
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 20 August 2021
Sec. Neurogenetics
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.720201
 Ashraf Yahia1,2,3,4†
Ashraf Yahia1,2,3,4† Liena E. O. Elsayed1,5*†
Liena E. O. Elsayed1,5*† Remi Valter3,4
Remi Valter3,4 Ahlam A. A. Hamed1Inaam N. Mohammed1
Ahlam A. A. Hamed1Inaam N. Mohammed1 Maha A. Elseed1
Maha A. Elseed1 Mustafa A. Salih6Typhaine Esteves3,4
Mustafa A. Salih6Typhaine Esteves3,4 Nicolas Auger3,4Rayan Abubaker7
Nicolas Auger3,4Rayan Abubaker7 Mahmoud Koko8
Mahmoud Koko8 Fatima Abozar1Hiba Malik1Rawaa Adil1Sara Emad1Mhammed Alhassan Musallam1Razaz Idris9
Fatima Abozar1Hiba Malik1Rawaa Adil1Sara Emad1Mhammed Alhassan Musallam1Razaz Idris9 Isra Z. M. Eltazi1Arwa Babai1
Isra Z. M. Eltazi1Arwa Babai1 Elhami A. A. Ahmed10Amal S. I. Abd Allah1Mathilde Mairey3
Elhami A. A. Ahmed10Amal S. I. Abd Allah1Mathilde Mairey3 Ahmed K. M. A. Ahmed1,11,12Mustafa I. Elbashir1Alexis Brice3
Ahmed K. M. A. Ahmed1,11,12Mustafa I. Elbashir1Alexis Brice3 Muntaser E. Ibrahim7Ammar E. Ahmed1Foudil Lamari13
Muntaser E. Ibrahim7Ammar E. Ahmed1Foudil Lamari13 Giovanni Stevanin3,4
Giovanni Stevanin3,4Introduction: Hereditary spastic paraplegia is a clinically and genetically heterogeneous neurological entity that includes more than 80 disorders which share lower limb spasticity as a common feature. Abnormalities in multiple cellular processes are implicated in their pathogenesis, including lipid metabolism; but still 40% of the patients are undiagnosed. Our goal was to identify the disease-causing variants in Sudanese families excluded for known genetic causes and describe a novel clinico-genetic entity.
Methods: We studied four patients from two unrelated consanguineous Sudanese families who manifested a neurological phenotype characterized by spasticity, psychomotor developmental delay and/or regression, and intellectual impairment. We applied next-generation sequencing, bioinformatics analysis, and Sanger sequencing to identify the genetic culprit. We then explored the consequences of the identified variants in patients-derived fibroblasts using targeted-lipidomics strategies.
Results and Discussion: Two homozygous variants in ABHD16A segregated with the disease in the two studied families. ABHD16A encodes the main brain phosphatidylserine hydrolase. In vitro, we confirmed that ABHD16A loss of function reduces the levels of certain long-chain lysophosphatidylserine species while increases the levels of multiple phosphatidylserine species in patient's fibroblasts.
Conclusion: ABHD16A loss of function is implicated in the pathogenesis of a novel form of complex hereditary spastic paraplegia.
Hereditary spastic paraplegia (HSP) is an entity that includes a heterogeneous group of disorders characterized clinically by spastic lower limbs, either alone (pure HSP) or in combination with other neurological and/or extra-neurological features (complex HSP) (1). Intellectual impairment, cerebellar ataxia, neuropathy, and skeletal deformities are common presentations complicating HSP (2). The advent of next-generation sequencing (NGS) has revolutionized HSP diagnosis and emphasized its extreme genetic heterogeneity broadening the list of the currently identified HSP-causing genes to include more than 80 genes (3). The HSP global prevalence has been estimated as 1.8/105(4), a value probably underestimated because of the lack of accessibility to new diagnostic technologies (3, 4).
Multiple pathological mechanisms contribute to the development of HSP. These mechanisms affect cellular processes involved in the corticospinal tract's neurodevelopment and maintenance, including dysfunctions of the endoplasmic reticulum (ER), mitochondria, cytoskeleton, cell signaling pathways, neuronal development, and cellular metabolism (5, 6). Accumulating evidence emphasizes the importance of maintaining lipid homeostasis for the health and function of neurons (7), particularly in HSP forms.
Lipids constitute approximately one- to two-thirds of the gray matter, white matter, and myelin in the human brain, where they have structural and signaling functions (8–12). Glycerophospholipids (GPL) constitute a major class of lipids, composed of a glycerol backbone linked at its sn-1 and sn-2 carbons to two fatty acid chains and at its sn-3 carbon to a polar head group (13, 14). GPL lacking one long-chain fatty acid attached to its glycerol backbone are called lysophospholipids (13).
The brain is highly enriched in GPL compared to other organs (15). The most abundant classes are phosphatidylethanolamine (55%), phosphatidylcholine (31%), and phosphatidylserine (PS, 7.7%) (15). Disturbances in the GPL biosynthesis and remodeling pathways are increasingly implicated in the pathogenesis of multiple HSP subtypes (7).
Herein we report a form of complicated HSP caused by homozygous variants in the ABHD16A gene involved in PS metabolism.
We used genetic and functional tools to reach the cause of the disease in the two consanguineous Sudanese families F37 and F69. Supplementary Figure 1 summarizes the approaches we followed in our quest; the detailed methodology is provided below.
This study was approved by local Ethics committees, and signed consent was obtained for all patients. The patients were evaluated at the Pediatric Neurology Clinic, Soba University Hospital (Khartoum, Sudan). We collected two ml of saliva from the patients and healthy related controls using OG-500 or OG-575 Oragene DNA kits (DNA Genotek Inc., Ottawa, ON, Canada). DNA was extracted according to the PrepIT L2P protocol provided by the manufacturer.
Skin punch biopsies were obtained from patients F37-314, F37-315, and F69-508 and unrelated healthy Sudanese controls using 4-mm-punches. Immediately after the biopsy, samples were preserved in 20 ml sterile clear glass vials filled with a culture medium composed of 50% Gibco™ DMEM (Dulbecco's Modified Eagle Medium), 10% Gibco™ L-Glutamine (l x solution), 10% Penicillin/Streptomycin (P 4333 Sigma-Aldrich, l x solution), 10% GibcoTM HEPES [4-(2-hydroxyethy1)-1-piperazineethanesulfonic acid], and 20% Gibco™ fetal bovine serum. Punches were sent at room temperature for fibroblasts culturing at the platform of biological resources of the Paris Brain Institute–ICM, Paris, France.
