
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Neurol. , 02 December 2021
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.734515
 Bin Chen1,2
Bin Chen1,2 Zaiqiang Zhang1,2
Zaiqiang Zhang1,2 Na Chen1,2Wei Li1,2,3Hua Pan1,2Xingao Wang1,2Yuting Ren1,2
Na Chen1,2Wei Li1,2,3Hua Pan1,2Xingao Wang1,2Yuting Ren1,2 Yuzhi Shi1,2
Yuzhi Shi1,2 Hongfei Tai1,2Songtao Niu1,2*
Hongfei Tai1,2Songtao Niu1,2*Mutations in the myelin protein zero gene are responsible for the autosomal dominant Charcot-Marie-Tooth disease (CMT). We summarized the genetic and clinical features of six unrelated Chinese families and the genetic spectrum of Chinese patients with myelin protein zero (MPZ) mutations. Our study reports data from a group of Chinese patients consisting of five males and one female with the age of disease onset ranging from 16 to 55 years. The initial symptom in all the patients was the weakness of the lower limbs. Electrophysiological presentations suggested chronic progressive sensorimotor demyelinating polyneuropathy. Overall six mutations were identified in the cohort, including four known mutations [c.103G>T (p.D35Y), c.233C>T (p.S78L), c.293G>A (p.R98H), and c.449-1G>T], and two novel mutations [c.67+4A>G with a mild CMT1B phenotype, and (c.79delG) p.A27fs with a rapidly progressive CMT1B phenotype]. According to the literature review, there are 35 Chinese families with 28 different MPZ mutations. The MPZ mutational spectrum in Chinese patients is very heterogeneous and differs from that of Japanese and Korean individuals, although they do share several common hot spot mutations.
Charcot-Marie-Tooth disease is the most common inherited disorder of the peripheral nervous system (PNS). It is characterized by slow-progressing weakness, muscle atrophy, and sensory impairment, with the symptoms being most evident in the distal part of the legs (1). Charcot-Marie-Tooth disease (CMT) is primarily classified into two types based on electrophysiological findings. A median motor nerve conduction velocity (NCV) of ≤38 m/s indicates CMT1 while NCV of >38 m/s with a reduced compound muscle action potential (cMAP) indicates CMT2 (1, 2). CMTs is genetically determined disorders involving nearly 100 genes (3).
Myelin protein zero (P0, MPZ) is the most abundant protein in peripheral myelin and is produced by Schwann cells (4). It is a member of the immunoglobulin supergene family and functions as an adhesion molecule that mediates the compaction of the PNS (5). MPZ mutations are responsible for autosomal dominant CMT, which can be divided into CMT1B, CMT2I, and Dejerine-Sottas syndrome, based on the clinical and electrophysiological characteristics. These are found in 4.1–5% of all CMT patients (6).
Almost 300 mutations in MPZ have been identified (7). The early onset (infantile and childhood) phenotypes likely represent developmentally impaired myelination, whereas the adult-onset phenotypes reflect axonal degeneration without antecedent demyelination (8). Previous studies have shown that the spectra and frequencies of MPZ mutations in Caucasian and Japanese cohorts are different (7). Several studies have reported Chinese patients with MPZ mutations, but it is currently unknown whether there are differences in the spectra of MPZ mutations between Chinese and other ethnicities (9–20). Herein, we report the mutational spectrum and clinical features of six unrelated Chinese families with MPZ in our hospital over a 7 years period.
We enrolled six probands from 76 unrelated Chinese families who visited the Beijing Tiantan Hospital from January 2012 to August 2019 with suspected CMT or related mutations in CMT genes as detected by targeted next-generation sequencing (NGS). All participants provided informed consent for this study, and ethical approval was obtained from the Human Research Ethics Committee of Beijing Tiantan Hospital. All patients were examined and evaluated by the participating neurologists. Their phenotypes were retrospectively defined based on clinical manifestations, family histories, and electrophysiological data, collected from the patient medical records. Nerve conduction studies were performed using standard techniques and a Medelec MS25 electromyograph (Mistro, Surrey, United Kingdom).
