 Jinfu Lin
Jinfu Lin Huan Li
Huan Li Ziyu Liao
Ziyu Liao Liang Wang
Liang Wang Cheng Zhang1,2*
Cheng Zhang1,2*- 1Department of Neurology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, China
- 2Guangdong Provincial Key Laboratory of Diagnosis and Treatment of Major Neurological Diseases, National Key Clinical Department and Key Discipline of Neurology, Guangzhou, China
Background: Duchenne and Becker muscular dystrophy (DMD/BMD) are X-linked recessively inherited neuromuscular disorders caused by deletions, duplications, or small mutations in the DMD gene. With advances in prenatal diagnosis decreasing the number of affected offspring from carrier mothers, the frequency of de novo variants could increase. Therefore, determining the differences between the carrier and de novo variants of the DMD gene, which are rarely explored, is important for trial planning and genetic diagnosis in the future.
Methods: A total of 440 patients, 349 of whom had DMD and 91 had BMD, diagnosed in our department between 2012 and 2019, along with their respective mothers, were included in this study. Multiplex ligation-dependent probe amplification was used to detected deletions and duplications in patients and their mothers. Small mutations were detected using next-generation sequencing in the patients, followed by Sanger sequencing in the mothers.
Results: Deletions, duplications, and small mutations were identified in 204, 46, and 99 of the 349 patients with DMD and in 50, 10, and 31 of the 91 patients with BMD, respectively. De novo deletions were more concentrated in hotspot regions than carrier deletions of DMD/BMD. No clear bias was observed in the variant distribution between carriers, de novo duplications, and small mutations in DMD/BMD. The carrier frequency of DMD (61.6%) was lower than that of BMD (69.2%), but the difference was not statistically significant. The carrier frequency of deletions of the DMD gene (51.2%) was significantly lower than those of duplications (75%) and small mutations (81.5%).
Conclusion: Compared to de novo deletions, deletions from carrier mothers had a wider distribution. Moreover, there was no significant difference between the carrier frequencies of DMD and BMD. Duplications and small mutations were more commonly inherited, while deletions were present de novo.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked recessively inherited fatal muscle disease, with an incidence of 15.9–21.9 for every 100,000 live newborn males (1–3). It is characterized by progressive weakness and muscle atrophy, accompanied by pseudohypertrophy of the gastrocnemius and a positive Gowers sign. Patients usually die of cardiorespiratory failure in the second or third decade of life. Pathogenic variants, such as deletions, duplications, and small mutations, in the DMD gene encoding dystrophin account for both DMD and Becker muscular dystrophy (BMD), a milder form of the disease with later onset and slower progression.
While DMD and BMD are usually diagnosed in men and rarely in women, many female carriers are asymptomatic but have a 50% risk of giving birth to male offspring with the disease. Previous studies have noted a different distribution of mutation types between de novo and carrier pathogenic variants (4, 5). With recent advances in molecular diagnostics that allow prenatal diagnosis, helping to identify female carriers before their first births, carrier mothers can be prevented from giving birth to offspring with DMD/BMD, consequently affecting the distribution of DMD/BMD pathogenic variants. Because of this, specific gene therapies, such as exon skipping therapy, could also be affected and might need to be changed. Therefore, more evidence on the difference between carrier and de novo pathogenic variants is needed. Hence, this study aimed to analyze the DMD gene variants in 440 patients with DMD/BMD, along with their respective mothers, and explore the difference.
Materials and Methods
Study Design and Participants
From January 2012 to June 2019, 440 Chinese male patients from independent families were diagnosed with DMD or BMD in our department based on clinical characteristics, serum creatine phosphokinase detection, and molecular genetic analysis. The diagnosis was based on disease severity, such as the age at which ambulation was lost (DMD <12 years old, BMD ≥12 years old). For patients with ambulation, those with obvious muscle weakness before 5 years of age were classified as having DMD, while those with a later onset, very mild motor dysfunction, and considerably longer survival were classified as having BMD. All clinical diagnoses were confirmed using molecular genetic analysis. The protocol of this study was approved by The Ethics Committee of the First Affiliated Hospital, Sun Yat-sen University.
Molecular Genetic Analysis
All patients and their mothers provided informed consent before molecular genetic analysis. Peripheral blood was drawn from the patients and their mothers, and DNA was extracted using a standard procedure. Multiplex ligation-dependent probe amplification (MLPA) was used to detect deletions and duplications in patients and their mothers (6). Patients with negative MLPA results were examined for small mutations using next-generation sequencing as previously described, and Sanger sequencing was then used to detect whether the mothers carried the small mutation of the DMD gene (6, 7).