DNA extracted from patients F37-314, F37-315, F69-508, and F69-509 was sent for NGS at the genotyping and sequencing core facility of the Paris Brain Institute -ICM, Paris, France. First, we screened the DNA for pathogenic variants in known HSP genes using a targeted next-generation sequencing panel surveillance of 65 genes as detailed in a previous report (16). Following negative panel surveillance, exome sequencing (ES) was used for the analysis of the DNA of three patients (F37-314, F69-508, and F69-509). ES data were generated and processed as described previously (17).
Sanger sequencing was performed on an ABI3730 sequencer (Applied Biosystems) using the Bigdye chemistry at GATC Biotech—Eurofins Genomics (Cologne, Germany). Segregation analysis of variants was performed in all available samples from the two families.
Fibroblasts were prepared at the density of 2 x 107 cells/100 mm culture plate, washed three times with 1 × Phosphate Buffer Saline (PBS), and then cultured for 4 h in 5 ml of High Glucose Dulbecco's Modified Eagle Medium (DMEM) 4.5 g.L-1 (Gibco) (for lipidomic studies, the medium contained neither phenol red nor serum to avoid interference). We centrifuged the cells-containing medium (4,500 g, 20°C, 5 min) and stored the supernatant at −80°C before lipids extraction. We detached the cells from the medium using trypsin, then centrifuged the mix (800 rpm, 20°C, 5 min), washed it with PBS three times, and froze (−80°C) the resulting pellet.
The cells were detached with 0,05% trypsin (Gibco) in PBS, centrifuged (1,000 rpm, 20 °C, 5 min), and washed with 1 × PBS. The resulting cell pellet was lysed in lysis buffer (100 mM NaCl, 10 mM Tris Hcl pH 7.4, 1% Triton X-100, and 1 × protease inhibitors–HaltTM Protease Inhibitor Cocktail 100 ×) (Thermo Scientific). After 30 min of incubation on ice, the lysates were centrifuged (13,000 rpm, 4 °C, 30 min), and the supernatants were collected.
The protein assay was performed with the “BCA Protein Assay Kit” (PierceTM), allowing the quantification of copper reduction caused by proteins in alkaline solution, with a colorimetric detection (absorbance at 562 nm) of the bicinchoninic acid reduced Cu+1 cation.
We compared the absorbance obtained to the absorbance values of a Bovine Serum Albumin (BSA) standard with a 0–20 μg.mL-1 range to deduce our samples' protein concentration.
Protein samples were mixed with Laemmli loading buffer (2% SDS, 10% Glycerol, 60 mM Tris-HCl pH 6.8, 0.01% Bromophenol blue, and 50 mM DTT), then denatured for 5 min at 95°C. Ten to fifteen micrograms of proteins were deposited on a precast acrylamide gel “NuPAGE™ 4 to 12%, Bis-Tris, 1.0 mm, Mini Protein Gel, 10-well” or “Novex 4–20% Tris-glycine mini protein gel” (ThermoFisher Scientific) with the molecular weight scale “PageRulerTM Plus Prestained Protein Ladder” (ThermoFisher Scientific). Migration was performed at 120–150 V constant in MOPS migration buffer (50 mM MOPS, 50 mM Tris-base, 1 mM EDTA and 0.1% SDS). A “semi-dry” transfer was carried out on a nitrocellulose membrane (Amersham™ Protran® 0.45 NC) for 60–90 min at 25 V constant and 0.22 A per membrane in transfer buffer (25 mM Tris-base, 292 mM Glycine, 20% ethanol, 0.1% SDS). The membrane was then incubated in Ponceau red to check transfer efficiency and washed by PBS.
The membrane was incubated in a saturation solution (1 × PBS, 5% milk, and 5% BSA) for 1 h with shaking at room temperature, then incubated overnight at 4°C in the saturation solution with the primary anti-ABHD16A (Abcam® EPR15463) or anti-alpha tubulin antibody (Abcam® ab7291). The membrane was then washed three times in 1 × PBS 0.1% Tween for 5 min, incubated for 1 h in saturation solution with anti-rabbit (IRDye® 800 Donkey anti-Rabbit IgG) or anti-mouse secondary antibodies (IRDye® 680 Goat anti-Mouse IgG) then washed three times with 1 × PBS. The detection was carried out by reading fluorescence with the Odyssey® Clx or with SuperSignal™ West Femto Maximum Sensitivity Substrate (ThermoFisher Scientific) at ChemiDoc™ system. The analysis of the images obtained was carried out with ImageJ software.
We extracted lipids from cells using the Bligh & Dyer method (18). The cell pellets obtained from the preparation steps were suspended in 1 ml of PBS, and 100 μl of each suspension was stored for protein quantification. The cell samples were placed in glass tubes and supplemented with 1 μg of LPS 17:1 as internal standard (Avanti® Polar Lipids, USA).
Lipids were extracted twice by adding 3 ml of chloroform/methanol (2:1, v/v). The lipid extract was evaporated to dryness under a stream of nitrogen, the residues were dissolved in 100 μl of chloroform/methanol (2:1, v/v), and 10 μl were injected into the LC-MSMS system. For PS quantification, a five-point calibration curve was made by a successive dilution of a solution mixture of Phosphatidyl-L-serine from bovine brain (Sigma Aldrich).
Separation of PS and LPS were carried on ultra-performance liquid chromatography (Acquity-Waters) equipped with a Waters-Acquity UPLC BEH C8 (1.7 μm, 2.1 ×100 mm) column at 60 °C. Mobile phases were: phase A: water/ methanol (95:5) (v/v) supplemented with 0.1% formic acid 0.028% ammonia and 5 μM phosphoric acid; mobile phase B: iPrOH/ MeOH/H2O (60:35:5, v/v/v) supplemented with 0.1% formic acid and 0.028% ammonia. PS and LPS detection and quantification were performed on a TQD mass spectrometer (Waters) equipped with electrospray ionization. The spectra were recorded in MRM, negative ion mode (Supplementary Table 1). The capillary voltage was set at 3 kV, cone voltage at −40 V, and source block temperature 130 °C.
