
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol., 24 December 2021
Sec. Epilepsy
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.743726
 Jiangwei Ding1,2
Jiangwei Ding1,2 Xinxiao Li3*
Xinxiao Li3* Haiyan Tian4Lei Wang1,5Baorui Guo1,2Yangyang Wang1,5
Haiyan Tian4Lei Wang1,5Baorui Guo1,2Yangyang Wang1,5 Wenchao Li1,5
Wenchao Li1,5 Feng Wang6*
Feng Wang6* Tao Sun1,2*
Tao Sun1,2*Background: SCN1A is one of the most common epilepsy genes. About 80% of SCN1A gene mutations cause Dravet syndrome (DS), which is a severe and catastrophic epileptic encephalopathy. More than 1,800 mutations have been identified in SCN1A. Although it is known that SCN1A is the main cause of DS and genetic epilepsy with febrile seizures plus (GEFS+), there is a dearth of information on the other related diseases caused by mutations of SCN1A.
Objective: The aim of this study is to systematically review the literature associated with SCN1A and other non-DS-related disorders.
Methods: We searched PubMed and SCOPUS for all the published cases related to gene mutations of SCN1A until October 20, 2021. The results reported by each study were summarized narratively.
Results: The PubMed and SCOPUS search yielded 2,889 items. A total of 453 studies published between 2005 and 2020 met the final inclusion criteria. Overall, 303 studies on DS, 93 on GEFS+, three on Doose syndrome, nine on the epilepsy of infancy with migrating focal seizures (EIMFS), six on the West syndrome, two on the Lennox–Gastaut syndrome (LGS), one on the Rett syndrome, seven on the nonsyndromic epileptic encephalopathy (NEE), 19 on hemiplegia migraine, six on autism spectrum disorder (ASD), two on nonepileptic SCN1A-related sudden deaths, and two on the arthrogryposis multiplex congenital were included.
Conclusion: Aside from DS, SCN1A also causes other epileptic encephalopathies, such as GEFS+, Doose syndrome, EIMFS, West syndrome, LGS, Rett syndrome, and NEE. In addition to epilepsy, hemiplegic migraine, ASD, sudden death, and arthrogryposis multiplex congenital can also be caused by mutations of SCN1A.
Voltage-gated sodium channel (VGSC) channels play an essential role in normal neurological function (1), especially in the initiation and propagation of action potential. To date, nine α subunits of sodium channels have been found and confirmed (Nav1.1–Nav1.9). These channels are composed of four homologous but distinct domains (DI–DIV), each of which contains six transmembrane segments (S1–S6) (2) (Figures 2, 3). SCN1A, a Nav1.1 α subunit composed of 26 coding exons and located in the 85-kb gene region, is the most common epileptic gene and the most common pathogenic gene in the Dravet syndrome (DS), a catastrophic and intractable epileptic encephalopathy (EE) (3). Phenotypes caused by de novo SCN1A pathogenic variants are very variable, ranging from the severely affected patients with DS to much milder cases of genetic epilepsy febrile seizures plus (GEFS+). In addition to gene mutations of SCN1A that can cause DS, other genes include PCDH19, SCN2A, SCN8A, SCN1B, GABRA1, GABRG2, GABRB3, STXBP1, HCN1, CHD2, and KCNA2 can also cause DS or DS-like phenotypes (4). They are also closely related to other epileptic diseases and nonepileptic diseases (5–10).
A systematic search was performed in PubMed and SCOPUS. The most recent search was performed on October 20, 2021, using the term “SCN1A” or “scn1a”.
All the articles with mutations of SCN1A associated with a particular disease were included in the criteria. We excluded articles not written in English or Chinese, nonoriginal work that has nothing to do with people, such as reviews, meta-analysis, animals or cells, experimental articles not adding information to the question posed in this review, and papers that could not be retrieved via PubMed or SCOPUS. The records were screened by JD and evaluated by XL with respect to the inclusion and exclusion criteria. Disagreements were resolved through a discussion between the two review authors (Figure 1).
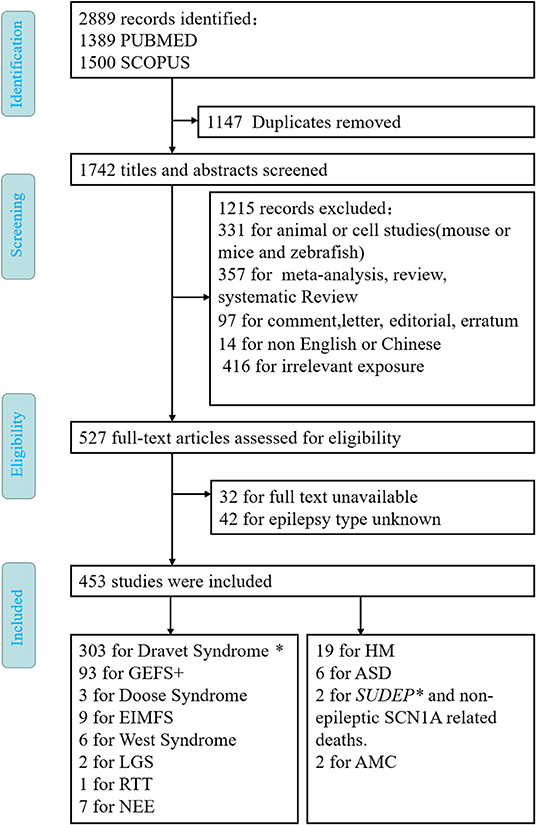
Figure 1. Flow diagram depicting search process and study selection. *means that SUDEP often occurs in Dravet syndrome.
After the elimination of duplicates (1,147 articles), the literature search yielded 1,742 articles (Figure 1). After screening all the abstracts, 1,215 records were excluded. Thus, 527 articles were included in the full-text analysis. Of these, 74 full-text articles were excluded. Articles were excluded based on the following exclusion criteria: animal or cell studies (n = 331); review, systematic review, and meta-analysis (n = 357); comment, letter, editorial, and erratum (n = 97); reports not in English or Chinese (n = 14); irrelevant exposure (n = 416); full text unavailable (n = 32); or epilepsy type unknown (n = 42). Finally, 453 studies met the inclusion and did not meet the exclusion criteria (Figure 1). It is well known that SCN1A is the main pathogenic cause of DS and GEFS+. Therefore, we only briefly describe SCN1A without discussing its specific mutation sites in detail.
The SCN1A gene is not only associated with DS and GEFS+, but can also cause other disorders, including epilepsy diseases such as Doose syndrome, epilepsy of infancy with migrating focal seizures (EIMFS), West syndrome, Lennox–Gastaut syndrome (LGS), Rett syndrome, and nonsyndromic epileptic encephalopathy (NEE), as well as nonepileptic diseases such as hemiplegia migraine, autism spectrum disorder (ASD), sudden death (sudden unexpected death in epilepsy [SUDEP] and nonepileptic SCN1A-related sudden deaths), and arthrogryposis multiplex congenita (AMC).
Mutations in the voltage-gated sodium channel subunit gene SCN1A are identified predominantly in patients with DS, also known as severe myoclonic epilepsy of infancy (SMEI), and in the families with GEFS+. However, SCN1A is less common in epileptic and nonepileptic disorders other than DS and GEFS+. Herein, we focus on these rare diseases with the exception of DS and GEFS+.
