 Haipo Yang
Haipo Yang Pan Gong
Pan Gong Xianru Jiao
Xianru Jiao Yue Niu
Yue Niu Qiujun Zhou
Qiujun Zhou Yuehua Zhang
Yuehua Zhang Zhixian Yang
Zhixian Yang- Department of Pediatrics, Peking University First Hospital, Beijing, China
Objective: The DYNC1H1 gene is related to a variety of diseases, including spinal muscular atrophy with lower extremity–predominant 1, Charcot–Marie–Tooth disease type 2O, and mental retardation, autosomal dominant13 (MRD13). Some patients with DYNC1H1 variant also had epilepsy. This study aimed to detect DYNC1H1 variants in Chinese patients with infantile spasms (ISs).
Methods: We reviewed clinical information, video electroencephalogram (V-EEG), and neuroimaging of a newly identified cohort of five patients with de novo DYNC1H1gene variants.
Results: Five patients with four DYNC1H1variants from four families were included. All patients had epileptic spasms (ESs), the median age at seizure onset was 7.5 months (range from 5 months to 2 years 7 months), and the interictal V-EEG results were hypsarrhythmia. Four of five patients had brain magnetic resonance imaging (MRI) abnormalities. Four de novo DYNC1H1 variants were identified, including two novel variants (p.N1117K, p.M3405L) and two reported variants (p.R1962C, p.F1093S). As for the variant site, two variants are located in the tail domain, one variant is located in the motor domain, and one variant is located in the stalk domain. All patients had tried more than five kinds of antiepileptic drugs. One patient has been controlled well by vigabatrin (VGB) for 4 years, and another patient by VGB and steroids for 1.5 years. The other three patients still had frequent ESs. All patients had severe intellectual disability and development delays.
Significance: IS was one of the phenotypes of DYNC1H1 variants. Most patients had non-specific brain MRI abnormality. Two of four DYNC1H1 variants were novel, expanding the variant spectrum. The IS phenotype was related to the variant's domains of DYNC1H1 variant sites. All patients were drug-refractory and showed development delays.
Introduction
The DYNC1H1 gene located in 14q32.31 encodes for dynein cytoplasmic one heavy chain 1. It is a large protein of 530 kDa and 4,646 amino acids (aa), which is highly conserved and has a few housekeeping roles (1). DYNC1H1comprises four major protein regions (Figure 1), that is, tail domains (aa residues 1–1,373 and 4,222–4,646), linker domain (aa 1,374–1,867), motor domains with AAA domains (ATPases associated with a variety of cellular activities, aa 1,868–3,168 and 3,553–4,221), and the stalk or microtubule-binding domain (MTBD, aa 3,169–3,552) (2).

Figure 1. The protein regions of DYNC1H1.
DYNC1H1 is highly intolerant to missense change (3). Heterozygous variants in the DYNC1H1 gene have been associated with a variety of diseases. In 2010, Harms et al. first described dominant spinal muscular atrophy (SMA) with lower extremity with DYNC1H1 variant (4). Weedon et al. identified a DYNC1H1 variant in a large pedigree with autosomal dominant axonal Charcot–Marie–Tooth disease in 2011 (5). Willemsen et al. and Poirier et al. reported DYNC1H1 variants caused severe intellectual disability with neuronal migration defects and malformations of cortical development (MCDs) (6, 7). Different DYNC1H1 gene variant sites were related to different phenotypes. However, patients with the same variant might also have different phenotypes; for example, p.Arg598Cys variant was found in one patient diagnosed as having SMA, and the other patient diagnosed with myopathy (8, 9).
Some patients with DYNC1H1 variants manifest epilepsy. In 2013, Poirier et al. reported eight patients with DYNC1H1 gene variant when studying the mutated genes of patients with MCD and microcephaly, among whom seven patients developed epileptic seizures, and one was diagnosed with Lennox-Gastaut syndrome (LGS) syndrome (7). In 2020, Amabile et al. summarized 103 DYNC1H1 variants in 200 patients with neurological developmental phenotypes across 143 unique families (10). Seizures were found in 18.5% (37/200) of patients. Here, we report five infantile spasm (IS) patients with DYNC1H1variants and characterize in detail the clinical phenotype, brain magnetic resonance imaging (MRI) features, and response to treatment and outcome.
