
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 18 October 2021
Sec. Pediatric Neurology
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.754045
This article is part of the Research TopicNeuronal Ceroid Lipofuscinosis: a Multidisciplinary UpdateView all 12 articles
 Emily Gardner
Emily Gardner Sara E. Mole*
Sara E. Mole*The neuronal ceroid lipofuscinoses (NCLs) are a group of inherited neurodegenerative disorders that affect children and adults. They share some similar clinical features and the accumulation of autofluorescent storage material. Since the discovery of the first causative genes, more than 530 mutations have been identified across 13 genes in cases diagnosed with NCL. These genes encode a variety of proteins whose functions have not been fully defined; most are lysosomal enzymes, or transmembrane proteins of the lysosome or other organelles. Many mutations in these genes are associated with a typical NCL disease phenotype. However, increasing numbers of variant disease phenotypes are being described, affecting age of onset, severity or progression, and including some distinct clinical phenotypes. This data is collated by the NCL Mutation Database which allows analysis from many perspectives. This article will summarise and interpret current knowledge and understanding of their genetic basis and phenotypic heterogeneity.
The neuronal ceroid lipofuscinoses (NCL), also known as Batten disease, are a group of inherited neurodegenerative life-limiting diseases that share some common clinical features including epileptic seizures, progressive psychomotor decline, and visual failure, and the accumulation of autofluorescent storage material. NCL usually begins in childhood, and most are inherited in an autosomal recessive manner. More than a dozen genes have been linked to families diagnosed with NCL (Table 1) (1). It is likely that most genes causing NCL have been identified.
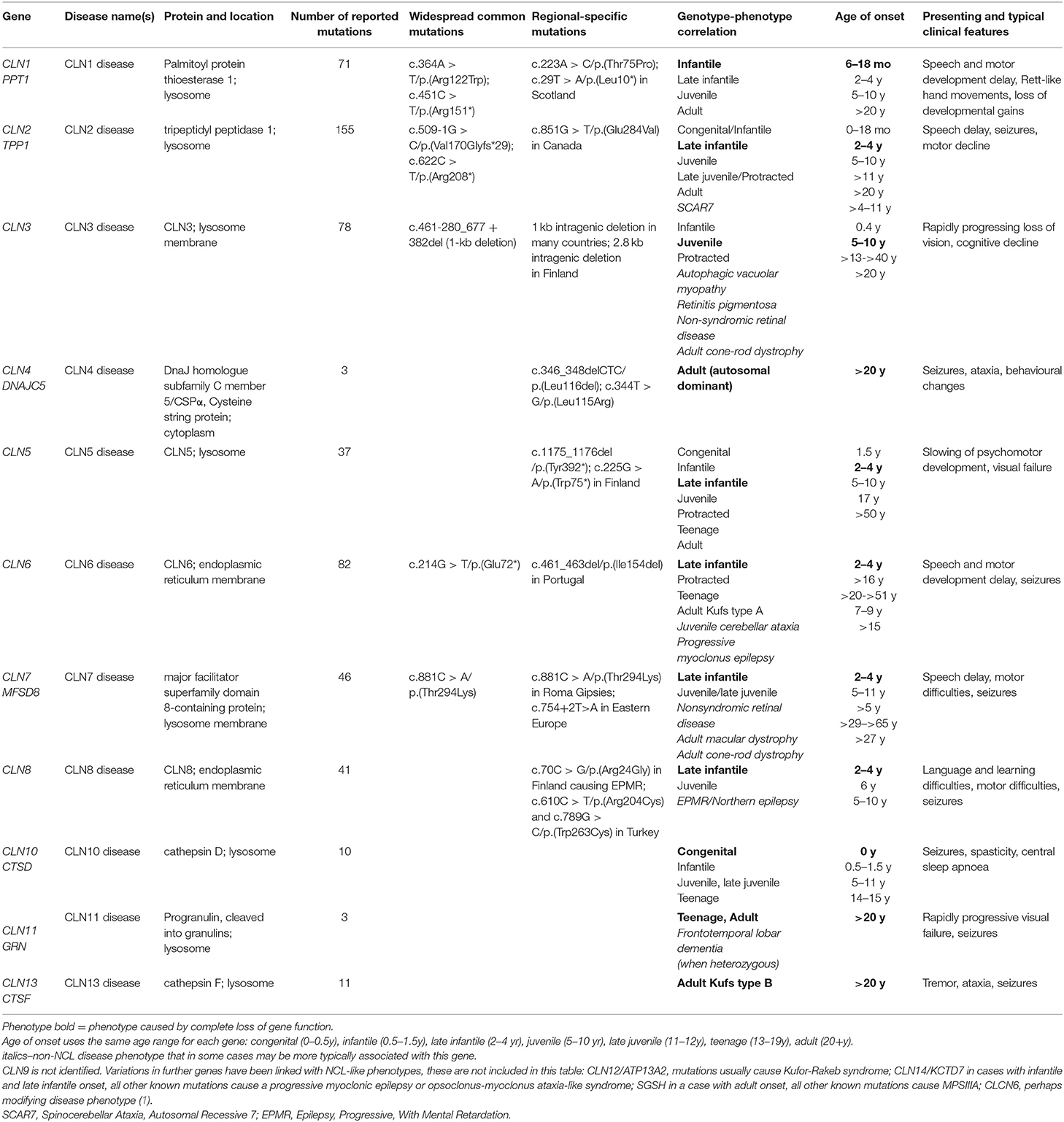
Table 1. Summary of genotype and phenotype in NCL.
This article summarises the genetic basis of NCL and discusses correlations with disease phenotype. All mutation details can be found in the freely accessible NCL mutation database (www.ucl.ac.uk/ncl-disease).
The concept of NCL as a group of inherited diseases first emerged in the 1960s (2), leading to classification into four broad ages of onset: infantile, late infantile, juvenile, and adult. At this time it was assumed that each of these types was caused by mutations in a different gene, named in advance as CLN1, CLN2, CLN3, and CLN4, respectively. The first genes to be identified, in the 1990s (CLN1, CLN2, CLN3), were responsible for the most common paediatric types. However, these first identified genes were not responsible for all childhood onset cases. For example, the genes CLN5, CLN6, CLN7, and CLN8 cause disease with onset in late infancy like CLN2, the first gene identified causing onset at that age (Table 1). CLN4 was not identified until 2011 (3).
