
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 22 September 2021
Sec. Pediatric Neurology
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.718808
This article is part of the Research Topic Pediatric Neurology - Case Report Collection 2021 View all 7 articles
 Ivana Frongia1†
Ivana Frongia1† Susanna Rizzi1*†
Susanna Rizzi1*† Margherita Baga1†
Margherita Baga1† Laura Maria Ceteroni1
Laura Maria Ceteroni1 Carlotta Spagnoli1Grazia Gabriella Salerno1Daniele Frattini1
Carlotta Spagnoli1Grazia Gabriella Salerno1Daniele Frattini1 Milja Kaare2
Milja Kaare2 Francesco Pisani3
Francesco Pisani3 Carlo Fusco1
Carlo Fusco1Background: Charcot–Marie–Tooth (CMT) is the most frequent group of inherited neuropathies and includes several heterogeneous phenotypes. Over 80 causative genes have been described so far. Variants in the microrchidia family CW-type zinc finger 2 (MORC2) gene have been described in several axonal polyneuropathy (CMT2) patients with childhood or adult onset. Occasionally more complex phenotypes with delayed milestones, severe hypotonia, intellectual disability, dystonic postures, pyramidal signs, and neuroimaging abnormalities have been reported.
Case Presentation: We report on a patient with a de novo MORC2 gene variant (c.1181A>G p.Tyr394Cys) with a history of developmental delay, axial hypotonia, progressive gait disorder with dystonic features, and intentional tremor. At the age of 8 years, he showed bilateral pyramidal signs (clonus, increased tendon reflexes, and Babinski sign) and bilateral pes cavus. The first neuroimaging performed at the age of 3 years demonstrated white matter abnormalities in the posterior periventricular zone, in the frontal lobes bilaterally and at the midbrain, stable during childhood and adolescence. Nerve conduction studies (NCS) were negative until the age of 15 years, when a sensory axonal neuropathy appeared. The association between pyramidal signs and neuropathy due to the MORC2 gene variant is increasingly being highlighted, although a neuroradiological correlate is evident only in about half of the cases. Longitudinal nerve conduction velocity (NCV) are helpful to identify late-onset features and provide useful information for diagnosis in patients with rare neurogenetic disorders.
Conclusions: Characterization of complex neurological disorders is important to delineate the expanding phenotypic spectrum of MORC2-related disease, to confirm if possible the pathogenicity of the variants and to deepen the genotype–phenotype correlation.
CMT disease is the most frequent inherited neuropathy, with an estimated prevalence in Europe of 10–28:100,000 (1). Over 80 causative genes have been described so far. Variants of the gene microrchidia family CW-type zinc finger 2 (MORC2) have been described in several axonal CMT patients, typically characterized by childhood or early adulthood onset of progressive weakness and sensory impairment. Occasionally delayed milestones, intellectual disability, dystonic attitude, pyramidal signs and brain magnetic resonance imaging (MRI) abnormalities have been reported (2). Here we report a patient with a de novo MORC2 gene variant highlighting the neurophysiological and neuroradiological findings.
Our patient is a 15-year-old boy with motor development characterized by gross motor delay, independent sitting at 9 months, and independent walking at 19 months. Language development was also reported as delayed; however, a formal neuropsychological profile is unavailable. No facial dysmorphism was observed. Transient internal rotation on the left superior limb was first observed at the age of 2 years and then disappeared. Subsequently, a complex neurological disorder became evident, with mild axial hypotonia, gait disorder, and intentional tremor. At the age of 8 years, bilateral pyramidal signs (unstained plantar clonus, increased tendon reflexes, and Babinski signs) appeared. His last neurological examination at 15 years of age showed asymmetrical distal dystonic posturing, spasticity, and bilateral pes cavus. Other neurological signs presented a stable course. Owing to dystonic features, levodopa and carbidopa therapy was set up, but it was withdrawn due to ineffectiveness. He followed an individualized physiotherapy program.
First brain MRI (1 Tesla), at the age of 3 years, demonstrated areas of white matter hypomyelinations stable during childhood and adolescence (Figure 1).