Methods
Patients
Five patients in four families with DYNC1H1 variants were retrospectively recruited from the Department of Pediatrics, Peking University First Hospital, from June 2017 to October 2020. This study was approved by the Peking University First Hospital Medical Ethics Committee. Information about the age at epileptic spasm (ES) onset, developmental milestones, neurological status, family history, video electroencephalogram (V-EEG), brain MRI results, treatment, and outcomes was collected in the clinic. Brain MRI and V-EEG were reviewed by a neuroradiologist and neurophysiologist, respectively. Patients were followed up at the pediatric neurology clinic or by telephone.
DNAs (3 μg) extracted from peripheral blood from probands and their parents were analyzed using whole-exome sequencing. Variants were checked with population databases gnomAD (http://gnomad.broadinstitute.org/) and evaluated using Polyphen2, SIFT, and Variant Taster. Variant pathogenicity was interpreted according to the American College of Medical Genetics (ACMG) guidelines (11). The variants were further confirmed by Sanger sequencing.
Literature Review
A systematic research of articles published in PubMed registered from 2012 to 2021, was performed. All the researched articles are based on the following terms “DYNC1H1.” The relevance of each result was determined, and references were reviewed to identify missing studies. All the epilepsy phenotype, epilepsy onset age, treatment, and prognosis are summarized in Supplementary Table 1.
Results
Clinical Features
Clinical features of affected individuals with DYNC1H1 variants are summarized in Table 1.
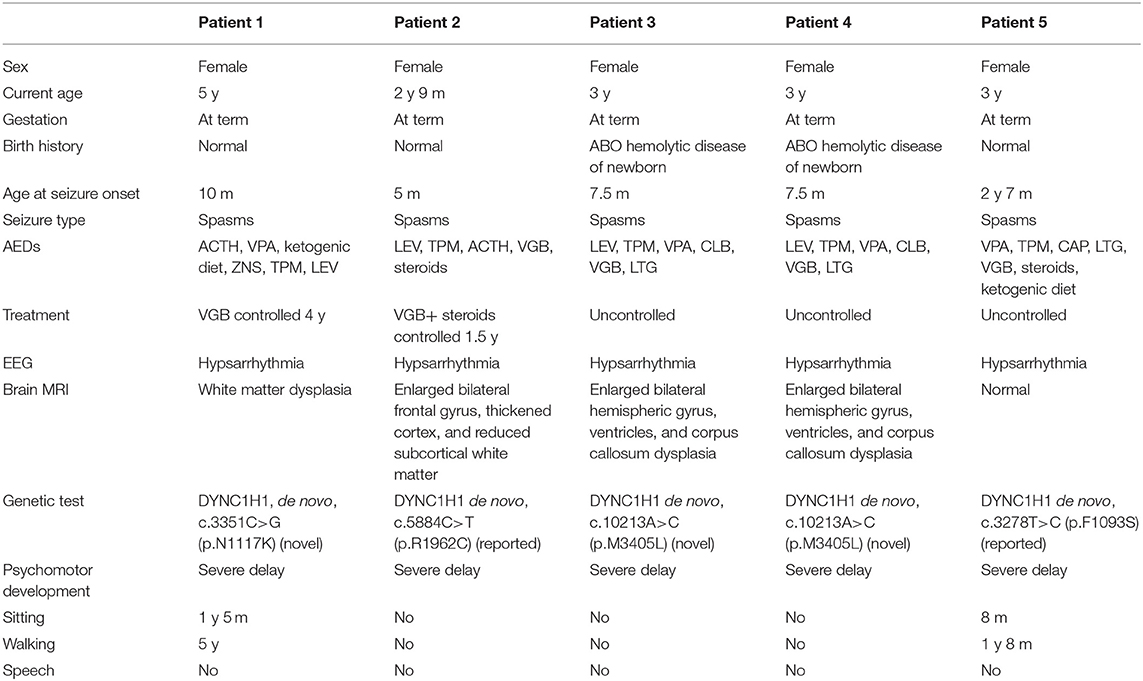
Table 1. The clinical information and gene variants of five patients.