Statistical Analysis
In the analysis of deletions, the starting and termination exons of a deletion were considered as the deleted ends. For example, for exon 45–52 deletion, the deleted ends were exons 45 and 52. The Chi-square test and Fisher's exact test were used for statistical analyses, and differences were considered statistically significant at P < 0.05.
Results
Distribution of Pathogenic Variants Among Patients With DMD and BMD
Among 349 patients with DMD and 91 with BMD, deletion was the most common variant type, with 204 and 50 deletions for DMD and BMD, respectively, followed by small mutations, with 99 and 31 mutations, respectively. Duplication was the least frequent type, with 46 and 10 duplications for DMD and BMD, respectively. The constitution of the DMD variants is similar to that of the BMD variants (Figure 1).
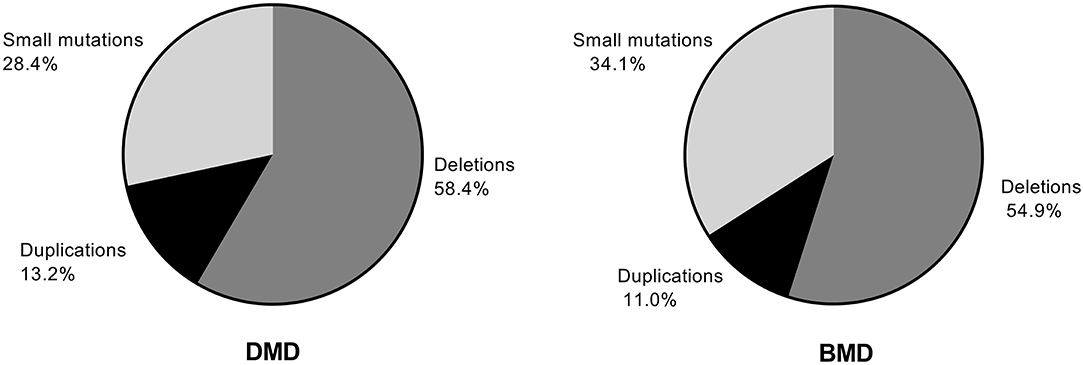
Figure 1. Distribution of deletions, duplications, and small mutations in patients with Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD).
Difference Between the Carrier and de novo Variants in Patients With DMD
We investigated the relationship between variant sites and the carrier status of the mothers of patients with DMD. Of the 204 deletions in the DMD gene observed in patients with DMD, 100 were carrier variants, and 104 were de novo variants. Deleted ends were mainly located at exons 3, 8, 12, 17, and 44–55, which account for 76 and 90.4% of the total deleted ends, respectively. However, the deleted ends of patients with carrier variants had a wider distribution than those with de novo variants (Figure 2A).
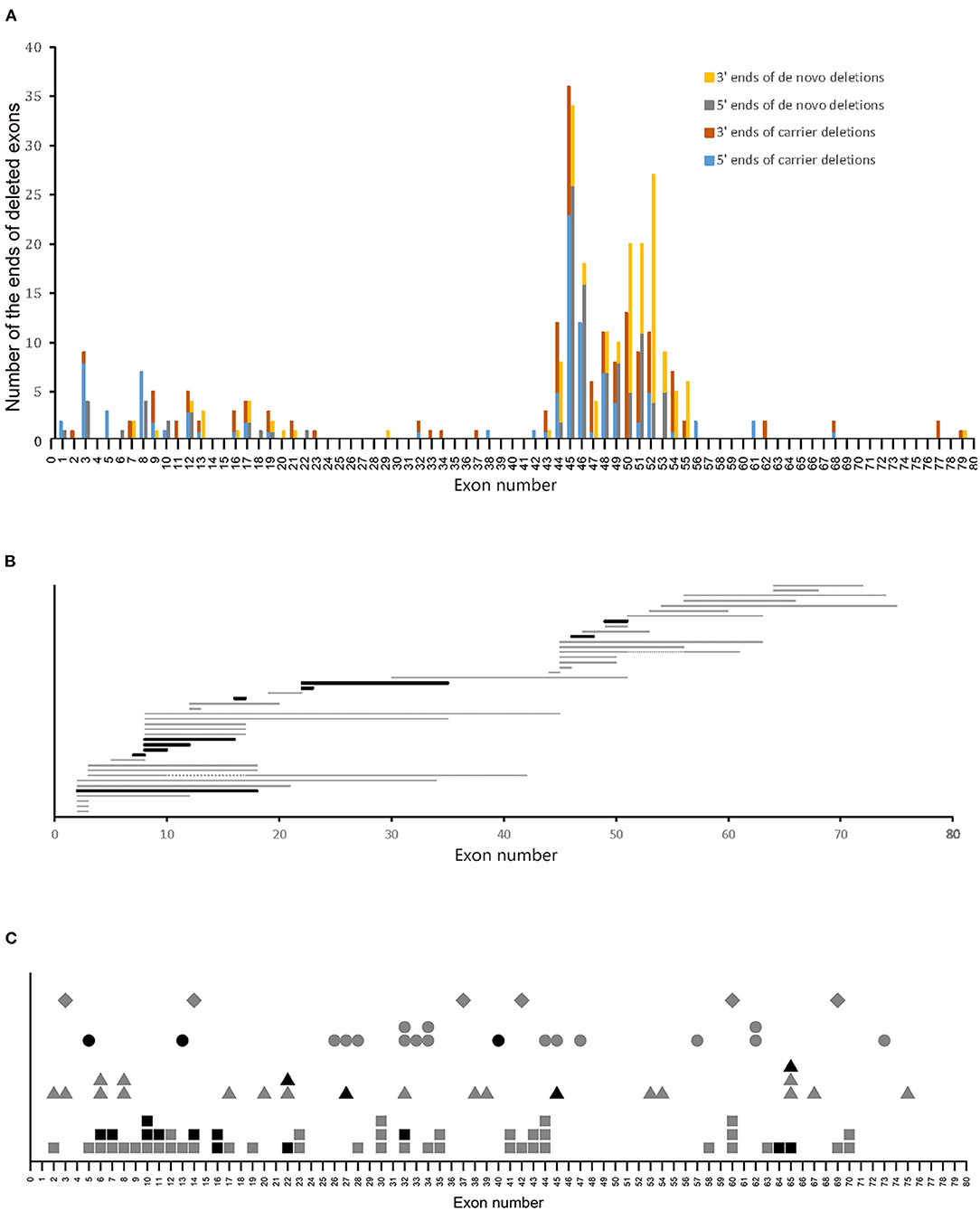
Figure 2. Variant sites in patients with DMD. (A) Distribution of ends of deletions in patients with DMD. (B) Distribution of duplications in patients with BMD. Horizontal bars represent the duplicated regions. Gray and black bars represent carrier and de novo variants, respectively. Two patients with carrier duplications had two duplicated regions each, and the two duplicated regions were connected with gray dotted lines. (C) Distribution of small mutations. Gray and black symbols represent carrier and de novo variants, respectively. Squares, triangles, circles, and rhombus represent non-sense mutations, splice site mutations, small deletions/insertions, and missense mutations, respectively.