Patients F37-314 (10 years) and F37-315 (7 years) were male siblings born to first-degree consanguineous Sudanese parents after uneventful pregnancies and deliveries (Figure 1). The two boys manifested a neurodevelopmental disorder with a comparable disease progression. Their disease was characterized by the absence of comprehensible speech, stiffening of their lower limbs observed after their first birthday, and intellectual impairment (Table 1).
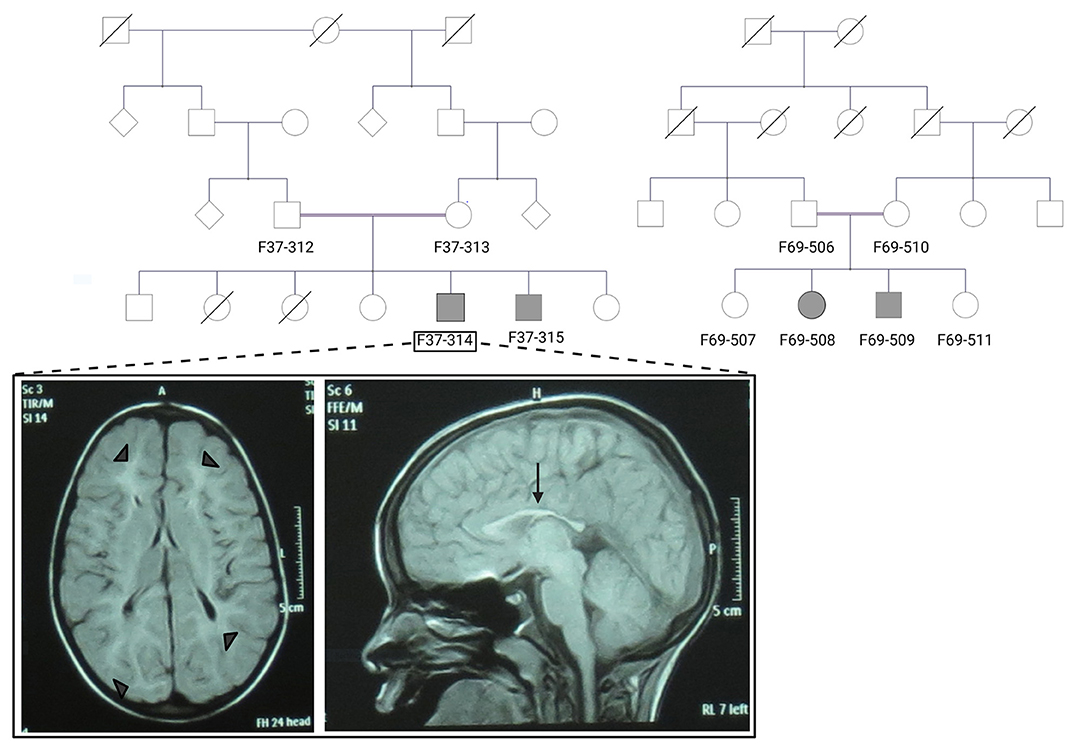
Figure 1. The pedigrees of families F37 and F69 and patient F37-314 brain MRI. The family pedigrees of family F37 (left) and F69 (right) show consanguinity in the two families. Axial T2 TIRM dark fluid brain MRI of patient F37-314 shows periventricular and subcortical white matter hyperintensities (left image: arrow heads), whereas sagittal T1 image shows thin corpus callosum (right image: arrow).
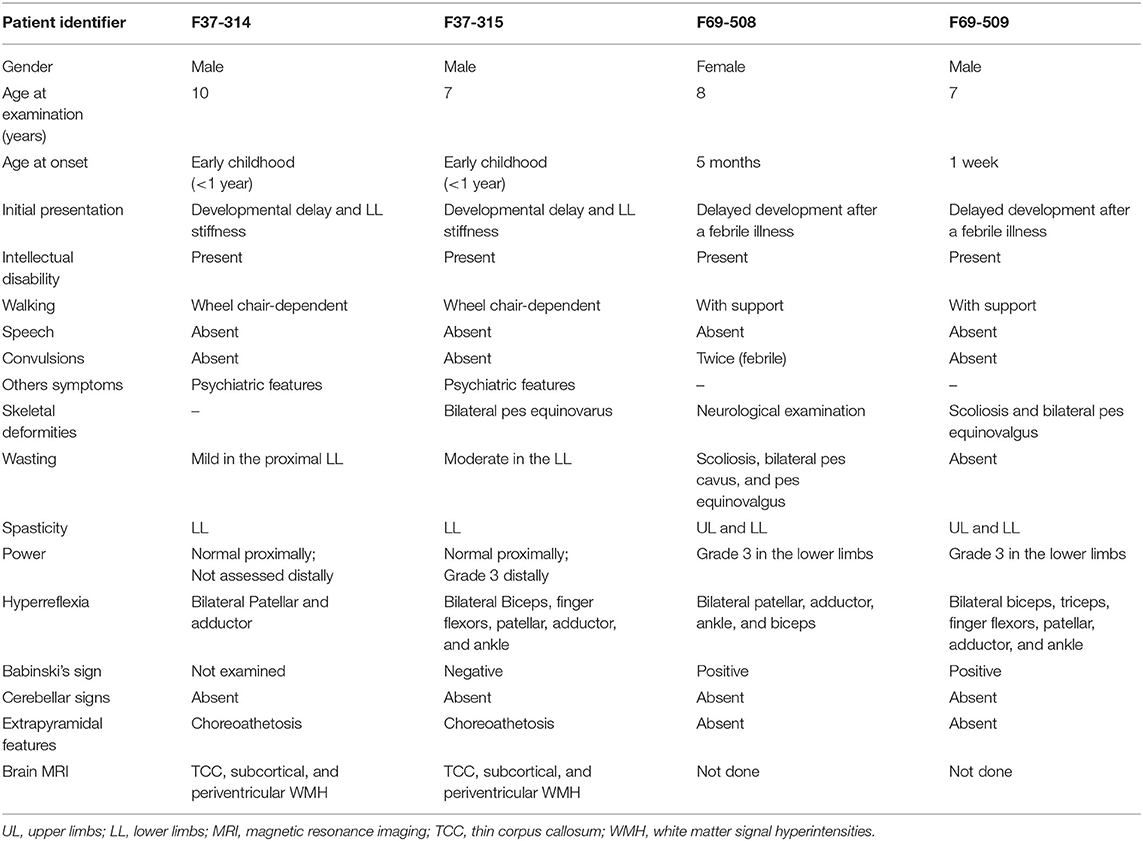
Table 1. Summary of the clinical presentation of the patients from the families F37 and F69.