Seizures, EEG, and Brain MRI Information
All five patients were from full-term birth. Patients 3 and 4 were dizygotic twins. Patient 5 had two febrile convulsions at 1½ and 2 years old. All patients had ES. For patients 1–4, the minimum seizure onset age was 5 months, and the maximum onset age was 7.5 months. As for patient 5, the first seizures occurred at the age of 2 years 7 months. All patients have tried at least five kinds of antiepileptic drugs (AEDs), including adrenocorticotropic hormone (ACTH), valproate (VPA), zonisamide (ZNS), topiramate (TPM), levetiracetam (LEV), vigabatrin (VGB), steroids, and a ketogenic diet. The ES of patient 1 was controlled by VGB at the age of 1.5 years for more than 4 years, albeit multifocal discharges on EEG were still observed until the last follow-up. For patient 2, ES was controlled by VGB and steroids at the age of 1 year 5 months for 1.5 years. The ES was not controlled for patients 3, 4, and 5.
EEG data for five patients all showed hypsarrhythmia (Figure 2). Brain MRI was abnormal in patients 1–4 and normal in patient 5. Brain MRI showed white matter dysplasia and developmental regression in patient 1; enlarged bilateral frontal gyrus, thickened cortex, and reduced subcortical white matter in patient 2; and enlarged bilateral hemispheric gyrus, ventricles, and corpus callosum dysplasia in patients 3 and 4 (Figure 3).
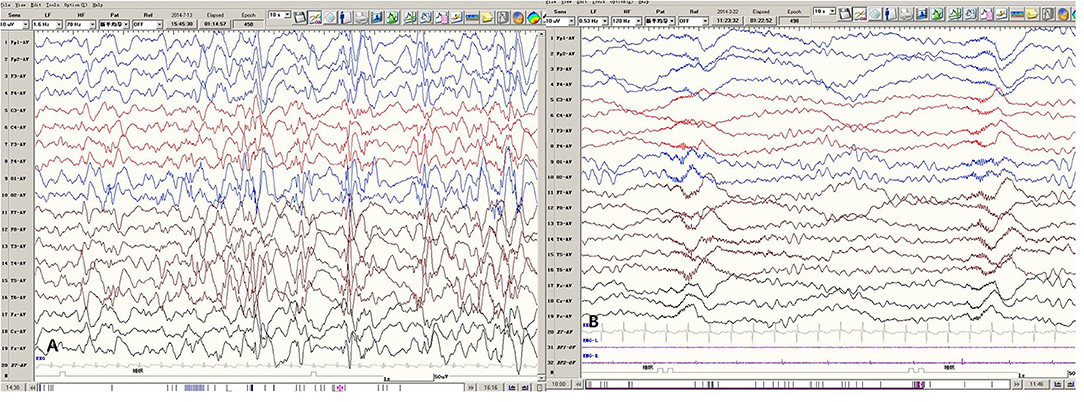
Figure 2. Representative EEGs of Patient 1. (A) The Interictal hypsarrhythmia EEG pattern. (B) The ES ictal EEG pattern.