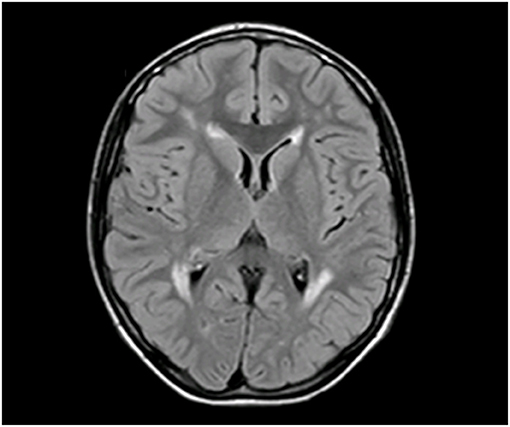
Figure 1. Brain MRI at 3 years, axial view: supratentorial white matter increased the T2 signal in the periventricular and frontal areas.
Nerve conduction studies (NCS) and electromyography (EMG) were performed at 4, 7, and 15 years. EMG was performed with a four-channel EMG machine (EBNeuro, Galileo NT PMS 2001, Mito II; Florence, Italy). EMG activity was amplified, and the filter band was set at the lower filter of 10 Hz to the upper filter of 10 kHz with a sweep duration of 100 ms. Sensitivity was assessed according to the potential amplitude varying between 100 and 500 mV. Standard techniques of electrophysiological studies were used for motor and sensory conduction velocities and amplitude measurements of the median, ulnar, peroneal, tibial, and sural nerves. Needle EMG of the bilateral anterior tibial, extensor indicis, and gastrocnemius muscles was also performed. The first two evaluations showed normal values. The last examination at the age of 15 years revealed axonal sensory polyneuropathy in all four limbs (Table 1). The patient has undergone serial diagnostic investigations, all unremarkable (Table 2). Considering the persistence of a complex neurological phenotype and assuming its genetic origin, whole-exome sequencing (WES) was performed including detection for both sequence variants (with mean coverage of 165× and 99.5% of the target nucleotides had a 0.20× read depth) and CNVs (sensitivity of 92% at one exon level) [Talevich E, Shain AH, Botton T, Bastian B. CCNV kit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. (2016) 12:e1004873] (https://blueprintgenetics.com/tests/whole-exomesequencing/whole-exome-family/). Informed consent was from parents obtained in accordance with international ethical standards. A heterozygous missense de novo variant c.1181A>G, p.(Tyr394Cys) in the MORC2 gene was identified. Mutation nomenclature is based on GenBank accession NM_001303256.2. This variant is absent in the large reference population cohorts of the Genome Aggregation Database (gnomAD, n > 120,000 exomes and >15,000 genomes). The variant is predicted as damaging by used in silico tools SIFT, PolyPhen, and MutationTaster. The variant was initially classified as a variant of uncertain significance (VUS), as there were not enough evidence to completely support its pathogenicity. Parental testing and recent publications identifying the same variant in other patients allowed reclassification of the variant to pathogenic. No other variants in known disease-causing genes were identified. Whole-exome data of the patient were also analyzed for secondary findings, according to recommendations of the American College of Medical Genetics and Genomics (ACMG; PMID 27854360); analysis was negative.
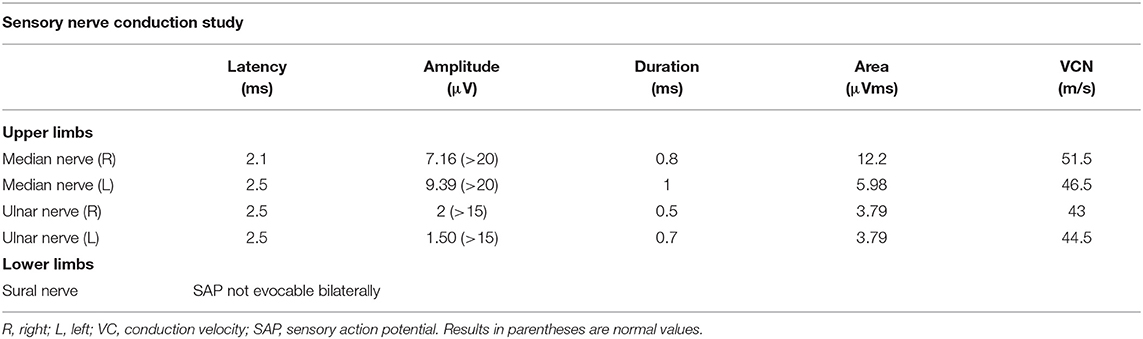
Table 1. Nerve conduction study performed at the age of 15 years.