A previous study revealed that most of the deletions in the DMD gene are located in exons 2–20 and 45–55, which were referred to as the proximal hotspot region and the distal hotspot region, respectively (8). In this study, 73% of the deletions in patients with carrier variants were located at the hotspot regions, and this was significantly lower than that in patients with de novo variants, of which 90.38% were in the hotspot regions (P < 0.05, using Chi-square test) (Table 1). Further analysis of the deletions in the hotspot regions revealed that compared to de novo deletions, carrier deletions tended to be more concentrated within the distal hotspot region. However, this difference was not statistically significant (P > 0.05, using the Chi-square test).

Table 1. Number of patients with DMD who had deletions located in the proximal, distal hotspot region and non-hotspot region.
Regarding duplications, no clear difference in distribution was found between patients with carrier and de novo variants (Figure 2B). Notably, we found 99 sites of small mutations scattered across the DMD gene. However, no clear discrepancy in the distribution of small mutation sites was observed with respect to the carrier status of the mothers (Figure 2C).
Difference Between the Carrier and de novo Variants in Patients With BMD
Of the 50 deletions in the DMD gene observed in patients with BMD, 31 were carrier variants, while 19 were de novo variants. The most frequent ends of deletions were exons 45, 47, 48, and 49, accounting for 48.4 and 76.3% of the total deleted ends in the carrier and de novo variants, respectively. Carrier deletions had a wider distribution than de novo deletions (Figure 3A). Moreover, 74.2% (23/31) of carrier deletions and 100% (19/19) of de novo deletions were in the hotspot regions (Table 2), and this difference was statistically significant (P < 0.05, using Chi-square test). Further analysis of these deletions in the hotspot regions revealed that compared to carrier deletions (78.3%), de novo deletions tended to be more concentrated in the distal hotspot region (94.7%). However, this difference was not statistically significant (P > 0.05, using Fisher's exact test).