A variety of experimental approaches reflecting the available technology were used to identify NCL genes. The first genes were identified using classic and time-consuming genetic linkage approaches requiring large numbers of similarly affected families followed by positional cloning of the genes [CLN1 (4) and CLN3 (5)]. A biochemical approach that detected a missing mannose-6-phosphate tagged lysosomal enzyme in a patient facilitated the identification of CLN2 (6), alongside ongoing genetic linkage studies. Availability of the human genome sequence meant that going forward fewer families were required to provide sufficient power for genetic linkage analysis, facilitating identification of CLN5 (7), CLN6 (8), CLN7 (9), and CLN8 (10). Some genes were identified by recognition of stretches of homozygosity in consanguineous families that narrowed the interval that contained the candidate gene. A gene first identified in an animal model led to identification of CLN10 (11, 12) in human disease. Improvements in sequencing technology later allowed fast and massively parallel sequencing of the whole exome in single families, and facilitated identification of the remaining disease genes [CLN4 (3), CLN11 (13), CLN12 (14), CLN13 (15), CLN14 (16)]. The few families suspected of carrying the putative CLN9 gene were later found to carry mutations in previously identified NCL genes (17, 18).
As monogenic disorders, each NCL is in effect a separate disease entity. All identified NCL genes lie on autosomes. Most cause disease though classic recessive inheritance, where deleterious mutations are present in disease gene alleles inherited from asymptomatic parents. However, adult onset CLN4 disease is dominantly inherited in the few families described with this disease (1, 3). There are three published reports of uniparental disomy in the NCLs, one in which a patient has complete isodisomy of chromosome 8, leading to homozygosity of a maternally-inherited deletion in CLN8 (19), and two patients for CLN1, both with paternal isodisomy of chromosome 1 (19, 20).
The majority of NCL genes encode proteins that reside in the endo/lysosomal pathways (1, 21–23). Most are lysosomal proteins—enzymes and soluble proteins (CLN1/PPT1, CLN2/TPP1, CLN5, CLN10/CTSD, CLN13/CTSF, CLN11/GRN,) or membrane proteins (CLN3, CLN7/MFSD8, CLN12/ATP13A2). Two encode endoplasmic reticulum membrane proteins (CLN6, CLN8). Other NCL proteins are cytoplasmic (CLN4/DNAJC5, CLN14/KCTD7) that peripherally associate with cellular membranes. The in vivo substrates for the lysosomal enzymes are incompletely defined, and much remains to be discovered around the functions of the membrane proteins. Nevertheless, recognition of the genetic basis of the NCLs enables the development of targeted therapies even though the underlying disease mechanism for each NCL is not yet fully delineated. It is unlikely that further NCL genes will be identified unless they cause disease in countries where little genetic analysis has been undertaken.
A gene-based classification system was codeveloped by international experts in the NCLs (24) that takes into account the full phenotypic consequences that have emerged over the years, and which includes secondary reference to the age of onset. This replaces the former age-based classification in use since the 1960s. It better supports ongoing gene-based therapeutic development.
There is a classic disease phenotype associated with complete loss of function for most NCL genes, with a typical age of onset and disease progression. The age at which first symptoms appear can be used to guide toward which gene(s) may be mutated. For example, clinically similar NCL disease arising from mutations in more than one gene (e.g., what was originally known as variant late infantile onset NCL) can be caused by loss-of-function mutations in CLN5, CLN6, CLN7, or CLN8.
Most NCL genes actually have a wide age of onset and varied disease courses determined by the underlying mutations (Table 1). The increasing implementation of next generation sequencing panels and exome sequencing in diagnosis is leading to more diagnoses of patients with atypical NCL and recognition of these broader phenotypes. These arise from mutations thought or known to have “milder” effects on NCL protein function; and these phenotypes can vary quite considerably. For example, classic CLN6 disease begins in early childhood (late infancy) (8, 25), but disease onset can be delayed as late as adulthood, which also has no associated visual failure (26, 27). Conversely, disease that presents in adulthood caused by mutations in CLN3 may have visual failure as its only or main sign, consistent with this being the presenting symptom for classic juvenile CLN3 disease. Mutations in CLN7 have been identified in cases of non-syndromic eye disease (28).
This broadening of phenotypes means that disease with a certain age of onset may be caused by loss of function of an NCL protein as well as milder mutations in a gene more usually associated with a younger age of onset. For example, disease beginning in the juvenile age range may be classic CLN3 disease or be juvenile CLN1 disease, or juvenile CLN2, CLN5, CLN6, CLN7, or CLN8 disease.
Some mutations cause distinct and varied disease that differs from the phenotypes arising from other mutations in the same gene. For example, a single recessive missense mutation in CLN8 [p.(Arg24Gly)] (10) causes the phenotype described as progressive epilepsy with mental retardation (EPMR) or Northern epilepsy that is found predominantly in Finland. This disease is very different to typical NCL as it is an intellectual developmental disorder that presents with seizures in the juvenile age range that cease in adulthood, and life expectancy is into late adulthood. It was the first genetic disease to be recognised for CLN8, with mutations that cause a more typical NCL described later. Similarly, a missense mutation in CLN2/TPP1 [p.(Val466Gly)] causes a phenotype first described as spinocerebellar ataxia SCAR7. This is a slowly progressing but not life-limiting disease with no ophthalmologic abnormalities or epilepsy, and without typical ceroid/lipofuscin storage (29). A single gain of function missense mutation in CLCN6 has recently been shown to cause very severe disease in children (30) that would not be classed as NCL, although the mouse model lacking the function of the homologous gene causes mild lysosomal storage disease and the CLCN6 gene was considered a candidate gene for mild NCL disease (31).