They sustained head control at 4 months, sat without support at 9 months, and had their first social smiles at 4 months. Both never spoke, except for some incomprehensible sounds. The older sibling (F37-314) started to walk at 1 year, while the younger walked at two, though their gait was abnormal due to spasticity of the lower limbs. At 3–4 years, their walking and speaking skills regressed; patient F37-314 reverted to crawling, while patient F37-315 only walked with support. They acquired sphincter control at 2 and 3 years of age (younger and older sib, respectively), and maintained sphincter control as their condition progressed. At the ages of seven and six, respectively, they became non-ambulant and wheelchair-dependent.
The upper limb examination of the two siblings was normal. Lower limb examination was significant for bilateral spasticity and hyperreflexia. We could not examine the older patient's ankle and distal lower limb because his legs were in a cast after corrective surgery for ankle deformity at the time of examination. The younger patient (F37-315) had talipes equinovarus, more pronounced on the left leg, and bilateral up-going plantar response. The younger patient had mild to moderate wasting of the lower limbs with preferential decrease in the strength distally and sparing of proximal lower limb motor function. Coordination was normal, we did not perform heel-on-shin test in the older sibling, and there was no oculomotor apraxia. Fundal examinations were normal. Due to a lack of cooperation, we could not examine their sensory system. We noted extrapyramidal signs in form of choreoathetosis in the two siblings. It was evident that both siblings had cognitive impairment and learning difficulties but no formal neuropsychology assessment was performed in this study. They were irritable, distractible, had bouts of crying, and had emotional lability. We also noted a stereotypic behavior in the form of hand clapping and head banging.
Brain Magnetic resonance imaging (MRI) showed thin corpus callosum and subcortical and periventricular white matter high signals in both cases (Figure 1).
The patients F69-508 and F69-509 were two Sudanese siblings aged 8 and 7 years, respectively (Table 1). They suffered from a neurodevelopmental disorder that was noted to start following a febrile illness. Both patients were outcomes of unremarkable pregnancies ending by normal vaginal deliveries. The parents were second-degree relatives; however, there was no family history of a similar condition (Figure 1).
Patient F69-508 was a healthy baby with normal development until she had attacks of febrile convulsions between the ages of 5 months and 1 year. After these episodes, all categories of developmental milestones regressed. She walked with support at the age of 4 years, and until the age of presentation, 8 years old, she could neither walk independently nor speak, apart from a few incomprehensive sounds. Her intellectual development was markedly delayed for her age, and she never controlled her sphincters. On examination, her head circumference was 51.5 cm, which is within the normal range for her age. Examination of her limbs was significant for spasticity, severe in the lower limbs and mild in the upper limbs, hyperreflexia, and bilateral up-going plantar response. Her patellar, adductor, ankle, and biceps reflexes were increased. She had normal finger flexor reflex and an absent jaw jerk. Hoffmann's sign was negative. She presented with skeletal abnormalities such as mild scoliosis, severe pes cavus, and pes equinovarus. Ocular motor and cerebellar examination were normal. We did not perform a fundus examination. Although a formal sensory examination was not possible, we did not identify any major sensory impairment.
The patient F69-509's disease started 1 week after birth with a febrile illness not complicated with convulsions. Following the febrile illness, the patient became hypotonic. Akin to his sister, patient F69-509 progressed to have global developmental impairment. He started to walk with support at 4 years and never walked unsupported. Although he could say more words compared to his sister, he never formulated a sentence. He had cognitive impairment and did not achieve sphincter control.
On clinical examination, he had severe spasticity in upper and lower limbs, hyperreflexia, and bilateral up-going plantar responses. The power was reduced in his lower limbs though he had a normal overall muscle bulk. The patient F69-509 had scoliosis and pes equinovalgus, and, similar to his sister, he had a normal head circumference for his age (51 cm). Ocular motor and cerebellar examinations were normal. Brain MRI was not available for the two affected children.
In the absence of genetic variants in targeted sequencing of 65 HSP genes (16), we performed ES on the patients from families F37 (F37-314) and F69 (F69-508 and F69-509) with a mean coverage of 30 and 100 × , respectively. Filtering of variants on ES data identified homozygous variants in ABHD16A as the only convincing variants.
In family F37, the nonsense variant NM_021160.2(ABHD16A):c.340C>T (p.Arg114*) was confirmed as homozygous in patients F37-314 and F37-315 using Sanger sequencing. We validated its segregation with the disease in the family F37 under an autosomal recessive model of inheritance. This variant was absent in the gnomAD v2.1.1 database and the Greater Middle East Variome project.
In family F69, the missense variant NM_021160.2(ABHD16A):c.1370G>A (p.Arg457Gln) (rs774259910) was confirmed homozygous in the patients F69-508 and F69-509 and segregated with the disease under an autosomal recessive model of inheritance. While this variant was absent in the Greater Middle East Variome project, it was found extremely rare in the gnomAD v2.1.1 population as it was reported only once in a heterozygous state in an African/African American male (its global gnomAD frequency was 0.000004). NM_021160.2(ABHD16A):c.1370G>A (p.Arg457Gln) was predicted to alter a highly conserved Arginine at position 457 (CADD score 36). Furthermore, NM_021160.2(ABHD16A):c.1370G was the last base before the splice site in ABHD16A's exon 16, and its disruption was predicted to alter ABHD16A mRNA splicing. We employed two splice-effect prediction algorithms, MaxEntScan and NNSPLICE to scan for novel splice sites. MaxEntScan uses the Maximum Entropy principle to identify splicing motifs (19). On the other hand, NNSPLICE employs a neural network model to recognize splice sites (20). The MaxENTScan score (0–12) was 8.51 for the wild-type variant and 2.69 for the mutant, while the NNSPLICE score (0–1) moved from 0.93 for the wild-type variant to 0 for the mutant.