Dravet syndrome is a refractory and catastrophic EE that is mainly caused by haploinsufficiency due to a loss-of-function mutation in the SCN1A gene (1, 11). About 80% of DS is caused by mutations in the SCN1A gene. To date, more than 1,800 mutations have been identified in SCN1A (12, 13). Heat-induced epilepsy, the most common type of epilepsy in DS, is often caused by fever, vaccinations, and hot baths (14–16). With aging, the incidence of heat-induced epilepsy decreases, turning into the refractory epilepsy. Meanwhile, the cognitive dysfunction continues to aggravate and stabilize. Photosensitive epilepsy can also be observed in some patients with DS (17). In addition to the epileptic seizures, DS and other comorbidities that can be combined include ataxia, premature death, language, and motor development delay, cognitive impairment, sleep disorders, ASD, and SUDEP, which seriously affect the quality of life of the patients and pose a heavy economic burden to the family and society (18–23).
Genetic epilepsy with febrile seizures plus, previously known as generalized epilepsy with the febrile seizures plus (FS+), was first discovered by Scheffer and Berkovic in an Australian family in 1997 (24). Since it was found that the phenotype of focal epilepsy can occur in the GEFS+ family, it was renamed genetic epilepsy with FS+. GEFS+ is an EE with a milder phenotype than DS; it is also related to the multiple gene mutations, including SCN1A, SCN2A, SCN1B, GABRD, SCN9A, STX1B, and Fgf13 (25, 26). We have previously found in animal models that GABRG2 mutations can also cause GEFS+ (27). Various clinical phenotypes can occur in the GEFS+ family, ranging from the most common febrile seizures (FS) and FS+ to the severe EE known as DS. In 2000, Escayg et al. first found mutations in the SCN1A gene (Thr875Met and Arg1648His) in GEFS+ families (28) (Figure 2, Table 1). Aside from DS, SCN1A gene mutations are the most common pathogenic genes for GEFS+. In fact, GEFS+ and DS are different manifestations of epilepsy caused by SCN1A mutations.
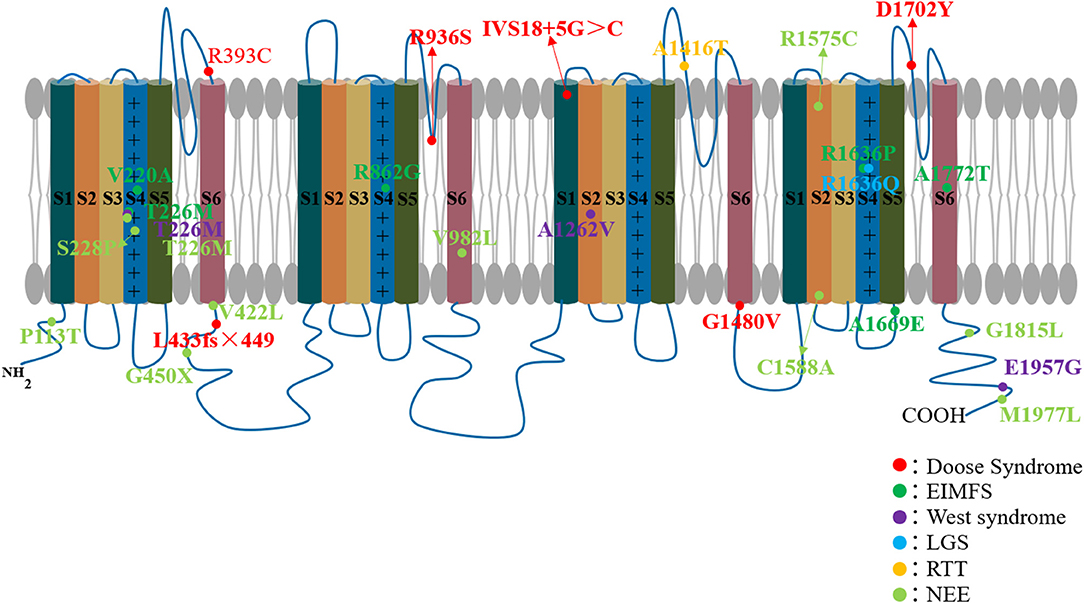
Figure 2. SCN1A mutations associated with epileptic encephalopathy. Each circular represents a patient's variant of the SCN1A gene.

Table 1. Clinical data and mutation sites or chromosomal deletions in SCN1A-associated non-dravet syndrome epilepsy.
Doose syndrome, also known as epilepsy with myoclonic atonic seizure (EMAS), was previously called myoclonic astatic epilepsy (MAE), a rare childhood EE (55). First reported by Doose in 1970, the International League Against Epilepsy (ILAE) in 2010 changed its name to epilepsy with myoclonus-atonic seizures based on the characteristics of epileptic seizures (56, 57). Usually, Doose syndrome develops from seven months to 6 years, and the peak age of onset is 2 to 4 years. Children usually have normal mental and motor development before the onset. Most children start with a generalized tonic–clonic seizure (GTCS). The initial seizures can be very frequent, followed by a variety of generalized seizures, including myoclonic seizures, dystonic seizures, myoclonic–dystonic seizures, and atypical absence; a small number of children may have tonic seizures in the later stages (55).
Doose syndrome is associated with mutations in a variety of epilepsy genes, including SCN1A, SCN1B, CACNA1H, SLC2A1, GABRG2, CHD2, SLC6A1, STX1B, GABRB3, SYNGAP1, and WDR45 (33). In 2005, Ebach et al. reported three cases of EMAS with SCN1A gene mutations (29). In 2007, Harkin et al. found one case of Doose syndrome due to SCN1A mutation in 188 patients with EE (30). Interestingly, Dimova et al. also found a case of EMAS caused by SCN1A gene mutation in a GEFS+ family. The patient started with a febrile seizure at the age of three, after which subsequent multiple myoclonic and myoclonic–astatic seizures appeared (31). Recently, Hinokuma et al. found one microdeletion at 2q24.2 involving SCN1A in 29 patients with Doose syndrome (33) (Figure 2, Table 1). All of the foregoing extends the phenotype of the SCN1A gene mutation to Doose syndrome.
Epilepsy of infancy with migrating focal seizures, previously known as infantile migratory partial epilepsy (MPSI) or infantile malignant migratory partial seizure (MMPSI), is a rare and early-onset developmental EE inherited in an autosomal dominant pattern. It is characterized by onset within 6 months of birth and mainly manifests in the form of frequent, migratory, and varying types of focal seizures. Epileptic seizures are related to the multifocal EEG discharge. Similar to DS, this disease is often associated with severe cognitive impairment and motor impairment. However, unlike DS, the most common causative gene is KCNT1 mutation.
Freilich et al. first identified the SCN1A mutation in a female infant diagnosed with MPSI. The female infant, who was delivered to term, developed epilepsy at 10 weeks after birth, accompanied by progressive hemiplegia, apnea, and progression of multifocal migratory partial epileptic seizures, leading to a recurrence of epileptic status and death at 9 months (34). In the same year, another case of SCN1A mutation was found in another patient with MPSI (35). In 2012, Shein et al. reported a case of SCN1A mutation-induced MPSI with good therapeutic effect assisted by hypothermia (36). In 2015, Lim et al. reported three cases with SCN1A mutation (MPSI) (38). In the same year, Zhang and colleagues found 46 cases of genetic mutations in 253 children with unexplained epilepsy and intellectual/developmental disabilities, of which only one was an SCN1A mutation causing malignant migrating partial seizures of infancy (37). In 2016, Shang et al. conducted genetic testing on nine cases of EIMFs and found that two (22.2%) carried an SCN1A mutation (39). Recently, Fang et al. also found one SCN1A mutation patient in five patients with EIMFS (41) (Figure 2, Table 1). SCN1A is currently considered to be the third most common type of genetic variation in EIMFS (58).