There is evidence that the most common and very widespread mutation in CLN3, a 1-kb deletion found worldwide accounting for ~ 90 percent of the affected alleles in CLN3 disease patients (32) does not completely abolish CLN3 function, indeed it may case a gain of function and therefore disease (33, 34). Due to this deletion dominating reports of CLN3 disease, this led to the suspicion that disease caused by complete loss of CLN3 function may not have been described in humans (33). Other distinct phenotypes have been associated with CLN3 mutations—these include retinitis pigmentosa without other clinical symptoms, even in mid-late adulthood (35) and a distinct disease described as autophagic myopathy associated with heart failure (36). As predicted (33) the phenotype of CLN3-associated disease maybe considerably broader (1). There are reports of other families with mutations in some NCL genes that also have predominantly visual problems (28).
Some mutations in NCL genes cause disease that overlaps with other recognised disease syndromes. This has been described for other rare diseases and more common neurological disorders, such as Niemann-Pick C disease with Alzheimer's disease (37), and type 1 Gaucher disease with Parkinson's disease (38).
Mutations in GRN cause diseases with different types of inheritance. A homozygous recessive (bi-allelic) mutation associated with rectilinear profiles, leads to CLN11 disease, whereas mutations present on one chromosome only cause frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) (13), which is the second most common type of early-onset dementia. The age of onset and neuropathology of FTLD-TDP and NCL are markedly different, yet there are some shared characteristics: there is autofluorescent, NCL-like storage material in the retina, postmortem brain and lymphoblasts of FTLD-TDP patients (39) and in induced pluripotent stem cells from FTLD-TDP patients (40). Progranulin-deficient mice (13) have features of both NCL and FTLP-TDP diseases (41–43). Therefore, autosomal dominant GRN mutations in FTLD-TDP patients cause disease through haploinsufficiency, and it is likely that there are shared disease mechanisms underlying disease in adult CLN11 and FTLD-TDP patients.
Some genes identified as causing NCL more commonly cause inherited diseases given different diagnoses. Mutations in CLN14/KCTD7 cause three different diseases (16, 44–46) classed as progressive myoclonic epilepsy (PME) (47, 48), and in rarer cases PME accompanied by vision loss and lysosomal storage and termed an NCL (16, 49). Mutations in ATP13A2 typically cause Kufor-Rakeb syndrome and also a late-onset autosomal recessive spastic paraplegia 78 (SPG78) and juvenile onset amyotrophic lateral sclerosis (ALS) (50–52), whereas one family was diagnosed with CLN12 disease (14, 53, 54). Fibroblasts from some SPG78 patients have lysosomal pathology (50). Atp13a2 knockout mice are reported to accumulate both NCL-type storage material and α-synuclein, and late-onset impairment in sensorimotor functioning. ATP13A2-related disease may therefore represent a disorder with features overlapping both NCL and Parkinson's disease (55). Mutations in SGSH usually underlie late infantile onset disease mucopolysaccharidosis type IIIA (MPSIIIA) (56), whereas a mutation in SGSH was described in a single case diagnosed with adult onset NCL. Thus, distinctions between inherited disease phenotypes may not be as clear cut as originally anticipated.
There are examples of disease including features of NCL. For example, CLCN7 underlies a severe autosomal recessive disease combining osteopetrosis, neurodegeneration and lysosomal storage disease (57–59).
The clear recessive nature of most NCL had always suggested that mutation carriers are healthy. Given that disease arises in those who are carriers or carrying compound heterozygous mutations in CLN11/GRN, it may be that carriers of mutations in other NCL genes also have deficits. If so, these are likely to be extremely mild or be very late onset and overlap with common features or ageing, and so have not been linked, even anecdotally.
CLN4 disease (Parry disease) is considered autosomal dominant, with disease manifesting in those carrying one of the three mutations in CLN4 so far described. Disease in humans caused by complete loss of CLN4 function is not known, although the severity of phenotype in animal models with no CLN4 function (60) would predict those carrying biallelic loss-of-function mutations would have very severe and early onset disease. Disease arising from mutations in CLN4/DNAJC5 may therefore be inherited recessively or dominantly.
There are a few reports of patients carrying changes in more than one NCL gene. One that was later found to be compound heterozygous for mutations in CLN5 also carries a single mutation in the CLCN6 gene that causes recessive NCL in animals (31). Another family is reported in which a single mutation in CLCN6 is the only described variation; a second heterozygous mutation may be present but not identified. In these two families the CLCN6 carrier parents were healthy. Some patients carry mutations in more than one gene that underlie variant late infantile NCL (47) (i.e., the mutation database lists changes in CLN5 that have been found alongside those in CLN6 or CLN7 or CLN8). These may be examples of a mutation or specific allele of one gene enhancing or ameliorating the NCL disease phenotype. In mouse NCL models, deletion of both cathepsin B and cathepsin L causes disease, but deletion of either gene alone does not (61).
A patient with disease that presented shortly after birth was found to carry heterozygous mutations in CLN5, together with a mutation in POLG1 that acts to maintain mitochondrial DNA integrity (62). Increased expression of CLN8 may act as a modifier of Gaucher disease (63). There may be connexions between the function of NCL genes; for example, GRN interacts with CTSD (40), CLN3 affects trafficking of enzymes to the lysosome (64); CLN5 interacts with CLN2 and CLN3 (65).
Human cancer cells acquire somatic mutations in the NCL genes which may confer a growth advantage (CLN1/PPT1, CLN2/TPP1, CLN3, CLN4/DNAJC5, CLN5, CLN6, CLN7/MFSD8, CLN8, CLN10/CTSD, CLN11/GRN, CLN12/ATP13A2, CLN13/CTSF, CLN14/KCTD7, as well as SGSH, and CLCN6) (www.sanger.ac.uk/genetics/CGP/cosmic/) (66). As more sequence variations are deposited through large-scale genome sequencing projects) (e.g., Exome Aggregation Consortium (ExAC): exac.broadinstitute.org/), further correlations may be revealed.
NCL are considered the most common inherited neurodegenerative disorder of childhood. They occur worldwide, with some forms first recognised in certain geographical regions. Some types are enriched in or absent from certain regions due to historical population (genetic) bottlenecks.