Table 2. Diagnostic investigations performed and resulted negative.
The MORC2 gene (OMIM 616661) encodes a member of the 92 MORC protein superfamily, a nuclear protein characterized by an N-terminal ATPase domain, a central zinc-finger CW domain, and a divergent C-terminal region with one or more coiled coils (2). It is involved in biological functions, such as regulation of heterochromatin condensation in DNA damage, transcriptional repression, lipid metabolism, cytoskeleton functions, and axonal transport (3, 4). Several studies pointed out the role and expression of MORC2 in both central and peripheral nervous systems (2, 5–7).
Up to now, a broad phenotypic spectrum associated with MORC2-related disease has been delineated. Primarily autosomal dominant CMT disease axonal type 2Z (OMIM 616688) with possible central nervous system involvement was described. Subsequently, both early-onset patients with severe and progressive SMA-like features and late-onset cases (usually less severe, with distal muscle weakness, and sensory impairment) have been reported (2, 7, 8). Recently, more complex MORC2-related clinical and neuroradiological pictures such as Leigh-like syndrome have been described. Microcephaly, seizures, cognitive impairment, pigmentary retinopathy, hearing loss, and diaphragmatic paralysis can be sometimes associated (2, 7–9). We report a patient with progressive neurological phenotype characterized by both peripheral and central nervous system (PNS, CNS) involvement due to a de novo MORC2 gene variant with stable neuroradiological picture and evolutive neurophysiological findings.
Typically, MORC2 variants cause early-onset length-dependent sensory-dominant axonal neuropathy. Rarely, some patients show normal NCV: they are often pediatric or young adult subjects, but no further information is available about the follow-up (10, 11). Our patient performed serial NCS: NCV was negative until the age of 15 when sensory axonal neuropathy appeared. According to our experience, instrumental evidence of neuropathy may appear later than the onset of the clinical picture.
Recent studies reveal that the MORC2 gene is expressed in both human and murine neural tissues, (neurons, axons, and Schwann cells) (5). Its role in the development, maturity, and regulation in PNS and CNS has been demonstrated in animal models. MORC2 dysfunction led to axonal swellings and altered expression of different neurotransmitter receptors (5). Brain MRI is often normal, but in recent years reports of brain abnormalities MORC2-related have been increasing such as cerebellar atrophy and white matter abnormalities (2, 7, 10–13). Finally, only a single case with cortical dysplasia has been described (10) and rarely Leigh-like neuroradiological features with bilateral symmetric abnormalities in the basal ganglia, brainstem, and/or cerebellum were detected (10). Of note, longitudinal neuroradiological studies are still limited but in our experience, white matter changes remain stable at subsequent controls performed during adolescence.
The c.1181A>G, p.(Tyr394Cys) variant in MORC2 has been identified in literature in other three patients with axonal sensory polyneuropathy and white matter abnormalities. Including our patient, pyramidal signs are present in 50% of individuals (Table 3).
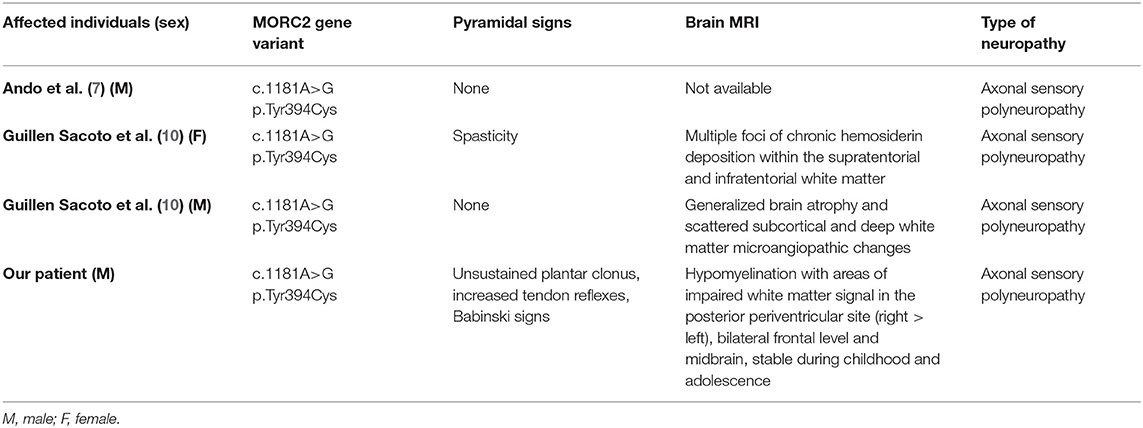
Table 3. Clinical, neuroradiological, and neurophysiological features of individuals with the p.(Tyr394Cys) variant on the MORC2 gene.