West syndrome, also known as infantile spasms (IS), is a refractory classic EE characterized by repetitive epileptic spasms (ES) and hypsarrhythmia (44). The etiology of the West syndrome is complex and varied. Genetic studies of individuals with unexplained IS have identified over 37 genes as pathogenic (59). However, SCN1A was not reported in a recent review of West syndrome, indicating its rarity in this disease (42, 59). Hattori et al. reported a case of partial epileptic seizures at four months and a West syndrome infant at 8 months with characteristic facial appearance, big toe abnormalities, and developmental delay. Chromosome and gene sequencing revealed the deletion of the SCN1A gene and 2q31.1 region [arr 2q24.3q31.3 (166,303,447–180,982.972) × 1 (build19)] (43). In 2003, Wallace et al. found one case of SCN1A mutation in 23 patients with West syndrome (42). Ilona et al. found only one SCN1A mutation in 45 patients clinically diagnosed with West syndrome by genetic testing (44). Most recently, Na et al. performed targeted gene panel sequencing for 150 early onset DEE infants aged ≤ 3 months and only one patient with SCN1A mutation was found. These findings indicate that the phenotypic heterogeneity of SCN1A mutation has extended to West syndrome (Figure 2, Table 1).
Lennox–Gastaut syndrome is a childhood EE whose main clinical features include multiple types of drug-resistant seizures, intellectual disability, and abnormal EEG with diffuse spines slow complex wave or paroxysmal fast activity. The etiology of LGS is also complex and varied; about 75% of cases have obvious causes such as cortical malformations, posthypoxic ischemic results, postmeningitis/encephalitis, or metabolic encephalopathy, while about 25% are cryptogenic (60). LGS is associated with a variety of genetic mutations, including ion channel genes (SCN2A, KCNT1, GABRA1, SCN8A, and GABRB3), transcription regulation genes (CHD2), neurocutaneous syndrome-related genes (TSC1 and TSC2), metabolic genes (Alg13), and others (45, 61, 62). However, SCN1A mutations rarely occur in LGS (30, 47). Harkin et al. found an SCN1A mutation in one out of 188 epileptic encephalopathy patients diagnosed with LGS (30). In 2009, Selmer and colleagues examined the SCN1A gene in 22 adult patients with LGS and found a mutation in one of them (47) (Figure 2, Table 1). In summary, SCN1A is rare, but it can still occur in LGS.
Rett syndrome is a rare single-gene disease that is more prevalent in females. RTT patients usually have an early stagnation period of onset 6–18 months after birth, and then enter a rapid regression period of development. The typical phenotype includes intractable epileptic seizures and severe mental retardation, particularly a rapid regression in language and limited progress in the psychomotor development. They may also be accompanied by the related complications such as autism, hand stereotypes, and respiratory pattern disorders (63). While more than 95% of patients carry de novo mutation(s) in the methyl-CpG-binding protein 2 (MECP2) gene (classical RTT), a small fraction of the patients (atypical RTT) may carry genetic mutations in other genes, such as the cyclin-dependent kinase-like 5 (CDKL5) and FOXG1 (64, 65).
The role of SCN1A dysfunction in RTT has also been highlighted by a few cases (46). Henriksen and colleagues (46) reported two patients with RTT caused by mutations in SCN1A. The first case is a 19-year-old female who developed febrile seizures at 5 months of age and subsequently developed afebrile focal seizures and intractable generalized seizures, including myotonic, tonic, and tonic–clonic. She also had several episodes with convulsive status epilepticus. She manifested normal hand functions and started to use a few words until she was 15 months old, but after that, her development slowed down. She stopped using her hands, her gait became broad and ataxic, and her speech disappeared. Between 1 and 2 years of age, she developed autism. At the age of 19, she still had dysmotility of hands and ataxia and suffered from breath holding and teeth grinding. Her height was only 141 cm. Her clinical signs and symptoms were consistent with classic RTT. Genetic testing showed that she was negative for MECP2, CDKL5, and FOXG1 genes, which are common to RTT, but SCN1A mutations were found. The second case occurred in a 32-year-old woman. She had her first febrile bilateral tonic–clonic seizure when she was 7 months old. The seizures worsened between the ages of one and two. Like the first patient, she grew normally until 12 to 15 months of age, but later acquired developmental disabilities and began to lose acquired skills. Her hand functions gradually declined, her speech disappeared, and she no longer seemed interested in her surroundings. She also suffered from bruxism and hand-washing stereotypes. At age of 32, she could walk for a few meters with support but still had ataxic and apraxic hand movements. She could not speak and had slight scoliosis. Epilepsy was always present. She also met the classic diagnostic criteria for RTT. Whole-exome sequencing unveiled the variant in SCN1A (Figure 2, Table 1).
Developmental and epileptic encephalopathies (DEEs), also known as early onset epileptic encephalopathies, early infantile epileptic encephalopathies (EIEEs), or early infantile-onset developmental and epileptic encephalopathies (EIODEEs) (45, 51, 66), comprise a kind of refractory epileptic encephalopathy that is mainly characterized by early-onset in neonates or infants, refractory epileptic seizures, and severe abnormal electroencephalogram discharge, psychomotor retardation, or regression. DEEs include early myoclonic encephalopathy (EME), Otahara syndrome, EIMFS, West syndrome, and DS (57). Nonsyndromic epileptic encephalopathy (NEE) can be referred to as clinically diagnosed epileptic encephalopathy without the inclusion of a specific syndrome or epileptic disorder (51).
In 2014 and 2016, Japanese scholars Ohashi and Kobayashi et al. described a distinct SCN1A phenotype called early infantile SCN1A encephalopathy, in which the patient had an apparent movement disorder (49, 51). Sadleir et al. also reported eight cases of SCN1A mutation with hyperkinetic movement disorder in 2017 (53) (Figure 2, Table 1). This may become a new type of epileptic encephalopathy shortly. Similarly, SCN1A mutations are rarely found in other cases of NEE (48, 52, 54).
Hemiplegic migraine is the most common neurological disorder that often presents with aura, which is associated with sensory and motor disturbances (67). Familial (FHM) and sporadic (SHM) hemiplegic migraines are severe subtypes of migraine associated with transient hemiparesis (68). FHM, a rare autosomal dominant genetic disorder, is a subtype of migraine with aura (MA) (69). The common classification and pathogenic genes are CACNAIA (FHM1), ATP1A2 (FHM2), and SCN1A (FHM3) (70). Familial hemiplegic migraine type 3 (FHM3) is seldom caused by mutations in SCN1A (71). Martin et al. first identified the SCN1A mutation in 2005 in the three familial migraine families (5). Subsequently, numerous SCN1A mutations have been found in FMH3 and, currently, about 60 patients carry SCN1A mutations (67, 72–85). In addition to FHM, SCN1A mutations are also found in a very small number of sporadic hemiplegic migraine patients (68, 86, 87) (Figure 3, Table 2). Therefore, it is confirmed that SCN1A is one of the pathogenic genes for hemiplegic migraine.