Incidence and prevalence rates are not available world-wide. Incidence rates are probably more robust than estimated prevalence rates, and generally reported between 1 in 14,000 (Iceland) up to 1 in 100,000 (67). The most common NCL in Northern Europe and the UK are juvenile CLN3 disease and late infantile CLN2 disease, but all types are present.
There is an urgency in making an NCL diagnosis now that disease modifying treatments are available or in the pipeline. Biomarkers that follow disease progression and allow the effectiveness of therapies to be monitored are likely to emerge in the near future (68, 69).
New comprehensive approaches are changing the order of diagnostic tests and removing the need for former investigations. Protocols for enzymatic and genetic testing are widely available, making rapid genetic and biochemical diagnosis of most forms of NCL increasingly straightforward (Table 2).
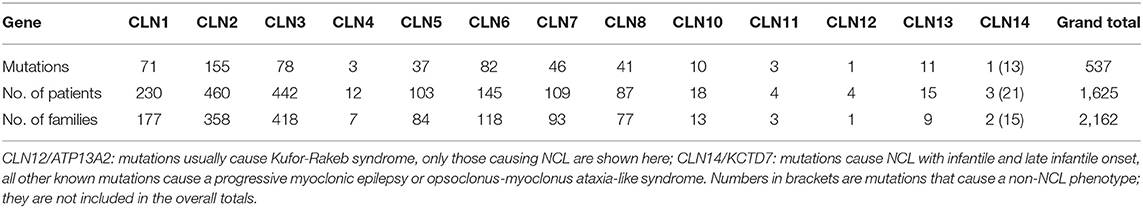
Table 2. Summary of NCL gene mutations, patients and families currently contained within the NCL Mutation Database.
Enzyme testing can rapidly confirm deficiencies of CTSD, PPT1, and TPP1 using saliva, blood samples and dried blood spots (70). These enzyme assays should always be applied in cases with an unusual presentation or later onset, and all diagnoses should be supported by DNA sequencing and mutation analysis where possible. For classic juvenile CLN3 disease, the vacuolated lymphocytes which are a common feature, can be visualised by blood film examination (71).
New DNA technologies now allow testing for many genes in a single step regardless of the presentation (70). NCL genes are part of panels designed to interrogate genes underlying a larger group of syndromic and non-syndromic inherited epilepsies. Some common mutations may be screened by DNA-based testing. This can speed earlier diagnosis of NCL before the appearance of other symptoms and also provides a genetic diagnosis for clinically milder or variant phenotypes. As DNA sequencing leads to the description of multiple genetic variation, the genetic cause of atypical disease for some cases will become clearer. Some patients that previously may have been given a diagnosis of NCL may be demonstrated to have atypical forms of other diseases, and vice versa. Carrier detection is not possible by histology and is unreliable by enzyme assay; it should always be based on mutation analysis.
Ultrastructural examination of a skin biopsy or blood sample may be helpful for confirmation of NCL disease for atypical forms that are not enzyme deficiencies or do not receive a genetic diagnosis (Table 1). Extracerebral storage is readily detected in childhood NCLs but not necessarily in NCL presenting in adulthood (27).
Prenatal diagnosis can be offered to families with a prior history of NCL disease. Preimplantation genetic diagnosis (72) or a combination of enzyme assay and mutational analysis, perhaps with ultrastructural examination of chorionic villus samples obtained at 12–15 weeks gestation, can provide a rapid diagnosis (70).
Some NCL genes are conserved in unicellular or simple organisms, indicating their fundamental function within eukaryotic cells (73). For example, yeasts contain homologous genes to CLN1/PPT1, CLN3 CLN10/CTSD, CLN12/ATP13A2. The slime mould Dictyostelium discoideum particularly expresses further NCL gene homologues or members of gene families (e.g., CLN2/TPP1, CLN4/DNAJC5, CLN5, CLN6, CLN7/MFSD8 family). NCL also occurs in animals (e.g., dogs, sheep, cows, monkey). Cell and animal models carrying mutations in genes equivalent to those causing human NCL are well used in research. These range from yeasts, up to rodents and other mammals (for clinical development). Some of these models are naturally occurring (e.g., mouse, dog, sheep), others are engineered models (e.g., mouse, pig). Some animal NCL disease is caused by mutations in genes not reported to cause similar disease in humans [ARSG in dogs (74), CLCN6 engineered in mice (31), CTSB/CTSL engineered in mice as double gene mutations (61)].
The NCL Mutation Database (www.ucl.ac.uk/ncl-disease) lists known disease-causing mutations and sequence variations in NCL genes by gene and by individual. Five hundred and thirty-seventh NCL disease-causing mutations are currently listed (Table 2) across >1,625 patients and >2,160 families. Where possible the age of onset, ethnic background and current location, are listed for each family. Data are gathered from case reports or larger collections in clinical or scientific publications, or referred directly, and updated periodically. These vary in detail according to the report source, e.g., case reports usually have more specifics than reports of large group genetic screens. Mutations are mostly described in single individuals or occasionally siblings from the same family. Some mutations are more common in certain populations due to local founder effects. Several NCL genes have widespread distribution across several continents due to ancient founder effects (Table 1).
An estimate of the proportion of cases caused by each mutation can be made, although there is a considerable under-representation of the occurrence of common mutations since the emphasis is on the collation of novel and rare mutations. The most prevalent mutations are the 1 kb deletion in CLN3 and two mutations in CLN2 (1).
Correlations can be drawn between genotype, phenotype and morphological changes in patients, and have been reviewed previously (47, 75), for example for CLN2 disease (76). These derived correlations can be used to predict the disease course in a newly diagnosed family.