Genomic DNA was extracted from peripheral venous blood samples from the six probands and available family members following standard procedures. All patients were negative for 17p12 (PMP22) duplication and deletion. The NGS panel covered all the exons and flanking sequences of genes that were known to be associated with hereditary neuropathies (21). Sanger sequencing of the variants and co-segregation analysis were conducted for all patients and available family members.
The identified variants were determined using the databases of genomic variants, including the 1,000 Genomes Project, the Genome Aggregation Database (gnomAD), and The Single Nucleotide Polymorphism Database (dbSNP). A database of 8,000 healthy controls of Chinese origin was also screened. The biological relevance of the novel amino acid changes was studied using the Mutation Taster (https://www.mutationtaster.org) and SIFT-Indels (https://sift.bii.a-star.edu.sg/www/SIFT_indels2.html). Splice site mutations were predicted using Human Splicing Finder (HSF) software 3 (http://www.umd.be/HSF3/HSF.shtml) and single nucleotide variants within splicing consensus regions (scSNV).
The probands were from six unrelated families and included five males and one female. The age of disease onset for five patients was >40 years. The first symptom in all patients was distal weakness in the lower limbs. Three patients (1, 5, and 6) had a family history of peripheral neuropathy, which is characterized by an autosomal dominant inheritance. At the age of 43 years, patient 1 started showing weakness in both the lower limbs. Three years after the first symptoms appeared, the patient had slower walking than before. The father of the patient also showed lower limb weakness in his 40s. Patient 5 had a history of abnormal gait for about 3 years. His mother and sister showed similar symptoms. Patient 6 suffered from ankle sprains at the age of 16 years, and their grandfather and father also developed an abnormal gait in their 40s. Family members of the remaining patients were not available for testing. Patient 2 had weakness in his lower limbs for approximately 14 years, with a rapid progression of the ailment in the past 2 years. Patient 3 had a history of weakness in both the lower limbs for approximately 11 years and started showing weakness in the hands for the past 2 years. Patient 4 has had a history of abnormal gait for the past 12 years. All probands displayed distal limb muscle atrophy with pes cavus.
Blood creatine kinase levels were normal in all the patients except patients 2 and 3 [patient 2 (360 U/L) and patient 3 (566 U/L); normal range 24–194 U/L] However, patient 2 had normal levels of cerebrospinal fluid protein [31 mg/dl (normal range 15–45 mg/dl)] and cell count 3/μl (normal range 0–5). The remaining five patients did not undergo lumbar puncture. The clinical data of six CMT patients with MPZ mutations are summarized in Table 1.

Table 1. Clinical features of six CMT patients with MPZ mutations.
The electrophysiological results from six patients demonstrated a sensorimotor demyelinating polyneuropathy with multiple motor nerves showing prolonged distal latencies. The F-wave persistence rate of the bilateral tibial nerves was 65%, with significantly prolonged latencies in patient 2. The nerve conduction studies of six patients are summarized in Table 2. Based on their clinical and electrophysiological features, all patients were diagnosed with CMT1 according to the diagnostic criteria of CMT.
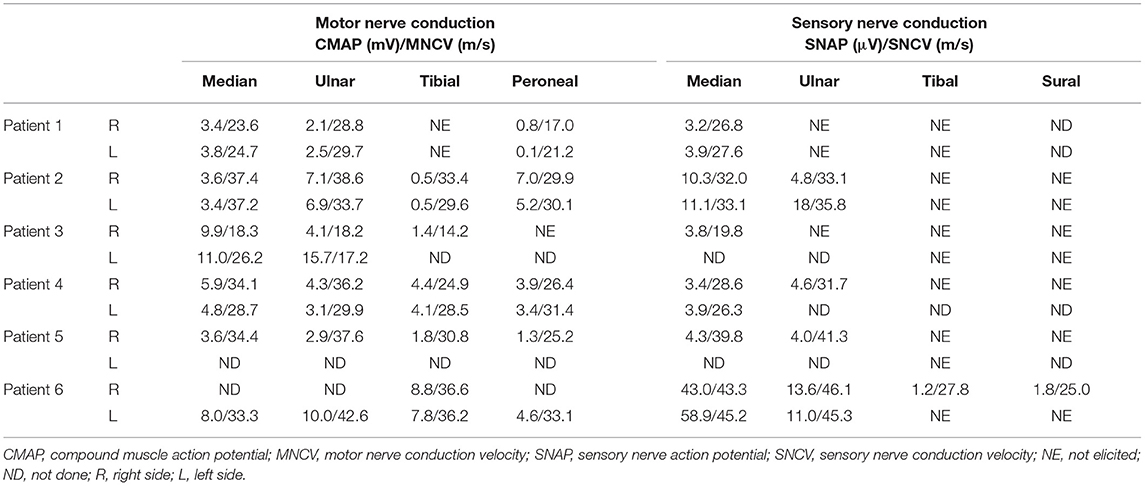
Table 2. Nerve conduction velocity results of six patients.