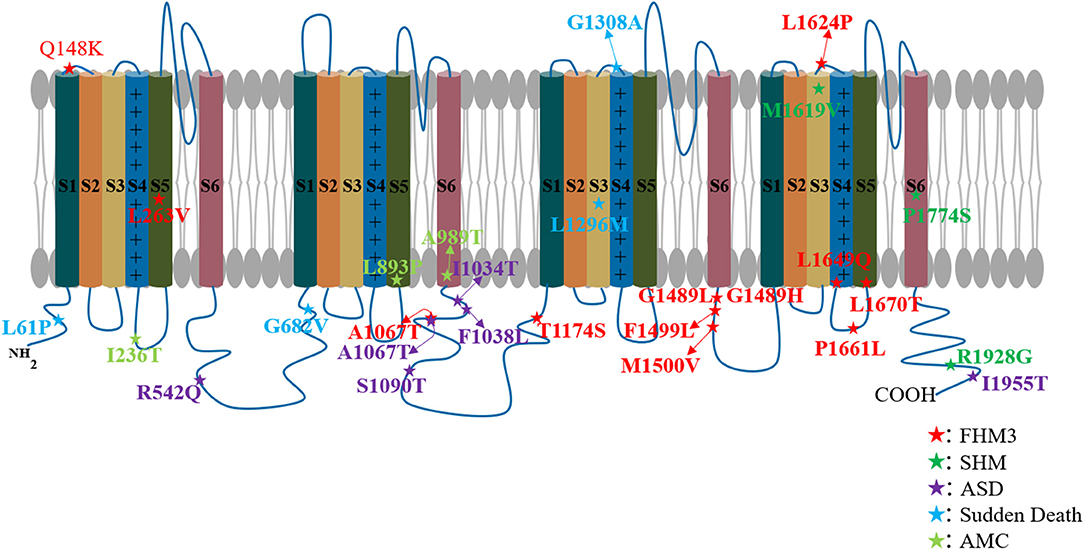
Figure 3. SCN1A mutations associated with non-epileptic disease. Each asterisk represents a patient's variant of the SCN1A gene.

Table 2. Clinical data and mutation sites or chromosomal deletions in SCN1A-associated non-epileptic disease.
Autism spectrum disorder is a complex psychiatric disorder characterized by impaired communication and social skills, and also restricted and repetitive behaviors (95). ASD can occur by itself or as a complication of epilepsy such as DS (6, 88, 96). DS caused by SCN1A gene mutation is associated with ASD (22, 96–98). Li et al. evaluated 37 patients with DS, nine of whom (24.3%) met autism criteria. They also found that people with autism had more severe intellectual disabilities than people without autism (97). Han et al. found an autism-like phenotype in SCN1A-mutated DS model mice (99). Interestingly, low-dose Clonazepam (a positive allosteric regulator of GABAAR) was used to mitigate this symptom, suggesting that GABAgic neurons may be directly related to ASD (99). Autism spectrum disorders can last from childhood to adulthood and even throughout life. Berkvens et al. conducted a follow-up on 13 patients with DS, among whom eight (61.5%) were classified as having ASD (96). Furthermore, ASD can occur in isolation from epilepsy. Weiss et al. found five missense mutations in patients with autism (6). Roak et al. also found one case of SCN1A missense mutation (p.Pro1894Leu) in 20 patients with ASD, and this mutation may be inherited from its parent (88). A recent study of 134 cases of autism identified 16 variants and 12 genes with evidence of pathogenicity, including three SCN1A mutations (91). In summary, SCN1A is closely related to ASD and has been considered as an ASD candidate gene (6, 7, 11, 89, 90) (Figure 3, Table 2).
Epilepsy-related deaths include seizures leading to asphyxia, injury, drowning, the occurrence of epileptic status, suicide, and SUDEP, which is a common cause of death in patients with epilepsy (100). SUDEP is a sudden, accidental death of a person with epilepsy, with or without witnesses, not from trauma or drowning, and with or without epileptic seizures; an epilepsy status must be ruled out and no structural or toxic cause of death is found at autopsy (101, 102). SUDEP generally occurs in 1.2 per 1,000 people with epilepsy per year (101). The sodium channels SCN1A, SCN1B, and SCN5A are considered as genes related to SUDEP (8, 103–106). DS, which is mainly caused by the SCN1A mutation gene, is the best model for studying the SCN1A gene (107). The mortality rate in patients with DS is about 20%, with SUDEP generally present in the deaths of children and adults with epileptic status (108). SUDEP occurs at a higher rate in DS than in other childhood epilepsies, accounting for up to about 50–60% of mortality (109, 110).
Sudden death associated with SCN1A mutations has also been reported in nonepileptic patients. In 2016, Halvorsen et al. (92) found one SCN1A mutant aged 20.8 months among nine children with sudden disease who died of the unknown causes. The child developed normally with a history of febrile convulsions but, interestingly, her siblings were diagnosed with DS. In 2018, Brownstein et al. (9) found an association between SCN1A mutation and sudden death in younger infants. The first case is a girl who died suddenly at the age of 2 months, with the cause of death recorded as sudden infant death syndrome (SIDS). Gene sequencing revealed an SCN1A mutation. Microscopic examination of the hippocampus revealed focal bilamination of the dentate gyrus. The other case occurred in a 7-week-old female with two SCN1A mutations (92) (Figure 3, Table 2). These results suggest that SCN1A mutations are not only closely related to SUDEP but also associated with nonepileptic-related sudden death.
The exact mechanism of SUDEP remains unclear. In systemically knockout heterozygous SCN1A+/- mice, severe arrhythmias were found to be characterized by prolonged PR interval, increased heart rate variability, and even atrioventricular block, suggesting that changes in the cardiac SCN1A may be related to SUDEP (111). In another study, paroxysmal chronic bradycardia and associated ventricular electrical dysfunction were found in heterozygous SCN1A+/− mice; notably, atropine and N-methyl scopolamine were effective in preventing sudden death in mice (112). In addition, respiratory dysfunction was also found in mouse models of DS, which may also be one of the causes of SUDEP in SCN1A mutant mice (109).
Arthrogryposis multiplex congenita refers to an etiologically heterogeneous condition that is characterized by the congenital joint contractures in two or more body areas (113). AMC is generally thought to be the downstream result of a reduction in the fetal movements. AMC has an overall incidence of one in 3,000 to 5,000 (114).
Although over 320 genes have been implicated, exemplifying the genetic heterogeneity of the condition (115), AMC is poorly related to SCN1A, with only two reports documented (93, 94). The first report described SCN1A mutations in three infants with AMC from three different families (93). During the fetal period, they are characterized by abnormal development of different joints and a lack of fetal movements (in family 1, bilateral flexion of both hands, hyperextension of knees, and reduced swallowing; in family 2, arthrogryposis of the upper limbs and microretrognathism; in family 3, bilateral camptodactyly, hyperextension of knees, and hallux valgus of feet). It is noteworthy that one of the infants (family 1) developed refractory epilepsy 2 days after birth, while the other two patients both died due to early termination of pregnancy. This suggests that in addition to peripheral joint dysplasia, AMC patients may also have abnormalities of the central nervous system, such as epilepsy, which may be similar to DS. The other description was reported by Laquerriere et al., who sequenced 315 patients with AMC and found 51 gene mutations in 166 (52.7%), including the rare SCN1A (94) (Figure 3, Table 2).
SCN1A not only causes DS and GEFS+; other epileptic encephalopathies, such as Doose syndrome, EIMFS, West syndrome, LGS, RTT, and NEE, are also directly related to SCN1A. In addition to epilepsy, FHM3, SHM, ASD, sudden death, and AMC can also be caused by SCN1A mutations (Figure 4). This review serves as a reminder to epilepsy specialists that gene sequencing is only an adjunct method for diagnosing DS. The diagnosis cannot only be made by gene sequencing but must be individualized according to the clinical manifestations of the patient to formulate a better management scheme.