We found no other HSP family carrying loss of function pathogenic variants in our in-house exome database of 450 exomes of ataxia and HSP cases and in the GENESIS platform (https://www.tgp-foundation.org/genesis-log-in) gathering 786 exomes of pure and complex spastic paraplegia.
We checked for the presence of the ABHD16A protein in the fibroblasts of three affected patients and two unrelated controls. While the positive control protein, alpha-tubulin, was present in all the examined fibroblasts, ABHD16A was absent in the fibroblasts derived from the patients F69-508, F37-314, and F37-315 and was present in the fibroblasts obtained from the two unrelated healthy controls (Supplementary Figure 2). This corroborated the predictions that both NM_021160.2(ABHD16A):c.340C>T (p.Arg114*) and NM_021160.2(ABHD16A):c.1370G>A (p.Arg457Gln) were null variants.
We then looked for the consequences of the absence of ABHD16A in fibroblasts of patients by measuring PS/LPS using UPLC\MS. The levels of the main detected PS species, expressed as arbitrary units/mg proteins, were systematically higher in fibroblasts from the patients F37-314, F37-315, and F69-508 compared to cells from unrelated healthy controls (Table 2). However, the difference between the patients and the controls' cells was statistically significant for only 34:1, 36:2, 36:1, and 42:1 species (Table 2, p = 0.014), likely due to the low sample size. In contrast, the levels of long-chain LPS (18:1, 20:4, 20:0, and 20:1) were reduced in the patient's fibroblasts compared to control cells (Figure 2, p < 0.0001). These results, suggest that ABHD16A pathogenic variants are loss of function mutations, leading to accumulation of several PS substrate species and reduction of several LPS product species.
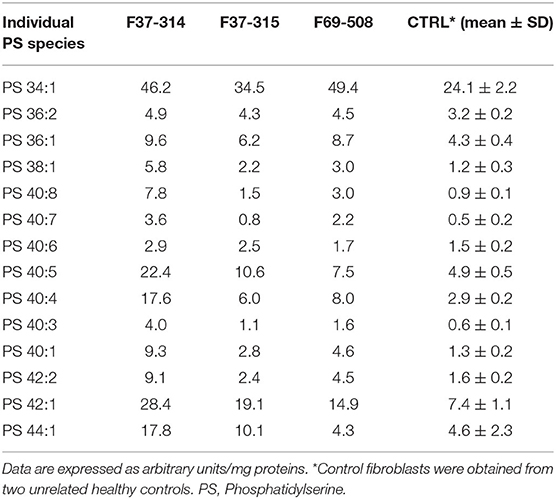
Table 2. Phosphatidylserine molecular species in fibroblasts derived from the patients F69-508, F37-314, and F37-315 and two unrelated healthy controls.
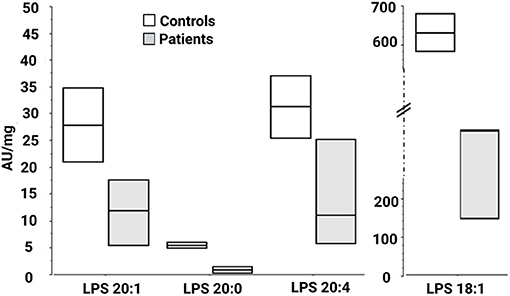
Figure 2. Lysophosphatidylserine molecular species in fibroblasts derived from patients F37-314, F37-315, and F69-508. LPS 18:1, 20:0, 20:1, and 20:4 are lower in the patients than in the healthy controls. AU, arbitrary units; LPS, lysophosphatidylserine.
We identified two homozygous null variants in the ABHD16A gene segregating in two Sudanese families, causing a novel neurodevelopmental disorder. The clinical presentation in the patients from the two families was severe and included developmental delay, intellectual impairment, spasticity, and skeletal deformities, reminiscent of complex forms of hereditary spastic paraplegia.
ABHD16A is a 21-exon gene which encodes for the α/β hydrolase domain-containing 16A (ABHD16A) enzyme, which is the main brain phosphatidylserine (PS) hydrolase (21). It has two protein-coding transcripts, NM_021160 (the most ubiquitous) and NM_001177515, which code for two functionally related isoforms of the ABHD16A enzyme, with 558 and 525 amino acids length, respectively (22, 23). NM_021160 is highly expressed in the brain, muscles, testes, and heart, while the highest expression of NM_001177515 is in the testes, with still a minimal expression in the brain (22, 23). The two variants identified in this study are predicted to have the same effect on the two ABHD16A transcripts, leading to their complete loss of function; we focused our study on the canonical and most ubiquitous transcript, NM_021160.
The ABHD16A enzyme is a member of the α/β hydrolase domain-containing protein family that participates in lipid metabolism and intracellular signaling (23). Pathogenic variants in genes coding for enzymes belonging to α/β hydrolases family have already been linked to inherited metabolic diseases. This is the case for ABHD5 and ABHD12 causing Chanarin-Dorfmann (OMIM # 275630) and polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract (PHARC; OMIM # 612674) syndromes, respectively (24, 25). ABHD16A is highly conserved in mammals (22, 23). ABHD16A has a heterogeneous expression in the murine brain and is enriched in the cerebellum, particularly in the granular layer and the olfactory bulb (26). It is an integral ER protein with a cytosolically oriented active site (26). In the ER, it colocalizes with the principle brain LPS hydrolase ABHD12, responsible for the PHARC syndrome (OMIM # 612674) (25–27). ABHD12, unlike ABHD16A, is ubiquitously expressed in the brain (26). These two enzymes, ABHD16A and ABHD12, are the main enzymes responsible for the brain LPS metabolism, as ABHD16A generates LPS while ABHD12 catabolizes them into fatty acid and phosphoserine (Figure 3).
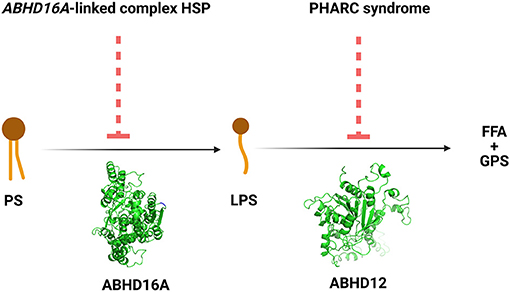
Figure 3. The reactions and the disorders across the ABHD16A-ABHD12 pathway. HSP, hereditary spastic paraplegia; PHARC syndrome, syndrome of polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract; PS, phosphatidylserine; LPS, lysophosphatidylserine; FFA, free fatty acid; GPS, glycerophosphoserine.