We identified six MPZ mutations in six probands which were verified by Sanger sequencing, these included four known mutations [c.103G>T (p.D35Y), c.233C>T (p.S78L), c.293G>A (p.R98H), and c.449-1G>T], (7, 8) a novel frameshift variant c.79delG (p. A27fs) in patient 2, and a novel intronic splice site variant (c.67+4A>G) in patient 1 and his father (Figure 1A). Both variants were absent from the controls (1,000 Genomes, gnomAD, dbSNP, and 8,000 healthy Chinese controls). The c.79delG in exon 2 leads to a shift in the open reading frame and an amino acid change at position 46 from valine (GTG) to a stop codon (TGA), which causes translation termination. p.A27 and adjacent amino acid residues are highly conserved among different animal species (Figure 1B). Mutation Taster and SIFT-Indels predicted that the c.79delG is probably a disease-causing mutation. According to the ACMG Standards, c.79delG is considered a pathogenic variant due to the evidence of pathogenicity for PVS1 (loss of protein function), PM2 (absent from controls), PP3 (harmful effects on gene or gene products). The c.67+4A>G variant is located within the highly conserved intron 1 and may most likely affect splicing by HSF and cause abnormal splicing by abolishing the donor splice site of exon 2 (Ada-score = 0.996, RF-score = 0.846) by scSNV. The c.67+4A>G variant was predicted to be a likely pathogenic variant due to PM1 (located in p0 C-terminal domain), PM2 (absent from controls), PP1 (coseparation in the family), PP3 (harmful effects on gene or gene products), according to ACMG.
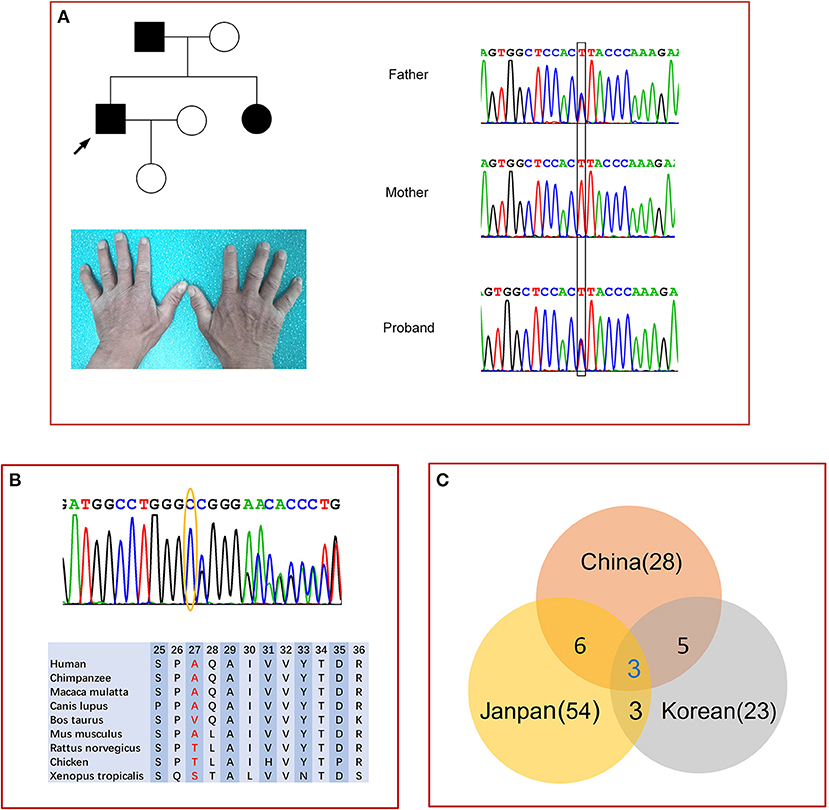
Figure 1. Genetic analysis of MPZ. (A) Pedigree and sequencing chromatograms of family 1 with c.67+4A>G variants. Circle, female; square, male; filled symbol, patient. The bases in the square frame are mutational sites. Mild muscle atrophy in the first dorsal interosseous muscle of patient 1. (B) Sanger sequencing results confirm the guanine deletion mutation at nucleotide position 79 (yellow oval) in MPZ exon 2 of patient 2, and the adjacent amino acid residues among different species. (C) Venn diagram of the MPZ mutational spectra in three countries.