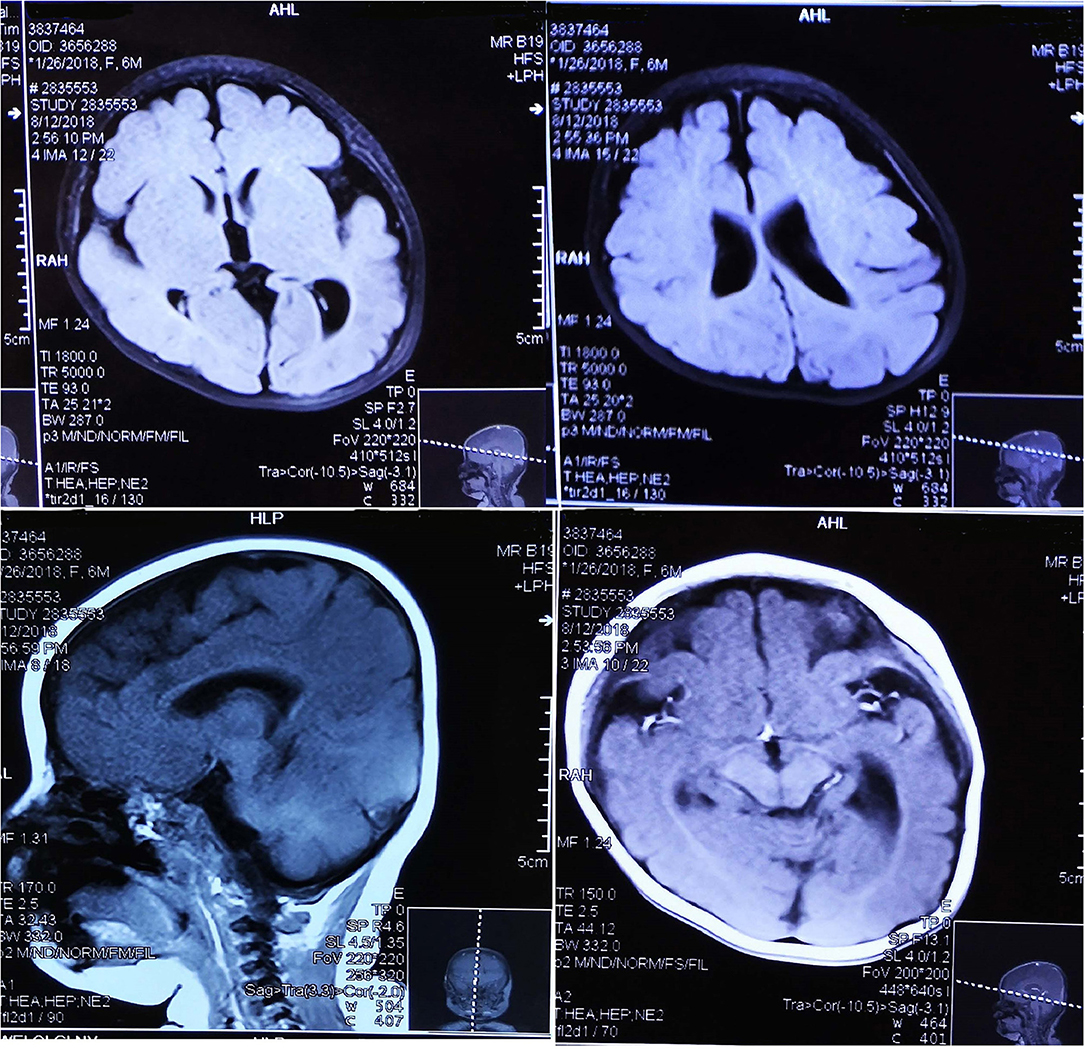
Figure 3. The brain MRI of patient 3 showed MCD.
Genetic Analysis
Four de novo variants were identified in five patients from four families. Two variants (p.N1117K, p.M3405L) were novel, and two (p.R1962C, p.F1093S) were previously reported (12, 13). All four variants located in different domains, including p.N1117K in the distal dimerization domain of the protein (tail domains), p.R1962C in theAAA1 of the protein (motor domain), p.M3405L in the AAA4 of the protein (stalk or MTBD, motor domain), and p.F1093S in the AAA1 of the protein (tail domains). All the detected variants were pathogenic according to the ACMG criteria.