The PS substrates for the ABHD16A-ABHD12 pathway are continuously replenished from other phospholipid precursors, particularly at the mitochondrial-associated membranes (28). PS is the major acidic phospholipid class in the brain, accounting for >7% of the total brain phospholipids (15). It participates in multiple neuronal processes, including synaptic formation, synaptic functions, and neuronal signaling (28–30). Pathogenic variants in genes coding certain enzymes involved in PS metabolism have been associated with human genetic disorders. For example, gain-of-function pathogenic variants in PTDSS1 coding phosphatidylserine synthase causes Lens-Majewski syndrome (OMIM # 151050) characterized by skeletal deformities and intellectual disability; similarly, pathogenic variants in PISD coding phosphatidylserine decarboxylase were associated with Liberfarb syndrome (OMIM # 618889), also characterized by skeletal deformities (31, 32).
LPS, the deacylated derivative of PS, participates in cellular signaling and has at least three receptors in humans, namely G-protein-coupled receptor (GPR) 34, GPR 174, and the purinergic receptor P2Y-G-Protein-Coupled 10 (33–37). In addition, LPS potentiates the effects of neuronal growth factor (NGF) in causing PC12 cells to differentiate into primary neurons (38). The combined actions of NGF and LPS are important for acquiring and maintaining the unipolar and bipolar morphology of the primary neurons (38). However, the mechanism by which LPS potentiates the effects of NGF is not determined yet (38).
PS and LPS levels are altered to variable extents across the brains of ABHD16A and ABHD12 knockout mice (26). Loss of ABHD16A activity in mice causes a decrease in brain LPS, particularly in the cerebellum and olfactory bulb (26). In this model, the reduced LPS species are mostly those with long-chain fatty acids (C18 to C22), and this is in line with what we observed in our patients' fibroblasts. Regarding the peripheral, more accessible tissues, a reduction in ABHD12 activity was documented in lymphoblastoid cells from a human patient with PHARC syndrome compared to healthy controls (39).
To explore the signatures of ABHD16A deficiency in human fibroblasts and its contribution to the novel phenotype identified in the patients from the families F37 and F69, we first, validated the lack of expression of ABHD16A in patients' fibroblasts vs. controls. We then measured PS and LPS in the fibroblasts derived from three patients with ABHD16A variants and two healthy controls using UPLC\MS. We found the levels of most PS species higher in the ABHD16A patients' cells, particularly those with long-chain fatty acids compared to healthy controls' cells. These results are in line with what has been reported in the ABHD16A knockout mice model and confirm the loss of function of the ABHD16A protein and its specificity against long chains PS.
In the ABHD16A−/− mouse model that was established by Kamat et al. they could identify sub-Mendelian production of the mice which was explained by presumed prenatal lethality (21).
The pathogenic mechanisms of ABHD16A deficiency could be due to the loss of LPS potentiation of NGF-mediated neuronal development or another yet unidentified LPS-mediated signaling. ABHD16A deficiency is associated with low brain LPS levels; thus, in contrast to PHARC syndrome, the pathogenesis of the ABHD16A-associated HSP is unlikely due to LPS-mediated activation of microglia and neuroinflammation (26). The pathogenic mechanism can also be related to PS metabolism. Indeed, gain-of-function pathogenic variants in PTDSS1 coding PS synthase cause sclerosing bone dysplasia and intellectual disability (Lens-Majewski syndrome; OMIM # 151050), and pathogenic variants in PISD1 coding PS decarboxylase cause Spondyloepimetaphyseal dysplasias (31, 32). Interestingly, the patients from both F37 and F69 families exhibited skeletal deformities and had intellectual disabilities. Further studies are needed to elucidate the pathogenesis of the ABHD16A-associated HSP and delineate its associated phenotype. Fibroblasts - and the study of their PS/LPS contents by MS–can provide a means for establishing the diagnosis of future patients with the novel ABHD16A-linked complex HSP, particularly those with missense variants.
Important therapeutic options may emerge based on the association of defects in ABHD16A with HSP and neurodevelopmental phenotype and low levels of brain LPS identified in our current study. The pathogenic mechanisms of ABHD16A-associated HSP are yet to be fully revealed; however, given the interplay of the two lipases along the ABHD16A-ABHD12 axis, normalization of low LPS level can be implemented in treating ABHD16A-associated HSP using selective ABHD12 inhibitors. Although a number of ABHD12 inhibitors are known, they are mostly non-selective. However, a study identified DO264 as an in-vivo selective inhibitor of ABHD12, elevating LPS in a dose dependent manner in human macrophages and mouse brain (21). Another study constructed a model for some triterpenes-based ABHD12 reversible inhibitors (40). The model was based on results of provisional screening that identified selective inhibition of ABHD12 by ursolic acid with negligible non-selectivity (40). The provisional structure-activity relationships that the study established can pave the way for discovery of more selective ABHD12 inhibitors that may be target for treatment of ABHD16A-associated HSP.
In conclusion, we identified a novel form of complicated HSP caused by ABHD16A loss of function pathogenic variants and the subsequent altered PS metabolism. Our study emphasizes the role of lipid metabolism and the ABHD16A-ABHD12 axis in HSP; though, the data on the pathogenesis of ABHD16A-associated HSP is still limited and requires functional experiments to link PS/LPS levels to neurodegeneration. We propose that lipidomics could be used to validate the functional effect of ABHD16A missense variants in the future, often a bottleneck in genetic studies.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/clinvar/, SCV001653694; SCV001653696.
