanisms from yeast, worms and flies. Development. (2013) 140:13–21. doi: 10.1242/dev.083634
13. Shi SH, Jan LY, Jan YN. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. (2003) 112:63–75. doi: 10.1016/S0092-8674(02)01249-7
14. Nishimura T, Kato K, Yamaguchi T, Fukata Y, Ohno S, Kaibuchi K. Role of the PAR-3-KIF3 complex in the establishment of neuronal polarity. Nat Cell Biol. (2004) 6:328–34. doi: 10.1038/ncb1118
15. Schwamborn JC, Puschel AW. The sequential activity of the GTPases Rap1B and Cdc42 determines neuronal polarity. Nat Neurosci. (2004) 7:923–9. doi: 10.1038/nn1295
16. Ohno S. Intercellular junctions and cellular polarity: the PAR-aPKC complex, a conserved core cassette playing fundamental roles in cell polarity. Curr Opin Cell Biol. (2001) 13:641–8. doi: 10.1016/S0955-0674(00)00264-7
17. Henrique D, Schweisguth F. Cell polarity: the ups and downs of the Par6/aPKC complex. Curr Opin Genet Dev. (2003) 13:341–50. doi: 10.1016/S0959-437X(03)00077-7
18. Ghosh S, Marquard T, Thaler JP, Carter N, Andrews SE, Pfaff SL, et al. Instructive role of aPKC zeta subcellular localization in the assembly of adherens junctions in neural progenitors. Proc Natl Acad Sci USA. (2008) 105:335–40. doi: 10.1073/pnas.0705713105
19. Parker SS, Mandell EK, Hapak SM, Maskaykina IY, Kusne Y, Kim J-Y, et al. Competing molecular interactions of aPKC isoforms regulate neuronal polarity. Proc Natl Acad Sci USA. (2013) 110:14450–5. doi: 10.1073/pnas.1301588110
20. Guglielmetti F, Rattray M, Baldessari S, Butelli E, Samanin R, Bendotti C. Selective up-regulation of protein kinase C epsilon in granule cells after kainic acid-induced seizures in rat. Brain Res Mol Brain Res. (1997) 3:1–2. doi: 10.1016/S0169-328X(97)00142-3
21. Gao R, Pratt CP, Yoon S, Martin-de-Saavedra MD, Forrest MP, Penzes P. CNTNAP2 is targeted to endosomes by the polarity protein PAR3. Eur J Neurosci. (2020) 51:1074–86. doi: 10.1111/ejn.14620
22. Rodenas-Cuadrado P, Ho J, Vernes SC. Shining a light on CNTNAP2: complex functions to complex disorders. Eur J Hum Genet. (2014) 22:171–8. doi: 10.1038/ejhg.2013.100
23. Thomas AM, Schwartz MD, Saxe MD, Kilduff TS. Cntnap2 knockout rats and mice exhibit epileptiform activity and abnormal sleep-wake physiology. Sleep. (2017) 40:zsw026. doi: 10.1093/sleep/zsw026
24. Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. (2000) 289:113–6. doi: 10.1126/science.289.5476.113
25. Zhang T, Hou C, Zhang S, Liu S, Li Z, Gao J. Lgl1 deficiency disrupts hippocampal development and impairs cognitive performance in mice. Genes Brain Behav. (2019) 18:e12605. doi: 10.1111/gbb.12605
26. Ravid S. The tumor suppressor Lgl1 regulates front-rear polarity of migrating cells. Cell Adh Migr. (2014) 8:378–83. doi: 10.4161/cam.29387
27. Wang T, Liu Y, Xu X, Deng C, Wu K, Zhu J, et al. Lgl1 Activation of Rab10 promotes axonal membrane trafficking underlying neuronal polarization. Dev Cell. (2011) 21:431–44. doi: 10.1016/j.devcel.2011.07.007
28. Betschinger J, Mechtler K, Knoblich JA. The Par complex directs asymmetric cell division by phosphorylating the cytoskeletal protein Lgl. Nature. (2003) 422:326–30. doi: 10.1038/nature01486
29. Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. (1972) 32:281–94. doi: 10.1016/0013-4694(72)90177-0