Neurodevelopment
All patients had severe developmental delays and intellectual disabilities, but no developmental scales were available. Among them, only two patients could walk independently at the last follow-up. The motor development of patients 3 and 4 underwent a retrograde process. Patients 3 and 4 could control their necks at the age of 5 months. After seizure onset, they could not hold their head well and could not sit until 3 years old. At the last follow-up, all patients still had language backward. Only patient 5 could understand simple instructions and speak a few words, but he had from significant language regression to unable to speak after ES onset. The other four patients could not speak any words.
Results on Literature Search
A total of 18 articles found DYNC1H1 gene associated with epilepsy. Forty-three patients were reported to have epilepsy. In these 43 patients, the epilepsy phenotype was described in 44.2% (19/43), including focal seizure, myoclonic seizures, tonic seizures, atonic seizures, generalized tonic–clonic seizures, and IS. EEG results were reported in 11.6% of patients (5/43), including multifocal epileptiform discharges; interictal EEG showed waxing and waning of waves in the frontal, temporal, and occipital areas, and high-amplitude rhythmic waves were frequently observed; generalized spike-and-wave complexes and irregular polyspikes and slow waves, predominantly in the left frontal area; an attenuated background of mixed theta and delta frequencies; and semiperiodic spike and slow wave activity in the right temporal occipital region and generalized slowing. Treatment was described in 48.8% of patients (21/43). Nine patients were seizure-free or had controlled epilepsy. Ten patients had uncontrolled epilepsy or were therapy-refractory. The other three patients did not have their therapy result.
Discussion
We reported five patients with IS from four families with de novo DYNC1H1 variants, including two novel and two previously reported variants. We also reviewed previous literature about DYNC1H1 gene–associated epilepsy.
In 2018, Palmer et al. reported one patient with p.R1962C variant had IS (12). His EEG revealed an abnormal background and multifocal epileptiform activity. His brain MRI showed pachygyria (12). This patient had severe developmental delay and intellectual disability, and he was diagnosed with an autism spectrum disorder (12). Patient 2, carrying the same p.R1962C variant, manifested ES, MCD, severe developmental delay, and intellectual disability, similar to the reported patient (12). However, Poirier et al. reported a 19-year-old patient with p.R1962C variant who had a focal seizure, severe developmental delay, intellectual disability, and the predominant postpachygyria (7). Therefore, patients with the same DYNC1H1 variant might have different seizure types. In 2016, Helbig et al. reported a patient diagnosed as having IS with the variant site p.F1093S. The same variant was found in patient 5 (13). Besides IS, several epileptic phenotypes have been reported in patients with DYNC1H1 variants, such as focal onset epilepsy, myoclonic epilepsy, and atonic seizures (2, 6, 7, 12–17). All the reported epileptic phenotypes are summarized in Supplementary Table 1.
Patients with pachygyria carrying DYNC1H1 variants manifested epilepsy (10, 14). Abnormal brain MRI was observed in our four patients, including three MCD and one white matter dysplasia. DYNC1H1 is known to bind both bicd cargo adaptor (BICD2), a microtubule motor adaptor associated with SMA lower extremity–predominant 2, and lissencephaly 1 (LIS1), a dynein regulator associated with MCD/lissencephaly (18), which might explain why the variant of DYNC1H1 could lead to pachygyria or MCD. For those patients with MCD, posterior predominant lesions were most common (7). In 2020, Amabile et al. found 37 patients had epileptic seizures, among whom 28 patients showed MCD (10). For our patients, MCD was mainly observed at the frontal lobe in one patient and bilateral hemispheres in two, which were different from the previous reports (7). Epileptogenic mechanisms linked to pathogenic variants of DYNC1H1 gene were not clear; MCD may be the reason; however, patients from previous and our studies without MCD could also have epilepsy (16). In 2017, Lin et al. reported an epileptic encephalopathy patient who had DYNC1H1 mutation through analysis of the interaction network of DYNC1H1, which showed that DYNC1H1 interacts with many epilepsy genes, such as TBC1D24, ALDH7A1, MECP2, DEPDC5, SGCE, GRIN2B, ATP1A2, MYO5A, NBEA, CLCN4, IFT172, UBE3A, PCDH19, KCNQ3, and so on. Thus, DYNC1H1 may cause epilepsy by affecting other epilepsy-related gene function, such as interaction with other mutations present in their genomes or environmental factor. In addition to the above speculation, the mutation of DYNC1H1 gene itself may also cause epilepsy, because DYNC1H1 is highly conserved and takes part in a variety of intracellular functions (17).