This database is important (1). The severity of mutations has implications for treatment. It may be important to know if residual protein or function remains. Treatments may be developed that do not fully compensate for complete loss of gene function and can reduce but not completely eliminate the disease burden—these may be sufficient to improve health in families carrying so-called mild mutations but not in individuals lacking all gene function (2). The location of mutations in the protein may highlight key residues and functional or regulatory domains, aiding understanding of protein function (3). The data reveals the relative frequency of mutations; as ultra-rare, found only within certain ethnic groups, or widespread (4). The data is freely available and contained in excel tables that can be downloaded and used by researchers. For example, there is increasing information on frequency of mutations or disease in specific ethnic groups (4). Efficacy of a new treatment may be demonstrated earlier or more robustly if the mutations and their effects on disease progression of the participants are understood. Going forward, functional data for each mutation can begin to be incorporated, as available.
Other databases exist through international cooperation, enabling collection of natural history data for all NCL types and genotype-phenotype data through databases DEM-CHILD (www.dem-child.eu) (77, 78). There are disease rating scales (79–81) to follow disease progression. This is increasing understanding of the genetic spectrum of NCL disease as well as provide necessary control data for use in future clinical trials (77).
Most genes that cause NCL disease in humans are probably identified. This, combined with the broader range of associated phenotypes now described, has shown that the genetic picture is considerably more complex than was first envisioned at the start of the genetic era of the NCL. The functions of all NCL genes and thereby disease mechanisms are not yet known. As understanding increases overlap with other rare and common diseases, such as retinal dystrophies may indicate shared disease mechanisms (82).
The gene dosage or the specific mutations show correlation with clinical phenotype. Some variation in clinical phenotype is therefore explained by differing levels of residual protein function. However, variation between families and even siblings shows that co-inheritance of other genetic variations could influence disease phenotype. It is still unclear whether the underlying pathogenic mechanisms are partly shared between classic NCL forms and the alternative disease forms.
The era of genomic medicine is approaching, where genomic information will be used to design the best clinical care for an individual. For the NCL, personalised treatment approaches will be tailored to the underlying mutation and the genetic background of each patient. An early example is the design and delivery of an oligonucleotide therapy for a child with CLN7 disease (83).
Therapeutic development beyond current palliative treatments is advancing slowly. This relies on continued collection of natural history data for the broadening NCL spectrum to provide a control cohort to aid design of future clinical trials. The first approved treatment is for children with classic late infantile CLN2 disease which delivers recombinant protein directly into the brain at regular intervals. For the best long-term clinical benefit for any NCL disease, treatment must begin as early as possible, before any symptoms, which requires rapid and earlier diagnosis using genotype. This may be facilitated by advances in DNA-based approaches that allow future newborn screening (84, 85).
SM devised, interpreted the data, and wrote the review. EG collated the data on mutations, genes and phenotype, and contributed to the writing. All authors contributed to the article and approved the submitted version.
This work was funded by the Medical Research Council (MRC), award MR/V033956. SM (ORCID:0000-0003-4385-4957) reports support by Biomarin for maintaining the NCL mutation database. The support of UCL and the MRC that provides funding to the MRC Laboratory for Molecular Cell Biology University Unit at UCL (award code MC_U12266B) toward lab and office space is acknowledged. All research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health is made possible by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank all those who contribute to the NCL mutation database, other genetic databases, and research publications and case reports, from which NCL mutation data is drawn.
1. Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim Biophys Acta. (2015) 1852:2237–41. doi: 10.1016/j.bbadis.2015.05.011
2. Zeman W, Dyken P. Neuronal ceroid-lipofuscinosis (Batten's disease): relationship to amaurotic familial idiocy? Pediatrics. (1969) 44:570–83.
3. Noskova L, Stranecky V, Hartmannova H, Pristoupilova A, Baresova V, Ivanek R, et al. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am J Hum Genet. (2011) 89:241–52. doi: 10.1016/j.ajhg.2011.07.003
4. Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P, et al. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. (1995) 376:584–7. doi: 10.1038/376584a0
5. Consortium TIBD. Isolation of a novel gene underlying batten disease, CLN3. Cell. (1995) 82:949–57. doi: 10.1016/0092-8674(95)90274-0
6. Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, et al. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. (1997) 277:1802–5. doi: 10.1126/science.277.5333.1802