The p.(Tyr394Cys) variant has also been identified in four individuals reported in ClinVar (ClinVar ID 432089), where its interpretation of pathogenicity is conflicting. However, based on the patients reported until now, the de novo occurrence of the variant, the absence of the variant in control population databases, and the damaging prediction of in silico tools used, we believe that the p.(Tyr394Cys) variant is to be considered as pathogenic. HGMD Professional (version 2021.1) lists altogether 25 variants in MORC2 in association with MORC2-related phenotypes. All of the reported variants are missense variants. The reported variants are located along the entire gene, and at the moment there seems to be no clear correlation between variant location and clinical phenotype caused by the variant. Guillen Sacoto et al. (10) recently highlighted a correlation between the degree of HUSH hyperactivation in cellular assays and the severity of central nervous system involvement. The variability of the genotype–phenotype correlation probably reflects the limitations of our understanding of the relationship between the biologic functions of the affected protein, the postulated modulator genes, and environmental factors.
In conclusion, the emerging phenotype of MORC2-related pathologies is not limited to the symptoms of polyneuropathy but extended through the involvement of the CNS. The association with pyramidal signs concomitant with neuropathy is increasingly being highlighted (14, 15), although a neuroradiological correlate is evident only in about half of the cases. Currently, pyramidal signs are reported in a minority of MORC2-patients; collecting a larger case study could define which variants are most associated with this feature (2). Longitudinal NCV is helpful in identifying late-onset features and providing useful information for the diagnosis and long-term follow-up of patients with rare progressive neurogenic disorders and previously normal NCV. Characterization of complex cases is important in delineating the expanding phenotype spectrum of MORC2-related clinical pictures and, if possible, in confirming the pathogenicity of the variants found and better deepening the genotype–phenotype correlation.
The datasets presented in this study can be found in online repositories. The names of the repository and accession number can be found at: https://www.ncbi.nlm.nih.gov/genbank/ (NM_001303256.2).
IF, MB, LMC, and FP contributed to manuscript development. SR, CS, GGS, DF, and CF contributed to manuscript development, patient management, and diagnostic definition. MK contributed to diagnostic definition. All authors contributed to the article and approved the submitted version.