30. Cilio MR, Sogawa Y, Cha BH, Liu XZ, Huang LT, Holmes GL. Long-term effects of status epilepticus in the immature brain are specific for age and model. Epilepsia. (2003) 44:518–28. doi: 10.1046/j.1528-1157.2003.48802.x
31. Sloviter RS, Zappone CA, Harvey BD, Frotscher M. Kainic acid-induced recurrent mossy fiber innervation of dentate gyrus inhibitory interneurons: possible anatomical substrate of granule cell hyperinhibition in chronically epileptic rats. J Comp Neurol. (2006) 494:944–60. doi: 10.1002/cne.20850
32. Song M, Tian F, Wang Y, Huang X, Guo J, Ding D. Potential roles of the RGMa-FAK-Ras pathway in hippocampal mossy fiber sprouting in the pentylenetetrazole kindling model. Mol Med Rep. (2015) 11:1738–44. doi: 10.3892/mmr.2014.2993
33. Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. (1985) 5:1016–22. doi: 10.1523/JNEUROSCI.05-04-01016.1985
34. Santhakumar V, Aradi I, Soltesz I. Role of mossy fiber sprouting and mossy cell loss in hyperexcitability: a network model of the dentate gyrus incorporating cell types and axonal topography. J Neurophysiol. (2005) 93:437–53. doi: 10.1152/jn.00777.2004
35. Shetty AK, Turner DA. Aging impairs axonal sprouting response of dentate granule cells following target loss and partial deafferentation. J Comp Neurol. (1999) 414:238–54.
36. Rao MS, Hattiangady B, Reddy DS, Shetty AK. Hippocampal neurodegeneration, spontaneous seizures, and mossy fiber sprouting in the F344 rat model of temporal lobe epilepsy. J Neurosci Res. (2006) 83:1088–105. doi: 10.1002/jnr.20802
37. Mathern GW, Babb TL, Leite JP, Pretorius K, Yeoman KM, Kuhlman PA. The pathogenic and progressive features of chronic human hippocampal epilepsy. Epilepsy Res. (1996) 26:151–61. doi: 10.1016/S0920-1211(96)00052-6
38. Song M, Tian F, Xia H, Xie Y. Repulsive guidance molecule a suppresses seizures and mossy fiber sprouting via the FAK-p120RasGAP-Ras signaling pathway. Mol Med Rep. (2019) 19:3255–62. doi: 10.3892/mmr.2019.9951
39. Paradiso B, Zucchini S, Su T, Bovolenta R, Berto E, Marconi P, et al. Localized overexpression of FGF-2 and BDNF in hippocampus reduces mossy fiber sprouting and spontaneous seizures up to 4 weeks after pilocarpine-induced status epilepticus. Epilepsia. (2011) 52:572–8. doi: 10.1111/j.1528-1167.2010.02930.x
40. Hapak SM, Rothlin CV, Ghosh S. PAR3-PAR6-atypical PKC polarity complex proteins in neuronal polarization. Cell Mol Life Sci. (2018) 75:2735–61. doi: 10.1007/s00018-018-2828-6
41. Buchser WJ, Slepak TI, Gutierrez-Arenas O, Bixby JL, Lemmon VP. Kinase/phosphatase overexpression reveals pathways regulating hippocampal neuron morphology. Mol Syst Biol. (2010) 6:391. doi: 10.1038/msb.2010.52
42. Yamanaka T, Tosaki A, Kurosawa M, Akimoto K, Hirose T, Ohno S, et al. Loss of aPKC lambda in differentiated neurons disrupts the polarity complex but does not induce obvious neuronal loss or disorientation in mouse brains. PLoS ONE. (2013) 8:e84036. doi: 10.1371/journal.pone.0084036
43. Hengst U, Deglincerti A, Kim HJ, Jeon NL, Jaffrey SR. Axonal elongation triggered by stimulus-induced local translation of a polarity complex protein. Nat Cell Biol. (2009) 11:1024–30. doi: 10.1038/ncb1916
44. Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol. (2000) 2:540–7. doi: 10.1038/35019582
45. Plant PJ, Fawcett JP, Lin DCC, Holdorf AD, Binns K, Kulkarni S, et al. A polarity complex of mPar-6 and atypical PKC binds, phosphorylates and regulates mammalian Lgl. Nat Cell Biol. (2003) 5:301–8. doi: 10.1038/ncb948