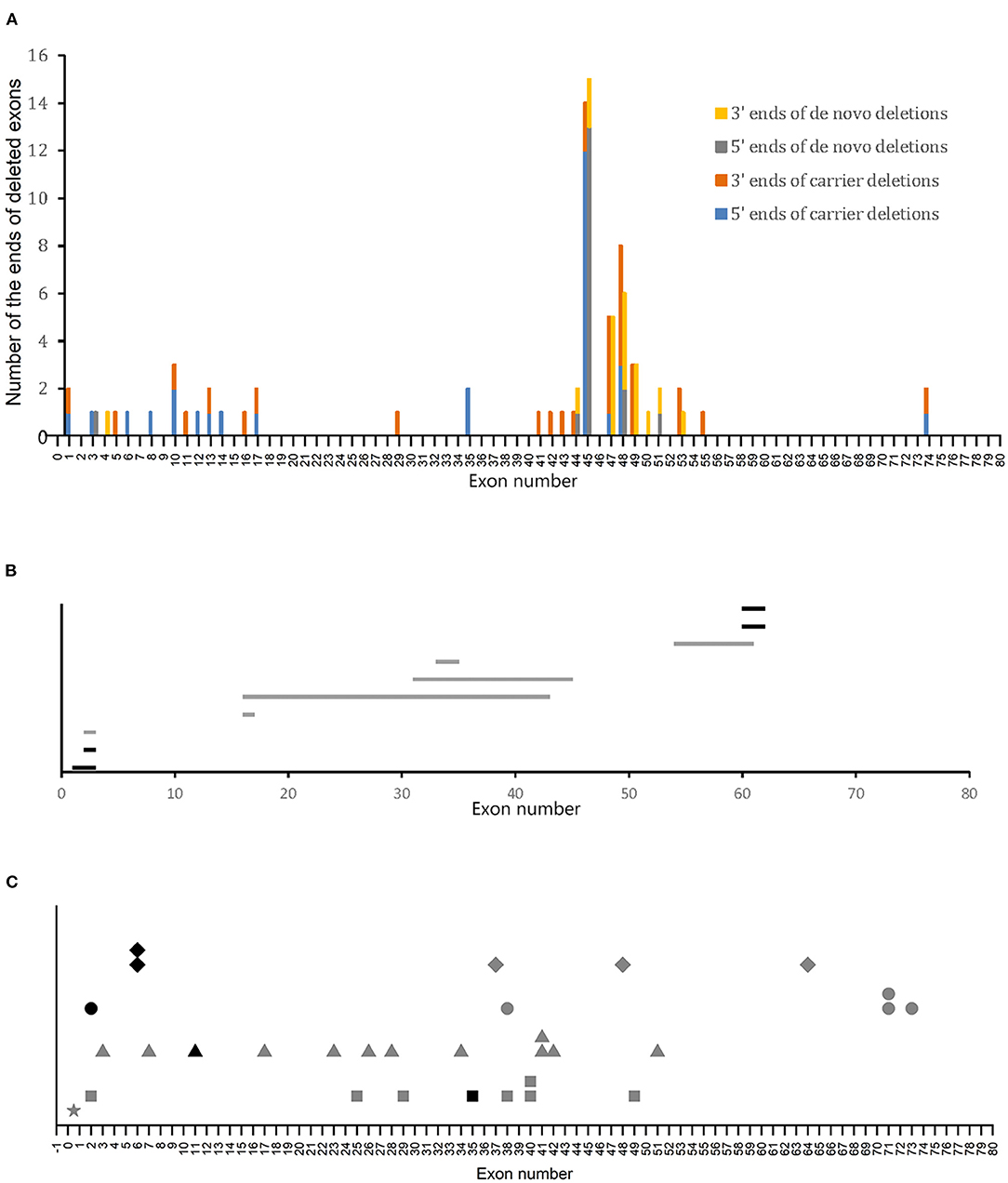
Figure 3. Variant sites in patients with BMD. (A) Distribution of deletion ends in patients with BMD. (B) Distribution of duplications in patients with BMD. Horizontal bars represent the duplicated regions. Gray and black bars represent carrier and de novo variants, respectively. (C) Distribution of small mutations. Gray and black symbols represent carrier and de novo variants, respectively. Squares, triangles, circles, and rhombus represent non-sense mutations, splice site mutations, small deletions/insertions, and missense mutations, respectively. The star represents a point mutation (c.-54T>A) in the 5′-untranslated region (UTR).

Table 2. Number of patients with BMD who had deletions located in the proximal, distal hotspot region and non-hotspot region.
We next investigated the sites of duplications and small mutations and their relationship with the carrier status of BMD. Six of the 10 duplications and 26 of the 31 small mutations were carrier variants, which included a point mutation (c.-54T>A) in the 5′-untranslated region. No clear bias of variant sites of either duplications or small mutations was indicated between the carrier and de novo variants (Figures 3B,C).