Several studies have reported the relationship between domain location of the variants and clinical phenotype (2, 10). A previous study reported that DYNC1H1 variants in eight patients with MCD were located in the stalk domain, AAA1, the linker region, and the tail domain (7). The study also reported that four unrelated patients with MCD disorder had de novo variants in the stalk domain who exhibited obvious clinical symptoms of early-onset epilepsy encephalopathy (7). Amabile et al. summarized 103 DYNC1H1 variants and concluded that among 26 neuromuscular patients with obvious central nervous system involvement (intellectual disability, MCD, or other brain MRI abnormalities), 23 had variants located in the stem or neck domains, and only three had variants located in the motor domain (10). In contrast, of the 59 patients classified as having a primarily intellectual disability, MCD, and autism, 18 had variants located within the stem domain, five in the neck/linker, and 36 in the motor domain (10). Beecroft et al. summarized that a majority of patients with MCD had variants in the stalk of the motor domain (9). Some studies reported that variants associated with central nervous system manifestations such as intellectual disability and MCD had clustered in the motor domains (4, 18). Variants from the patients with seizures were mostly reported in the motor domain, whereas variants from the patients with behavioral abnormalities were largely reported in the beginning tail, linker, and motor domains (2). Variants from the patients with MRI abnormalities, specifically, pachygyria, were largely reported in the motor domain (2). In our study, the patient numbers were too small to conclude the relationship between the variant domain and the clinical phenotype. The de novo p.N1117K and p.F1093S variant of DYNC1H1 identified in patients 1 and 5 were located in the tail domain and the stem domain of the protein. Both patients had obvious intellectual disability and development delay and severe epilepsy. The brain MRI revealed white matter dysplasia in patient 1, whereas it was normal in patient 5. As indicated earlier, the variants in patients with epilepsy were usually located in the motor domain of the protein; thus, these two patients were not consistent with the previous study (7, 10). The p.R1962C variant identified in patient 2 was located in the motor domain. She had ES and MCD, which was consistent with the previous reports (7, 19). The p.M3405L variant identified in patients 3 and 4 was in the stalk domain (MTBD), which belonged to the motor domains. Both patients had early-onset epilepsy and MCD, consistent with the previous report (2, 7, 19–21).
Until now, few cases have described the epilepsy treatment of patients with DYNC1H1 variant in detail. In 2020, Becker et al. reported four patients with DYNC1H1 variants had epilepsy and found that most patients remained seizure-free with single or combined anticonvulsive medication, whereas one patient had a therapy-refractory course (2). Di Donato et al. reported 13 patients with DYNC1H1 variants complicated with epilepsy, among which seven cases had seizures controlled by drugs (14). Matsumoto et al. reported two patients with DYNC1H1 variants who had refractory epilepsy. The patients were treated with more than five kinds of AEDs, but their epilepsy was not controlled successfully (16). In our study, all five patients tried at least five AEDs, and two patients were controlled by VGB and steroids.
In our study, all five patients had severe developmental delay and intellectual disability; in addition, three patients had development or language retrograde after seizure onset. In the previous studies, patients with intellectual disability were usually diagnosed with MRD13, who had the intellectual disability and cortical malformations (6). Four of our patients had abnormal brain MRI, which was consistent with the previous report (6). Some studies indicated that variants involved in central nervous system manifestations were clustered in the motor domains (4, 19). As for patient 5, the DYNC1H1 variant was located in the tail domain, but her brain MRI was normal, which was not consistent with the previous reports (4, 19). Therefore, the mechanism of intellectual disability in patients with DYNC1H1 variants still needs further research.