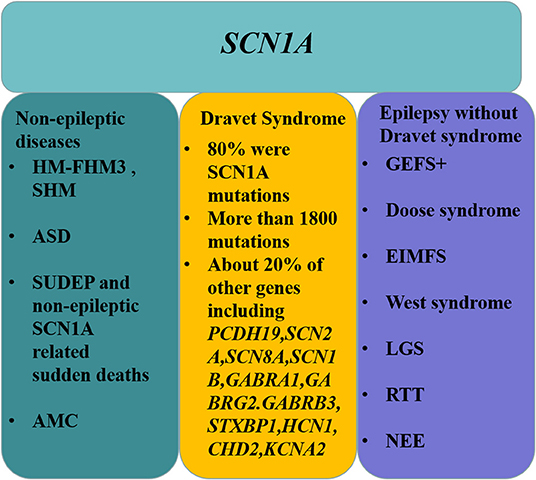
Figure 4. Outline of diseases associated with the SCN1A gene.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This study was supported by the National Natural Science Foundation of China, Grant/Award Number: 81971085, and the Advantages Discipline Group Project of Ningxia Medical University, Grant/Award Number: XY201511.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.743726/full#supplementary-material
1. Marini C, Scheffer I, Nabbout R, Suls A, De Jonghe P, Zara F, et al. The genetics of Dravet syndrome. Epilepsia. (2011) 52 Suppl 2:24–9. doi: 10.1111/j.1528-1167.2011.02997.x
2. Sato C, Ueno Y, Asai K, Takahashi K, Sato M, Engel A, et al. The voltage-sensitive sodium channel is a bell-shaped molecule with several cavities. Nature. (2001) 409:1047–51. doi: 10.1038/35059098
3. Lopez-Santiago L, Isom LL. Dravet syndrome: a developmental and epileptic encephalopathy. Epilepsy Currents. (2019) 19:153575971882203. doi: 10.1177/1535759718822038
4. Steel D, Symonds J, Zuberi S, Brunklaus A. Dravet syndrome and its mimics: beyond SCN1A. Epilepsia. (2017) 58:1807–16. doi: 10.1111/epi.13889
5. Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. (2005) 366:371–7. doi: 10.1016/S0140-6736(05)66786-4
6. Weiss L, Escayg A, Kearney J, Trudeau M, MacDonald B, Mori M, et al. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol Psychiatry. (2003) 8:186–94. doi: 10.1038/sj.mp.4001241
7. D'Gama A, Pochareddy S, Li M, Jamuar S, Reiff R, Lam A, et al. Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron. (2015) 88:910–7. doi: 10.1016/j.neuron.2015.11.009
8. Rochtus A, Goldstein R, Holm I, Brownstein C, Pérez-Palma E, Haynes R, et al. The role of sodium channels in sudden unexpected death in pediatrics. Mol Genet Genomic Med. (2020) 8:e1309. doi: 10.1002/mgg3.1309
9. Brownstein C, Goldstein R, Thompson C, Haynes R, Giles E, Sheidley B, et al. SCN1A variants associated with sudden infant death syndrome. Epilepsia. (2018) 59:e56–62. doi: 10.1111/epi.14055
10. Coll M, Allegue C, Partemi S, Mates J, Del Olmo B, Campuzano O, et al. Genetic investigation of sudden unexpected death in epilepsy cohort by panel target resequencing. Int J Legal Med. (2016) 130:331–9. doi: 10.1007/s00414-015-1269-0
11. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P, et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. (2001) 68:1327–32. doi: 10.1086/320609
12. Mantegazza M, Broccoli V. SCN1A/Na 1.1 channelopathies: mechanisms in expression systems, animal models, and human iPSC models. Epilepsia. (2019) 60 Suppl 3:S25–S38. doi: 10.1111/epi.14700
13. Mei D, Cetica V, Marini C, Guerrini R. Dravet syndrome as part of the clinical and genetic spectrum of sodium channel epilepsies and encephalopathies. Epilepsia. (2019) 60 Suppl 3:S2–S7. doi: 10.1111/epi.16054
14. Auvin S, Jeljeli M, Desnous B, Soussi-Yanicostas N, Dournaud P, Sterkers G. Altered vaccine-induced immunity in children with Dravet syndrome. Epilepsia. (2018) 59:e45–50. doi: 10.1111/epi.14038
15. Scheffer I. Diagnosis and long-term course of Dravet syndrome. EJPN. (2012) 16 Suppl 1:S5–8. doi: 10.1016/j.ejpn.2012.04.007
16. Xu X, Zhang Y, Sun H, Liu X, Yang X, Xiong H, et al. Early clinical features and diagnosis of Dravet syndrome in 138 Chinese patients with SCN1A mutations. Brain Dev. (2014) 36:676–81. doi: 10.1016/j.braindev.2013.10.004
17. Verbeek N, Kasteleijn-Nolst Trenité D, Wassenaar M, van Campen J, Sonsma A, Gunning W, et al. Photosensitivity in Dravet syndrome is under-recognized and related to prognosis. Clin Neurophysiol. (2017) 128:323–30. doi: 10.1016/j.clinph.2016.11.021
18. Brown A, Arpone M, Schneider A, Micallef S, Anderson V, Scheffer I. Cognitive, behavioral, and social functioning in children and adults with Dravet syndrome. Epilepsy Behav. (2020) 112:107319. doi: 10.1016/j.yebeh.2020.107319
19. Licheni S, Mcmahon J, Schneider A, Davey M, Scheffer I. Sleep problems in Dravet syndrome: a modifiable comorbidity. Dev Med Child Neurol. (2018) 60:192–8. doi: 10.1111/dmcn.13601
20. Selvarajah A, Zulfiqar-Ali Q, Marques P, Rong M, Andrade D. A systematic review of adults with Dravet syndrome. Seizure. (2021) 87:39–45. doi: 10.1016/j.seizure.2021.02.025
21. Jansson J, Hallböök T, Reilly C. Intellectual functioning and behavior in Dravet syndrome: A systematic review. Epilepsy Behav. (2020) 108:107079. doi: 10.1016/j.yebeh.2020.107079
22. Ouss L, Leunen D, Laschet J, Chemaly N, Barcia G, Losito E, et al. Autism spectrum disorder and cognitive profile in children with Dravet syndrome: delineation of a specific phenotype. Epilepsia open. (2019) 4:40–53. doi: 10.1002/epi4.12281
23. Cooper M, Mcintosh A, Crompton D, McMahon J, Schneider A, Farrell K, et al. Mortality in Dravet syndrome. Epilepsy Res. (2016) 128:43–7. doi: 10.1016/j.eplepsyres.2016.10.006
24. Scheffer I, Berkovic S. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain. (1997) 120:479–90. doi: 10.1093/brain/120.3.479
25. Myers K, Scheffer I, Berkovic S. Genetic literacy series: genetic epilepsy with febrile seizures plus. Epileptic Disord. (2018) 20:232–8. doi: 10.1684/epd.2018.0985
26. Puranam R, He X, Yao L, Le T, Jang W, Rehder C, et al. Disruption of Fgf13 causes synaptic excitatory-inhibitory imbalance and genetic epilepsy and febrile seizures plus. J Neurosci. (2015) 35:8866–81. doi: 10.1523/JNEUROSCI.3470-14.2015
27. Li X, Guo S, Xu S, Chen Z, Wang L, Ding J, et al. Neocortex- and hippocampus-specific deletion of Gabrg2 causes temperature-dependent seizures in mice. Cell Death Dis. (2021) 12:553. doi: 10.1038/s41419-021-03846-x
28. Escayg A, MacDonald B, Meisler M, Baulac S, Huberfeld G, An-Gourfinkel I, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. (2000) 24:343–5. doi: 10.1038/74159
29. Ebach K, Joos H, Doose H, Stephani U, Kurlemann G, Fiedler B, et al. SCN1A mutation analysis in myoclonic astatic epilepsy and severe idiopathic generalized epilepsy of infancy with generalized tonic-clonic seizures. Neuropediatrics. (2005) 36:210–3. doi: 10.1055/s-2005-865607
30. Harkin L, McMahon J, Iona X, Dibbens L, Pelekanos J, Zuberi S, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. (2007) 130:843–52. doi: 10.1093/brain/awm002