The studies involving human participants were reviewed and approved by The Ethical Committee of Medical Campus, University of Khartoum, Sudan and The Ethical Committee of National University, Sudan (approval number NU-RECG200). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
AY, LE, MIE, FL, and GS: conceptualization. AY, LE, RAb, MK, RI, IE, ABa, EA, AAb, MMa, AAh, FA, HM, RAd, SE, and MMu: data curation. AY, LE, and RV: formal analysis. ABr, MIE, AEA, and GS: funding acquisition. AY, LE, AH, IM, MAE, MS, TE, NA, RAb, MK, RI, IE, ABa, EA, AAb, MMa, AAh, FA, HM, RAd, SE, MMu, and FL: investigation. AY, LE, RV, TE, NA, FL, and GS: methodology. ABr, MIE, MI, AEA, and GS: project administration. ABr, AH, IM, MAE, MS, FL, and GS: resources. MIE, MI, AEA, and GS: supervision. AY, FL, and GS: validation. AY, LE, RV, TE, NA, FL, and GS: writing—original draft. AH, IM, MAE, MS, RAb, MK, RI, IE, ABa, EA, AAb, MMa, AAh, FA, HM, RAd, SE, MMu, ABr, MIE, MI, and AEA: writing—review and editing. All authors contributed to the article and approved the submitted version.
This study was financially supported by the European Union through the H2020 programme (SOLVE-RD, to GS). AY and LE were supported by the Ministry of Higher Education, Sudan; the University of Khartoum, Sudan, and the French Embassy, Khartoum, Sudan. MS was supported by Researchers Supporting Project (grant RSP-2020/38), King Saud University, Riyadh, Saudi Arabia.
GS declares to have received funding support from Biogen, unrelated to this work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors are grateful to the I-GenSeq ICM facility for exome processing, the Center for biological resources of ICM for cell culture, as well as Dr. Fanny Mochel for helpful advice.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.720201/full#supplementary-material
1. Fink JK. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. (2013) 126:307–28. doi: 10.1007/s00401-013-1115-8
2. Erfanian Omidvar M, Torkamandi S, Rezaei S, Alipoor B, Omrani MD, Darvish H, et al. Genotype–phenotype associations in hereditary spastic paraplegia: a systematic review and meta-analysis on 13,570 patients. J Neurol. (2019) 268:2065–82. doi: 10.1007/s00415-019-09633-1
3. Yahia A, Stevanin G. The history of gene hunting in hereditary spinocerebellar degeneration: lessons from the past and future perspectives. Front Genet. (2021) 12:638730. doi: 10.3389/fgene.2021.638730
4. Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology. (2014) 42:174–83. doi: 10.1159/000358801
5. Blackstone C. Converging cellular themes for the hereditary spastic paraplegias. Curr Opin Neurobiol. (2018) 51:139–46. doi: 10.1016/j.conb.2018.04.025
6. Boutry M, Morais S, Stevanin G. Update on the genetics of spastic paraplegias. Curr Neurol Neurosci Rep. (2019) 19:18. doi: 10.1007/s11910-019-0930-2
7. Darios F, Mochel F, Stevanin G. Lipids in the physiopathology of hereditary spastic paraplegias. Front Neurosci. (2020) 14:74. doi: 10.3389/fnins.2020.00074
8. O'Brien JS, Sampson EL. Lipid composition of the normal human brain: gray matter, white matter, and myelin. J Lipid Res. (1965) 6:537–44. doi: 10.1016/S0022-2275(20)39619-X
9. Krause MR, Regen SL. The structural role of cholesterol in cell membranes: from condensed bilayers to lipid rafts. Acc Chem Res. (2014) 47:3512–21. doi: 10.1021/ar500260t
10. Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol. (2017) 18:361–74. doi: 10.1038/nrm.2017.16
11. Grassi S, Giussani P, Mauri L, Prioni S, Sonnino S, Prinetti A. Lipid rafts and neurodegeneration: structural and functional roles in physiologic aging and neurodegenerative diseases. J Lipid Res. (2020) 61:636–54. doi: 10.1194/jlr.TR119000427
12. Ouweneel AB, Thomas MJ, Sorci-Thomas MG. The ins and outs of lipid rafts: functions in intracellular cholesterol homeostasis, microparticles, cell membranes. J Lipid Res. (2020) 61:676–86. doi: 10.1194/jlr.TR119000383
13. Lordan R, Tsoupras A, Zabetakis I. Phospholipids of animal and marine origin: structure, function, anti-inflammatory properties. Molecules. (2017) 22:1964. doi: 10.3390/molecules22111964
14. Colin LA, Jaillais Y. Phospholipids across scales: lipid patterns and plant development. Curr Opin Plant Biol. (2020) 53:1–9. doi: 10.1016/j.pbi.2019.08.007
15. Choi J, Yin T, Shinozaki K, Lampe JW, Stevens JF, Becker LB, et al. Comprehensive analysis of phospholipids in the brain, heart, kidney, and liver: brain phospholipids are least enriched with polyunsaturated fatty acids. Mol Cell Biochem. (2018) 442:187–201. doi: 10.1007/s11010-017-3203-x
16. Elsayed LEO, Mohammed IN, Hamed AAA, Elseed MA, Johnson A, Mairey M, et al. Hereditary spastic paraplegias: identification of a novel SPG57 variant affecting TFG oligomerization and description of HSP subtypes in Sudan. Eur J Hum Genet. (2016) 25:100–110. doi: 10.1038/ejhg.2016.108
17. Elsayed LEO, Mohammed IN, Hamed AAA, Elseed MA, Salih MAM, Yahia A, et al. Novel homozygous missense mutation in the arg1 gene in a large sudanese family. Front Neurol. (2020) 11:569996. doi: 10.3389/fneur.2020.569996
18. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. (1959) 37:911–7. doi: 10.1139/o59-099
19. Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. (2004) 11:377–94. doi: 10.1089/1066527041410418
20. Reese MG. Improved splice site detection in genie. J Comput Biol. (1997) 20:311–23. doi: 10.1145/267521.267766