7. Savukoski M, Klockars T, Holmberg V, Santavuori P, Lander ES, Peltonen L. CLN5, a novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis. Nat Genet. (1998) 19:286–8. doi: 10.1038/975
8. Wheeler RB, Sharp JD, Schultz RA, Joslin JM, Williams RE, Mole SE. The gene mutated in variant late-infantile neuronal ceroid lipofuscinosis (CLN6) and in nclf mutant mice encodes a novel predicted transmembrane protein. Am J Hum Genet. (2002) 70:537–42. doi: 10.1086/338708
9. Siintola E, Topcu M, Aula N, Lohi H, Minassian BA, Paterson AD, et al. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am J Hum Genet. (2007) 81:136–46. doi: 10.1086/518902
10. Ranta S, Zhang Y, Ross B, Lonka L, Takkunen E, Messer A, et al. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8. Nat Genet. (1999) 23:233–6. doi: 10.1038/13868
11. Siintola E, Partanen S, Strömme P, Haapanen A, Haltia M, Maehlen J, et al. Cathepsin D deficiency underlies congenital human neuronal ceroid lipofuscinosis. Brain. (2006) 129:1438–45. doi: 10.1093/brain/awl107
12. Steinfeld R, Reinhardt K, Schreiber K, Hillebrand M, Kraetzner R, Brück W, et al. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am J Hum Genet. (2006) 78:988–98. doi: 10.1086/504159
13. Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. (2012) 90:1102–7. doi: 10.1016/j.ajhg.2012.04.021
14. Bras J, Verloes A, Schneider SA, Mole SE. Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet. (2012) 21:2646–50. doi: 10.1093/hmg/dds089
15. Smith KR, Dahl HH, Canafoglia L, Andermann E, Damiano J, Morbin M, et al. Cathepsin F mutations cause Type B Kufs disease, an adult-onset neuronal ceroid lipofuscinosis. Hum Mol Genet. (2013) 22:1417–23. doi: 10.1093/hmg/dds558
16. Staropoli JF, Karaa A, Lim ET, Kirby A, Elbalalesy N, Romansky SG, et al. A homozygous mutation in KCTD7 links neuronal ceroid lipofuscinosis to the ubiquitin-proteasome system. Am J Hum Genet. (2012) 91:202–8. doi: 10.1016/j.ajhg.2012.05.023
17. Haddad SE, Khoury M, Daoud M, Kantar R, Harati H, Mousallem T, et al. CLN5 and CLN8 protein association with ceramide synthase: biochemical and proteomic approaches. Electrophoresis. (2012) 33:3798–809. doi: 10.1002/elps.201200472
18. Schulz A, Dhar S, Rylova S, Dbaibo G, Alroy J, Hagel C, et al. Impaired cell adhesion and apoptosis in a novel CLN9 Batten disease variant. Ann Neurol. (2004) 56:342–50. doi: 10.1002/ana.20187
19. Travaglini L, Aiello C, Alesi V, Loddo S, Novelli A, Tozzi G, et al. Uniparental disomy of chromosome 1 unmasks recessive mutations of PPT1 in a boy with neuronal ceroid lipofuscinosis type 1. Brain Dev. (2016) 9:182–3. doi: 10.1016/j.braindev.2016.08.010
20. Niida Y, Yokoi A, Kuroda M, Mitani Y, Nakagawa H, Ozaki M, et al. A Girl with infantile neuronal ceroid lipofuscinosis caused by novel PPT1 mutation and paternal uniparental isodisomy of chromosome 1. Brain Dev. (2016) 38:674–7. doi: 10.1016/j.braindev.2016.01.004
21. Huber RJ, Mathavarajah S. Cln5 is secreted and functions as a glycoside hydrolase in Dictyostelium. Cell Signal. (2018) 42:236–48. doi: 10.1016/j.cellsig.2017.11.001
22. Cárcel-Trullols J, Kovács AD, Pearce DA. Cell biology of the NCL proteins: what they do and don't do. Biochim Biophys Acta. (2015) 1852:2242–55. doi: 10.1016/j.bbadis.2015.04.027
23. Butz ES, Chandrachud U, Mole SE, Cotman SL. Moving towards a new era of genomics in the neuronal ceroid lipofuscinoses. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165571. doi: 10.1016/j.bbadis.2019.165571
24. Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. (2012) 79:183–91. doi: 10.1212/WNL.0b013e31825f0547
25. Gao H, Boustany RM, Espinola JA, Cotman SL, Srinidhi L, Antonellis KA, et al. Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am J Hum Genet. (2002) 70:324–35. doi: 10.1086/338190
26. Arsov T, Smith KR, Damiano J, Franceschetti S, Canafoglia L, Bromhead CJ, et al. Kufs disease, the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6. Am J Hum Genet. (2011) 88:566–73. doi: 10.1016/j.ajhg.2011.04.004
27. Berkovic SF, Staropoli JF, Carpenter S, Oliver KL, Kmoch S, Anderson GW, et al. Diagnosis and misdiagnosis of adult neuronal ceroid lipofuscinosis (Kufs disease). Neurology. (2016) 87:579–84. doi: 10.1212/WNL.0000000000002943
28. Roosing S, van den Born LI, Sangermano R, Banfi S, Koenekoop RK, Zonneveld-Vrieling MN, et al. Mutations in MFSD8, encoding a lysosomal membrane protein, are associated with nonsyndromic autosomal recessive macular dystrophy. Ophthalmology. (2015) 122:170–9. doi: 10.1016/j.ophtha.2014.07.040
29. Sun Y, Almomani R, Breedveld GJ, Santen GW, Aten E, Lefeber DJ, et al. Autosomal recessive spinocerebellar ataxia 7 (SCAR7) is caused by variants in TPP1, the gene involved in classic late-infantile neuronal ceroid lipofuscinosis 2 disease (CLN2 disease). Hum Mutat. (2013) 34:706–13. doi: 10.1002/humu.22292
30. Polovitskaya MM, Barbini C, Martinelli D, Harms FL, Cole FS, Calligari P, et al. A recurrent gain-of-function mutation in CLCN6, encoding the ClC-6 Cl(-)/H(+)-Exchanger, causes early-onset neurodegeneration. Am J Hum Genet. (2020) 107:1062–77. doi: 10.1016/j.ajhg.2020.11.004
31. Poët M, Kornak U, Schweizer M, Zdebik AA, Scheel O, Hoelter S, et al. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc Natl Acad Sci USA. (2006) 103:13854–9. doi: 10.1073/pnas.0606137103
32. Mitchison HM, O'Rawe AM, Taschner PE, Sandkuijl LA, Santavuori P, de Vos N, et al. Batten disease gene, CLN3: linkage disequilibrium mapping in the Finnish population, and analysis of European haplotypes. Am J Hum Genet. (1995) 56:654–62.