Conclusion
Here we summarized the clinical features, treatment, and outcomes of five patients with DYNC1H1 variants. IS was one of the phenotypes of DYNC1H1 variants. Most patients had nonspecific brain MRI abnormalities. DYNC1H1-associated diseases had heterogeneity of gene variants and clinical phenotypes. All patients were drug-refractory, although some could be controlled lately. All patients had severe developmental delay and intellectual disability. For the present study, the data were too small to conclude the relationship between the clinical phenotypes and the variants' domain location; further study may be needed to draw a more accurate conclusion.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: BankIt2488557 Seq1 MZ733295; BankIt2489627 Seq1 MZ736872; BankIt2489989 Seq1 MZ754976, and BankIt2490423 Seq1 MZ781301.
Ethics Statement
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
HY and ZY designed the study, drafted the initial manuscript, and revised the manuscript. PG, XJ, QZ, YN, and YZ helped to collect and summarize data and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (81771393 and 82171436), Beijing Natural Science Foundation (7202210), and Capital's Funds for Health Improvement and Research (2020-2-4077).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the patients and their families for participating. We thank Dr. Xiaodong Wang to help us corrected the spelling and grammar of our manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.733178/full#supplementary-material
References
1. Schiavo G, Greensmith L, Hafezparast M, Fisher EM. Cytoplasmic dynein heavy chain: the servant of many masters. Trends Neurosci. (2013) 36:641–51. doi: 10.1016/j.tins.2013.08.001
2. Becker LL, Dafsari HS, Schallner J, Abdin D, Seifert M, Petit F, et al. The clinical-phenotype continuum in DYNC1H1-related disorders—genomic profiling and proposal for a novel classification. J Hum Genet. (2020) 65:1003–17. doi: 10.1038/s10038-020-0803-1
3. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of function intolerance across human protein-coding genes. Nature. (2020) 581:434−43. doi: 10.1038/s41586-020-2308-7
4. Harms MB, Allred P, Gardner R Jr, Filho JAF, Florence J, Pestronk A, et al. Dominant spinal muscular atrophy with lower extremity predominance: linkage to 14q32. Neurology. (2010) 75:539–46. doi: 10.1212/WNL.0b013e3181ec800c
5. Weedon MN, Hastings R, Caswell R, Xie W, Paszkiewicz K, Antoniadi T, et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal charcot-marie-tooth disease. Am J Hum Genet. (2011) 89:308–12. doi: 10.1016/j.ajhg.2011.07.002
6. Willemsen MH, Vissers LE, Willemsen MA, van Bon BW, Kroes T, de Ligt J, et al. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J Med Genet. (2012) 49:179–83. doi: 10.1136/jmedgenet-2011-100542
7. Poirier K, Lebrun N, Broix L, Tian G, Saillour Y, Boscheron C, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet. (2013) 45:439–47. doi: 10.1038/ng.2613
8. Antoniadi T, Buxton C, Dennis G, Forrester N, Smith D, Lunt P, et al. Application of targeted multi-gene panel testing for the diagnosis of inherited peripheral neuropathy provides a high diagnostic yield with unexpected phenotype-genotype variability. BMC Med Genet. (2015) 16:84. doi: 10.1186/s12881-015-0224-8
9. Beecroft SJ, McLean CA, Delatycki MB, Koshy K, Yiu E, Haliloglu G, et al. Expanding the phenotypic spectrum associated with mutations of DYNC1H1. Neuromuscul Disord. (2017) 27:607–15. doi: 10.1016/j.nmd.2017.04.011