46. Yamanaka T, Horikoshi Y, Izumi N, Suzuki A, Mizuno K, Ohno S. Lgl mediates apical domain disassembly by suppressing the PAR-3-aPKC-PAR-6 complex to orient apical membrane polarity. J Cell Sci. (2006) 119(Pt 10):2107–18. doi: 10.1242/jcs.02938
47. Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. (2000) 2:531–9. doi: 10.1038/35019573
48. Wan Q, Liu J, Zheng Z, Zhu H, Chu X, Dong Z, et al. Regulation of myosin activation during cell-cell contact formation by Par3-Lgl antagonism: entosis without matrix detachment. Mol Biol Cell. (2012) 23:2076–91. doi: 10.1091/mbc.e11-11-0940
49. Bailey MJ, Prehoda KE. Establishment of par-polarized cortical domains via phosphoregulated membrane motifs. J Neurochem. (2015) 35:199–210. doi: 10.1016/j.devcel.2015.09.016
50. Yamanaka T, Horikoshi Y, Sugiyama Y, Ishiyama C, Suzuki A, Hirose T, et al. Mammalian LgI forms a protein complex with PAR-6 and aPKC independently of PAR-3 to regulate epithelial cell polarity. Curr Biol. (2003) 13:734–43. doi: 10.1016/S0960-9822(03)00244-6
51. Soriano EV, Ivanova ME, Fletcher G, Riou P, Knowles PP, Barnouin K, et al. aPKC inhibition by Par3 CR3 flanking regions controls substrate access and underpins apical-junctional polarization. Dev Cell. (2016) 38:384–98. doi: 10.1016/j.devcel.2016.07.018
52. Morais-de-Sa E, Mirouse V, St Johnston D. aPKC phosphorylation of bazooka defines the apical/lateral border in drosophila epithelial cells. Cell. (2010) 141:509–23. doi: 10.1016/j.cell.2010.02.040
53. Liu Z, Yang Y, Gu A, Xu J, Mao Y, Lu H, et al. Par complex cluster formation mediated by phase separation. Nat Commun. (2020) 11:2266. doi: 10.1038/s41467-020-16135-6
54. Baluchnejadmojarad T, Roghani M. Coenzyme Q10 ameliorates neurodegeneration, mossy fiber sprouting, and oxidative stress in intrahippocampal kainate model of temporal lobe epilepsy in rat. J Mol Neurosci. (2013) 49:194–201. doi: 10.1007/s12031-012-9886-2
55. Vivash L, Tostevin A, Liu DSH, Dalic L, Dedeurwaerdere S, Hicks RJ, et al. Changes in hippocampal GABA(A)/cBZR density during limbic epileptogenesis: relationship to cell loss and mossy fibre sprouting. Neurobiol Dis. (2011) 41:227–36. doi: 10.1016/j.nbd.2010.08.021
56. Schmeiser B, Zentner J, Prinz M, Brandt A, Freiman TM. Extent of mossy fiber sprouting in patients with mesiotemporal lobe epilepsy correlates with neuronal cell loss and granule cell dispersion. Epilepsy Res. (2017) 129:51–8. doi: 10.1016/j.eplepsyres.2016.11.011
Keywords: temporal lobe epilepsy, atypical protein kinase C, partitioning defective 3, lethal giant larvae 1, mossy fiber sprouting
Citation: Zhang C, Tian F, Tan Z, Du J and Long X (2021) Altered Expression of Par3, aPKC-λ, and Lgl1 in Hippocampus in Kainic Acid-Induced Status Epilepticus Rat Model. Front. Neurol. 12:780042. doi: 10.3389/fneur.2021.780042
Received: 20 September 2021; Accepted: 08 November 2021;
Published: 08 December 2021.
Edited by:
Dinesh Upadhya, Manipal Academy of Higher Education, IndiaReviewed by:
Carmen Rubio, Manuel Velasco Suárez Instituto Nacional de Neurología y Neurocirugía, MexicoBinglin Zhu, University at Buffalo, United States
Copyright © 2021 Zhang, Tian, Tan, Du and Long. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyan Long, NDA5Njg1NjU2QHFxLmNvbQ==; bG9uZ3h5YW5AcXEuY29t