Difference in Carrier Frequencies Between Mothers of Patients With DMD and BMD
Regarding carrier frequencies, a higher frequency of DMD mutation was observed in the mothers of patients with BMD, as only 215 of the 349 (61.6%) mothers of patients with DMD were causative mutation carriers, while 63 (69.2%) of the 91 mothers of patients with BMD carried the mutations. However, this difference between the carrier frequencies of DMD and BMD was not statistically significant (Figure 4).

Figure 4. Comparison of the carrier frequency of DMD and BMD. No significant difference between the carrier frequencies of DMD and BMD was observed using the Chi-square test (P > 0.05).
Carrier Frequencies in the Mothers of Patients With DMD/BMD According to Variant Types
The carrier frequencies of deletions, duplications, and small mutations in mothers of patients with DMD/BMD were 51.2% (130/254), 75% (42/56), and 81.5% (106/130), respectively. The carrier frequency of deletions was significantly lower than those of duplications and small mutations (Figure 5).
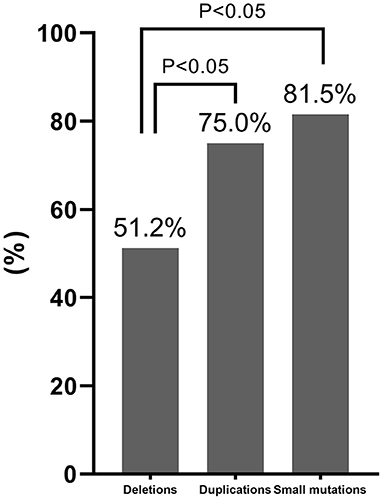
Figure 5. Comparison of the carrier frequency of different variant types. The carrier frequency of deletions was significantly lower than those of duplications and small mutations based on Chi-square test results (P < 0.05).
Discussion
In the present study, deletions were the most frequent mutations detected in both DMD and BMD, with small mutations being the second and duplications being the least frequent. The proportions of the three types of mutations in DMD and BMD were in accordance with previous reports (8, 9). In addition, the frequency of de novo mutations was 38.4 and 30.8% for DMD and BMD, respectively, with a 36.8% total frequency, which is consistent with previously reported frequencies ranging from 24 to 39.5% (6, 8, 10).
The present study revealed that compared to carrier variants, de novo variants were more concentrated in hotspot regions in both patients with DMD and BMD who had deletions. Furthermore, deletions from carrier mothers were scattered across the DMD gene. Previous reports by Ma et al. (10) and Lee et al. (11) revealed no significant difference in the distribution of deletion mutations from carrier and non-carrier mothers, perhaps owing to conclusions drawn from diagrams rather than quantitative and statistical analyses. Our study also indicated no significant difference in carrier frequency between deletion mutations in the proximal (58.6%, 17/29) and distal hotspot regions (40.6%, 56/138) in patients with DMD (Table 2), which is consistent with the report of Lee et al. (11). However, a trend of the carrier frequency of deletion mutations in the proximal being higher than that in distal hotspot regions was observed. Further research with a larger database is needed.
The theoretical frequency of DMD patients inheriting the mutations from carrier mothers is 2/3, as most patients with DMD do not survive long enough to produce offspring (12). In our study, the carrier frequency of DMD was 61.6%, which is slightly lower than the theoretical frequency. This may have resulted from including patients from independent families in the present study and the overall decrease in numbers of patients from carrier mothers due to advances in prenatal diagnosis. Meanwhile, the carrier frequency of BMD was 69.2% in the present study. As patients with BMD have milder symptoms, they can usually raise offspring; hence, mutations are more likely to pass on from a male patient to his daughters and on to his grandsons. Therefore, theoretically, the carrier frequency of BMD is higher than that of DMD. Nonetheless, in our study, the difference in carrier frequency between DMD and BMD was not statistically significant. This result is consistent with the report of Zimowski et al. (5) in a Polish population, in which the carrier frequency was 61.9% for DMD and 68.1% for BMD. A report by Toksoy et al. (13) in a Turkish population also revealed no statistically significant difference between the carrier frequency of DMD and BMD, with 45.5% for DMD and 42.9% for BMD. In contrast, Lee et al. (11) reported a significant difference in the carrier frequencies of DMD (57.6%) and BMD (89.5%) in a Japanese population, probably due to the small number of tested mothers (Table 3). In addition, the non-significant difference observed in our study may have been due to the recent advances in genetic diagnosis. BMD can be diagnosed more easily than ever before. While the clinical phenotype of BMD is highly variable and the onset of its symptoms varies from childhood to adulthood (14, 15), nearly 90% of patients with BMD will show the first symptoms by the age of 20 and are diagnosed by the age of 35 (16). Therefore, most patients with BMD will have been diagnosed before their daughters reach childbearing age. Moreover, with the help of prenatal diagnosis, the pathogenic variants will likely not be passed on to the grandsons.