In this study, we investigated six probands with inherited peripheral neuropathies associated with MPZ variants, who visited a single medical center. We detected two novel variants that are likely to induce a pathogenic phenotype. The c.67+4A>G variant was confirmed in an affected proband and cosegregation with similar peripheral neuropathy in the affected father. The p.A27fs mutation resulted in premature truncation of the protein, which led to a significant change in the protein structure and function. Both novel variants were not seen in healthy individuals with no known history of neurogenetic diseases or in multiple databases, which further suggested that c.67+4A>G and p.A27fs are disease-causing rather than normal variants of MPZ.
Our study further confirmed that MPZ mutations are associated with specific phenotypes, especially at the age of onset (8). In general, CMT1B patients in this study with MPZ mutations had relatively mild clinical phenotypes. The patient with the p.A27fs mutation showed a relatively rapid progression in the late stage of the disease, although a report showed frameshift mutation in MPZ caused a mild clinical phenotype (22). We found that the age of onset of patients with S78L and R98H mutations was during adulthood rather than during childhood or infancy (8). The clinical manifestations of MPZ mutations varied among the enrolled families. We also found mild elevations in serum CK levels in some patients and a normal CSF protein level in the CMT1B patient (patient 2) (7).
We searched the PubMed database using the terms “MPZ” and “Chinese” and excluded patients who may have been associated with previous reports based on the research team and publication time. We then summarized the MPZ mutational spectra and clinical characteristics of 35 families of unrelated Chinese origin (9–20). The total mutations summarized in the Chinese patients with MPZ mutations are shown in Table 3. According to the literature review, there were 35 known cases of unrelated Chinese families from mainland China and Taiwan presenting these mutations. To date, 28 different MPZ mutations have been identified in Chinese individuals. Overall, mutations in MPZ are heterogeneous in Chinese individuals without founder mutations. The most common type of MPZ mutation in Chinese individuals is the missense mutation (21/28,75%) followed by frameshift mutation (5/28,17.8%), and lastly, splice site mutation in introns (2/28,7.2%). The findings showed that in Chinese patients, 46% of the mutations occurred in exon 3, and 31% in exon 2. The R98H and R98C mutations have been found in 14% (5/35) of Chinese families, from North China and South China, along with those from Taiwan (9, 10, 16).