33. Kitzmuller C, Haines RL, Codlin S, Cutler DF, Mole SE. A function retained by the common mutant CLN3 protein is responsible for the late onset of juvenile neuronal ceroid lipofuscinosis. Hum Mol Genet. (2008) 17:303–12. doi: 10.1093/hmg/ddm306
34. Minnis CJ, Townsend S, Petschnigg J, Tinelli E, Bahler J, Russell C, et al. Global network analysis in Schizosaccharomyces pombe reveals three distinct consequences of the common 1-kb deletion causing juvenile CLN3 disease. Sci Rep. (2021) 11:6332. doi: 10.1038/s41598-021-93446-8
35. Wang F, Wang H, Tuan HF, Nguyen DH, Sun V, Keser V, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. (2014) 133:331–45. doi: 10.1007/s00439-013-1381-5
36. Cortese A, Tucci A, Piccolo G, Galimberti CA, Fratta P, Marchioni E, et al. Novel CLN3 mutation causing autophagic vacuolar myopathy. Neurology. (2014) 82:2072–6. doi: 10.1212/WNL.0000000000000490
37. Erickson RP, Larson-Thome K, Weberg L, Szybinska A, Mossakowska M, Styczynska M, et al. Variation in NPC1, the gene encoding Niemann-Pick C1, a protein involved in intracellular cholesterol transport, is associated with Alzheimer disease and/or aging in the Polish population. Neurosci Lett. (2008) 447:153–7. doi: 10.1016/j.neulet.2008.09.046
38. Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med. (2004) 351:1972–7. doi: 10.1056/NEJMoa033277
39. Ward ME, Chen R, Huang HY, Ludwig C, Telpoukhovskaia M, Taubes A, et al. Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci Transl Med. (2017) 9:eaah5642. doi: 10.1126/scitranslmed.aah5642
40. Valdez C, Wong YC, Schwake M, Bu G, Wszolek ZK, Krainc D. Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum Mol Genet. (2017) 26:4861–72. doi: 10.1093/hmg/ddx364
41. Gotzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, et al. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. (2014) 127:845–60. doi: 10.1007/s00401-014-1262-6
42. Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol. (2010) 177:311–24. doi: 10.2353/ajpath.2010.090915
43. Yin F, Dumont M, Banerjee R, Ma Y, Li H, Lin MT, et al. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. (2010) 24:4639–47. doi: 10.1096/fj.10.161471
44. Blumkin L, Kivity S, Lev D, Cohen S, Shomrat R, Lerman-Sagie T, et al. A compound heterozygous missense mutation and a large deletion in the KCTD7 gene presenting as an opsoclonus-myoclonus ataxia-like syndrome. J Neurol. (2012) 259:2590–8. doi: 10.1007/s00415-012-6545-z
45. Van Bogaert P, Azizieh R, Desir J, Aeby A, De Meirleir L, Laes JF., et al. Mutation of a potassium channel-related gene in progressive myoclonic epilepsy. Ann Neurol. (2007) 61:579–86. doi: 10.1002/ana.21121
46. Kousi M, Anttila V, Schulz A, Calafato S, Jakkula E, Riesch E., et al. Novel mutations consolidate KCTD7 as a progressive myoclonus epilepsy gene. J Med Genet. (2012) 49:391–9. doi: 10.1136/jmedgenet-2012-100859
47. Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. (2012) 33:42–63. doi: 10.1002/humu.21624
48. Metz KA, Teng X, Coppens I, Lamb HM, Wagner BE, Rosenfeld JA, et al. KCTD7 deficiency defines a distinct neurodegenerative disorder with a conserved autophagy-lysosome defect. Ann Neurol. (2018) 84:766–80. doi: 10.1002/ana.25351
49. Mastrangelo M, Sartori S, Simonati A, Brinciotti M, Moro F, Nosadini M, et al. Progressive myoclonus epilepsy and ceroidolipofuscinosis 14: the multifaceted phenotypic spectrum of KCTD7-related disorders. Eur J Med Genet. (2019) 62:103591. doi: 10.1016/j.ejmg.2018.11.025
50. Estrada-Cuzcano A, Martin S, Chamova T, Synofzik M, Timmann D, Holemans T, et al. Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain. (2017) 140:287–305. doi: 10.1093/brain/aww307
51. Spataro R, Kousi M, Farhan SMK, Willer JR, Ross JP, Dion PA, et al. Mutations in ATP13A2 (PARK9) are associated with an amyotrophic lateral sclerosis-like phenotype, implicating this locus in further phenotypic expansion. Hum Genomics. (2019) 13:19. doi: 10.1186/s40246-019-0203-9
52. Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP., et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. (2006) 38:1184–91. doi: 10.1038/ng1884
53. Tome FM, Brunet P, Fardeau M, Hentati F, Reix J. Familial disorder of the central and peripheral nervous systems with particular cytoplasmic lamellated inclusions in peripheral nerves, muscle satellite cells, and blood capillaries. Acta Neuropathol. (1985) 68:209–17. doi: 10.1007/BF00690197
54. De Volder AG, Cirelli S, de Barsy T, Brucher JM, Bol A, Michel C, et al. Neuronal ceroid-lipofuscinosis: preferential metabolic alterations in thalamus and posterior association cortex demonstrated by PET. J Neurol Neurosurg Psychiatry. (1990) 53:1063–7. doi: 10.1136/jnnp.53.12.1063
55. Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. (2013) 22:2067–82. doi: 10.1093/hmg/ddt057
56. Sleat DE, Ding L, Wang S, Zhao C, Wang Y, Xin W, et al. Mass spectrometry-based protein profiling to determine the cause of lysosomal storage diseases of unknown etiology. Mol Cell Proteomics. (2009) 8:1708–18. doi: 10.1074/mcp.M900122-MCP200
57. Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A, et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. (2001) 104:205–15. doi: 10.1016/S0092-8674(01)00206-9
58. Kasper D, Planells-Cases R, Fuhrmann JC, Scheel O, Zeitz O, Ruether K, et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. (2005) 24:1079–91. doi: 10.1038/sj.emboj.7600576
59. Di Zanni E, Palagano E, Lagostena L, Strina D, Rehman A, Abinun M, et al. Pathobiologic mechanisms of neurodegeneration in osteopetrosis derived from structural and functional analysis of 14 ClC-7 mutants. J Bone Miner Res. (2021) 36:531–45. doi: 10.1002/jbmr.4200
60. Fernandez-Chacon R, Wolfel M, Nishimune H, Tabares L, Schmitz F, Castellano-Munoz M, et al. The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron. (2004) 42:237–51. doi: 10.1016/S0896-6273(04)00190-4
61. Stahl S, Reinders Y, Asan E, Mothes W, Conzelmann E, Sickmann A, et al. Proteomic analysis of cathepsin B- and L-deficient mouse brain lysosomes. Biochim Biophys Acta. (2007) 1774:1237–46. doi: 10.1016/j.bbapap.2007.07.004
62. Staropoli JF, Xin W, Barone R, Cotman SL, Sims KB. An atypical case of neuronal ceroid lipofuscinosis with co-inheritance of a variably penetrant POLG1 mutation. BMC Med Genet. (2012) 13:50. doi: 10.1186/1471-2350-13-50
63. Zhang CK, Stein PB, Liu J, Wang Z, Yang R, Cho JH, et al. Genome-wide association study of N370S homozygous Gaucher disease reveals the candidacy of CLN8 gene as a genetic modifier contributing to extreme phenotypic variation [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. Am J Hematol. (2012) 87:377–83. doi: 10.1002/ajh.23118
64. Metcalfe DJ, Calvi AA, Seamann MNJ, Mitchison HM, Cutler DF. Loss of the Batten disease gene CLN3 prevents exit from the TGN of the mannose 6-phosphate receptor. Traffic. (2008) 11:1905–14. doi: 10.1111/j.1600-0854.2008.00807.x
65. Vesa J, Chin MH, Oelgeschlager K, Isosomppi J, DellAngelica EC, Jalanko A, et al. Neuronal ceroid lipofuscinoses are connected at molecular level: interaction of CLN5 protein with CLN2 and CLN3. Mol Biol Cell. (2002) 13:2410–20. doi: 10.1091/mbc.e02-01-0031
66. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer [Research Support, Non-U.S. Gov't]. Nucl Acids Res. (2011) 39:D945–50. doi: 10.1093/nar/gkq929
67. Mole SE, Williams RE, Goebel HH, (editors.). The Neuronal Ceroid Lipofusinoses (Batten disease). 2nd ed. Oxford: Oxford University Press (2011). p. 444.