10. Amabile S, Jeffries L, McGrath JM, Ji W, Spencer-Manzon M, Zhang H, et al. DYNC1H1-related disorders: a description of four new unrelated patients and a comprehensive review of previously reported variants. Am J Med Genet A. (2020) 182:2049–57. doi: 10.1002/ajmg.a.61729
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
12. Palmer EE, Schofield D, Shrestha R, Kandula T, Macintosh R, Lawson JA, et al. Integrating exome sequencing into a diagnostic pathway for epileptic encephalopathy: evidence of clinical utility and cost effectiveness. Mol Genet Genomic Med. (2018) 6:186–99. doi: 10.1002/mgg3.355
13. Helbig KL, Hagman KDF, Shinde DN, Mroske C, Powis Z, Li S, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. (2016) 18:898–905. doi: 10.1038/gim.2015.186
14. Di Donato N, Timms AE, Aldinger KA, Mirzaa GM, Bennett JT, Collins S, et al. Analysis of 17 genes detects mutations in 81% of 811 patients with lissencephaly. Genet Med. (2018) 20:1354–64. doi: 10.1038/gim.2018.8
15. Epilepsy Phenome/Genome Project, Epi4K Consortium. Diverse genetic causes of polymicrogyria with epilepsy. Epilepsia. (2021) 62:973–83. doi: 10.1111/epi.16854
16. Matsumoto A, Kojima K, Miya F, Miyauchi A, Watanabe K, Iwamoto S, et al. Two cases of DYNC1H1 mutations with intractable epilepsy. Brain Dev. (2021) 43:857–62. doi: 10.1016/j.braindev.2021.05.005
17. Lin Z, Liu Z, Li X, Li F, Hu Y, Chen B, et al. Whole-exome sequencing identifies a novel de novo mutation in DYNC1H1 in epileptic encephalopathies. Sci Rep. (2017) 7:258. doi: 10.1038/s41598-017-00208-6
18. Carter AP, Garbarino JE, Wilson-Kubalek EM, Shipley WE, Cho C, Milligan RA, et al. Structure and functional role of dynein's microtubule-binding domain. Science. (2008) 322:1691–95. doi: 10.1126/science.1164424
19. Hoang HT, Schlager MA, Carter AP, Bullock SL. DYNC1H1 mutations associated with neurological diseases compromise processivity of dynein-dynactin-cargo adaptor complexes. Proc Natl Acad Sci U.S.A. (2017) 114:E1597–606. doi: 10.1073/pnas.1620141114
20. Scoto M, Rossor AM, Harms MB, Cirak S, Calissano M, Robb S, et al. Novel mutations expand the clinical spectrum of DYNC1H1-associated spinal muscular atrophy. Neurology. (2015) 84:668–79. doi: 10.1212/WNL.0000000000001269
21. Chan SHS, van Alfen N, Thuestad IJ, Ip J, Chan AO, Mak C, et al. A recurrent de novo DYNC1H1 tail domain mutation causes spinal muscular atrophy with lower extremity predominance, learning difficulties and mild brain abnormality. Neuromuscul Disord. (2018) 28:750–56. doi: 10.1016/j.nmd.2018.07.002
Keywords: infantile spasms, epilepsy, malformations of cortical development, DYNC1H1 gene, intellectual disability
Citation: Yang H, Gong P, Jiao X, Niu Y, Zhou Q, Zhang Y and Yang Z (2021) De Novo Variants in the DYNC1H1 Gene Associated With Infantile Spasms. Front. Neurol. 12:733178. doi: 10.3389/fneur.2021.733178
Received: 30 June 2021; Accepted: 06 September 2021;
Published: 05 November 2021.
Edited by:
Vincenzo Salpietro, University College London, United KingdomReviewed by:
Ruzica Kravljanac, The Institute for Health Protection of Mother and Child Serbia, SerbiaMario Mastrangelo, Umberto 1 Polyclinic, Italy
Copyright © 2021 Yang, Gong, Jiao, Niu, Zhou, Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhixian Yang, emhpeGlhbi55YW5nQDE2My5jb20=