21. Kamat SS, Camara K, Parsons WH, Chen DH, Dix MM, Bird TD, et al. Immunomodulatory lysophosphatidylserines are regulated by ABHD16A and ABHD12 interplay. Nat Chem Biol. (2015) 11:164–71. doi: 10.1038/nchembio.1721
22. Savinainen JR, Patel JZ, Parkkari T, Navia-Paldanius D, Marjamaa JJT, Laitinen T, et al. Biochemical and pharmacological characterization of the human lymphocyte antigen B-associated transcript 5 (BAT5/ABHD16A). PLoS ONE. (2014) 9:e109869. doi: 10.1371/journal.pone.0109869
23. Xu J, Gu W, Ji K, Xu Z, Zhu H, Zheng W. Sequence analysis and structure prediction of ABHD16A and the roles of the ABHD family members in human disease. Open Biol. (2018) 8:180017. doi: 10.1098/rsob.180017
24. Lefèvre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in chanarin-dorfman syndrome. Am J Hum Genet. (2001) 69:1002–12. doi: 10.1086/324121
25. Fiskerstrand T, H'Mida-Ben Brahim D, Johansson S, M'Zahem A, Haukanes BI, Drouot N, et al. Mutations in ABHD12 cause the neurodegenerative disease PHARC: an inborn error of endocannabinoid metabolism. Am J Hum Genet. (2010) 87:410–7. doi: 10.1016/j.ajhg.2010.08.002
26. Singh S, Joshi A, Kamat SS. Mapping the neuroanatomy of ABHD16A, ABHD12, and lysophosphatidylserines provides new insights into the pathophysiology of the human neurological disorder PHARC. Biochemistry. (2020) 59:2299–311. doi: 10.1021/acs.biochem.0c00349
27. Blankman JL, Long JZ, Trauger SA, Siuzdak G, Cravatt BF. ABHD12 controls brain lysophosphatidylserine pathways that are deregulated in a murine model of the neurodegenerative disease PHARC. Proc Natl Acad Sci USA. (2013) 110:1500–5. doi: 10.1073/pnas.1217121110
28. Kim HY, Huang BX, Spector AA. Phosphatidylserine in the brain: metabolism and function. Prog. Lipid Res. (2014) 56:1–18. doi: 10.1016/j.plipres.2014.06.002
29. Newton AC, Keranen LM. Phosphatidyl-l-serine is necessary for protein kinase C's high-affinity interaction with diacylglycerol-containing membranes. Biochemistry. (1994) 33:6651–8. doi: 10.1021/bi00187a035
30. Scott-Hewitt N, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, et al. Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J. (2020) 39:e105380. doi: 10.15252/embj.2020105380
31. Sousa SB, Jenkins D, Chanudet E, Tasseva G, Ishida M, Anderson G, et al. Gain-of-function mutations in the phosphatidylserine synthase 1 (PTDSS1) gene cause lenz-majewski syndrome. Nat Genet. (2014) 46:70–6. doi: 10.1038/ng.2829
32. Peter VG, Quinodoz M, Pinto-Basto J, Sousa SB, Di Gioia SA, Soares G, et al. The Liberfarb syndrome, a multisystem disorder affecting eye, ear, bone, brain development, is caused by a founder pathogenic variant in thePISD gene. Genet Med. (2019) 21:2734–43. doi: 10.1038/s41436-019-0595-x
33. Sugo T, Tachimoto H, Chikatsu T, Murakami Y, Kikukawa Y, Sato S, et al. Identification of a lysophosphatidylserine receptor on mast cells. Biochem Biophys Res Commun. (2006) 341:1078–87. doi: 10.1016/j.bbrc.2006.01.069
34. Inoue A, Ishiguro J, Kitamura H, Arima N, Okutani M, Shuto A, et al. TGFα shedding assay: an accurate and versatile method for detecting GPCR activation. Nat Methods. (2012) 9:1021–29. doi: 10.1038/nmeth.2172
35. Shinjo Y, Makide K, Satoh K, Fukami F, Inoue A, Kano K, et al. Lysophosphatidylserine suppresses IL-2 production in CD4 T cells through LPS3/GPR174. Biochem Biophys Res. Commun. (2017) 494:332–8. doi: 10.1016/j.bbrc.2017.10.028
36. Kim HJ, Sim MS, Lee DH, Kim C, Choi Y, Park HS, et al. Lysophosphatidylserine induces eosinophil extracellular trap formation and degranulation: implications in severe asthma. Allergy Eur J Allergy Clin Immunol. (2020) 75:3159–70. doi: 10.1111/all.14450
37. Yanagida K, Valentine WJ. Druggable lysophospholipid signaling pathways. Adv. Exp. Med. Biol. (2020) 1274:137–76. doi: 10.1007/978-3-030-50621-6_7
38. Lourenssen S, Blennerhassett MG. Lysophosphatidylserine potentiates nerve growth factor-induced differentiation of PC12 cells. Neurosci Lett. (1998) 248:77–80. doi: 10.1016/S0304-3940(98)00275-4
39. Chen DH, Naydenov A, Blankman JL, Mefford HC, Davis M, Sul Y, et al. Two novel mutations in ABHD12: expansion of the mutation spectrum in pharc and assessment of their functional effects. Hum Mutat. (2013) 34:1672–8. doi: 10.1002/humu.22437
Keywords: ABHD16A, hereditary spastic paraplegia, next-generation sequencing, targeted-metabolomics, lipid metabolism, phosphatidylserine
Citation: Yahia A, Elsayed LEO, Valter R, Hamed AAA, Mohammed IN, Elseed MA, Salih MA, Esteves T, Auger N, Abubaker R, Koko M, Abozar F, Malik H, Adil R, Emad S, Musallam MA, Idris R, Eltazi IZM, Babai A, Ahmed EAA, Abd Allah ASI, Mairey M, Ahmed AKMA, Elbashir MI, Brice A, Ibrahim ME, Ahmed AE, Lamari F and Stevanin G (2021) Pathogenic Variants in ABHD16A Cause a Novel Psychomotor Developmental Disorder With Spastic Paraplegia. Front. Neurol. 12:720201. doi: 10.3389/fneur.2021.720201
Received: 07 June 2021; Accepted: 28 July 2021;
Published: 20 August 2021.
Edited by:
Valeria Tiranti, Fondazione IRCCS Istituto Neurologio Carlo Besta, ItalyReviewed by:
Yair Anikster, Sheba Medical Center, IsraelCopyright © 2021 Yahia, Elsayed, Valter, Hamed, Mohammed, Elseed, Salih, Esteves, Auger, Abubaker, Koko, Abozar, Malik, Adil, Emad, Musallam, Idris, Eltazi, Babai, Ahmed, Abd Allah, Mairey, Ahmed, Elbashir, Brice, Ibrahim, Ahmed, Lamari and Stevanin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liena E. O. Elsayed, ZG9jdG9ybGJvQGhvdG1haWwuY29t; TGVlbHNheWVkQHBudS5lZHUuc2E=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.