31. Dimova P, Yordanova I, Bojinova V, Jordanova A, Kremenski I. Generalized epilepsy with febrile seizures plus: novel SCN1A mutation. Pediatr Neurol. (2010) 42:137–40. doi: 10.1016/j.pediatrneurol.2009.09.007
32. Angione K, Eschbach K, Smith G, Joshi C, Demarest S. Genetic testing in a cohort of patients with potential epilepsy with myoclonic-atonic seizures. Epilepsy Res. (2019) 150:70–7. doi: 10.1016/j.eplepsyres.2019.01.008
33. Hinokuma N, Nakashima M, Asai H, Nakamura K, Akaboshi S, Fukuoka M, et al. Clinical and genetic characteristics of patients with Doose syndrome. Epilepsia open. (2020) 5:442–50. doi: 10.1002/epi4.12417
34. Freilich E, Jones J, Gaillard W, Conry J, Tsuchida T, Reyes C, et al. Novel SCN1A mutation in a proband with malignant migrating partial seizures of infancy. Arch Neurol. (2011) 68:665–71. doi: 10.1001/archneurol.2011.98
35. Carranza Rojo D, Hamiwka L, McMahon J, Dibbens L, Arsov T, Suls A, et al. De novo SCN1A mutations in migrating partial seizures of infancy. Neurology. (2011) 77:380–3. doi: 10.1212/WNL.0b013e318227046d
36. Shein S, Reynolds T, Gedela S, Kochanek P, Bell M. Therapeutic hypothermia for refractory status epilepticus in a child with malignant migrating partial seizures of infancy and SCN1A mutation: a case report. Ther Hypothermia Temp Manag. (2012) 2:144–9. doi: 10.1089/ther.2012.0013
37. Zhang Y, Kong W, Gao Y, Liu X, Gao K, Xie H, et al. Gene mutation analysis in 253 Chinese children with unexplained epilepsy and intellectual/developmental disabilities. PLoS ONE. (2015) 10:e0141782. doi: 10.1371/journal.pone.0141782
38. Lim B, Hwang H, Kim H, Chae J, Choi J, Kim K. et al. Epilepsy phenotype associated with a chromosome 2q243 deletion involving SCN1A: Migrating partial seizures of infancy or atypical Dravet syndrome? Epilepsy Res. (2015) 109:34–9. doi: 10.1016/j.eplepsyres.2014.10.008
39. Shang K, Zhang Y, Yang X, Liu A, Yang Z, Liu X, et al. Clinical features and gene mutations in epilepsy of infancy with migrating focal seizures. Zhonghua er ke za zhi. (2016) 54:735–9. doi: 10.3760/cma.j.issn.0578-1310.2016.10.005
40. Gokben S, Onay H, Yilmaz S, Atik T, Serdaroglu G, Tekin H, et al. Targeted next generation sequencing: the diagnostic value in early-onset epileptic encephalopathy. Acta Neurol Belg. (2017) 117:131–8. doi: 10.1007/s13760-016-0709-z
41. Fang Z, Xie L, Yan L, Lin H, Pan Y, Liu B, et al. Clinical and genetic characteristics of epilepsy of infancy with migrating focal seizures in Chinese children. Epilepsy Res. (2021) 174:106669. doi: 10.1016/j.eplepsyres.2021.106669
42. Wallace R, Hodgson B, Grinton B, Gardiner R, Robinson R, Rodriguez-Casero V, et al. Sodium channel alpha1-subunit mutations in severe myoclonic epilepsy of infancy and infantile spasms. Neurology. (2003) 61:765–9. doi: 10.1212/01.WNL.0000086379.71183.78
43. Hattori Y, Kawawaki H, Horino A, Thuji H, Nukui M, Kuki I, et al. A case of West syndrome with a deletion at chromosome 2q243-q313. Brain Dev. (2017) 49:131–5. doi: 10.11251/ojjscn.49.131
44. Krey I, Krois-Neudenberger J, Hentschel J, Syrbe S, Polster T, Hanker B, et al. Genotype-phenotype correlation on 45 individuals with West syndrome. EJPN. (2020) 25:134–8. doi: 10.1016/j.ejpn.2019.11.010
45. Na J, Shin S, Yang D, Kim B, Kim H, Kim S, et al. Targeted gene panel sequencing in early infantile onset developmental and epileptic encephalopathy. Brain Dev. (2020) 42:438–48. doi: 10.1016/j.braindev.2020.02.004
46. Henriksen M, Ravn K, Paus B, von Tetzchner S, Skjeldal O De. novo mutations in SCN1A are associated with classic Rett syndrome: a case report. BMC Med Genet. (2018) 19:184. doi: 10.1186/s12881-018-0700-z
47. Selmer K, Lund C, Brandal K, Undlien D, Brodtkorb E. SCN1A mutation screening in adult patients with Lennox-Gastaut syndrome features. Epilepsy Behav. (2009) 16:555–7. doi: 10.1016/j.yebeh.2009.08.021
48. Saitoh M, Shinohara M, Hoshino H, Kubota M, Amemiya K, Takanashi J, et al. Mutations of the SCN1A gene in acute encephalopathy. Epilepsia. (2012) 53:558–64. doi: 10.1111/j.1528-1167.2011.03402.x
49. Ohashi T, Akasaka N, Kobayashi Y, Magara S, Kawashima H, Matsumoto N, et al. Infantile epileptic encephalopathy with a hyperkinetic movement disorder and hand stereotypies associated with a novel SCN1A mutation. Epileptic Disord. (2014) 16:208–12. doi: 10.1684/epd.2014.0649
50. Mercimek-Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner E, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia. (2015) 56:707–16. doi: 10.1111/epi.12954
51. Kobayashi Y, Tohyama J, Kato M, Akasaka N, Magara S, Kawashima H, et al. High prevalence of genetic alterations in early-onset epileptic encephalopathies associated with infantile movement disorders. Brain Dev. (2016) 38:285–92. doi: 10.1016/j.braindev.2015.09.011
52. Kwong A, Ho A, Fung C, Wong V. Analysis of mutations in 7 genes associated with neuronal excitability and synaptic transmission in a cohort of children with non-syndromic infantile epileptic encephalopathy. PLoS ONE. (2015) 10:e0126446. doi: 10.1371/journal.pone.0126446
53. Sadleir L, Mountier E, Gill D, Davis S, Joshi C, DeVile C, et al. Not all epileptic encephalopathies are Dravet syndrome: early profound Thr226Met phenotype. Neurology. (2017) 89:1035–42. doi: 10.1212/WNL.0000000000004331
54. Spagnoli C, Frattini D, Rizzi S, Salerno G, Fusco C. Early infantile SCN1A epileptic encephalopathy: expanding the genotype-phenotype correlations. Seizure. (2019) 65:62–4. doi: 10.1016/j.seizure.2019.01.002
55. Kelley S, Kossoff E. Doose syndrome (myoclonic-astatic epilepsy): 40 years of progress. Dev Med Child Neurol. (2010) 52:988–93. doi: 10.1111/j.1469-8749.2010.03744.x
56. Doose H, Gerken H, Leonhardt R, Völzke E, Völz C. Centrencephalic myoclonic-astatic petit mal. Clinical and genetic investigation. Neuropadiatrie. (1970) 2:59–78. doi: 10.1055/s-0028-1091841
57. Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE commission on classification and terminology, 2005-2009. Epilepsia. (2010) 51:676–85. doi: 10.1111/j.1528-1167.2010.02522.x