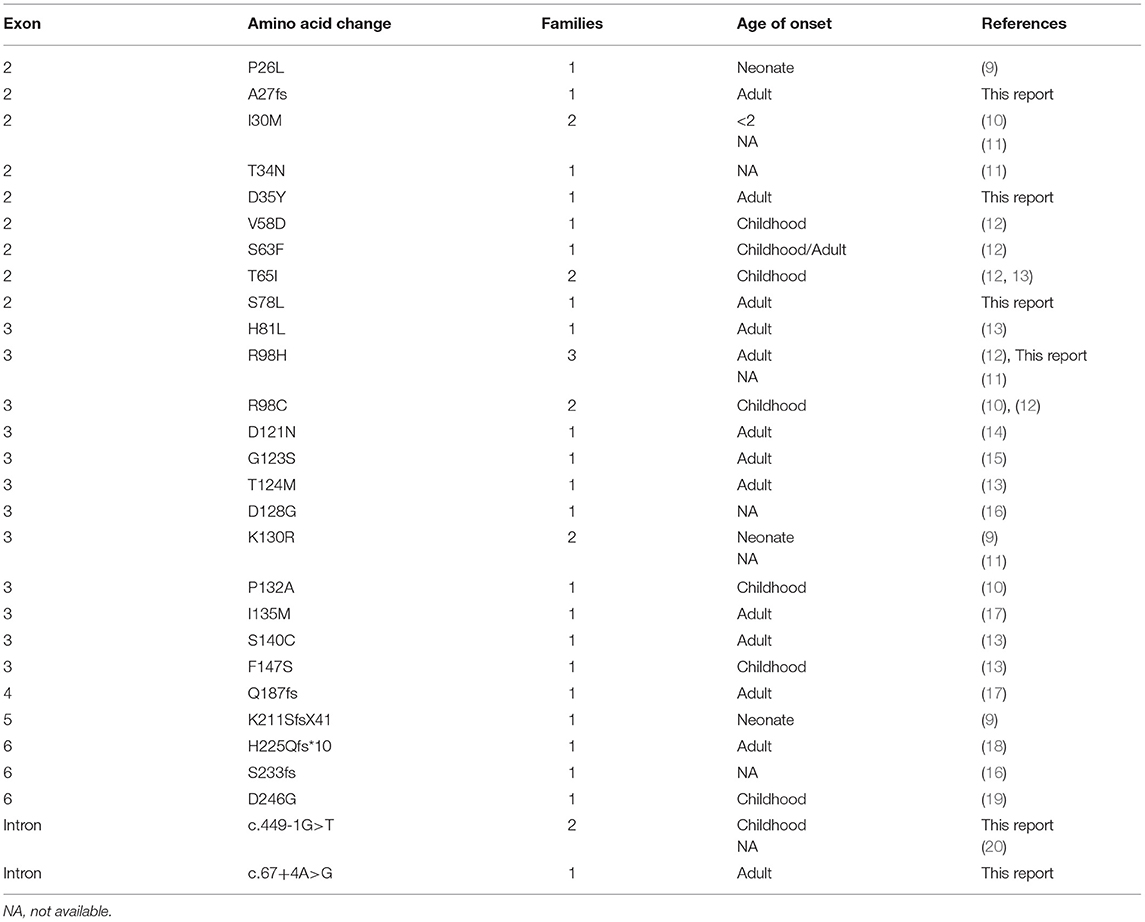
Table 3. MPZ mutations in the 35 Chinese families.
Fifty-four different mutations in the MPZ have been reported in Japanese individuals (1, 7, 23–26) and 23 mutations in Korean individuals (27, 28). Among them, only six identical mutations (D35Y, S78L, R98C, R98H, T124M, K130R) were found in both Chinese and Japanese patients, (7) and five identical mutations (S78L, R98C, T124M, P132A, c.449- 1G>T) were found in both Chinese and Korean families (Figure 1C) (27, 28). T124M in MPZ could be found in 8–17% of Japanese CMT families, but only in 3% of Chinese families. Interestingly, the R98H mutation has the highest MPZ mutation frequency in Japan (7). Mutations that were not detected in Japan could also not be found in China. The c.449-1G>T mutation was identified in 13.8% (5/36) of the affected Korean families and 5.7% (2/35) of Chinese families (27, 28).
In conclusion, we identified six MPZ mutations including two novel mutations in this study. Patients with c.67+4A>G mutations tended to have relatively milder clinical manifestations. Patients with p.A27fs mutation tended to have a rapid progression in the late stage of the disease. MPZ mutations in Chinese individuals were very heterogeneous. The frequency and spectrum of MPZ mutations in Chinese individuals are different from those in Japanese and Koreans, although common hot spot mutations exist among these ethnic groups. This report will help with genetic and clinical studies of Chinese CMT patients with MPZ mutations.
The datasets presented in this article are not readily available due to ethical or privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Human Research Ethics Committee of Beijing Tiantan Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
BC, SN, and ZZ designed the study and wrote the paper. SN, XW, HT, YR, YS, and BC analyzed the clinical data. NC and HP analyzed the electrophysiological data. BC and WL analyzed the genetic results. All authors have read and approved the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the patients and their families for their cooperation.