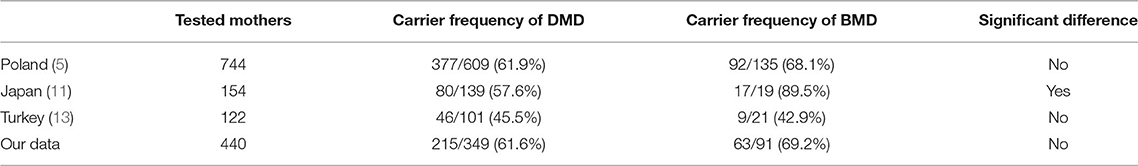
Table 3. Comparison of carrier frequencies of DMD and BMD in different countries.
In the present study, the carrier frequency of deletions was significantly lower than those of duplications and small mutations in mothers of patients with DMD/BMD, consistent with the results of previous studies in Polish, Chinese, and Japanese populations, which also revealed a lower carrier frequency of deletions than those of duplications and small mutations (5, 10, 11, 17, 18). However, the difference in the reports by Lee et al. and Zhang et al. (11, 18) showed no statistical significance, probably due to the relatively small sample sizes. Recent studies from India and Turkey have also revealed that the carrier frequency of small mutations is significantly higher than that of deletions (4, 13) (Table 4). Duplications and small mutations in the DMD gene are more common during spermatogenesis, while deletions appear more commonly during oogenesis (19–21); hence, duplications and small mutations from spermatogenesis are inherited by daughters and then passed on to the grandsons and appear to be carrier mutations. However, deletions from oogenesis are either directly passed on to sons or are indirectly passed on to grandsons through carrier daughters and appear to be de novo or carrier mutations. This may account for the difference in the carrier frequencies of the three variant types.
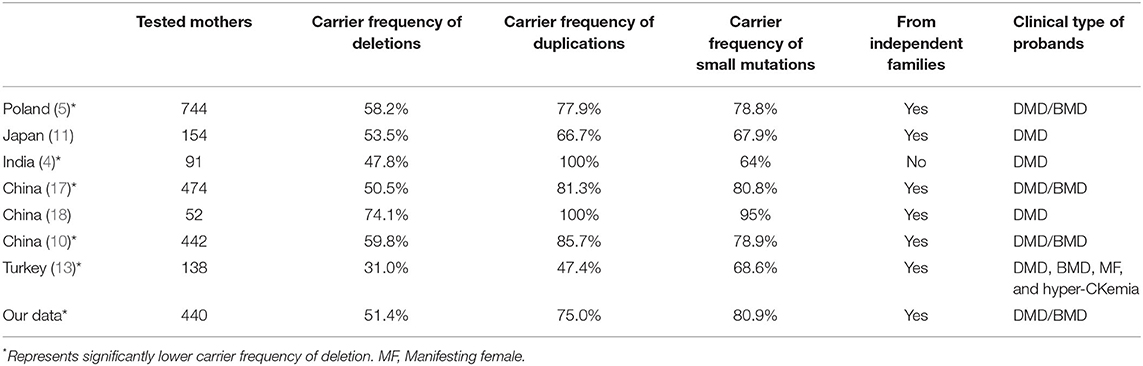
Table 4. Carrier frequency of DMD/BMD based on different variant types in different countries.
Notably, deletion ends were distributed more widely in the DMD gene carrier mutations than in de novo mutations. Additionally, 79.1% (170/215) and 71.4% (45/63) of carrier mothers had no significant family history in the DMD and BMD carrier groups, respectively. We discovered a difference in DMD gene mutations between the carriers with family history and the carriers without family history but found it statistically insignificant (Supplementary Figures 1, 2; Supplementary Table 1). As de novo mutations are derived from the oogenesis of mothers and carrier mutations are derived from spermatogenesis or oogenesis of maternal ancestors, there may be some differences between the distribution of deletions derived from spermatogenesis and oogenesis. However, further research is needed to explore the differences and their origins.