MK was employed by company Blueprint Genetics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pareyson D, Saveri P, Pisciotta C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr Opin Neurol. (2017) 30:471–80. doi: 10.1097/WCO.0000000000000474
2. Albulym OM, Kennerson ML, Harms MB, Drew AP, Siddell AH, Auer-Grumbach M, et al. MORC2 mutations cause axonal Charcot-Marie-Tooth disease with pyramidal signs. Ann Neurol. (2016) 79:419–27. doi: 10.1002/ana.24575
3. Douse CH, Bloor S, Liu Y, Shamin M, Tchasovnikarova IA, Timms RT, et al. Neuropathic MORC2 mutations perturb GHKL ATPase dimerization dynamics and epigenetic silencing by multiple structural mechanisms. Nat Commun. (2018) 9:651. doi: 10.1038/s41467-018-03045-x
4. Tchasovnikarova IA, Timms RT, Douse CH, Roberts RC, Dougan G, Kingston RE, et al. Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2. Nat Genet. (2017) 49:1035–44. doi: 10.1038/ng.3878
5. Sancho P, Bartesaghi L, Miossec O, García-García F, Ramírez-Jiménez L, Siddell A, et al. Characterization of molecular mechanisms underlying the axonal Charcot-Marie-Tooth neuropathy caused by MORC2 mutations. Hum Mol Genet. (2019) 28:1629–44. doi: 10.1093/hmg/ddz006
6. Shao Y, Li Y, Zhang J, Liu D, Liu F, Zhao Y, et al. Involvement of histone deacetylation in MORC2-mediated down-regulation of carbonic anhydrase IX. Nucleic Acids Res. (2010) 38:2813–24 doi: 10.1093/nar/gkq006
7. Ando M, Okamoto Y, Yoshimura A, Yuan JH, Hiramatsu Y, Higuchi Y, et al. Clinical and mutational spectrum of Charcot-Marie-Tooth disease type 2Z caused by MORC2 variants in Japan. Eur J Neurol. (2017) 24:1274–82. doi: 10.1111/ene.13360
8. Sevilla T, Lupo V, Martínez-Rubio D, Sancho P, Sivera R, Chumillas MJ, et al. Mutations in the MORC2 gene cause axonal Charcot-Marie-Tooth disease. Brain. (2016) 139:62–72. doi: 10.1093/brain/awv311
9. Hyun YS, Hong YB, Choi BO, Chung KW. Clinico-genetics in Korean Charcot-Marie-Tooth disease type 2Z with MORC2 mutations. Brain. (2016) 139:e40. doi: 10.1093/brain/aww082
10. Guillen Sacoto MJ, Tchasovnikarova IA, Torti E, Forster C, Andrew EH, Anselm I, et al. De novo variants in the ATPase module of MORC2 cause a neurodevelopmental disorder with growth retardation and variable craniofacial dysmorphism. Am J Hum Genet. (2020) 107:352–63. doi: 10.1016/j.ajhg.2020.06.013
11. Schottmann G, Wagner C, Seifert F, Stenzel W, Schuelke M. MORC2 mutation causes severe spinal muscular atrophy-phenotype, cerebellar atrophy, and diaphragmatic paralysis. Brain. (2016) 139(Pt 12):e70. doi: 10.1093/brain/aww252
12. Zanni G, Nardella M, Barresi S, Bellacchio E, Niceta M, Ciolfi A, et al. De novo p.T362R mutation in MORC2 causes early onset cerebellar ataxia, axonal polyneuropathy and nocturnal hypoventilation. Brain. (2017) 140:e34. doi: 10.1093/brain/awx083
13. Stettner G, Knirsch U, Berger W, Graf U, Hendricks B, Seidl R, et al. Infantile-onset CMT2Z is caused by two MORC2 gene mutations and is associated with a distinct phenotype. Neuromusc Disor. (2019) 29:S200–S1. doi: 10.1016/j.nmd.2019.06.571
14. Fusco C, Frattini D, Pisani F, Spaggiari F, Ferlini A, Della Giustina E. Coexistent central and peripheral nervous system involvement in a Charcot-Marie-Tooth syndrome X-linked patient. J Child Neurol. (2010) 25:759–63. doi: 10.1177/0883073809344119
Keywords: Charcot–Marie–Tooth, axonal neuropathy, pyramidal signs, white matter abnormalities, MORC2 gene
Citation: Frongia I, Rizzi S, Baga M, Ceteroni LM, Spagnoli C, Salerno GG, Frattini D, Kaare M, Pisani F and Fusco C (2021) Infantile-Onset Charcot–Marie–Tooth Disease With Pyramidal Features and White Matter Abnormalities Due to a De novo MORC2 Gene Variant: A Case Report and Brief Review of the Literature. Front. Neurol. 12:718808. doi: 10.3389/fneur.2021.718808
Received: 01 June 2021; Accepted: 19 August 2021;
Published: 22 September 2021.
Edited by:
Brahim Tabarki Melaiki, University of Sousse, TunisiaReviewed by:
Anna Rubegni, Fondazione Stella Maris (IRCCS), ItalyCopyright © 2021 Frongia, Rizzi, Baga, Ceteroni, Spagnoli, Salerno, Frattini, Kaare, Pisani and Fusco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susanna Rizzi, c3VzYW5uYS5yaXp6aUBhdXNsLnJlLml0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.