58. Burgess R, Wang S, McTague A, Boysen K, Yang X, Zeng Q, et al. The genetic landscape of epilepsy of infancy with migrating focal seizures. Ann Neurol. (2019) 86:821–31. doi: 10.1002/ana.25619
59. Pavone P, Polizzi A, Marino S, Corsello G, Falsaperla R, Marino S, et al. West syndrome: a comprehensive review. Neurol Sci. (2020) 41:3547–62. doi: 10.1007/s10072-020-04600-5
60. Al-Banji M, Zahr D, Jan M. Lennox-Gastaut syndrome. Management update. Neurosciences. (2015) 20:207–12. doi: 10.17712/nsj.2015.3.20140677
61. Mastrangelo M. Lennox-Gastaut Syndrome: a state of the art review. Neuropediatrics. (2017) 48:143–51. doi: 10.1055/s-0037-1601324
62. Jia Y, Lin Y, Li J, Li M, Zhang Y, Hou Y, et al. KCNT1Quinidine Therapy for Lennox-Gastaut Syndrome With Mutation. A Case Report and Literature Review. Front Neurol. (2019) 10:64. doi: 10.3389/fneur.2019.00064
63. Yang L, Jiang M, Yu R, Hu R, Xiong F, Li J, et al. A case report of precocious puberty related to Rett syndrome and a literature review. Pharmazie. (2021) 76:559–61. doi: 10.1691/ph.2021.1747
64. Pejhan S, Rastegar M. Role of DNA Methyl-CpG-Binding protein MeCP2 in rett syndrome pathobiology and mechanism of disease. Biomolecules. (2021) 11:75. doi: 10.3390/biom11010075
65. Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of Neurological Defects in a Mouse Model of Rett Syndrome. Science. (2007) 315:1143–7. doi: 10.1126/science.1138389
66. Liu X, Shen Q, Zheng G, Guo H, Lu X, Wang X, et al. Gene and Phenotype Expansion of Unexplained Early Infantile Epileptic Encephalopathy. Front Neurol. (2021) 12:633637. doi: 10.3389/fneur.2021.633637
67. Khaiboullina SF, Mendelevich EG, Martynova EV, Davidyuk YN, Giniatullin RA, Rizvanov AA. Testing Genes Implicated in the Novel Case of Familial Hemiplegic Migraine. Bionanoscience. (2017) 7:1–4. doi: 10.1007/s12668-016-0314-x
68. de Vries B, Freilinger T, Vanmolkot K, Koenderink J, Stam A, Terwindt G, et al. Systematic analysis of three FHM genes in 39 sporadic patients with hemiplegic migraine. Neurology. (2007) 69:2170–6. doi: 10.1212/01.wnl.0000295670.01629.5a
69. Omata T, Takanashi JI, Wada T, Arai H, Tanabe Y. Genetic diagnosis and acetazolamide treatment of familial hemiplegic migraine. Brain Dev. (2011) 33:332–4. doi: 10.1016/j.braindev.2010.05.006
70. Pietrobon D. Familial hemiplegic migraine. Neurotherapeutics. (2007) 4:274–84. doi: 10.1016/j.nurt.2007.01.008
71. Shao N, Zhang H, Wang X, Zhang W, Yu M, Meng H. Familial hemiplegic migraine type 3 (FHM3) with an SCN1A mutation in a Chinese family: a case report. Front Neurol. (2018) 9:976. doi: 10.3389/fneur.2018.00976
72. Kowalska M, Prendecki M, Kapelusiak-Pielok M, Grzelak T, Łagan-Jedrzejczyk U, Wiszniewska M, et al. SCN1AAnalysis of Genetic Variants in SCN1A, SCN2A, KCNK18, TRPA1 and STX1A as a possible marker of migraine. Curr Genomics. (2020) 21:224–36. doi: 10.2174/1389202921666200415181222
73. Zhang Y, Chen N, Zhou M, Guo J, Guo J, He L, et al. A novel SCN1A mutation identified in a Chinese family with familial hemiplegic migraine: A case report. Cephalalgia. (2017) 37:1294–8. doi: 10.1177/0333102416677049
74. Gargus JJ, Tournay A. Novel Mutation Confirms Seizure Locus SCN1A is Also Familial Hemiplegic Migraine Locus FHM3. Pediatr Neurol. (2007) 37:407–10. doi: 10.1016/j.pediatrneurol.2007.06.016
75. Scheffer I, Nabbout R. SCN1A-related phenotypes: Epilepsy and beyond. Epilepsia. (2019) S17–24. doi: 10.1111/epi.16386
76. Vanmolkot K, Babini E, de Vries B, Stam A, Freilinger T, Terwindt G, et al. The novel p.L1649Q mutation in the SCN1A epilepsy gene is associated with familial hemiplegic migraine: genetic and functional studies. Hum Mutat. (2007) 28:522. doi: 10.1002/humu.9486
77. Vahedi K, Depienne C, Le Fort D, Riant F, Chaine P, Trouillard O, et al. Elicited repetitive daily blindness: a new phenotype associated with hemiplegic migraine and SCN1A mutations. Neurology. (2009) 72:1178–83. doi: 10.1212/01.wnl.0000345393.53132.8c
78. Castro M, Stam A, Lemos C, de Vries B, Vanmolkot K, Barros J. et al. First mutation in the voltage-gated Nav11 subunit gene SCN1A with co-occurring familial hemiplegic migraine and epilepsy. Cephalalgia. (2009) 29:308–13. doi: 10.1111/j.1468-2982.2008.01721.x
79. Frosk P, Mhanni A, Rafay M. SCN1A mutation associated with intractable myoclonic epilepsy and migraine headache. J Child Neurol. (2013) 28:389–91. doi: 10.1177/0883073812443309
80. Barros J, Ferreira A, Brandão A, Lemos C, Correia F, Damásio J, et al. Familial hemiplegic migraine due to L263V SCN1A mutation: discordance for epilepsy between two kindreds from Douro Valley. Cephalalgia. (2014) 34:1015–20. doi: 10.1177/0333102414527015
81. Schubert V, Auffenberg E, Biskup S, Jurkat-Rott K, Freilinger T. Two novel families with hemiplegic migraine caused by recurrent SCN1A mutation p.F1499L. Cephalalgia. (2017) 38:1503–8. doi: 10.1177/0333102417742365
82. Pelzer N, Haan J, Stam A, Vijfhuizen L, Koelewijn S, Smagge A, et al. Clinical spectrum of hemiplegic migraine and chances of finding a pathogenic mutation. Neurology. (2018) 90:e575–e82. doi: 10.1212/WNL.0000000000004966
83. Weller C, Pelzer N, de Vries B, López M, De Fàbregues O, Pascual J, et al. Two novel SCN1A mutations identified in families with familial hemiplegic migraine. Cephalalgia. (2014) 34:1062–9. doi: 10.1177/0333102414529195
84. Domitrz I, Kosiorek M, Zekanowski C, Kamińska A. Genetic studies of Polish migraine patients: screening for causative mutations in four migraine-associated genes. Hum Genomics. (2016) 10:3. doi: 10.1186/s40246-015-0057-8
85. Fan C, Wolking S, Lehmann-Horn F, Hedrich U, Freilinger T, Lerche H, et al. Early-onset familial hemiplegic migraine due to a novel SCN1A mutation. Cephalalgia. (2016) 36:1238–47. doi: 10.1177/0333102415608360
86. Chastan N, Lebas A, Legoff F, Parain D, Guyant-Marechal L. Clinical and electroencephalographic abnormalities during the full duration of a sporadic hemiplegic migraine attack. Neurophysiologie clinique. (2016) 46:307–11. doi: 10.1016/j.neucli.2016.03.004
87. Dube M, Lakhotia AN, Yadav V, Jain R. Sporadic hemiplegic migraine with SCN1A gene mutation—a case report. US Neurology. (2018) 14:108. doi: 10.17925/USN.2018.14.2.108