Distinguishing carrier status and prenatal diagnosis are of great importance to reduce DMD/BMD patient numbers. Mothers in our study were unaware of their carrier status until they and their affected children came to our department and went through genetic detection. The cases of three mothers with negative DMD mutation detection results of peripheral blood DNA were suspected as mosaicism because they gave birth to or were pregnant with more than one affected child or fetus with the same DMD mutation. However, the suspected mosaicism was not confirmed at the molecular level. There were 40 families in our study with multiple affected siblings in which the carrier mothers were unaware of their carrier status and gave birth to another one or more affected children without a prenatal diagnosis. Of the 31 follow-up prenatal diagnoses in our study, eight affected male fetuses were distinguished and aborted. A previous study in the Netherlands revealed through prenatal diagnosis, the percentage of the first affected boys increased from 62% in 1961–1974 to 88% in 1993–2002 (22). To prevent carrier mothers from giving birth to more affected children, it is essential to make an early diagnosis of DMD/BMD probands, for instance, by introducing DMD newborn screening (1, 23). Most of the DMD and BMD carrier mothers did not have a family history in our study, and it was difficult to establish their carrier status before their first birth. Therefore, it is still difficult to prevent carrier mothers without a family history from giving birth to a first affected child unless newborn screening of DMD/BMD carriers is introduced in the future.
The limitations of our study were that it was a single-center study revealing only the experience of our hospital. Moreover, the sample size is relatively small, and further researches with larger databases are needed.
In conclusion, our study revealed a lower carrier frequency of deletions compared to duplications and small mutations of the DMD gene. Moreover, compared to carrier variants, de novo variants were more concentrated within the hotspot regions in DMD/BMD patients who had deletions. The results highlight the variability of the mutation spectrum of the DMD gene attributable to a decreasing number of affected offspring from carrier mothers. Up to date research focusing on pathogenic variants spectrum of DMD/BMD patients is of vital importance for research in specific genetic therapy.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by The Ethics Committee of the First Affiliated Hospital, Sun Yat-sen University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
JL designed the study, analyzed the data, and drafted the manuscript. HL, ZL, and LW assisted in data acquisition. CZ assisted in data analysis and in revising the manuscript critically for important intellectual content. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the National Natural Science Foundation of China (81,771,359, 81,471,280, and 81,271,401), Science and Technology Planning Project of Guangzhou (1,561,000,153/201,508,020,012), Southern China International Cooperation Base for Early Intervention and Functional Rehabilitation of Neurological Diseases (2015B050501003), Guangdong Provincial Engineering Centre for Major Neurological Disease Treatment, Guangdong Provincial Translational Medicine Innovation Platform for Diagnosis and Treatment of Major Neurological Disease, and Guangdong Provincial Clinical Research Centre for Neurological Diseases.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We appreciated the participation of the patients and their mothers in this study. Meanwhile, we would like to thank Editage (www.editage.cn) for English language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.714677/full#supplementary-material
Abbreviations
BMD, Becker muscular dystrophy; DMD, Duchenne muscular dystrophy; MLPA, Multiplex ligation-dependent probe amplification.
References
1. Moat SJ, Bradley DM, Salmon R, Clarke A, Hartley L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). Eur J Hum Genet. (2013) 21:1049–53. doi: 10.1038/ejhg.2012.301
2. Mendell JR, Shilling C, Leslie ND, Flanigan KM, Al-Dahhak R, Gastier-Foster J, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. (2012) 71:304–13. doi: 10.1002/ana.23528
3. Ryder S, Leadley RM, Armstrong N, Westwood M, de Kock S, Butt T, et al. The burden, zepidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis. (2017) 12:79. doi: 10.1186/s13023-017-0631-3
4. Nagabushana D, Polavarapu K, Bardhan M, Arunachal G, Gunasekaran S, Preethish-Kumar V, et al. Comparison of the carrier frequency of pathogenic variants of DMD gene in an Indian Cohort. J Neuromuscul Dis. (2021). doi: 10.3233/JND-210658. [Epub ahead of print].