68. Kline RA, Wishart TM, Mills K, Heywood WE. Applying modern omic technologies to the neuronal ceroid lipofuscinoses. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165498. doi: 10.1016/j.bbadis.2019.06.012
69. Iwan K, Clayton R, Mills P, Csanyi B, Gissen P, Mole SE, et al. Urine proteomics analysis of patients with neuronal ceroid lipofuscinoses. iScience. (2021) 24:102020. doi: 10.1016/j.isci.2020.102020
70. Mole SE, Anderson G, Band HA, Berkovic SF, Cooper JD, Kleine Holthaus SM, et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. (2019) 18:107–16. doi: 10.1016/S1474-4422(18)30368-5
71. Anderson G, Smith VV, Malone M, Sebire NJ. Blood film examination for vacuolated lymphocytes in the diagnosis of metabolic disorders; retrospective experience of more than 2,500 cases from a single centre. J Clin Pathol. (2005) 58:1305–10. doi: 10.1136/jcp.2005.027045
72. Shen J, Cram DS, Wu W, Cai L, Yang X, Sun X, et al. Successful PGD for late infantile neuronal ceroid lipofuscinosis achieved by combined chromosome and TPP1 gene analysis. Reprod Biomed Online. (2013) 27:176–83. doi: 10.1016/j.rbmo.2013.04.011
73. Minnis CJ, Thornton CD, FitzPatrick LM, McKay TR. Cellular models of Batten disease. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165559. doi: 10.1016/j.bbadis.2019.165559
74. Abitbol M, Thibaud JL, Olby NJ, Hitte C, Puech JP, Maurer M, et al. A canine Arylsulfatase G (ARSG) mutation leading to a sulfatase deficiency is associated with neuronal ceroid lipofuscinosis. Proc Natl Acad Sci USA. (2010) 107:14775–80. doi: 10.1073/pnas.0914206107
75. Mole SE, Williams RE, Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenet. (2005) 6:107–26. doi: 10.1007/s10048-005-0218-3
76. Gardner E, Bailey M, Schulz A, Aristorena M, Miller N, Mole SE. Mutation update: review of TPP1 gene variants associated with neuronal ceroid lipofuscinosis CLN2 disease. Hum Mutat. (2019) 40:1924–38. doi: 10.1002/humu.23860
77. Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, et al. Study of intraventricular cerliponase alfa for CLN2 disease. N Engl J Med. (2018) 378:1898–907. doi: 10.1056/NEJMoa1712649
78. Nickel M, Simonati A, Jacoby D, Lezius S, Kilian D, Van de Graaf B, et al. Disease characteristics and progression in patients with late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: an observational cohort study. Lancet Child Adolesc Health. (2018) 2:582–90. doi: 10.1016/S2352-4642(18)30179-2
79. Steinfeld R, Heim P, von Gregory H, Meyer K, Ullrich K, Goebel HH, et al. Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations. Am J Med Genet. (2002) 112:347–54. doi: 10.1002/ajmg.10660
80. Kwon JM, Adams H, Rothberg PG, Augustine EF, Marshall FJ, Deblieck EA, et al. Quantifying physical decline in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology. (2011) 77:1801–7. doi: 10.1212/WNL.0b013e318237f649
81. Marshall FJ, de Blieck EA, Mink JW, Dure L, Adams H, Messing S, et al. A clinical rating scale for Batten disease: reliable and relevant for clinical trials. Neurology. (2005) 65:275–9. doi: 10.1212/01.wnl.0000169019.41332.8a
82. Cotman SL, Mole SE, Kohan R. Future perspectives: moving towards NCL treatments. Biochim Biophys Acta. (2015) 1852:2336–8. doi: 10.1016/j.bbadis.2015.04.001
83. Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Larson A, et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N Engl J Med. (2019) 381:1644–52. doi: 10.1056/NEJMoa1813279
84. Barcenas M, Xue C, Marushchak-Vlaskin T, Scott CR, Gelb MH, Turecek F. Tandem mass spectrometry assays of palmitoyl protein thioesterase 1 and tripeptidyl peptidase activity in dried blood spots for the detection of neuronal ceroid lipofuscinoses in newborns. Anal Chem. (2014) 86:7962–8. doi: 10.1021/ac501994b
Keywords: neuronal ceroid lipofuscinosis, batten disease, NCL, CLN, mutation, gene, lysosomal disease
Citation: Gardner E and Mole SE (2021) The Genetic Basis of Phenotypic Heterogeneity in the Neuronal Ceroid Lipofuscinoses. Front. Neurol. 12:754045. doi: 10.3389/fneur.2021.754045
Received: 05 August 2021; Accepted: 20 September 2021;
Published: 18 October 2021.
Edited by:
Ruth Elizabeth Williams, Evelina London Children's Hospital, United KingdomReviewed by:
Maria Gogou, Aristotle University of Thessaloniki, GreeceCopyright © 2021 Gardner and Mole. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sara E. Mole, cy5tb2xlQHVjbC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.