88. O'Roak B, Deriziotis P, Lee C, Vives L, Schwartz J, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. (2011) 43:585–9. doi: 10.1038/ng.835
89. Koshimizu E, Miyatake S, Okamoto N, Nakashima M, Tsurusaki Y, Miyake N, et al. Performance comparison of bench-top next generation sequencers using microdroplet PCR-based enrichment for targeted sequencing in patients with autism spectrum disorder. PLoS ONE. (2013) 8:e74167. doi: 10.1371/journal.pone.0074167
90. Alvarez-Mora M, Calvo Escalona R, Puig Navarro O, Madrigal I, Quintela I, Amigo J, et al. Comprehensive molecular testing in patients with high functioning autism spectrum disorder. Mutation research. (2016) 784–5:46–52. doi: 10.1016/j.mrfmmm.2015.12.006
91. Yin J, Chun CA, Zavadenko NN, Pechatnikova NL, Naumova OY, Doddapaneni HV, et al. Next Generation Sequencing of 134 Children with Autism Spectrum Disorder and Regression. Genes (Basel). (2020) 11:853. doi: 10.3390/genes11080853
92. Halvorsen M, Petrovski S, Shellhaas R, Tang Y, Crandall L, Goldstein D, et al. Mosaic mutations in early-onset genetic diseases. Genet Med. (2015) 18:746–9. doi: 10.1038/gim.2015.155
93. Jaber D, Gitiaux C, Blesson S, Marguet F, Buard D, Varela Salgado M, et al. De novo mutations of SCN1A are responsible for arthrogryposis broadening the SCN1A-related phenotypes. J Med Genet. (2021) 58:737–42. doi: 10.1136/jmedgenet-2020-107166
94. Laquerriere A, Jaber D, Abiusi E, Maluenda J, Mejlachowicz D, Vivanti A, et al. Phenotypic spectrum and genomics of undiagnosed arthrogryposis multiplex congenita. J Med Genet. (2021). doi: 10.1136/jmedgenet-2020-107595. [Epub ahead of print].
96. Berkvens J, Veugen I, Veendrick-Meekes M, Snoeijen-Schouwenaars F, Schelhaas H, Willemsen M, et al. Autism and behavior in adult patients with Dravet syndrome (DS). Epilepsy Behav. (2015) 47:11–6. doi: 10.1016/j.yebeh.2015.04.057
97. Li B, Liu X, Yi Y, Deng Y, Su T, Zou X, et al. Autism in Dravet syndrome: prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation. Epilepsy Behav. (2011) 21:291–5. doi: 10.1016/j.yebeh.2011.04.060
98. Xiong Z, Yi L, Cao D, He W, Chen J, Gao S, et al. Dravet syndrome with autism inherited from a paternal mosaic heterozygous mutation on SCN1A. J Neurol Sci. (2016) 369:53–6. doi: 10.1016/j.jns.2016.07.038
99. Han S, Tai C, Westenbroek R, Yu F, Cheah C, Potter G, et al. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature. (2012) 489:385–90. doi: 10.1038/nature11356
100. McGuone D, Crandall L, Devinsky O. Sudden unexplained death in childhood: a neuropathology review. Front Neurol. (2020) 11:582051. doi: 10.3389/fneur.2020.582051
101. Lee B. Predicting sudden unexpected death in epilepsy. Neurology. (2021) 96:e2670–e2. doi: 10.1212/WNL.0000000000012003
102. Nashef L. Sudden unexpected death in epilepsy: terminology and definitions. Epilepsia. (1997) 38:S6–8. doi: 10.1111/j.1528-1157.1997.tb06130.x
103. Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. (2012) 491:56–65. doi: 10.1038/nature11632
104. Baruteau AE, Tester DJ, Kapplinger JD, Ackerman MJ, Behr ER. Sudden infant death syndrome and inherited cardiac conditions. Nature Reviews Cardiology. (2017) 14:715–26. doi: 10.1038/nrcardio.2017.129
105. Neubauer J, Lecca MR, Russo G, Bartsch C, Medeiros-Domingo A, Berger W, et al. Post-mortem whole-exome analysis in a large sudden infant death syndrome cohort with a focus on cardiovascular and metabolic genetic diseases. Eur J Hum Genet. (2017) 25:404–9. doi: 10.1038/ejhg.2016.199
106. Wedekind H, Smits J, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, et al. De Novo mutation in the SCN5A gene associated with early onset of sudden infant death. Circulation. (2001) 104:1158–64. doi: 10.1161/hc3501.095361
107. Hata Y, Oku Y, Taneichi H, Tanaka T, Igarashi N, Niida Y, et al. Two autopsy cases of sudden unexpected death from Dravet syndrome with novel de novo SCN1A variants. Brain Dev. (2020) 42:171–8. doi: 10.1016/j.braindev.2019.10.005
108. Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia. (2011) 52(Supplement s2):44–9. doi: 10.1111/j.1528-1167.2011.03001.x
109. Kalume F. Sudden unexpected death in Dravet syndrome: respiratory and other physiological dysfunctions. Respir Physiol Neurobiol. (2013) 189:324–8. doi: 10.1016/j.resp.2013.06.026
110. Shmuely S, Sisodiya SM, Gunning WB, Sander JW. Mortality in Dravet syndrome: a review. Epilepsy Behav. (2016) 64:69–74. doi: 10.1016/j.yebeh.2016.09.007
111. Maier SK, Westenbroek RE, Yamanushi TT, Dobrzynski H, Boyett MR, Catterall WA, et al. An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc Natl Acad Sci USA. (2003) 100:3507–12. doi: 10.1073/pnas.2627986100
112. Kalume F, Westenbroek RE, Cheah CS, Yu FH, Catterall WA. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. (2013) 123:1798–808. doi: 10.1172/JCI66220
113. Niles K, Blaser S, Shannon P, Chitayat D. Fetal arthrogryposis multiplex congenita/fetal akinesia deformation sequence (FADS)-Aetiology, diagnosis, and management. Prenat Diagn. (2019) 39:720–31. doi: 10.1002/pd.5505
114. Lowry RB, Sibbald B, Bedard T, Hall JG. Prevalence of multiple congenital contractures including arthrogryposis multiplex congenita in Alberta, Canada, and a strategy for classification and coding. Birth Defects Res A Clin Mol Teratol. (2011) 88:1057–61. doi: 10.1002/bdra.20738
Keywords: SCN1A, Dravet syndrome, GEFS+, migraine, autism spectrum disorder
Citation: Ding J, Li X, Tian H, Wang L, Guo B, Wang Y, Li W, Wang F and Sun T (2021) SCN1A Mutation—Beyond Dravet Syndrome: A Systematic Review and Narrative Synthesis. Front. Neurol. 12:743726. doi: 10.3389/fneur.2021.743726
Received: 19 July 2021; Accepted: 29 November 2021;
Published: 24 December 2021.
Edited by:
David M. Labiner, University of Arizona, United StatesReviewed by:
Yam Nath Paudel, Monash University Malaysia, MalaysiaCopyright © 2021 Ding, Li, Tian, Wang, Guo, Wang, Li, Wang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinxiao Li, bHh4OTg1QDE2My5jb20=; Feng Wang, bnh3d2FuZ0AxNjMuY29t; Tao Sun, c3VudGFvX254bXVAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.