5. Zimowski JG, Pawelec M, Purzycka JK, Szirkowiec W, Zaremba J. Deletions, not duplications or small mutations, are the predominante new mutations in the dystrophin gene. J Hum Genet. (2017) 62:885–8. doi: 10.1038/jhg.2017.70
6. Chen WJ, Lin QF, Zhang QJ, He J, Liu XY, Lin MT, et al. Molecular analysis of the dystrophin gene in 407 Chinese patients with Duchenne/Becker muscular dystrophy by the combination of multiplex ligation-dependent probe amplification and Sanger sequencing. Clin Chim Acta. (2013) 423:35–8. doi: 10.1016/j.cca.2013.04.006
7. Wang L, Chen M, He R, Sun Y, Yang J, Xiao L, et al. Serum creatinine distinguishes Duchenne muscular dystrophy from Becker muscular dystrophy in patients aged ≤ 3 years: a retrospective study. Front Neurol. (2017) 8:196. doi: 10.3389/fneur.2017.00196
8. Tuffery-Giraud S, Béroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L, et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat. (2009) 30:934–45. doi: 10.1002/humu.20976
9. Tong YR, Geng C, Guan YZ, Zhao YH, Ren HT, Yao FX, et al. A comprehensive analysis of 2013 dystrophinopathies in china: a report from National Rare Disease Center. Front Neurol. (2020) 11:572006. doi: 10.3389/fneur.2020.572006
10. Ma P, Zhang S, Zhang H, Fang S, Dong Y, Zhang Y, et al. Comprehensive genetic characteristics of dystrophinopathies in China. Orphanet J Rare Dis. (2018) 13:109. doi: 10.1186/s13023-018-0853-z
11. Lee T, Takeshima Y, Kusunoki N, Awano H, Yagi M, Matsuo M, et al. Differences in carrier frequency between mothers of Duchenne and Becker muscular dystrophy patients. J Hum Genet. (2014) 59:46–50. doi: 10.1038/jhg.2013.119
12. Lane RJ, Robinow M, Roses AD. The genetic status of mothers of isolated cases of Duchenne muscular dystrophy. J Med Genet. (1983) 20:1–11. doi: 10.1136/jmg.20.1.1
13. Toksoy G, Durmus H, Aghayev A, Bagirova G, Sevinc RB, Basaran S, et al. Mutation spectrum of 260 dystrophinopathy patients from Turkey and important highlights for genetic counseling. Neuromuscul Disord. (2019) 29:601–13. doi: 10.1016/j.nmd.2019.03.012
14. Angelini C, Marozzo R, Pegoraro V. Current and emerging therapies in Becker muscular dystrophy (BMD). Acta Myol. (2019) 38:172–9.
15. Clemens PR, Niizawa G, Feng J, Florence J, D'Alessandro AS, Morgenroth LP, et al. The CINRG Becker natural history study: baseline characteristics. Muscle Nerve. (2020) 62:369–76. doi: 10.1002/mus.27011
16. Bushby KM, Gardner-Medwin D. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. I. Natural history. J Neurol. (1993) 240:98–104. doi: 10.1007/BF00858725
17. Kong X, Zhong X, Liu L, Cui S, Yang Y, Kong L. Genetic analysis of 1051 Chinese families with Duchenne/Becker muscular dystrophy. BMC Med Genet. (2019) 20:139. doi: 10.1186/s12881-019-0873-0
18. Zhang J, Ma D, Liu G, Wang Y, Liu A, Li L, et al. Genetic analysis of 62 Chinese families with Duchenne muscular dystrophy and strategies of prenatal diagnosis in a single center. BMC Med Genet. (2019) 20:180. doi: 10.1186/s12881-019-0912-x
19. Grimm T, Meng G, Liechti-Gallati S, Bettecken T, Müller CR, Müllerl B. On the origin of deletions and point mutations in Duchenne muscular dystrophy: most deletions arise in oogenesis and most point mutations result from events in spermatogenesis. J Med Genet. (1994) 31:183–6. doi: 10.1136/jmg.31.3.183
20. Kawamura J, Kato S, Ishihara T, Hiraishi Y, Kawashiro T. [Difference of new mutation rates in dystrophin gene between deletion and duplication mutation in Duchenne and Becker muscular dystrophy]. Rinsho Shinkeigaku. (1997) 37:212–7.
21. Helderman-van DEA, de Jong R, den Dunnen JT, Houwing-Duistermaat JJ, Kneppers AL, Ginjaar HB, et al. Recurrence risk due to germ line mosaicism: Duchenne and Becker muscular dystrophy. Clin Genet. (2009) 75:465–72. doi: 10.1111/j.1399-0004.2009.01173.x
22. Helderman-van DEA, Madan K, Breuning MH, van der Hout AH, Bakker E, de Die-Smulders CE, et al. An urgent need for a change in policy revealed by a study on prenatal testing for Duchenne muscular dystrophy. Eur J Hum Genet. (2013) 21:21–6. doi: 10.1038/ejhg.2012.101
Keywords: Duchenne muscular dystrophy, Becker muscular dystrophy, DMD gene, carrier variants, de novo variants, carrier frequency
Citation: Lin J, Li H, Liao Z, Wang L and Zhang C (2021) Comparison of Carrier and de novo Pathogenic Variants in a Chinese DMD/BMD Cohort. Front. Neurol. 12:714677. doi: 10.3389/fneur.2021.714677
Received: 25 May 2021; Accepted: 14 July 2021;
Published: 05 August 2021.
Edited by:
Xin-Ming Shen, Mayo Clinic, United StatesReviewed by:
Shiwen Wu, Chinese PLA General Hospital, ChinaFlorian Barthelemy, University of California, Los Angeles, United States
Copyright © 2021 Lin, Li, Liao, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheng Zhang, emhhbmdjaDZAbWFpbC5zeXN1LmVkdS5jbg==