1. Hattori N, Yamamoto M, Yoshihara T, Koike H, Nakagawa M, Yoshikawa H, et al. Demyelinating and axonal features of Charcot-Marie-Tooth disease with mutations of myelin-related proteins (PMP22, MPZ and Cx32): a clinicopathological study of 205 Japanese patients. Brain. (2003) 126:134–51. doi: 10.1093/brain/awg012
2. Harding AE, Thomas PK. The clinical features of hereditary motor sensory neuropathy types I II. Brain. (1980) 103:259–80. doi: 10.1093/brain/103.2.259
3. Pipis M, Rossor AM, Laura M, Reilly MM. Next-generation sequencing in Charcot-Marie-Tooth disease: opportunities and challenges. Nat Rev Neurol. (2019) 15:644–56. doi: 10.1038/s41582-019-0254-5
4. Lemke G, Axel R. Isolation and sequence of a cDNA encoding the major structural protein of peripheral myelin. Cell. (1985) 40:501–8. doi: 10.1016/0092-8674(85)90198-9
5. Shy ME. Peripheral neuropathies caused by mutations in the myelin protein zero. J Neurol Sci. (2006) 242:55–66. doi: 10.1016/j.jns.2005.11.015
6. Pareyson D, Saveri P, Pisciotta C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr Opin Neurol. (2017) 30:471–80. doi: 10.1097/WCO.0000000000000474
7. Taniguchi T, Ando M, Okamoto Y, Yoshimura A, Higuchi Y, Hashiguchi A, et al. Genetic spectrum of Charcot-Marie-Tooth disease associated with myelin protein zero gene variants in Japan. Clin Genet. (2021) 99:359–75. doi: 10.1111/cge.13881
8. Sanmaneechai O, Feely S, Scherer SS, Herrmann DN, Burns J, Muntoni F, et al. Genotype-phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain. (2015) 138:3180–92. doi: 10.1093/brain/awv241
9. Wang Y, Peng W, Guo HY, Li H, Tian J, Shi YJ, et al. Next-generation sequencing-based molecular diagnosis of neonatal hypotonia in Chinese Population. Sci Rep. (2016) 6:29088. doi: 10.1038/srep29088
10. Xu JL, Zhang Y, Zhao CY, Jiang PF, Yuan ZF, Yu YL, et al. A genotyping study of 13 cases of early-onset Charcot-Marie-Tooth disease. Zhongguo Dang Dai Er Ke Za Zhi. (2019) 21:670–5.
11. Chen CX, Dong HL, Wei Q, Li LX, Yu H, Li JQ, et al. Genetic spectrum and clinical profiles in a southeast Chinese cohort of Charcot-Marie-Tooth disease. Clin Genet. (2019) 96:439–48. doi: 10.1111/cge.13616
12. Lee YC, Soong BW, Liu YT, Lin KP, Kao KP, Wu ZA. Median nerve motor conduction velocity is concordant with myelin protein zero gene mutation. J Neurol. (2005) 252:151–5. doi: 10.1007/s00415-005-0621-6
13. Liu L, Li X, Zi X, Huang S, Zhan Y, Jiang M, et al. Two novel MPZ mutations in Chinese CMT patients. J Peripher Nerv Syst. (2013) 18:256–60. doi: 10.1111/jns5.12040
14. Duan X, Gu W, Hao Y, Wang R, Wen H, Sun S, et al. A novel Asp121Asn mutation of myelin protein zero is associated with late-onset axonal charcot-marie-tooth disease, hearing loss and pupil abnormalities. Front Aging Neurosci. (2016) 8:222. doi: 10.3389/fnagi.2016.00222
15. Lee YC, Yu CT, Lin KP, Chang MH, Hsu SL, Liu YF, et al. MPZ mutation G123S characterization: evidence for a complex pathogenesis in CMT disease. Neurology. (2008) 70:273–7. doi: 10.1212/01.wnl.0000296828.66915.bf
16. Hsu YH, Lin KP, Guo YC, Tsai YS, Liao YC, Lee YC. Mutation spectrum of Charcot-Marie-Tooth disease among the Han Chinese in Taiwan. Ann Clin Transl Neurol. (2019) 6:1090–101. doi: 10.1002/acn3.50797
17. Lin KP, Soong BW, Chang MH, Chen WT, Lin JL, Lee WJ, et al. Clinical and cellular characterization of two novel MPZ mutations, p.I135M and p.Q187PfsX63. Clin Neurol Neurosurg. (2012) 114:124–9. doi: 10.1016/j.clineuro.2011.09.015
18. He J, Guo L, Xu G, Xu L, Lin S, Chen W, et al. Clinical and genetic investigation in Chinese patients with demyelinating Charcot-Marie-Tooth disease. J Peripher Nerv Syst. (2018) 23:216–26. doi: 10.1111/jns.12277
19. Tsang MHY, Chiu ATG, Kwong BMH, Liang R, Yu MHC, Yeung KS, et al. Diagnostic value of whole-exome sequencing in Chinese pediatric-onset neuromuscular patients. Mol Genet Genomic Med. (2020) 8:e1205. doi: 10.1002/mgg3.1205
20. Song S, Zhang Y, Chen B, Zhang Y, Wang M, Wang Y, et al. Mutation frequency for Charcot-Marie-Tooth disease type 1 in the Chinese population is similar to that in the global ethnic patients. Genet Med. (2006) 8:532–5. doi: 10.1097/01.gim.0000232481.96287.89
21. Chen B, Niu S, Wang X, Li W, Chen N, Zhang Z. Clinical, electrophysiological, genetic, and imaging features of six Chinese Han patients with hereditary neuropathy with liability to pressure palsies (HNPP). J Clin Neurosci. (2018) 48:133–7. doi: 10.1016/j.jocn.2017.10.069
22. Howard P, Feely SME, Grider T, Bacha A, Scarlato M, Fazio R, et al. Loss of function MPZ mutation causes milder CMT1B neuropathy. J Peripher Nerv Syst. (2021) 26:177–83. doi: 10.1111/jns.12452
23. Abe A, Numakura C, Kijima K, Hayashi M, Hashimoto T, Hayasaka K. Molecular diagnosis and clinical onset of Charcot-Marie-Tooth disease in Japan. J Hum Genet. (2011) 56:364–8. doi: 10.1038/jhg.2011.20
24. Shimizu H, Oka N, Kawarai T, Taniguchi K, Saji N, Tadano M, et al. Late-onset CMT2 associated with a novel missense mutation in the cytoplasmic domain of the MPZ gene. Clin Neurol Neurosurg. (2010) 112:798–800. doi: 10.1016/j.clineuro.2010.07.020
25. Yonekawa T, Komaki H, Saito Y, Takashima H, Sasaki M. Congenital hypomyelinating neuropathy attributable to a de novo p.Asp61Asn mutation of the myelin protein zero gene. Pediatr Neurol. (2013) 48:59–62. doi: 10.1016/j.pediatrneurol.2012.09.011
26. Nishiyama S, Sugeno N, Tateyama M, Aoki M. Late-onset Charcot-Marie-Tooth disease type 1B due to a novel mutation in the extracellular disulfide bridge of MPZ gene. Clin Neurol Neurosurg. (2013) 115:208–9. doi: 10.1016/j.clineuro.2012.04.016
27. Choi BO, Kim SB, Kanwal S, Hyun YS, Park SW, Koo H, et al. MPZ mutation in an early-onset Charcot-Marie-Tooth disease type 1B family by genome-wide linkage analysis. Int J Mol Med. (2011) 28:389–96.
Keywords: myelin protein zero, Charcot-Marie-Tooth disease, spectrum, Chinese, Japanese, Koreans
Citation: Chen B, Zhang Z, Chen N, Li W, Pan H, Wang X, Ren Y, Shi Y, Tai H and Niu S (2021) Two Novel Myelin Protein Zero Mutations in a Group of Chinese Patients. Front. Neurol. 12:734515. doi: 10.3389/fneur.2021.734515
Received: 01 July 2021; Accepted: 01 November 2021;
Published: 02 December 2021.
Edited by:
Angelo Schenone, University of Genoa, ItalyReviewed by:
Gianluigi Mancardi, University of Genoa, ItalyCopyright © 2021 Chen, Zhang, Chen, Li, Pan, Wang, Ren, Shi, Tai and Niu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Songtao Niu, bml1X3Nvbmd0YW9AMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.